
Assembling the Genome
Below is a basic guide to how the GRC maintains genomes.
The GRC only maintains genomes that have been generated using a hierarchical (clone) based assembly method. Typically, these projects are considered complete in that most of the genome is well represented and typically the funding for the main genome project has come to an end. Currently, the only genomes supported are human, mouse and zebrafish.
In this phase of genome assembly, the GRC focuses on the following:
- identifying and correcting assembly errors
- identifying regions of allelic complexity that require the addition of a partial assembly for that locus.
- working with the research community to address questions and concerns
- producing updated assemblies on a regular cycle
A set of TPF files are maintained for each assembled chromosome and partial assembly. These files are stored in a central database that manages TPF tracking and validation. A series of checks and analyses are performed and regions of the genome needing manual curation are identified and cataloged for further review.
Sequences (also known as components) which are adjacent on the TPF are expected to have a specific type of sequence alignment known as a full dovetail (Figure 1).
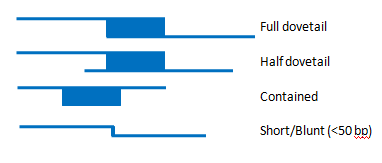
Figure 1. The top picture is a full dovetail alignment. This is the type of join expect between two sequences that will be used to construct a contig sequence. Below that is a half-dovetail alignment. In this case, the end of one of the sequences does not align. This can be used to build a contig, but may indicate that the two sequences do not actually represent adjacent loci, but rather overlap in a repetitive sequence. The third picture shows an alignment where one clone is contained within another. This typically happens because one clone has an assembly problem that the smaller clone is correcting. The last picture shows a Short/Blunt alignment. These types of alignments must always have external support and be certified by a curator.
A program call 'find_overlaps' assesses all adjacent component sequences to determine if they have an appropriate overlap. If an alignment can be identified, it is evaluated to ensure it meets our criteria for joining two sequences in a contig. We use this evaluation to prioritize overlaps that need further investigation. Alignments can fall into the following categories:
- Green: Excellent alignment, meets all defined criteria.
- Yellow: Minor alignment problem.
- Red: Serious alignment problem requiring review.
- Black: An alignment certificate providing external evidence for accepting the join has been submitted, but not approved.
- Purple: An alignment certificate has been approved for this join
The criteria for binning the overlaps is organism-specific and can evolve over time. The criteria are readily available on our Overlap View pages (see below). Overlaps that fall into the red category can be resolved in multiple ways. In a small number of cases, our automatic procedure will fail to find the correct overlap. In these cases, the curator may produce the alignment using other software and then load this alignment to the database. In other cases, the alignment may be poor but the reasons for the poor alignment are well understood and there is additional, external evidence that the two clones should be joined to build a contig sequence. In these cases, the curator may submit a certificate validating the join. The external evidence for certification must be provided in the certificate. Certificates are then approved by a separate member of the consortium. Certificate information is available through over Overlap View page (see below).
In addition to evaluating the overlaps, contig sequences are assembled based on information in the TPF files and the overlaps generated above. In some cases, contigs will fail to assemble due to inconsistent order within the TPF. Contig sequences are built on a daily basis to further evaluate the genome build process. At the time of a genome release, all contigs described on the TPF must be able to be built in a consistent fashion.
The current TPFs for human and mouse are available from our TPF Overview page. The 'View' menu provides different views of these data. The 'tpf view' has one line per clone and mimics the TPF file, with some additional information added (Figure 2). In contrast, the 'join view' option displays the data organized so there are two clones per line.
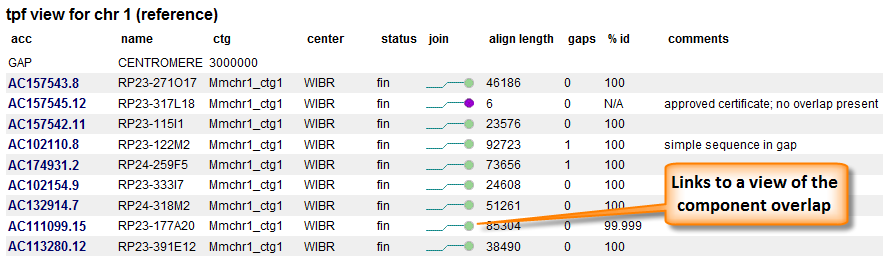
Figure 2. TPF view. The first three columns are taken from the TPF. Some additional information concerning the clone are added. The join status of the component with the component after it is represented by a colored ball. Ball colors represent either alignment evaluation or certificate statue. Additional alignment information is provided as well as any comments associated with the join.
In order to further confirm joins between adjacent components additional evaluation is performed. The most common procedure is to perform a pair end analysis. This involves sequencing both ends of a genomic clone with a known (or approximate size) and aligning these ends to your sequence of interest. If the end sequences align on opposite strands and the alignments are separated by a distance similar to the clone insert size, the paired ends are considered concordant and provide additional evidence for the join. In some cases, the paired end alignments will deviate from expectation. This is an indication of an invalid join or polymorphic sequence between the component sequences and the clone sequences. Paired end analysis as well as alignment and certificate information can be seen in the Overlap View pages (Figure 3).
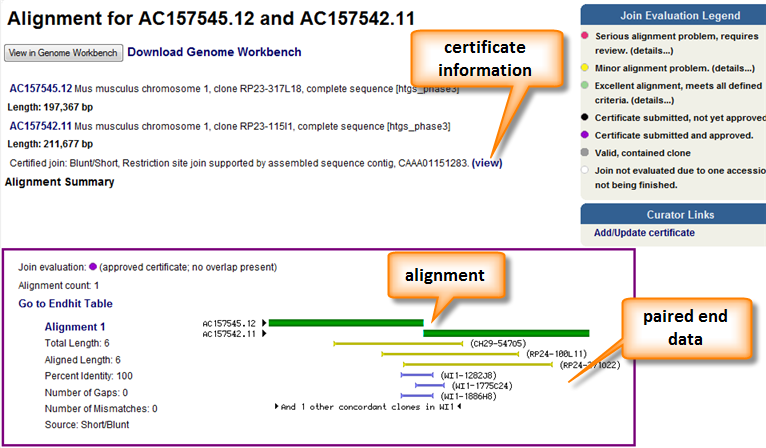
Figure 3. The overlap view page provides a graphical representation of the alignment between two components as well as any additional supporting information. Any certificate information is also provided. A pairwise alignment is available below the graphic. The alignment itself can be download and viewed in Genome Workbench.