Clinical Description
Individuals with Okur-Chung neurodevelopmental syndrome (OCNDS) frequently have nonspecific clinical features, delayed language development, motor delay, intellectual disability, generalized hypotonia starting in infancy, difficulty feeding, and nonspecific dysmorphic facial features.
To date, 51 individuals have been identified with a pathogenic variant in CSNK2A1 [Okur et al 2016, Trinh et al 2017, Akahira-Azuma et al 2018, Chiu et al 2018, Colavito et al 2018, Owen et al 2018, Angione et al 2019, Duan et al 2019, Nakashima et al 2019, Martinez-Monseny et al 2020, Wu et al 2020, Seo et al 2020, Wu et al 2021]. The following description of the phenotypic features associated with this condition is based on 36 individuals with clinical data from these reports.
Table 2.
Select Features of Okur-Chung Neurodevelopmental Syndrome
View in own window
| Feature | Proportion of Persons w/Feature | Comment |
|---|
Developmental delay /
intellectual disability | 35/35 1 | Generally mild-to-moderate, w/language development most affected domain |
| Dysmorphic facial features | 29/36 | Round face & short, broad nasal tip |
| Behavioral issues | 27/36 | |
| Hypotonia | 22/36 | Generally mild |
| Brain MRI abnormalities | 11/20 2 | Nonspecific |
| Musculoskeletal findings | 15/36 | Kyphoscoliosis, loose joints, hernia |
| Difficulty feeding | 14/36 | From infancy to childhood |
| Postnatal short stature | 14/36 | Generally 2-3 SD below mean |
| Difficulty gaining weight | 13/36 | |
| Sleep issues | 13/36 | Disrupted circadian rhythm |
| Microcephaly / smaller head (absolute or relative) | 12/36 | |
| Seizures | 11/36 | No specific type |
Ataxia / gait difficulties /
poor coordination | 9/36 | |
SD = standard deviation(s)
- 1.
2 The number of individuals reported to have had brain MRI performed
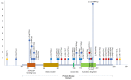
Developmental delay (DD) and intellectual disability (ID). The majority of affected individuals have DD affecting all areas of development. However, language development is more impaired than gross motor skills in most individuals. ID has also been reported in about three quarters of individuals. The average age of acquisition of selected early developmental milestones is as follows ():
Average age at which affected individuals achieved three developmental milestones: unsupported sitting, walking, and first words. The n values below the bar plots indicate the number of individuals for whom data were available for a given milestone. Data (more...)
Sitting: 11 months (n=20)
Walking: 28.8 months (n=25)
First meaningful words: 38.3 months (n=18)
Behavioral findings. Affected individuals have been reported to be affectionate and happy when they are able to communicate. The most common behavior issues reported:
Stereotypic movements (~1/3 of affected individuals)
Autism spectrum disorder (~1/4)
Aggressiveness and tantrums in part related to inability to communicate needs (~1/4)
Attention-deficit/hyperactivity disorder (~1/5)
Seizures/epilepsy. About one third of affected individuals have been reported to have had a seizure at least once. Some individuals with only a single seizure have not been treated with anti-seizure medication; those with multiple unprovoked seizures have been treated (see Management).
Other neurologic features
Gastrointestinal
Infant feeding difficulties are common and manifest as poor suck in early infancy or difficulty transitioning to solid foods later in infancy.
Feeding and swallowing difficulties may require gastrostomy tube placement.
Constipation is common.
Growth. Some affected individuals are smaller at birth, although true intrauterine growth restriction is rare. About one third of affected individuals have been reported to have short stature (14/36; 38%) or failure to thrive (13/36; 36%), with height and weight usually measuring between 2 and 3 SD below the mean for either growth domain in those individuals.
Only one reported individual had congenital microcephaly. Outside of the newborn period, most affected individuals have an occipitofrontal circumference that is below the mean but still within the normal range for age and sex.
Musculoskeletal features. The most common musculoskeletal issues are scoliosis/kyphosis and loose/hyperextensible joints associated with hypotonia. Umbilical and inguinal hernias have also been reported.
Sleep. Sleep issues mainly stemming from circadian rhythm disturbance in early childhood may pose challenges for families. Rarely, individuals may also experience sleep apnea [Chiu et al 2018, Nakashima et al 2019].
Neuroimaging. More than half of the individuals for whom brain MRI results were reported had nonspecific abnormalities. Abnormalities reported in more than one individual include delayed myelination (n=3), smaller anterior pituitary gland (n=2), and thin corpus callosum (n=2). Other reported abnormal brain findings are simple gyral pattern, cerebellar vermis hypoplasia, cerebral atrophy, mega cisterna magna, solid lesion of the pineal gland with minor cystic inclusions, duplication of the pituitary gland, Rathke cleft cysts, and absent olfactory bulbs.
Other associated features (rare)
Ophthalmologic involvement is nonspecific (e.g., strabismus) and infrequent.
Immunologic. Individuals with OCNDS tend to have frequent minor infections, and a few individuals have documented IgG deficiency (hypogammaglobulinemia) and/or IgA deficiency.
Genitourinary abnormalities. Pelvicaliectasia, duplicated renal collection system, ectopic kidney, and labial adhesions have rarely been reported.
Cardiovascular findings. Atrial septal defect has been reported in three unrelated individuals; pulmonary valve abnormality and tetralogy of Fallot have each been reported in one individual.
Skin. Dry skin, eczema, palmar erythema, and cutis marmorata have each been reported in one individual.
Facial features. No specific dysmorphic features have been observed. If present, dysmorphic features include round facies and short, broad nasal tip.
Prognosis. OCNDS is typically not a progressive disease, and individuals achieve many developmental milestones; however, speech difficulties may persist long term. It is unknown whether life span in OCNDS is abnormal; based on current data, life span is not limited by this condition. Data on possible progression are still limited.