Clinical Description
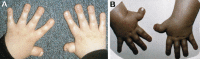
Rubinstein-Taybi syndrome (RSTS) is a multisystem disorder characterized by short stature, variable structural abnormalities, characteristic facial appearance, broad thumbs and halluces, and variable degrees of intellectual disability. The most consistent craniofacial features are microcephaly, highly arched eyebrows, downslanted palpebral fissures, convex nasal ridge, low-hanging columella, and grimacing smile. The thumbs and halluces are broad and often angulated [Hennekam et al 1990, Stevens et al 1990].
RSTS is frequently recognized at birth or in infancy because of the striking facial features and characteristic hand and foot findings. Problems in early life include respiratory difficulties, feeding issues, poor weight gain, recurrent infections, and severe constipation.
To date, at least 600 individuals have been identified with a pathogenic variant in CREBBP or EP300 [Fergelot et al 2016, Pérez-Grijalba et al 2019, Cross et al 2020, Douzgou et al 2022]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Rubinstein-Taybi Syndrome
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| Growth deficiency | 73% | |
| Eye findings | 80% | |
| Hearing loss | 30% | Mostly conductive, but can also be sensorineural |
| Respiratory features | Common | Infections, aspiration |
| Cardiac features | 33% | |
| Genitourinary anomalies | 27% | Cryptorchidism most common (~78%-100% of affected males) |
| Gastrointestinal features | 88% | Feeding difficulties, constipation |
| Skeletal abnormalities | Common | 20% scoliosis, 92% thumb/hallux anomalies |
| Neurologic issues | 21% | |
| Dental anomalies | 73% | Talon cusps, enamel hypoplasia |
| Skin findings | 24% | Keloids, pilomatrixomas |
| Recurrent infections | 17% | Primarily respiratory |
| Tumors | 30% | Benign and malignant |
| Developmental delays | 98% | |
| Behavioral issues | 41% autism/autistic features, 27%-64% anxiety | |
| Brain MRI abnormalities | 74% | Various findings |
Growth. Although prenatal growth is usually normal, growth deficiency begins in the first year of life. There is typically an absence of a growth spurt in adolescence. A higher incidence of microcephaly and growth restriction has been noted in infants with EP300 pathogenic variants, possibly related to the increased incidence of preeclampsia.
While body mass index is normal for males at age 21, it is increased for females at this age. Many adults develop obesity of unclear etiology [Stevens et al 2011].
Average height for adult males is 162.6 cm and for adult females is 151.0 cm [Beets et al 2014]. Beets et al [2014] published growth charts for RSTS.
Eye findings include strabismus, refractory errors, ptosis, nasolacrimal duct obstruction, cataracts, coloboma, nystagmus, congenital glaucoma, and corneal and retinal abnormalities.
Hearing loss. Recurrent or refractory middle ear disease can result in conductive hearing loss. Sensorineural hearing loss may also be seen.
Respiratory. Obstructive sleep apnea commonly occurs and may be caused by the combination of a narrow palate, micrognathia, hypotonia, obesity, and easy collapsibility of the laryngeal walls. There are reported incidences of intubation problems due to facial anatomy and laryngo-, tracheo-, and bronchomalacia and anesthesia complications. These include arrhythmias and obstructive symptoms occurring post extubation. Aspiration, asthma, and recurrent upper respiratory infections may also occur. Interstitial lung disease has also been reported in several individuals [Bradford et al 2022].
Cardiac. Approximately one third of affected individuals have congenital heart defects (e.g., atrial septal defect, ventricular septal defect, patent ductus arteriosus, coarctation of the aorta, pulmonary stenosis, bicuspid aortic valve, pseudotruncus arteriosus, aortic stenosis, vascular ring, conduction abnormalities).
Genitourinary. Renal abnormalities, including hydronephrosis and duplications of the renal system, are very common. Other genitourinary complications include hypospadias, vesicoureteral reflux, nephrolithiasis, and urinary tract infections. Most boys have undescended testes.
Gastrointestinal. Feeding problems, gastroesophageal reflux, and constipation are very common. Intestinal malrotation should be suspected if there is bilious vomiting, recurrent abdominal pain, failure to pass stool, or bloody stools [Stevens 2015]. Anorectal malformations have also been reported [Belanger Deloge et al 2023].
Orthopedic. In addition to angulated thumbs and duplicated halluces, orthopedic issues include dislocated patellae, lax joints, spine curvatures, Legg-Perthes disease, increased fracture risk, and cervical vertebral abnormalities.
Neurologic. The most common intracranial malformation involves abnormal structure of the corpus callosum. Occasional craniospinal and posterior fossa abnormalities including Chiari malformation, Dandy-Walker malformation, syringomyelia, os odontoideum, and cervical cord compression have been reported [Marzuillo et al 2013]. There may also be spinal cord tethering or lipoma. Seizures or abnormal EEG findings can occur.
Dental. Dental problems include crowding of teeth, malocclusion, multiple caries, hypodontia, hyperdontia, natal teeth, and talon cusps (most commonly on the upper incisors of the secondary dentition).
Skin. Keloids may occur with only minimal trauma to the skin. Pilomatrixomas (sometimes multiple) are relatively common [Boot et al 2018]. Ingrown nails are common, especially in the partially duplicated thumbs and halluces.
Recurrent infection is reported in some individuals; infections include otitis media, pneumonia, and other respiratory infections. There are reports of individuals with humoral or cellular immunodeficiency.
Tumors. There are early reports of various benign and malignant tumors in individuals with RSTS including neuroblastoma, rhabdomyosarcoma, medulloblastoma, and hematologic malignancies. A study of Dutch individuals with RSTS did not confirm an increased risk for malignancies. However, the incidence of meningiomas and pilomatrixomas was significantly elevated [Boot et al 2018]. There are currently no recommendations for additional surveillance for malignancy before the age of 40 years.
Endocrine. Persistent hyperinsulinemic hypoglycemia has been reported in a few children with RSTS, primarily in those with EP300 pathogenic variants [Welters et al 2019].
Puberty. Puberty and sexual development are typically normal.
Development and intellect. Delayed development is typical in children with RSTS. In one study, the average age of walking was 30 months, first words 25 months, and toilet training 62 months [Stevens et al 1990]. Speech delay occurs in 90% of children and some remain largely nonverbal. Waite et al [2016] noted deficits in verbal and visuospatial working memory.
The average IQ of affected individuals in one study was 51 and in another study 36 [Stevens et al 1990, Hennekam et al 1992]. IQ scores range from 25 to 79. Performance IQ is usually higher than verbal IQ [Stevens et al 1990, Hennekam et al 1992]. Some individuals with EP300-related RSTS have normal intellect [Fergelot et al 2016].
In one study of adults with RSTS, families reported a decline in developmental abilities over time in 32%, including decreased social interaction, more limited speech, and worsening stamina and mobility [Stevens et al 2011].
Behavior. Impulsivity, distractibility, instability of mood, and stereotypies are frequently observed [Verhoeven et al 2010]. Other abnormal behaviors include attention problems, hyperactivity, overfriendliness, increased pain threshold, self-injurious behaviors, and aggressive behaviors. Approximately 62% of adults with RSTS were reported to have autistic-like behaviors and one third had unreasonable fears or anxiety [Stevens et al 2011]. There may be an insistence on sameness and repetitive questioning [Waite et al 2015]. Crawford et al [2017] noted higher levels of panic attack, agoraphobia, and obsessive-compulsive disorder.
Brain MRI findings. The most common feature on brain imaging is a dysmorphic or dysplastic corpus callosum (73.6%) with or without minor dysplasia of the cerebellar vermis, periventricular posterior white matter hyperintensity, and other less common anomalies. Other infrequent findings include Chiari malformation, Dandy-Walker malformation, and underdeveloped pituitary gland.
Prognosis. More than 90% of individuals with RSTS survive into adulthood [Milani et al 2015].
It is unknown whether life span in RSTS is abnormal. One reported individual is alive at age 67 years [Stevens et al 2011], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.