Clinical Description
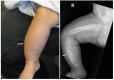
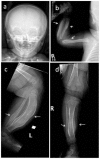
Caffey disease is characterized by massive subperiosteal new bone formation (hyperostosis) usually involving the diaphyses of the long bones, as well as the ribs, mandible, scapulae, and clavicles [Caffey & Silverman 1945, Caffey 1957].
Onset. The clinical findings most often appear at age two months (typically between birth and age five months). Rarely, hyperostosis can be detected by ultrasound examination late in the third trimester of pregnancy [Schweiger et al 2003]. One report describes prenatal periosteal inflammation in a fetus with heterozygous COL1A1 pathogenic variant p.Arg1014Cys [Kamoun-Goldrat et al 2008].
Skeletal manifestations. Typically the skeletal manifestations of Caffey disease first appear with soft-tissue swelling and pain over the affected bones between birth and age five months. Massive subperiosteal new bone formation usually involving the diaphyses of the long bones can be seen on imaging. Hyperostosis of the long bones is typically asymmetric, although symmetric hyperostosis has been reported [Tilva et al 2023].
Hyperostosis can also involve the ribs, mandible, scapulae, and clavicles. The hyperostosis resolves before age two years [Kamoun-Goldrat & le Merrer 2008, Cerruti-Mainardi et al 2011, Ranganath et al 2011].
Constitutional manifestations. Skeletal manifestations are accompanied by fever and elevated serum biochemical markers of inflammation (e.g., white blood cell count, erythrocyte sedimentation rate, C-reactive protein) [Gensure et al 2005].
Recurrence of hyperostosis, joint swelling, pain, and fever have been reported multiple times, until late adolescence in individuals with the typical infantile presentation [Borochowitz et al 1991; Navarre et al 2013; ALBagshi & ALZoayed 2015; A Guerin, unpublished data]. Etiology and precipitating factors for recurrence remain unclear [Navarre et al 2013].
Additional findings reported. In one reported family, an individual with COL1A1 pathogenic variant p.Arg1014Cys had a history of Caffey disease as a child and developed joint laxity, skin hyperextensibility, hernias, and multiple fractures in adulthood [Gensure et al 2005]. Additional individuals in the family with the COL1A1 pathogenic variant p.Arg1014Cys had varying degrees of joint laxity and skin hyperextensibility. Skin biopsy of affected individuals showed collagen fibrils that were larger, more variable in shape, and less densely packed than age- and sex-matched controls. Granulofilamentous material was also visible in the matrix along the collagen fibrils. Cultured fibroblasts showed a mix of normal type I collagen and abnormal disulfide crosslinking, either within or between abnormal collagen fibrils. These findings have not been identified in other individuals/families with the same COL1A1 pathogenic variant [Cho et al 2008, Cerruti-Mainardi et al 2011, Ranganath et al 2011].
Other
Tumoral calcinosis (1 individual); thought to be due to constant remodeling after repeated inflammatory events [
Issa El Khoury et al 2012]
Prognosis. In many individuals, the manifestations of Caffey disease resolve spontaneously by age two years and do not predispose to long-term bone abnormalities. Affected individuals from one family had short stature in adulthood and residual bone deformities [Suphapeetiporn et al 2007]. Fractures have been reported in some individuals [Gensure et al 2005, Suphapeetiporn et al 2007].
Histopathology. Bone and muscle biopsy of affected sites in a few individuals have demonstrated an inflammatory reaction [Katz et al 1981].
Nomenclature
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], Caffey disease known to be caused by a heterozygous COL1A1 pathogenic variant is referred to as COL1A1-related Caffey disease and included in the osteosclerotic disorders group.
"Prenatal lethal forms of hyperostosis," also referred to as "prenatal Caffey disease" or "Caffey dysplasia" [Nemec et al 2012], are distinct from Caffey disease (also known as infantile cortical hyperostosis) (see Differential Diagnosis).