Clinical Description
Progressive pseudorheumatoid dysplasia (PPD) is a skeletal dysplasia characterized by predominant involvement of articular cartilage with progressive joint stiffness and enlargement, and the absence of signs of inflammation [Dalal et al 2012]. Progression of the disease severely affects gait and posture and causes significant morbidity.
PPD does not have any extraskeletal manifestations, such as craniofacial features or cognitive involvement.
To date, approximately 215 families with members affected with PPD have been identified with biallelic pathogenic variants in CCN6 [Dalal et al 2012, Garcia Segarra et al 2012, Bhavani et al 2015]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Progressive Pseudorheumatoid Dysplasia: Frequency of Select Features
View in own window
| Feature | % of Persons with Feature | Comment |
|---|
|
Interphalangeal joint involvement
| 100% | Giving appearance of swollen joints that are painless |
|
Large joint involvement
| 100% | Typically knees, hips, wrists, & elbows |
|
Spine involvement
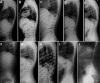
| 93% | Typically platyspondyly |
|
Short stature
| 50% | Not all have short stature. |
Onset. Children with PPD are normal at birth and during infancy. Onset is typically between ages three and six years [Wynne-Davies et al 1982, Garcia Segarra et al 2012]; the range of onset age is from one year to 16 years [Delague et al 2005, Dalal et al 2012].
Initial presenting features in the majority are interphalangeal joint swelling, pain, and gait abnormalities. Joint deformities become manifest over time. Joint pain is rarely the presenting symptom and is disproportionately mild compared to the severity of arthropathy.
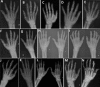
Joints. Enlargement, stiffness, and restricted range of movement in the hands start in the proximal interphalangeal joints and progress to the distal interphalangeal joints () [Garcia Segarra et al 2012]. These joints develop progressive contractures.
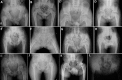
The joint enlargement, stiffness, and restricted range of movement gradually involve all large joints (e.g., knees, hips, wrists, and elbows) [Dalal et al 2012, Garcia Segarra et al 2012]. Hip involvement commonly manifests as coxa vara later in the disease course. The knees show either genu varum or genu valgum. Typically, the shoulder joints are not severely affected [Dalal et al 2012].
Spine. Scoliosis and/or kyphosis are noted in a majority of affected individuals during adolescence. Lordosis may also be seen. The neck is only occasionally involved [Dalal et al 2012].
Height. Height is initially normal; however, short stature becomes evident in some individuals as the skeletal changes progress. Adult height is typically below the third centile [Garcia Segarra et al 2012]. Flexion deformities at the hips and knees as well as spinal changes contribute in part to the short stature.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been observed.
Mild variation in age of onset, severity, and progression noted in different families is not explained by the type of pathogenic variants or their locations.
Intrafamilial variation has been observed [Bhavani et al 2015].
Prevalence
The prevalence of PPD has been estimated at one per million in the United Kingdom (prevalence category of <1-9:1,000,000) [Wynne-Davies et al 1982]. However, the disease may be underdiagnosed due to the overlap of clinical features with juvenile idiopathic arthritis. To date more than 215 families with molecularly confirmed PPD have been reported.
PPD is more frequent in communities with a high rate of consanguinity [Delague et al 2005, Dalal et al 2012]. The largest series has been published from India [Dalal et al 2012, Bhavani et al 2015].
PPD has also been observed among populations with a high rate of consanguinity in Kuwait, Lebanon, Iran, Jordan, Saudi Arabia, Syria, Palestine, and Morocco.