

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Atlanta (GA): Centers for Disease Control and Prevention (US); 2014.

The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General.
Show detailsIntroduction
Smoking has long been linked to adverse effects on the respiratory system, causing malignant and nonmalignant diseases, exacerbating chronic lung diseases, and increasing the risk for respiratory infections. The observational evidence showing associations with multiple diseases of the respiratory tract is extensive as is the evidence supporting the biological plausibility of smoking as a cause of these associations (U.S. Department of Health and Human Services [USDHHS] 2004, 2010). In addition to finding that smoking caused lung cancer, the 1964 Surgeon General's report also determined that: “Cigarette smoking is the most important of the causes of chronic bronchitis in the United States, and increases the risk of dying from chronic bronchitis and emphysema” (U.S. Department of Health, Education, and Welfare [USDHEW] 1964, p. 31).
This chapter updates previous reviews on smoking and respiratory health, covering chronic obstructive pulmonary disease (COPD), asthma, and idiopathic pulmonary fibrosis (IPF)—a form of interstitial lung disease (see Chapter 4, “Advances in Knowledge of the Health Consequences of Smoking: From 1964–2014,” for a detailed description of the conclusions). It also addresses tuberculosis, an infectious disease that has not been previously covered in the reports of the Surgeon General. The understanding of COPD, a leading cause of premature mortality and morbidity, has evolved substantially over the past 5 decades with advances related to its pathogenesis, genetic basis, natural history, and underlying structural changes in the lung. Asthma is the most common chronic disease of childhood and is also very common among adults. This chapter considers the effect of smoking on the incidence and exacerbation of asthma in children and adolescents, and in adults. It also updates the evidence on smoking and IPF, a fibrotic disease of the lung. The emerging evidence on a role for smoking in increasing the risk of developing tuberculosis and for unfavorably affecting its clinical course is examined.
Smokefree policies have now been implemented in many jurisdictions in the United States and other countries (see Chapter 14, “Current Status of Tobacco Control”). This chapter considers the evidence on the benefits of such polices for respiratory illness.
Chronic Obstructive Pulmonary Disease
Perspectives on the epidemiology, genetics, and pathogenesis of COPD have changed profoundly since the 1964 Surgeon General's report (USDHEW 1964). Smoking and chronic respiratory diseases were subsequently covered in numerous reports of the Surgeon General (see Chapter 4 for detailed descriptions of these reports and their conclusions for respiratory illness). This chapter updates previous reviews on COPD, emphasizing how the evidence on smoking and COPD has progressed during the last 50 years and examines how advances in the understanding of the epidemiology, genetics, pathogenesis, and heterogeneity of COPD in relation to smoking will alter disease prevention, management, and prognosis in the future. This chapter does not address the interrelationships among COPD and other common comorbid diseases caused by smoking—cardiovascular diseases and cancer in particular. These comorbidities are strong determinants of outcome for those with COPD (Decramer and Janssens 2013). For each of the primary topics in this chapter, some of the most significant articles were selected to document the progress made over the past 50 years.
Epidemiology of Chronic Obstructive Pulmonary Disease
By the time of publication of the 1964 Surgeon General's report, several key studies had already linked chronic bronchitis to cigarette smoking; and community surveys had characterized the frequency of chronic bronchitis and chronic respiratory symptoms (Short et al. 1939; Stuart-Harris 1954; Higgins 1974; USDHHS 1984). These and other key studies set the stage for the findings of the 1964 report.
COPD, as defined today, was not recognized as a distinct clinical entity in 1964 (Fletcher et al. 1959). Clinicians tended to use the terms “chronic bronchitis” and “emphysema” to refer to the disease constellation now termed COPD; however, the clinical classification lacked specificity and differed across countries. That era preceded the widespread use of spirometry in clinical settings and the availability of computed tomography (CT) to noninvasively determine the presence of emphysema. The time period before, and shortly following, the 1964 report was important in the development of hypotheses related to the pathogenesis of COPD and the role of cigarette smoking in its causation. Two hypotheses were extant: one attributed susceptibility to develop COPD to bronchial hyperresponsiveness, the “Dutch” hypothesis (Orie et al. 1961), and the other to respiratory infections, the “British” hypothesis (Fletcher 1959). The landmark cohort study of London men carried out by Fletcher and Peto (1977) in the 1960s described the progressive decline of lung function with aging and the acceleration of this decline in smokers. In this study, smoking was the dominant determinant of decline beyond that expected from aging alone and infection did not affect the rate of decline.
A key discovery in the early 1960s was the increased risk for COPD associated with α1-antitrypsin deficiency (AAT), a consequence of genetic mutations that increase risk for COPD, particularly in smokers (Eriksson 1965), discussed in more detail below. That discovery identified one genetic factor that increased risk for COPD in smokers and launched substantial research on underlying mechanisms.
Even before the 1964 report, the complexity and overlap of various chronic obstructive lung diseases—chronic bronchitis, asthma, and irreversible obstructive lung disease (primarily emphysema)—were recognized (Fletcher et al. 1959). In its 1995 guidelines, the American Thoracic Society (ATS) proposed a conceptual framework that captured the overlap of the major chronic lung diseases associated with airflow obstruction (Figure 7.1) (ATS 1995). The framework comprises a Venn diagram with overlapping circles indicating chronic bronchitis, emphysema, and asthma, overlaid by a rectangle representing airflow obstruction (Figure 7.1). In this schema, “COPD” comprises persons with chronic bronchitis and/or emphysema, who also have evidence of airflow obstruction (indicated by shading in Figure 7.1). ATS' 1995 definition of COPD referred to chronic airflow obstruction caused by chronic bronchitis or emphysema. The development of guidelines by the Global Initiative for Chronic Obstructive Lung Disease (GOLD) in 2001 changed the approach to the classification of COPD by focusing on the airflow obstruction component (the rectangle in Figure 7.1) rather than symptoms or a clinical diagnosis (Pauwels et al. 2001). GOLD guidelines recommended that lung function be measured after administration of a bronchodilator (to help identify and exclude from the diagnosis of COPD, those people whose primary problem is asthma, although some people with COPD respond to a bronchodilator). The definition of COPD referred to airflow obstruction that is not fully reversible. Thus, since 1964 the concept of permanent airflow obstruction has become central to the identification of COPD, although symptoms and structural changes documented by imaging are considered relevant to clinical management (GOLD 2013).

Figure 7.1
Venn diagram with overlapping circles indicating chronic bronchitis, emphysema, and asthma, overlaid by a rectangle indicating airflow obstruction. Source: American Thoracic Society 1995. Reprinted with permission from American Thoracic Society, © (more...)
Given the changes in clinical approaches and definitions during the past 50 years, the prevalence of COPD cannot be readily tracked. The 1964 report did include the findings of several population-based studies that incorporated lung function measures. A study by Ferris and Anderson (1962) in a New Hampshire town determined that “obstruction” (defined as a forced expiratory volume in 1 second [FEV1]/forced vital capacity [FVC] ratio of less than 60%) was found in 24.9% of male smokers, 7.3% of male nonsmokers, 17.5% of female smokers, and 9.4% of female nonsmokers. Data from the Ferris and Anderson study were reanalyzed to give population-based estimates of what would now be considered “chronic bronchitis,” defined as bouts of cough and phlegm for 3 weeks for more than 3 winters (Reid et al. 1964). This symptom pattern was reported by 13% of men and 8% of women. In addition, 72% of men and 30% of women in the study community reported current cigarette, pipe, or cigar smoking (Reid et al. 1964).
By 1979, nationally representative estimates of disease burden became available based on data from the National Health Interview Survey (NHIS). Estimates of the prevalence of COPD (which included physician-diagnosed chronic and unqualified bronchitis [International Classification of Diseases (ICD)-9 490–491]; emphysema [ICD-9 492]; asthma [ICD-9 493]; and other chronic obstructive pulmonary diseases and allied conditions [ICD-9 494–496]), for 55–84-year-olds in the United States from 1979–1985, ranged from 9.4% in 1979 to 11.0% in 1985 among men, and 8.8% in 1979 to 11.9% in 1985 among women (Feinleib et al. 1989). For the U.S. adult population 25 years of age and older, estimates of disease burden based on report of a diagnosis of emphysema or chronic bronchitis were between 5.5% and 6.5% during the period 1980–2000 (Mannino et al. 2002). The most recent national data (2011 Behavioral Risk Factor Surveillance System Data) yields a 6.3% overall estimate of the prevalence of self-reported, physician-diagnosed COPD, chronic bronchitis, or emphysema among adults 18 years of age and older (Figure 7.2) (Centers for Disease Control and Prevention [CDC] 2012a). At the time of this survey, the prevalence of smoking among U.S. adults had decreased to 19% of the population (CDC 2012b).

Figure 7.2
Age-adjusteda prevalence of chronic obstructive pulmonary disease (COPD)b among adults—Behavioral Risk Surveillance System, United Statesc, 2011. a Age-adjusted to the 2000 U.S. standard population, using 5 age groups: 18–44 years, 45–54 (more...)
These figures are likely underestimates, even considering the most recent estimates. COPD is frequently underdiagnosed clinically (Mannino et al. 2000); consequently, the disease burden is likely to be underestimated when based on data from questionnaire-based surveys. The National Health and Nutrition Examination Survey (NHANES) includes lung function data on U.S. adults 25 years of age and older. During 1971–1975, the estimated prevalence of moderate or worse obstruction (FEV1/FVC <70% and FEV1 <80% predicted) was 7.7%, and the estimated prevalence of mild obstruction (FEV1/FVC <70% and FEV1 ≥80% predicted) was 7.4%, for a total prevalence of 15.1% (Mannino et al. 2002). By 1988–1994, these estimates had decreased to 6.6% and 6.9%, respectively, with an overall prevalence of 13.5% (Mannino et al. 2002). Spirometry was performed without administration of a bronchodilator so that some of those NHANES participants meeting criteria for obstruction may have had asthma.
Although the prevalence figures do not document a trend of increasing COPD burden since 1964, mortality from COPD has risen progressively in recent decades (Figure 7.3). With regard to the disease now termed COPD, the 1964 Surgeon General's report referred to non-neoplastic respiratory diseases (chronic bronchitis and emphysema, in particular). In 1964, the general category of “other bronchopulmonic diseases” (ICD-7 525–524) was the 10th leading cause of death. In 2010, the category of “chronic lower respiratory diseases” (ICD-10 J40–47), in which COPD predominates, was the third leading cause of death in the United States (National Center for Health Statistics 2012). This figure highlights the steep rise in women and the recent overtaking of men by women in terms of the total number of COPD deaths. While the mortality counts are affected by changes in classification, the trends are nonetheless clear.
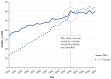
Figure 7.3
Deaths due to chronic obstructive pulmonary disease among adult men and women, 1979–2007, United States. Source: Dance 2012. Reprinted with permission from Macmillan Publishers Ltd., © 2012.
A separate “COPD” ICD code was introduced in the early 1970s, and by the mid-1980s this became the main code used for COPD deaths (Feinleib et al. 1989). In 1985, 74,662 deaths were attributed to COPD (Feinleib et al. 1989), a number that increased to 119,054 in 2000 (Mannino et al. 2002) and 133,575 in 2010 (Ford et al. 2013). Between the years 2000–2005, the age-adjusted mortality rate for COPD (standardized to the 2000 U.S. standard population) for adults 25 years of age and older remained stable, between 60 and 65 per 100,000 population.
COPD mortality has increased dramatically over the past several decades. Part of this increase is related to changes in how COPD is characterized and classified, but a substantial part is due to male and female birth cohorts with high smoking rates advancing to ages where death from COPD is more common (older than 70 years of age). Other factors, such as decreasing mortality from other chronic diseases including cardiovascular disease may have contributed. In recent years, the COPD-related mortality rate has stabilized and may show some signs of decreasing in certain age and race-ethnicity groups, reflecting declines in smoking that began several decades ago.
Gender Effects in COPD
The 1964 Surgeon General's report focused on men because of the limited data available on women and smoking at the time. As the disease increased among women, researchers addressed whether risk for COPD from smoking differed by gender. The findings with regard to gender and susceptibility and severity of COPD are mixed. By the time of the 1984 Surgeon General's report, which focused on COPD, the evidence synthesis resulted in the clearly inclusive statement that “Cigarette smoking is the major cause of chronic obstructive lung disease in the United States for both men and women” (USDHHS 1980, p. 8). The 1980 Surgeon General's report, The Health Consequences of Smoking for Women, noted that an epidemic of chronic obstructive lung disease among women had started. In the preface, this report tackled head-on the apparent “Fallacy of Women's Immunity” to the harmful effects of smoking, and highlighted as a key theme that “women are not immune to the damaging effect of smoking already documented for men. The apparently lower susceptibility to smoking-related disease among women smokers is an illusion reflecting the fact that women lagged one-quarter century behind men in their widespread use of cigarettes” (p. v). Whether or not there are gender differences for COPD susceptibility and severity continues to be debated, but the weight of recent evidence does indicate that: (1) smoking is the key risk factor for COPD in men and women, although the dose-response effects may vary, with women potentially more susceptible at lower exposure; (2) women appear to develop severe COPD at younger ages than men and with lower cumulative cigarette smoke exposure; and (3) men and women now have similar relative risk (RR) of death from COPD.
Prevalence by Gender
During the period between NHANES I (1971–1975) and NHANES III (1988–1994), the prevalence of moderate COPD increased in women (from 50.8 to 58.2 per 1,000 population, not a statistically significant change), while the prevalence decreased in men (from 108.1 to 74.3 per 1,000 population, a statistically significant decrease), but male rates remained higher than those for women (Mannino et al. 2002).
Gender-Specific Manifestations of COPD
The 2001 Surgeon General's report noted the extensive pathologic evaluation of the lungs of male and female smokers performed by Thurlbeck and colleagues (1974) showing that male smokers had higher emphysema scores, and a greater prevalence of emphysema, when compared to lung sections from female smokers. In a paper by Martinez and colleagues (2007), an analysis of CT data from the National Emphysema Treatment Trial (NETT) revealed that women had significantly less emphysema, despite similar severity of COPD as measured by the level of FEV1; on histologic section, the airways of women with COPD had smaller lumens and thicker walls. Although some of these differences may represent baseline gender differences between the lungs of men and women, they may also support differences in how COPD develops and progresses in men versus women.
A series of observations during the past decade indicate that women seem to develop more severe COPD at an earlier age, in comparison with men who smoked the same cumulative number of cigarettes. In the NETT study (Martinez et al. 2007), women reported less pack-years1 of cigarette smoking, but had similarly severe spirometrically defined COPD as men, raising the question of heightened susceptibility in women to the lung-damaging effects of smoking (Gan et al. 2006). Gan and colleagues observed that beyond 45 years of age, female current smokers had a faster annual decline in FEV1 compared to male smokers.
Silverman and colleagues (1998) noted a high prevalence of women (almost 80%) in a group with severe, early-onset COPD (FEV1 <40% of predicted at younger than 53 years of age) recruited for a genetic study. In addition, Sorheim and colleagues (2010) observed that in people with COPD before 60 years of age, women had lower FEV1 and more severe COPD with lower cigarette smoking exposure. In this study, women had greater reductions in FEV1 than men in the less than 20 pack-year range; after 25–30 pack-years of smoking, the dose-response relationship was similar to that for men.
The COPDGene study enrolled smokers with and without COPD at 21 clinical centers throughout the United States (ClinicalTrials.gov 2013). Participants were self-classified non-Hispanic Whites and African Americans 45-80 years of age with at least 10 pack-years of lifetime smoking. During the study visit, participants underwent spirometry, before and after inhaled bronchodilator, and completed detailed questionnaires on respiratory disease, medical history, and medications. Foreman and colleagues (2011) analyzed data from the first 2,500 individuals in the COPDGene study. Severe, early-onset COPD participants were predominantly women (66%), with proportionally higher rates in African American smokers; in addition to race and gender, maternal smoking and maternal COPD were also associated with severe, early-onset COPD.
Gender-Specific Morbidity and Mortality
Hospitalization rates for COPD have been approximately equal for men and women since 1995 (Akinbami and Liu 2011). However, during the period 1980–2000, annual death rates from COPD increased in men until about 1995 and then stabilized (Figure 7.4). Among women, death rates during this timeframe tripled and continued to increase. In 2000, 59,936 women died from COPD compared to 59,118 men (Mannino et al. 2002). In a more recent assessment of changes in mortality rates due to COPD (Ford et al. 2012), 5,185 individuals from NHANES I were evaluated at follow-up through 1993 (baseline exam 1971–1975, follow-up 1992–1993), and 10,954 participants from the NHANES III Linked Mortality Study (baseline 1988–1994) were followed up through 2006. Age-adjusted mortality rates among participants with moderate to severe COPD, compared to persons with normal spirometry, were higher in both NHANES I and NHANES III, although there was an overall decrease in mortality rates due to COPD in NHANES III compared to NHANES I. Specifically, in NHANES I and NHANES III, respectively, the age-adjusted mortality rates for COPD were 29.9 and 20.2 per 100,000, compared to the age-adjusted mortality rates of 10.4 and 6.2 in participants with normal spirometry. However, further highlighting previous epidemiologic trends, there was a decrease in the mortality rate among men with moderate or severe COPD (decreased by 17.8%) in contrast to the 3% increase in the mortality rate in women (Figure 7.5) (Ford et al. 2012).
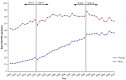
Figure 7.4
Age-adjusted mortality rate from chronic obstructive pulmonary disease among adults 25 years of age or older, by gender, United States, 1968–2010. Source: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention (more...)

Figure 7.5
Age-adjusted all-cause mortality rates per 1,000 person-years (95% confidence interval [CI]) for men and women 25–74 years of age in the United States by survey and Global Initiative for Chronic Obstructive Lung Disease classification. Source: (more...)
The most recent and comprehensive assessment of smoking-related mortality in the United States (Thun et al. 2013) evaluated temporal trends in gender-specific smoking-related mortality across three time periods (1959–1965, 1982–1988, 2000–2010) in seven large cohorts (see Chapter 12, “Smoking-Attributable Morbidity, Mortality, and Economic Costs”). In the “contemporary” cohort that encompassed the years 2000–2010, male and female current smokers had similar RRs for mortality from COPD (26.61 for men, 22.35 for women), with this RR for women representing almost a doubling of risk when compared to the 1982–1988 time period (Figure 7.6).
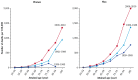
Figure 7.6
Changes in rates of death from chronic obstructive pulmonary disease over time among current women and men smokers in three time periods. Source: Thun et al. 2013. Reprinted with permission from Massachusetts Medical Society, © 2013. Note: Data (more...)
Genetics and Genomics of COPD
The first significant understanding of the role of genetics in the pathogenesis of COPD began with the 1963 discovery of the increased risk of obstructive lung disease associated with AAT (Laurell and Eriksson 1963). The role of genetic factors in COPD and susceptibility to cigarette smoke was reviewed in the 2010 Surgeon General's report (USDHHS 2010). This section focuses on recent advances in genetics and genomics made possible by the rapid advances in technology since the 2010 report.
Rare Genetic Syndromes with COPD
A small percentage of COPD patients (estimated at 1–2%) have severe AAT deficiency, a Mendelian syndrome often presenting as severe, early-onset COPD (Silverman and Sandhaus 2009). The genetic basis for severe AAT deficiency is well-understood; a relatively rare alteration in a single DNA nucleotide base in the SERPINA1 gene sequence causes a single amino acid change in the protein sequence of AAT at amino acid 342. This genetic variant is referred to as the PI*Z allele. Other even less common severe AAT deficiency variants lead to very low expression of the normal M protein (e.g., Mheerlen) or the absence of any AAT protein (e.g., Null alleles). Individuals that inherit two severe deficiency variants—most commonly genotype PI ZZ—are at substantially increased risk for early-onset COPD. Persons with the PI ZZ variant have been described as having lower lobe predominant panlobular emphysema, but a substantial fraction of those affected do not develop an emphysema distribution in this classic pattern (Parr et al. 2004). Severe AAT deficiency is found in approximately 1 in 3,000 Americans, with an increased prevalence in Whites (Silverman et al. 1989). Thus, the genetic variants explain little of the variation in risk for COPD among smokers in the population, but studies in affected individuals have provided important insights into how smoking causes COPD. The discovery of AAT deficiency was a major factor in the formulation of the protease-antiprotease hypothesis for the pathogenesis of COPD (Janoff 1985; USDHHS 2010). This hypothesis relates the development of COPD to an imbalance between the increased proteolytic activity in the lungs of smokers and the diminished activity of the opposing antiproteases, primarily AAT. The primary mechanism for increased COPD risk is likely the reduced plasma levels of circulating AAT in severely deficient persons, which relates to polymerization of the Z protein in the endoplasmic reticulum of hepatocytes (Lomas et al. 1992). However, the Z polymers, which are also detectable within the plasma and in lung tissue samples, appear to have pro-inflammatory activity, which could also contribute to the pathogenesis of COPD in persons with PI ZZ (Mahadeva et al. 2005).
Another SERPINA1 variant, the PI*S allele, leads to a moderate reduction in AAT protein levels; persons that inherit one PI*S allele and one PI*Z allele (PI SZ) are likely at increased risk for COPD (Turino et al. 1996)—although not to the same magnitude of risk as persons with PI ZZ. Whether persons with one normal and one severe deficiency SERPINA1 variant (e.g., PI MZ) are at increased risk for COPD has been a controversial issue for decades. In a meta-analysis published in 2004, Hersh and colleagues found that studies, which compared risk in COPD cases to controls, often showed an increased risk for the PI MZ genotype, while population-based studies did not demonstrate reduced spirometric values in persons with the PI MZ variant. Only a minority of the studies included in this meta-analysis provided results adjusted for cigarette smoking intensity. More recently, Sorheim and colleagues (2010) assessed COPD risk associated with PI MZ risk in the Genetics of Chronic Obstructive Lung Disease (GenKOLS) case-control study in Norway and in the International COPD Genetics Network, a family-based study. Although PI MZ was not associated with significantly increased risk for COPD, persons with the PI MZ variant had a significantly lower FEV1/FVC ratio in both populations, and an increased risk for CT-defined emphysema was observed in the GenKOLS Study.
Although AAT deficiency is the most widely recognized Mendelian syndrome, which increases risk for COPD, other rare Mendelian syndromes have been studied as well. People with cutis laxa, a very rare syndrome with marked dermatologic manifestations related to skin laxity, can also develop early-onset emphysema due to mutations in several genes, including ELN (Corbett et al. 1994) and FBLN5 (Loeys et al. 2002). Although mutations in these cutis laxa genes do not commonly cause COPD, a rare functional variant in the ELN gene has been reported in several severe early-onset pedigrees (Kelleher et al. 2005; Cho et al. 2009).
Common Genetic Determinants of COPD
Several types of studies have suggested that genetic factors other than AAT deficiency and cutis laxa influence COPD susceptibility. Aggregation of pulmonary function and airflow obstruction was demonstrated beginning in the 1970s in studies in the general population and with twins (Lewitter et al. 1984; Redline et al. 1989). Based on data from familial aggregation analyses, linkage analysis studies were performed using panels of short tandem repeat markers in families from the Boston Early-Onset COPD study for both categorical and quantitative COPD-related phenotypes. These linkage analysis approaches, which have been highly successful in Mendelian syndromes, implicated several genomic regions that may contain susceptibility genes for COPD (Silverman et al. 2002a,b; Palmer et al. 2003). Within these linkage regions, SERPINE2 (DeMeo et al. 2006) and XRCC5 (Hersh et al. 2010) on chromosome 2q and SOX5 (Hersh et al. 2011) on chromosome 12p were suggested as possible COPD susceptibility genes. However, as with many other complex diseases, convincing replication of these linkage-based findings in multiple studies by different investigative groups has thus far been lacking. Therefore, the role of these linkage analysis regions and positional candidate genes remains to be determined.
Similarly, the results of many candidate gene association studies, which compared COPD cases and controls for the distribution of genetic variants within genes, typically selected based on their hypothesized roles in COPD pathogenesis, have been largely inconsistent (Castaldi et al. 2010). By contrast, the application of genome-wide association studies (GWAS) has unequivocally associated common variants in several genetic loci with COPD susceptibility (Table 7.1). Pillai and colleagues (2009) found genome-wide significant associations between COPD and single nucleotide polymorphisms (SNPs that are used as markers across the genome) in the CHRNA3/CHRNA5/IREB2 region on chromosome 15q25. Of interest, DeMeo and colleagues (2009) performed gene expression studies comparing normal and COPD lung tissues and identified SNPs to be tested for association with COPD in several studies. The association analyses identified IREB2 as a COPD susceptibility gene. In a GWAS based in the Framingham Heart Study (Wilk et al. 2009), the HHIP region was associated with FEV1/FVC ratio, and this same region nearly reached genome-wide significance with COPD susceptibility in the study by Pillai and colleagues (2009). The FAM13A locus has been strongly associated with COPD susceptibility in multiple populations (Cho et al. 2010). In a collaborative GWAS, including four study populations (GenKOLS, Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints [ECLIPSE], National Emphysema Treatment Trial/Normative Aging study, and COPDGene), a fourth statistically significant link was found for a region on chromosome 19q (Cho et al. 2012).
Table 7.1
Genome-wide association studies of chronic obstructive pulmonary disease (COPD) and COPD-related phenotypes.
Several of these associations for COPD risk have been replicated in other studies. For example, participants with airflow obstruction were identified in the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) and SpiroMeta consortium studies, and compared to controls with normal spirometry; a genome-wide significant association to the chromosome 15q25 region was found (Wilk et al. 2012). The HHIP and FAM13A associations with COPD have also been replicated in multiple studies (Van Durme et al. 2010; Young et al. 2010a,b; Soler Artigas et al. 2011). Thus, the frustration of inconsistent genetic association results in COPD over the past decade has been replaced by optimism regarding the likely importance of these GWAS-identified loci in COPD susceptibility.
A complicating issue in COPD genetic studies is the overwhelming influence of cigarette smoking on COPD susceptibility. In light of this large effect, most COPD genetics studies have only involved current or former cigarette smokers; however, this approach may have limited detection of gene-by-smoking interactions and hence the identification of those genes determining susceptibility in smokers. In a collaborative study in CHARGE and SpiroMeta of more than 50,000 individuals, genome-wide association analyses of FEV1 and FEV1/FVC ratio were performed with joint assessment of main SNP effects and SNP-by-smoking effects. Three novel genome-wide significant regions, on chromosomes 2, 6, and 17, were identified (Hancock et al. 2012).
It is possible that genetic determinants of COPD risk may act through genetic effects which may increase nicotine addiction or smoking intensity. In fact, both the chromosome 15q25 region (which includes genes of several components of the nicotinic acetylcholine receptor, i.e., CHRNA3 and CHRNA5) and the chromosome 19q region (which includes CYP2A6) have been associated with smoking pattern (Thorgeirsson et al. 2010; Tobacco and Genetics Consortium 2010). Additional research will be required to determine whether nicotine addiction is the mechanism that links these genetic loci to COPD susceptibility.
Genetic Determinants of COPD-Related Phenotypes
Studies of quantitative disease-related phenotypes may provide increased power to detect significant associations for genetic determinants for a complex disease. Several large-scale collaborative GWAS (CHARGE and SpiroMeta) have been highly successful at identifying multiple genomic regions that influence lung function levels in population-based samples (Hancock et al. 2010; Repapi et al. 2010). A combined analysis of CHARGE and SpiroMeta has identified 26 genome-wide significant regions associated with spirometric measures (Soler Artigas et al. 2011). Of interest, both the HHIP and FAM13A loci have been associated with lung function values in these general population samples; it is not yet clear how many of the other genomic regions, associated with lung function levels in the general population, also influence COPD susceptibility in smokers (Silverman 2012).
Kong and colleagues (2011) performed GWAS of CT-defined emphysema in three sets of COPD cases (Gen-KOLS, ECLIPSE, and NETT), using both radiologists' assessments of emphysema severity and quantitative densitometric measures of emphysema. Although the quantitative densitometric measurements of emphysema did not demonstrate any genome-wide significant associations, borderline genome-wide significant associations near the BICD1 gene were observed for the endpoint of the radiologists' visual assessment of emphysema severity.
Several GWAS of the rate of FEV1 decline have been reported. Imboden and colleagues (2012) performed GWAS of FEV1 decline in a collaborative study of general population participants, including separate analyses in 1,441 persons with asthma and 2,667 in persons who were not asthmatics. However, no significant associations with FEV1 decline were found across the genome. When Hansel and colleagues (2012) studied FEV1 decline within 4,048 persons with mild-to-moderate COPD in the Lung Health Study, two genome-wide significant regions were identified, but they were not replicated in other populations. Genetic association analyses in the ECLIPSE and International COPD Genetics Network studies suggested that the CHRNA3/CHRNA5/IREB2, HHIP, and FAM13A COPD GWAS loci influence different aspects of the COPD syndrome (Pillai et al. 2010). For example, the CHRNA3/5 locus was most strongly associated with emphysema, while the HHIP locus was associated with COPD exacerbation frequency.
Gene Expression Studies in COPD
In order to provide insight into the biological pathways involved in COPD pathogenesis, multiple studies have assessed genome-wide gene expression using RNA extracted from lung tissue samples in persons with COPD and controls. Four key lung tissue gene expression studies were compared by Zeskind and colleagues (2008). Ning and colleagues (2004) used both serial analysis of gene expression and microarray analysis in 14 smokers with moderate COPD and 12 smokers with normal spirometry. Golpon and colleagues (2004) studied gene expression using microarrays in 11 COPD cases (6 with AAT deficiency) and 5 controls. Spira and colleagues (2004a) compared lung tissue samples from 18 severe emphysema cases undergoing lung volume reduction surgery to samples from 12 persons with normal spirometry or mild airflow obstruction undergoing resection of a pulmonary nodule. Wang and colleagues (2008) obtained 48 lung tissue samples from persons with a range of spirometric abnormalities, who were undergoing surgical resection of a pulmonary nodule. All four of these studies identified multiple genes that showed significant differential expression between lung tissue samples from persons with COPD and controls, but only minimal overlap in the specific differentially expressed genes was observed across the four studies. This limited degree of replication could be related to several factors, including the small sample sizes of the studies, differences in the analytical approaches, and variation in inclusion and exclusion criteria for participant selection. Nonetheless, Zeskind and colleagues (2008) used pathway analysis to demonstrate that many of the differentially expressed genes observed in these four studies represented similar biological processes. Further evidence for concordance between different lung tissue gene expression studies was provided by Bhattacharya and colleagues (2009). They assessed lung tissue gene expression using both the presence and absence of COPD (15 COPD cases and 18 smoking controls) and quantitative spirometric measures (56 total persons), and found 254 differentially expressed gene biomarkers. A subset of 84 of these gene biomarkers was also assessed in the Spira and colleagues study (2004a), and this subset of biomarkers generated by Bhattacharya and colleagues was able to differentiate COPD cases versus controls in the Spira data set with 97% predictive accuracy.
Wang and colleagues (2008) showed that some of the variability in lung tissue gene expression resulted from variation in the cellular profiles included within the lung tissue sample. One approach to overcome this source of variability is to focus on a particular cell type, and several studies have focused on airway epithelial cells obtained at bronchoscopy. Spira and colleagues (2004b) pioneered this approach, and they identified genes that were differentially expressed within airway epithelial cells in response to smoking. Ammous and colleagues (2008) found substantial variability in gene expression within the small airways of smokers, suggesting that this distal sampling site could provide especially useful information about COPD pathogenesis.
Protein Biomarkers of COPD
The identification of biomarkers of lung destruction and inflammation in COPD has been a major research focus during the past 50 years (Yoon and Sin 2011; Rosenberg and Kalhan 2012). Yoon and Sin (2011) discussed the optimal characteristics of a COPD biomarker, which include having a close relationship to relevant health outcomes, playing an important biological role in disease, and demonstrating modifiability with effective treatment interventions. Since cigarette smoking induces lung inflammation, ideal COPD biomarkers would differentiate smokers with and without COPD.
This section briefly summarizes some of the key evidence related to the potential of several proteins and protein breakdown products as COPD biomarkers. Because COPD often involves destruction of lung parenchyma, development of biomarkers based on breakdown products of lung extracellular matrix components has been pursued. Desmosine and isodesmosine, specific degradation products of elastin, can be measured in the plasma and urine. Small studies have suggested that urinary desmosine levels are increased in response both to current smoking and to COPD (Stone et al. 1995). Clinical trials of AAT augmentation therapy in AAT-deficient persons did not demonstrate consistent effects on urinary desmosine levels (Luisetti et al. 2008). Part of the inconsistency in the results of these interventional trials could be related to technical difficulties in desmosine assays, and more recently developed mass spectrometry approaches may be more reliable (Ma et al. 2003). Mass spectrometry analysis was performed in a total of 390 individuals, including those with stable COPD, individuals with COPD during an exacerbation, and controls with normal spirometry (smokers and nonsmokers) (Huang et al. 2012). Significantly higher desmosine levels were found in blood samples from persons with stable COPD, compared to control smokers or nonsmokers, but urinary desmosine levels were not different when stable COPD cases and controls were compared. During COPD exacerbations, elevated urinary desmosine levels were observed. A more recently characterized degradation product of collagen, proline-glycine-proline, is chemotactic for neutrophils and has been suggested to have higher levels in both serum and induced sputum of persons with COPD than in nonsmoking controls (O'Reilly et al. 2009).
A variety of systemic markers of inflammation have been studied as potential COPD biomarkers. For example, with adjustment for ever smoking status and pack-years of smoking, elevated C-reactive protein (CRP) levels were associated with a significantly increased risk of incident COPD in the Rotterdam Study (van Durme et al. 2009). Of interest, the greatest effect of elevated CRP on COPD risk was observed among former smokers; nonsmokers were not at increased risk if CRP was elevated. Among 34 bloodstream biomarkers assessed in 201 COPD cases and 37 smoking controls from the ECLIPSE study, fibrinogen demonstrated a significantly higher mean level in COPD cases, compared with controls, and was the most reproducible biomarker in blood samples collected 3 months apart (Dickens et al. 2011). However, the relevance of these nonspecific inflammatory markers for COPD pathogenesis, and their response to treatment interventions, remains to be demonstrated.
Proteins synthesized within the lungs may have greater potential to serve as specific COPD biomarkers. Significantly increased serum levels of surfactant protein D (Lomas et al. 2009), decreased serum levels of Clara cell secretory protein 16 (CC16) (Lomas et al. 2008), and increased serum pulmonary and activation-regulated chemokine/chemokine liand-18 (PARC/CCL-18) (Sin et al. 2011) have been found in persons with COPD compared to smokers with normal spirometry. Of interest, treatment of persons with COPD with oral corticosteroids reduced serum surfactant protein D (Lomas et al. 2009) and PARC levels (Sin et al. 2011). In the longitudinal evaluation of lung function in the ECLIPSE study, CC16 levels were associated with both baseline FEV1 level and change in FEV1 over 3 years of observation (Vestbo et al. 2011).
Pathogenesis of COPD
Changes in Views of COPD Pathogenesis During the Past 50 Years
Small airway disease and emphysema form the basis for the largely irreversible airway obstruction that characterizes COPD. Emphysema is defined pathologically as the destruction of alveolar tissue with coalescence and enlargement of airspaces. As mentioned above, although these terms were not used then, two observations made around the time of the 1964 Surgeon General's report established the elastase:antielastase hypothesis as the basis for the lung injury that results in emphysema. These seminal findings were: (1) elastases instilled into the lungs of experimental animals resulted in airspace destruction and enlargement (Gross et al. 1965); and (2) persons with deficient AAT are at increased risk for the development of emphysema (Laurell 1963). With many additional concepts added to this basic premise over time, the elastase:antielastase hypothesis, which was extensively discussed in the 2010 Surgeon General's report, remains a central component of our understanding of emphysema 50 years later.
Because AAT is the main inhibitor of neutrophil elastase (NE), this led to the understanding of the inflammatory nature of COPD with neutrophils and NE receiving most attention initially. NE is not only a potent elastase capable of causing experimental emphysema (Janoff et al. 1977; Senior et al. 1977; Snider et al. 1984) but it is also a secretagogue (Nadel 2000; Kohri et al. 2002). In addition, neutrophils produce other serine proteinases with elastolytic activity, as well as matrix metalloproteinase (MMPs), including the elastolytic MMP-9.
Macrophages are the main inflammatory cell patrolling the normal lung parenchyma and their numbers are greatly expanded with long-term smoking (Niewoehner et al. 1974; Merchant et al. 1992). They also produce elastases, including MMP-9 and MMP-12. Results from gene-targeted knockout mice, combined with cigarette smoking models, demonstrated an interaction between MMP-12 (Hautamaki et al. 1997) and NE (Shapiro et al. 2003) contributing to emphysema in mice. MMPs and serine proteinases work together to degrade the inhibitor of the other and, thus, lead to lung destruction. Proteolytic cleavage products of elastin also serve as a macrophage chemokine (Senior et al. 1984; Houghton et al. 2006) and collagen fragments are chemotactic for neutrophils (Weathington et al. 2006). In addition to matrix destruction, which fuels the positive inflammatory feedback loop, a variety of traditional CC and CXC chemokines have also been implicated in generating the complex inflammatory–immune network in COPD (see the “Immune Function and Autoimmune Disease” section in Chapter 10, “Other Specific Outcomes”).
Macrophages also regulate the inflammatory response in COPD. For example, cigarette smoke alters the macrophage phenotype via oxidant-induced inactivation of histone deacetylase-2, shifting the balance toward acetylated or loose chromatin, exposing NF-κB sites, and resulting in transcription of MMPs, pro-inflammatory cytokines such as IL-8, and TNF-α; this leads to neutrophil recruitment (Ito et al. 2006).
Recently, the role of adaptive immunity in COPD has been appreciated (see Chapter 10). CD8+ T cells are also recruited in response to cigarette smoke and release interferon inducible protein-10 that, in turn, leads to macrophage production of MMPs (Grumelli et al. 2004; Maeno et al. 2007). In further support of T cell involvement, inducible transgenic mice overexpressing interferon gamma (IFN-γ) developed emphysema (Wang et al. 2000). Of note, interferon IFN-γ transgenic mice develop proteinase-mediated emphysema, but not airway disease (“British mice” per the British hypothesis) (Wang et al. 2000), while overexpression of IL-13 produces both emphysema and airway remodeling (“Dutch mice” per the Dutch hypothesis) (Zheng et al. 2000).
The role of B cells and auto-immunity to promote progression of COPD is an emerging concept. B cells accumulate in bronchus-associated lymphoid tissue (BALT) in persons with COPD, particularly those with advanced disease (Hogg et al. 2004). Antibodies have been found against elastin fragments (Lee et al. 2007), as well as immunoglobulin G autoantibodies with avidity for pulmonary epithelium and the potential to mediate cytotoxicity (Feghali-Bostwick et al. 2008).
In summary, cigarette smoke initiates an inflammatory process that later becomes more complex and independent of smoking over time. For example, matrix fragments themselves can continue to drive the inflammation. In the airway, colonization by microorganisms may sustain inflammation. Hence, smoking cessation, although critical, may not totally reverse progression of advanced COPD.
Over the past decade, the role of structural cell apoptosis, particularly in the vascular endothelial cell, has become recognized as a driver of emphysema (Kasahara et al. 2000). Ceramide released from one apoptotic structural cell can also cause the death of neighboring cells (Petrache et al. 2005). Clearly, the loss of an alveolar unit includes both the cells and matrix. Emphysema could be established either by inflammatory cell-mediated matrix destruction followed by cell detachment and death, or alternatively, cigarette smoke oxidant-mediated structural cell death via a variety of mechanisms, including Rtp801 inhibition of mTOR that leads to inflammation and proteolysis (Yoshida et al. 2010). Likely, both mechanisms are operable.
COPD is characterized by its irreversible nature, raising the important issue of lung repair capacity in COPD. Cigarette smoke has a variety of effects that inhibit repair, ranging from impairing elastin and collagen synthesis and cross-linking (Laurent et al. 1983; Osman et al. 1985) to enhancing epithelial cell death and inhibiting epithelial cell migration and repair (Cantral et al. 1995; Nakamura et al. 1995; Carnevali et al. 1998; Wang et al. 2001; Kotton et al. 2005). There have been hints that repair may be possible. For example, retinoic acid clearly reversed elastase-induced emphysema in rats (Massaro and Massaro 1997), but unfortunately retinoic acid had no effect in human trials (Roth et al. 2006; Stolk et al. 2012). The complex process of elastic fiber production appears to be inefficient, if even possible, following growth and development (Mecham et al. 1995; Shifren and Mecham 2006). Collagen turnover is equally complicated with loss of collagen in the airspace and excess collagen accumulation around the airways (Wright 1995; Wright and Churg 1995). In addition to the complexity of restoring the lung's intricate network of interwoven fibers composed of extracellular matrix components, the role of cell death and regeneration remains uncertain. It is unlikely that lung repair recapitulates lung development, where large airspaces septate to form alveoli. Identification of stem cells residing in the distal small airways or alveoli, and their role in COPD, is an area of active investigation and therapeutic interest.
Current Models of COPD Pathogenesis from Murine and Human Studies
The evolution of the current understanding of COPD presented above is largely derived from observations made in human lung tissue and cells. These observations led to hypotheses on causal relationships that have been tested in animal models. Animal models have played a major role in our current understanding of the pathogenesis of emphysema, beginning with the classic study by Gross and colleagues demonstrating that pulmonary instillation of an elastase resulted in emphysema (Gross et al. 1965; Snider et al. 1992a,b). Although no animal model replicates all aspects of human disease, COPD has an advantage over many other disease models because the primary causal agent is known—cigarette smoke—and chronic cigarette smoke exposure in experimental animals results in inflammation very similar to that in humans followed by several pathologic changes characteristic of COPD. Such models have been far less useful for drug discovery.
The mouse has been most extensively used in the past two decades because the ability to genetically engineer mice allows for the performance of controlled experiments in mammals. While the airspace of the mouse replicates humans fairly well, airway structure differs greatly. Mice still develop aspects of airway remodeling including inflammation, fibrosis, and mucus hypersecretion, but the findings are much more subtle; because mice lack extensive airway branching, they really do not have small airways, a major site of obstruction in humans. Hence, the understanding of emphysema is much more advanced than that of small airway disease.
The current dominant paradigm of the pathogenesis of emphysema comprises four interrelated events: (1) chronic exposure to cigarette smoke leads to inflammatory and immune cell recruitment within the terminal airspaces of the lung; (2) these inflammatory cells release proteinases that damage the extracellular matrix of the lung; (3) endothelial cells and other structural cells undergo apoptosis due to oxidant stress and loss of matrix-cell attachment; and (4) ineffective repair of elastin and other extracellular matrix components result in airspace enlargement.
Unfortunately, despite strong evidence supporting these basic concepts, a drug therapy has yet to be developed that halts the underlying process that leads to COPD. In part, developing such therapeutic interventions is hindered by insufficient understanding of airway disease and a lack of validated endpoints for short-term trials that are predictive of major clinical endpoints for the long-term. The understanding of the pathogenesis of emphysema is much greater than for airway disease, but emphysema is a less attractive therapeutic target due to its protracted and irreversible nature. Further, human confirmation of disease mechanisms is starting to emerge from genetic studies. For example, although candidate gene association studies are fraught with hazards, a large, well-controlled candidate gene study using multiple replication populations supported a role for MMP-12 in promoting both emphysema and asthma (Hunninghake et al. 2009). The main utility of powerful new genetic approaches has been to define new candidate genes in an unbiased manner, allowing both the confirmation and broadening of understanding of the mechanisms that lead to specific COPD phenotypes.
Lessons from Imaging Studies
The evolution of chest CT imaging, over the past several decades, has created a robust technology for deriving image-based biomarkers that can be used to both visualize and quantify major COPD subtypes. Quantitative volumetric CT is now well-established as a method to assess three critical components of COPD: emphysema (Bankier et al. 2002; Madani et al. 2006, 2008), airway wall thickening (Orlandi et al. 2005; Coxson 2008; Kim et al. 2009b; Washko et al. 2009), and expiratory air trapping (Eda et al. 1997; Matsuoka et al. 2007, 2008). These measures correlate quite well with pathologic measures of emphysema (Bankier et al. 2002; Madani et al. 2006, 2008) and small airway disease (Nakano et al. 2005; McDonough et al. 2011). The particular advantages of CT in phenotypic characterization of COPD include the ability to provide anatomic lobar and sublobar information regarding the distribution and severity of parenchymal abnormalities (Hasegawa et al. 2006; Revel et al. 2008) and the ability to follow abnormalities over time with sequential CT imaging (Shaker et al. 2004; Matsuoka et al. 2006; Dirksen 2008; Stoel et al. 2008). Current cigarette smoking does increase the lung density measurements assessed by CT. This increase is, presumably, related to the accumulation of smoking-related toxins and the associated inflammatory response (Ashraf et al. 2011), which can impact the assessment of emphysema using quantitative densitometric approaches.
Chest CT can be used to subclassify COPD into either emphysema or airway-predominant disease, and to assess the severity and unique patterns of these different expressions of disease (Gevenois et al. 1995, 1996). The degree of emphysema and the degree of gas trapping can be estimated as continuous variables from CT imaging (Kubo et al. 1999; Matsuoka et al. 2008; Gorbunova et al. 2010). In addition, chest CT can be used to define the extent and severity of pulmonary vascular disease, which may be a primary or secondary component of the development of disabling COPD (Barr et al. 2010; Matsuoka et al. 2010). Wells and colleagues (2012) have shown that pulmonary artery enlargement, as detected on chest CT, is associated with enhanced risk for severe exacerbations of COPD.
Researchers have found that the magnitude of emphysema, the severity of image-defined airway inflammation, and gas trapping do not directly correlate with GOLD grade for COPD severity, showing that CT-defined characteristics related to COPD are independent variables from physiologic obstruction (FEV1). The extent and severity of CT-defined emphysema has been shown to correlate well with clinical parameters such as Modified Medical Research Council dyspnea score, 6-minute walk distance, and number of annual exacerbations; and several groups have documented that emphysema is associated with a greater decrease in FEV1 over time and increased mortality (McDonough et al. 2011; Nishimura et al. 2012). Increased lung emphysema and airway wall thickness have been positively associated with enhanced risk for COPD exacerbations, independent of the severity of airflow obstruction (Han et al. 2011).
CT imaging can define significant structural and functional lung abnormalities in persons having a substantial smoking history, but who have normal spirometry (FEV1). These abnormalities include emphysema, evidence of airway wall thickening, and excessive gas trapping on expiratory CT. It is also common for persons with a history of smoking (with or without obstruction) to show substantial gas trapping on expiratory CT scans, although they have no CT evidence of emphysema. Both abnormal gas trapping and physiologic obstruction can be used to define these persons as having COPD; however, their pathophysiology and disease expression are substantially different than that of persons who have an emphysema-predominant form of COPD.
Visual analysis of the pattern and extent of emphysema can identify small amounts of centrilobular emphysema, usually in the upper lobes, in persons who have minimal quantitative emphysema. This appears to be one of the earliest manifestations of lung structural change associated with COPD. Both visual analysis and a quantitative texture-based analysis can be used to identify specific patterns, distributions, and the severity of emphysema including centrilobular, panlobular, and paraseptal patterns.
Evidence Synthesis
This section has reviewed a wide range of evidence related to COPD and smoking. Since the causal conclusion in the 1964 report related to “chronic bronchitis,” a term that can be considered equivalent to COPD, there have been great gains in the understanding of the pathogenesis of COPD and the clinical phenotype of COPD, and an understanding of genetic basis of susceptibility to COPD is emerging. Prior reports have advanced the conclusions of the 1964 report and affirmed that cigarette smoking is by far the leading cause of COPD in the United States.
This report addresses additional aspects of the COPD epidemic caused by tobacco smoking: trends in disease prevalence and mortality; gender differences in risk of COPD associated with smoking; advancing understanding of pathogenesis; emerging findings on the genetics of COPD; and phenotypic characterization of COPD using new approaches. Compared with 1964, COPD is a far more prominent cause of death. Age-adjusted mortality rates have risen sharply since 1964 and are only now beginning to drop in men. In contrast, prevalence data do not show a trend of increase, although methods have not been uniform over time and approaches for diagnosis and classification have changed as well.
The epidemiologic and clinical information suggests differences in COPD when comparing men and women. Studies involving the examination of pathology specimens and use of lung imaging suggest that men have more emphysema than women (Thurlbeck et al. 1974; Martinez et al. 2007) and women may be at greater risk than men for early onset COPD (Silverman et al. 1998; Martinez et al. 2007). Additionally, the COPD mortality rate for women has risen more steeply than that for men. A potential biological basis for such gender differences is uncertain at present. New approaches for characterizing COPD using imaging and molecular signatures may provide further insights.
The pathogenesis of COPD has been covered extensively in previous reports, most comprehensively in the 1984 and 2010 reports. Understanding continues to deepen through use of the ever-more powerful tools of molecular biology and animal models. Advances in understanding of the genetic basis of susceptibility to tobacco smoke will provide further insights and perhaps a basis for preventive strategies.
Conclusions
- The evidence is sufficient to infer that smoking is the dominant cause of chronic obstructive pulmonary disease (COPD) in men and women in the United States. Smoking causes all elements of the COPD phenotype, including emphysema and damage to the airways of the lung.
- Chronic obstructive pulmonary disease (COPD) mortality has increased dramatically in men and women since the 1964 Surgeon General's report. The number of women dying from COPD now surpasses the number of men.
- The evidence is suggestive but not sufficient to infer that women are more susceptible to develop severe chronic obstructive pulmonary disease at younger ages.
- The evidence is sufficient to infer that severe α1-antitrypsin deficiency and cutis laxa are genetic causes of chronic obstructive pulmonary disease.
Implications
Despite substantial progress in the epidemiology, genetics, imaging, and pathogenesis of COPD during the past 50 years, additional research in all of these areas will be required to provide a comprehensive understanding of COPD. A major focus will be to understand the heterogeneity of COPD, which is likely a syndrome of multiple diseases that share the common physiological manifestation of chronic airflow obstruction. Identification of specific subtypes of COPD, which will almost certainly have unique epidemiologic, genetic, and pathobiologic characteristics, has the potential to dramatically alter the approaches to the diagnosis and treatment of COPD. Smoking avoidance will remain the key primary prevention approach for COPD, but for the millions of already affected individuals, translation of the advances in pathogenesis, genetics, and imaging to improved clinical care will provide important challenges for decades to come.
Asthma
Asthma is one of the most common chronic respiratory diseases, affecting approximately 5–10% of the U.S. population (Moorman et al. 2007). The disease usually begins during childhood, but can start at any age. Childhood asthma may go into remission and then recur later in life. Asthma is characterized by variable airflow obstruction, which results in the symptoms of wheezing and dyspnea with exertion (National Asthma Education and Prevention Program 2007). In the modern conceptualization of asthma, chronic airway inflammation is the main underlying pathophysiologic abnormality that causes increased constriction of smooth muscles and decreased airway caliber and there can be an overlap between asthma and COPD (Figure 7.1). Chronic changes in the airway, referred to as airway remodeling, can lead to irreversible loss of lung function.
Exposures to allergens and environmental pollutants have long been recognized as factors that can cause or exacerbate asthma, particularly in vulnerable populations (Matsui et al. 2008). Allergic reactions to the antigens of dust mites, cockroaches, and cats have been widely cited as adverse factors in asthma, and both outdoor and indoor exposures to the byproducts of combustion have been linked to poor asthma control (National Heart, Lung, and Blood Institute 2007). This section reviews the accumulating evidence that active cigarette smoking contributes to both the incidence of asthma and its exacerbation.
Conducting epidemiologic studies of the effects of smoking and other environmental factors on asthma is a challenging undertaking. One of these challenges is the nature of asthma itself, which often remits and relapses over time. Indeed, many asthma patients have long periods of symptom-free intervals, only to have the disease return later in life. Consequently, establishing the clear temporal sequence between smoking and the initiation or exacerbation of asthma can be difficult. Moreover, a bias termed the healthy smoker effect may complicate epidemiologic studies of asthma and active smoking (Becklake and Lalloo 1990). This bias occurs when persons who have increased susceptibility to the health effects of smoking quit with relatively greater frequency than less susceptible persons, causing an overrepresentation in the remaining cohort of current smokers of those who are less likely to be affected. The topic of active smoking and asthma in children and adults was reviewed in the 2004 report of the Surgeon General (USDHHS 2004) and is updated in this section.
Biologic Mechanisms
The mechanisms by which active smoking could contribute to the causation of asthma include chronic airways inflammation, impaired mucociliary clearance, impaired growth of the lungs during childhood, and increased bronchial hyperresponsiveness (USDHHS 2004, 2006, 2010). Immunologic mechanisms include effects on T cell function (increased development of T helper cell 2 [Th2] pathways relative to Th1 pathways and a higher ratio of Th2/Th1), increased production of IgE, and greater allergic sensitization (see Chapter 10).
Since the publication of the 2004 and 2006 Surgeon General's reports, increasing evidence supports the role of Th2 cells and the related cytokines IL-4, IL-5, and IL-13 in the pathogenesis of asthma, especially severe asthma (Levine and Wenzel 2010). In addition, more studies have been published that support the impact of active smoking on increased Th2 pathway activation and allergic sensitization (Nouri-Shirazi and Guinet 2006; Broide 2008; Nakamura et al. 2008; Van Hove et al. 2008; Baena-Cagnani et al. 2009; Robays et al. 2009). Consequently, greater activation of the Th2 pathway may be one mechanism by which active smoking increases the incidence of asthma and the frequency and severity of exacerbations.
Emerging data suggest that cigarette smoke may increase neurogenic inflammation in the bronchial airway (Bessac et al. 2008; Simon and Liedtke 2008). The human airways are innervated by peripheral sensory neurons with specific receptors that are activated by inhaled noxious agents; neuronal activation may cause neurogenic inflammation of the airway. In particular, cigarette smoke can activate TRPA1s (transient receptor potential cation channel, subfamily A, member 1) in airway sensory neurons and result in inflammation and hyperresponsiveness of the airway (Andre et al. 2008; Bessac et al. 2008; Simon and Liedtke 2008; Lin et al. 2010). The TRPA1 is likely activated by oxidants contained within cigarette smoke.
In experiments with knockout mice, TRPA1-deficient mice, after a challenge with ovalbumin, experienced (1) markedly reduced airway inflammation and eosinophilia, (2) much lower levels of Th2 cytokines (IL-5 and IL-13) and pro-inflammatory cytokines (TNF-α and eotaxin), (3) greatly decreased production of mucous, and (4) a far lower incidence of airway hyperresponsiveness (Caceres et al. 2009). Pharmacologic inhibition of the receptor produced similar results. Although more research is needed, activation of the TRPA1 pathway is a plausible mechanism for the impact of cigarette smoke on inflammation and hyperresponsiveness of the airway.
Description of the Literature Review
For the present review, PubMed was searched for studies that focused on active smoking and asthma and were published from January 1, 2002, to December 31, 2009. The literature review obtained and reviewed studies that evaluated active smoking and the incidence of asthma, asthma status, or exacerbation of asthma in children, adolescents, or adults. The review did not include studies that focused on only respiratory symptoms and did not use a specific definition of asthma. For this review, the evidence cited in the 2004 Surgeon General's report on smoking was synthesized with newly available evidence to formulate revised conclusions.
Epidemiologic Evidence
Smoking and the Incidence of Asthma in Children and Adolescents
Because most asthma begins during childhood and adolescence, exposure to environmental risk factors during this period is of particular interest to researchers who are studying this disease. The 2004 Surgeon General's report on smoking and health reviewed 6 relevant studies; the current literature review identified 12 additional studies. Of these 12, 6 were cross-sectional studies that indicated an association between active smoking and the incidence of asthma during adolescence (Annesi-Maesano et al. 2004; Sturm et al. 2004; Avila et al. 2005; Fernandez-Benitez et al. 2007; Mallol et al. 2007; Gomez et al. 2009); no studies were found that explicitly evaluated smoking in childhood. Cross-sectional studies, however, cannot clearly separate the temporal sequence of initiating smoking and incidence of asthma, a concern because the presence of undiagnosed asthma or airway hyperresponsiveness might make adolescents less likely to smoke.
Three of the 12 new studies used population-based cohorts to evaluate the effect of active smoking on the risk of incident asthma during adolescence (Genuneit et al. 2006; Gilliland et al. 2006; Van de Ven et al. 2007), and a fourth used such a cohort to evaluate incident wheeze (Table 7.2S) (Vogelberg et al. 2007). Each of the first 3 studies associated active smoking with a higher risk of developing new-onset asthma during adolescence. However, none of these 3 studies began at birth; thus, some of the apparent incident asthma in adolescence could represent recurrence. Three of the 4 studies controlled for multiple potential confounders including socioeconomic status (SES) (Gilliland et al. 2006; Van de Ven et al. 2007; Vogelberg et al. 2007). Two studies (Genuneit et al. 2006; Gilliland et al. 2006) found strong evidence of an exposure-response relationship that involved either duration or intensity of smoking. In the analysis of the Children's Health Study in Southern California by Gilliland and colleagues (2006), the selection of different lags between smoking and asthma did not change the association of smoking with the onset of that disease.
Evidence Synthesis
The 2004 Surgeon General's report concluded that the evidence was inadequate to infer the presence or absence of a causal relationship between active smoking and asthma during childhood or adolescence; the available studies were judged to be inconsistent and to be without adequate control for potential confounders. The new evidence reviewed in the present report links active smoking to an increased risk of developing adolescent asthma; the finding of greater risk is consistent across geographic locations, study designs, and study years. Furthermore, the cohort studies reviewed convincingly demonstrate a temporal association between active smoking and onset of asthma during adolescence, although no study followed a cohort of subjects from birth. The findings are coherent even with a variety of definitions for asthma. The evidence for an exposure-response relationship is convincing, and several studies controlled for key potential confounders. However, the number of studies is limited. As detailed in the previous section, a biologically plausible relationship exists between active smoking and new-onset asthma. The evidence is consistent with the literature on active smoking and the incidence of asthma in adults.
Conclusion
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and incidence of asthma in adolescents.
Smoking and the Exacerbation of Asthma Among Children and Adolescents
Although extensive evidence implicates exposure to secondhand smoke as a cause of exacerbation of asthma among children and adults, there is less information about active smoking. Smoking is normally initiated during adolescence, and as asthma is usually first seen during childhood, active smoking during the teen years could adversely influence the clinical course of asthma soon after its onset. The 2004 Surgeon General's report on smoking and health concluded there was suggestive evidence of a causal relationship between active smoking and exacerbation of asthma among children and adolescents.
The results of three cross-sectional studies provide evidence that active smoking adversely affects control of asthma and leads to more exacerbations, as defined by asthma symptoms or use of health care for asthma (Yarnell et al. 2003; Austin et al. 2005; Navon et al. 2005). Because asthma is frequently characterized by relapse and remission, cross-sectional studies cannot definitively determine the temporal relationship between active smoking and exacerbation of the disease. Prospective cohort studies demonstrate that active smoking is associated with a higher risk of persistence of asthma in adolescence and early adulthood, but they have not explicitly examined exacerbations as an outcome (Sears et al. 2003; Bacopoulou et al. 2009).
Evidence Synthesis
Since the 2006 Surgeon General's report, only modest additional evidence has emerged for evaluating the impact of active smoking on the risk of exacerbating asthma during childhood and adolescence. Most of the data are cross-sectional and thus, temporality is in question. The evidence from adults is substantially more abundant. The additional evidence related to children and adolescents does not warrant a change in the conclusion reached in the 2004 Surgeon General's report, which is updated below.
Conclusion
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and exacerbation of asthma among children and adolescents.
Smoking and the Incidence of Asthma in Adults
The 2004 Surgeon General's report on smoking and health reviewed 15 cross-sectional and 6 cohort studies that evaluated the relationship between active smoking and adult asthma (USDHHS 2004). Since that report, 10 cross-sectional studies, 2 case-control studies, and 6 cohort studies have been added to the evidence base.
In considering asthma in adults who smoke tobacco, the overlap between asthma and COPD needs to be taken into account (Figure 7.1) (GOLD 2011). Smoking is the dominant cause of COPD (USDHHS 2004), and the clinical features of COPD and asthma can overlap.
Cross-sectional studies can provide information about the association between smoking and asthma, but their findings are subject to potential limitations, including both information bias, presumably from recall bias, and the inability to clearly establish a temporal relationship between smoking and asthma. With regard to recall bias, because the presence of asthma and smoking are assessed at the same time in these cross-sectional studies, persons living with asthma may be more likely than other study participants to remember and report past smoking behavior. In addition, because asthma is often characterized by relapses and remissions, cross-sectional studies cannot conclusively establish a causal connection between active smoking and the onset of asthma. In fact, the precise point when incident asthma develops may be difficult to identify.
Although cross-sectional studies have these limitations, they can still provide relevant evidence. Of the 10 cross-sectional studies, 9 provided evidence of an association between active smoking and prevalent asthma among adults (Chan-Yeung et al. 2002; Zhang et al. 2002; Gwynn 2004; Tutor and Campbell 2004; Aggarwal et al. 2006; Carter et al. 2006; Frank et al. 2006; Rose et al. 2006; Rahimi-Rad et al. 2008), and 1 revealed no clear association (Raherison et al. 2003). These studies represented a broad range of geographic locations, including China, Europe, India, the Middle East, the United Kingdom, and the United States.
Two case-control studies (one each from Sweden and Finland) of incident asthma found evidence of a relationship between active smoking and asthma (Table 7.3S). The Swedish study, a population-based examination of adult-onset asthma, found that current smoking was associated with a higher risk of adult-onset asthma (Toren et al. 2002). The study was limited, however, by its definition of adult-onset asthma, which relied on a self-reported physician diagnosis of asthma and no reported history of wheeze before 16 years of age. Also, past smoking was not evaluated as a risk factor for asthma, and SES, which was not statistically controlled, could have confounded the relationship between active smoking and asthma. Indeed, low SES has many correlates (e.g., poor diet, exposure to allergens, occupational exposure to dust or irritants, and exposure to ambient pollutants) that could act as confounders in the relationship between smoking and asthma in adults. The case-control study from Finland used a more rigorous clinical definition of adult-onset asthma that was based on physician diagnosis (using standardized clinical criteria). The study linked active smoking to a greater incidence of adult-onset asthma (Piipari et al. 2004). The study's conclusions were strengthened by controlling for a broad range of potential confounders, including SES and occupational exposures.
In addition to the two case-control studies, six cohort studies (Table 7.3S) have supported an association between active smoking and incident adult asthma. In Norway, a population-based cohort study of a population 15–70 years of age found that smoking was not associated with a greater incidence of asthma during an 11-year follow-up interval (Eagan et al. 2002). In a study of 1,139 New Zealand children born in 1972 and 1973 (Sears et al. 2003), smoking at 21 years of age was associated with self-reported persistent wheezing at age 21, but SES was not controlled and asthma was not specifically assessed. Butland and Strachan (2007) followed a cohort of English children born during 1 week in 1958 by interview at 17, 33, and 42 years of age. Incident asthma was higher in former smokers and never smokers than in current smokers as would be expected from reverse causation; but odds ratios (ORs) adjusted for gender, atopy, and IgE levels were increased among smokers when wheezing or asthma were examined together as an outcome. This was largely due to the association found in the group with wheezing without asthma. In two other cohort studies, both current and past active smoking were associated with a greater risk of incident adult asthma at 10-year follow-up (Hedlund et al. 2006; Polosa et al. 2008). Hedlund and colleagues (2006), in a Swedish cohort study, controlled extensively for confounders, including SES, and demonstrated a nonsignificant OR for persistent smoking with significant ORs for former smokers, again demonstrating the effect of reverse causation in the population. A clinical study (Polosa et al. 2008) with careful diagnosis of incident asthma in a population with allergic rhinitis, but no asthma at the start of follow-up, demonstrated a significant increase in new diagnosis of asthma after 10 years of follow-up with an increasing odds ratio with longer duration of smoking. In a 10-year follow-up of a Japanese cohort (Nakamura et al. 2009), a statistically significant increase in self-reported, physician-diagnosed asthma was reported, which was higher than the nonsignificant association demonstrated for former smokers.
Evidence Synthesis
The 2004 Surgeon General's report concluded that evidence linking active smoking to the incidence of asthma in adults was inadequate to infer a causal relationship. Since that time, the evidence base on the impact that active smoking has on the incidence of adult asthma has expanded with two of six cohort studies showing a statistically significant effect with current smoking for incident asthma and several studies having higher rates among former smokers, raising the question of reverse causation.
Conclusion
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and the incidence of asthma in adults.
Smoking and the Exacerbation of Asthma in Adults
The 2004 Surgeon General's report concluded that the evidence was sufficient to infer a causal relationship between active smoking and poor asthma control among adults, a conclusion that was based on studies demonstrating that active smoking increased the severity of asthma, the frequency of attacks, and the use of emergency health care (i.e., emergency department visits or hospitalizations) for exacerbations. Since the 2004 report, the evidence base has grown substantially, and new reports support this conclusion.
Numerous cross-sectional studies have examined the association between active smoking and exacerbation of asthma among adults, and only one of these studies did not find such an association (Gaga et al. 2005). Multiple cross-sectional studies have found an association of active smoking (compared with not smoking) with undesirable outcomes, including more respiratory symptoms, poorer asthma control, more severe asthma, worse quality of life, greater restriction of activity, more work disability, and higher risk of acute exacerbation requiring emergency health care (Suzuki et al. 2003; Ford et al. 2004; de Vries et al. 2005; Boulet et al. 2006, 2008; Ikaheimo et al. 2006; Laforest et al. 2006; Shavit et al. 2007; Stallberg et al. 2007; Strine et al. 2007; Chaudhuri et al. 2008; Meng et al. 2008; Peters et al. 2008; Seabra et al. 2008; Jang et al. 2009; Kim et al. 2009a). In addition, the baseline comparison in a clinical trial found that persons living with asthma who smoked had more respiratory symptoms, poorer quality of life, and lower daily peak expiratory flow rates (Lazarus et al. 2007) than their counterparts who were nonsmokers.
In 1998, the Copenhagen City Heart Study found that active smokers with asthma had a greater longitudinal decline in lung function, as measured by FEV1, than nonsmokers with asthma (Lange et al. 1998). Two other prospective cohort studies have confirmed that adult asthmatics who actively smoke have a more rapid decline of FEV1 than nonsmoking adults with asthma (Table 7.4S) (Apostol et al. 2002; James et al. 2005). In addition, a short-term follow-up study observed greater improvement in lung function among adults with asthma who quit smoking than among those who continued to smoke (Chaudhuri et al. 2006). Long-term decline in lung function does not necessarily reflect acute exacerbations, but these studies do establish that active smoking adversely affects the long-term natural history of adult asthma.
Other cohort studies provide evidence that active smoking confers a higher risk of exacerbation (Table 7.4S). Two studies found that active smoking was related to a higher risk of acute exacerbation that required emergency health care (Diette et al. 2002; Eisner and Iribarren 2007), and one of these studies (Eisner and Iribarren 2007) found that current smoking was related to greater severity of asthma. In another report from the same cohort used in the study by Eisner and Iribarren (2007), active smoking was associated with a higher risk of complete work disability (Eisner et al. 2006). Elsewhere, a small study found that active smoking was associated with a lower likelihood of asthma remission (Ronmark et al. 2007). One other cohort study (de Marco et al. 2006), however, found no association between change in smoking habits and severity of asthma at follow-up, as evidenced by the Global Initiative for Asthma (GINA) classification; but that study was underpowered, and the GINA classification is not a validated measure of disease severity for epidemiologic studies.
Cohort studies of adults with asthma have linked active smoking to death from respiratory causes (Omachi et al. 2008) and all-cause mortality (Bellia et al. 2007; Omachi et al. 2008). Although death was not clearly from asthma in these studies, these data indicate that smoking adversely affects life span in adult asthma.
Several randomized controlled trials have evaluated the differential efficacy of asthma therapy in smokers and nonsmokers. Of three trials that evaluated the impact of inhaled corticosteroids in smokers and non-smokers, two were placebo-controlled (Chalmers et al. 2002; Lazarus et al. 2007), and one compared high- and low-dose therapy (Tomlinson et al. 2005). In the placebo-controlled trials, inhaled corticosteroids had no clinical benefit among smokers, as measured by the primary study endpoint (postbronchodilator FEV1 and morning peak expiratory flow rate, respectively) (Chalmers et al. 2002; Lazarus et al. 2007). In addition, these trials did not find any benefits for secondary outcomes among nonsmokers, including bronchial hyperresponsiveness (Chalmers et al. 2002; Lazarus et al. 2007) and quality of life (Lazarus et al. 2007). Another trial, which compared high-dose and low-dose beclomethasone (2,000 micrograms [mcg] vs. 400 mcg per day), also found that smoking attenuated the efficacy of inhaled corticosteroids (Tomlinson et al. 2005). Low-dose therapy improved the primary outcome (morning peak expiratory flow rate) among nonsmokers only. Furthermore, the efficacy of high-dose beclomethasone was greatly attenuated in smokers. Elsewhere, a randomized controlled trial of oral corticosteroids (prednisolone 40 milligrams daily) versus placebo found that active smokers had no improvement in FEV1, daily peak expiratory flow rate, and asthma control, but nonsmokers experienced improvements in all three of these outcomes (Chaudhuri et al. 2003).
Evidence Synthesis
The 2004 Surgeon General's report concluded that the evidence was sufficient to infer a causal relationship between active smoking and both poor asthma control and exacerbation of disease among adults. Subsequently, the evidence base has grown and continues to support this conclusion. Currently, there is substantive evidence of coherence across a broad range of study outcomes, including such diverse endpoints as lung function, severity of disease, use of emergency health care, and quality of life. Randomized controlled trials provide strong evidence that smoking attenuates the therapeutic response to inhaled and systemic treatment with corticosteroids. Together, evidence from observational and clinical trials shows that active smoking adversely affects the natural history of adult asthma.
Conclusion
- The evidence is sufficient to infer a causal relationship between active smoking and exacerbation of asthma in adults.
Implications
Asthma is one of the most common chronic diseases of childhood. Asthma is also common among adults. Incidence of asthma is generally highest during childhood, but new cases occur among adults, too. The evidence reviewed in this chapter identifies active smoking as a possible cause of new-onset asthma among adolescents. The chapter also concludes that smoking is a cause of exacerbation of asthma among adults.
The evidence reviewed in this chapter shows that smoking should be considered an avoidable cause of asthma and that people who smoke should be counseled on the potential risk for developing asthma, should they continue to smoke.
Asthma is a chronic disease with a course marked by exacerbations—that is, deterioration—of the disease. Such exacerbations may lead to substantial morbidity and to economic costs from absenteeism, and can result in death. The evidence reviewed in this chapter is sufficient to conclude that active smoking exacerbates asthma in adults. The clinical implications are clear: people with asthma should not smoke.
Tuberculosis
Tuberculosis (TB) was a leading cause of death in the United States at the start of the twentieth century, a time when cigarette smoking was just beginning to become popular among men. Although rates of TB and smoking are both continuing to decline in the United States, the two epidemics continue globally. More than 1.3 billion people worldwide smoke (World Health Organization [WHO] 2010), and estimates indicate that each year sees almost 9 million cases of incident TB and 1.3 million deaths from the disorder (Mathers and Loncar 2006). Moreover, an estimated one-third of the world's population is infected with Mycobacterium tuberculosis (M. tuberculosis) and, therefore, is at risk of active TB disease. Annually, more than 30% of TB cases worldwide are diagnosed in China and India. These two countries account for more than 40% of the world's smokers (WHO 2008).
Smoking has long been considered a potential risk factor for TB mortality (Doll and Hill 1956), but only recently have large case-control and cohort studies shown that strikingly high rates of TB mortality are attributable to smoking (Jha et al. 2008). Several systematic reviews have assembled evidence of the association between smoking and TB (Davies et al. 2006; Bates et al. 2007; Chiang et al. 2007; Lin et al. 2007; Pai et al. 2007; Slama et al. 2007; WHO and International Union Against Tuberculosis and Lung Disease 2007). Each review found that cigarette smoking is associated with an approximate doubling of risk for TB infection, for having clinical evidence of TB disease, and for TB mortality. An analysis from WHO (2010) of the role of risk factors and social determinants in driving the global TB epidemic concluded that in the 22 countries experiencing 80% of the global TB burden, 23% of the cases can be attributed to smoking. The smoking-attributable burden of TB varies with the epidemiologic characteristics of the population, with the high attributable risks for smoking found in China and India, while HIV is the primary driver for the TB burden in sub-Saharan Africa (Lonnroth et al. 2010). To date, the series of Surgeon General's reports has not systematically assessed the association between smoking and TB.
Biologic Mechanisms
Given its effects on host defenses and the structure and function of the lungs, smoking is a biologically plausible cause of morbidity and mortality from TB (Pai et al. 2007; Stampfli and Anderson 2009; USDHHS 2010). The section on “Immune Function and Autoimmune Disease” in Chapter 10 of this report more fully describes the effects of smoking on the immune system, indicating multiple underlying mechanisms that may increase the risk for TB in smokers. However, the specific mechanisms by which cigarette smoking may influence risk of infection by M. tuberculosis and reactivation of latent TB infection are not completely understood.
Natural History of Tuberculosis
Figure 7.7 shows the natural history of TB from exposure to the organism and the initial infection through death from the disease. Figure 7.8 offers a closer look at the progression to active TB disease among persons exposed to M. tuberculosis. In brief, TB follows a two-stage process: infection of the host with M. tuberculosis and then the development of active disease (Golub et al. 2013). Infection occurs in an estimated 20–30% of close contacts of people with active disease, and within 2 years, active primary TB develops in 5–10% of those infected (Figure 7.8A) (Comstock et al. 1974; Comstock 1975). However, TB is a unique pathogen in that the first stage of this process can last a lifetime. In what is commonly referred to as latent TB infection, an intact immune system contains the primary infection in a dormant state. Over the course of an infected person's life, the risk that the dormant bacilli will progress to reactivation TB is 5–10% (Comstock et al. 1974; Comstock 1975). People who are immunocompromised have an altered prognosis after infection, with more than 40% experiencing early progression (Figure 7.8B) and the majority reactivating to TB disease later in life if untreated.
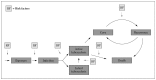
Figure 7.7
Natural history of tuberculosis from exposure to mortality. Source: Adapted from Rieder 1995 by the Center for Teaching and Learning with Technology, Johns Hopkins Bloomberg School of Public Health with permission from Springer Science & Business (more...)

Figure 7.8
Progression to active tuberculosis disease among persons exposed to M. tuberculosis. Source: Adapted from Parrish et al. 1998 with permission Elsevier, © 1998. Note: HIV = human immunodeficiency virus. Progression to active tuberculosis disease (more...)
Upon developing active TB disease, a small proportion of persons will spontaneously heal without treatment, but without treatment, the majority will ultimately die. Among persons who are treated for TB, treatment failure and/or recurrent disease are serious risks. Recurrence can result from endogenous reactivation of persistent TB bacilli (relapse) or from exogenous reinfection with a new TB strain.
Description of the Literature Review
An initial search of English publications in PubMed was conducted using the key terms “smoking” OR “tobacco” AND “tuberculosis.” These broad terms were searched in titles and abstracts. For purposes of reviewing the evidence of smoking as a risk factor for TB infection, TB disease, recurrence, and/or mortality, articles were excluded if they addressed other aspects of the effects of tobacco use on TB severity, treatment, or outcomes without providing evidence of risk. Such articles are discussed in this chapter as appropriate but are not included in the evidence tables (Tables 7.5S–7.8S). In addition to the PubMed search, references that were included in the systematic reviews and meta-analyses were reviewed, and any publications not previously identified were included. The literature search extended through December 2009.
Epidemiologic Evidence
Risk Factors: Potential Confounders and Modifiers
Three of the major risk factors for TB exposure and infection are predominantly related to SES: immunocompromising diseases, malnutrition, and alcohol consumption. SES itself is another important risk factor, serving as a proxy for many correlates. Attention to these four risk factors is critical in investigating smoking and risk of TB and in ensuring that confounding is considered and interactions with smoking are addressed.
TB and Alcohol Consumption and Tobacco Use
Alcohol consumption and tobacco use have been consistently linked over time, and an association between alcohol use and TB has long been observed. Lonnroth and colleagues (2008) and Rehm and colleagues (2009) reported a strong association between heavy alcohol use and incident TB, citing the pathogenic impact of alcohol on the immune system, which increases risk of reactivation TB. Based on patterns of increased risk ratios for TB, early studies (Brown and Campbell 1961; Lewis and Chamberlain 1963) suggested that alcohol use was the most important risk factor and that tobacco use was only important because most alcoholics in the studies also smoked. The majority of subsequent studies have shown an association between tobacco use and TB disease even when alcohol is considered in the analysis, but the association may be diminished by controlling for alcohol (Lin et al. 2007). Among the prospective cohort studies, one in Korea found alcohol use to be a strong independent risk factor for TB (Jee et al. 2009), and in a study in Taiwan, alcohol use was a stronger risk factor than smoking (Lin et al. 2009a). In both studies, however, smoking was a strong independent risk factor after adjusting for alcohol consumption.
The extent to which alcohol use has been considered as a potential confounding factor for the risk of TB has varied greatly among studies. Despite a lack of uniformity in how alcohol use is addressed, most studies have found a consistent positive association between smoking and TB after adjusting for alcohol consumption. For example, Gajalakshmi and Peto (2009) conducted a comprehensive investigation of smoking and alcohol use and the risk of TB in India and found that the risk of incident TB was 3.5 times greater for people who were both drinkers and smokers than for those who were not smokers or drinkers. Nondrinking smokers had an RR of 2.6, and nonsmoking drinkers had an RR of 2.1. In a retrospective multilevel analysis of data from the South African Demographic and Health Survey, Harling and colleagues (2008) reported high rates of TB among smokers and alcohol users after adjusting for many factors, including a multilevel adjustment for SES.
TB and Socioeconomic Status
Although poverty is often cited as a risk factor for TB infection and disease, it is best seen as a proxy for multiple other relevant factors, including population density, race/ethnicity, nutritional status, and access to health care. In many countries, those who are poor are more likely to both smoke and to have higher rates of TB infection and disease than those of a higher SES (Chapman and Dyerly 1964; Kuemmerer and Comstock 1967). Harling and colleagues (2008) conducted a multilevel analysis of the impact of demographic, behavioral, and socioeconomic individual risk factors and of group-level measures of SES on risk for TB. This study combined data from the 1998 South African Demographic and Health Survey with data from the 1996 South African national census. Although the study relied on the potentially biased outcome of self-reported TB and linked two cross-sectional studies, it provided a comprehensive analysis of the SES–TB relationship. After adjusting for SES, both alcohol abuse and cigarette smoking were associated with risk for TB. In a meta-analysis, Lin and colleagues (2007) found that the association between smoking and TB mortality was stronger in studies that adjusted for SES than in those that did not, suggesting that not considering SES may lead to an underestimation of measures of association. After adjusting for SES, this same review did not find a difference in risk for pulmonary TB disease associated with smoking. However, the authors did not assess the quality of the SES measurements in each study, thus potentially limiting the interpretation of this summary adjustment.
TB and Gender
Some studies suggest that smoking may account for differences observed between men and women in TB incidence and mortality, primarily because, compared with women, smoking is substantially more common among men, and on average men smoke a greater number of cigarettes. In one of the first studies to investigate the relationship between smoking and TB disease, Lowe (1956) reported a greater proportion of heavy smokers among men than women, but TB cases were more likely to be observed in heavy smokers of either gender compared with controls. Lowe did not observe a significant difference between the smoking habits of cases and controls among persons younger than 30 years of age; this evidence suggests that smoking is a strong contributor to the reactivation of TB in older ages but a weaker contributor to the reactivation at younger ages. Yu and colleagues (1988) also identified differences in risk for TB by age and gender that were attributed to tobacco use, and Nisar and colleagues (1993) suggested that smoking may be a key factor in higher TB disease rates among men because (a) men smoke more than women, and (b) smoking likely increases risk of TB infection, which is a necessary precursor to actual TB disease. More recently, Crampin and colleagues (2004) assessed differences among risk factors for men and women in Malawi. Finally, Lin and colleagues (2009b), in their systematic review and meta-analysis, suggested that smoking may be the cause of gender differences in TB disease because the odds ratio (OR) for men (vs. women) decreased from 1.62 to 1.06 after adjusting for current smoking.
Tobacco and TB
Evidence on tobacco and the natural history of TB suggests that tobacco may affect disease risk at each stage of the disease process, as reviewed below. Consequently, the evidence is considered separately for each stage of TB disease.
Tobacco and TB Infection
To date, evidence on the association between tobacco use and TB infection is limited (Table 7.5S). Establishing a temporal relationship between exposure to tobacco smoke and the onset of M. tuberculosis is difficult because determining when a person becomes infected is almost impossible—unless this is determined as part of a contact investigation of a person with active TB disease. Only one study, a case-control study of prison inmates in South Carolina carried out by Anderson and colleagues (1997), used smoking data that were collected before assessing the acquisition of M. tuberculosis. This study compared two groups. The case group was composed of those who were tuberculin skin test (TST) negative upon incarceration but were found to be TST positive at a follow-up reading. The control group was composed of those with a TST reading that remained negative. Those who developed a positive TST were assumed to have become infected with TB while incarcerated. After adjusting for age and living conditions, current smokers were more likely to have converted their TST (OR = 1.78; 95% confidence interval [CI], 0.98–3.21) than the reference group (never and past smokers). In addition, inmates who had smoked for more than 15 years had twice the odds of converting their skin tests (OR = 2.12; 95% CI, 1.03–4.36) as the reference group (nonsmokers), suggesting that the cumulative effects of long-term smoking have a greater impact on risk than the number of cigarettes smoked per day. One major limitation of this study was that cases were more likely than controls to have been previously exposed to people with TB, a risk factor for infection that was not controlled for in the analysis.
Only three studies had the primary objective of investigating the association between smoking and latent TB infection (Anderson et al. 1997; Plant et al. 2002; den Boon et al. 2005). The study in South Africa by den Boon and colleagues (2005) reported an almost twofold increased risk for latent TB infection among adults who were ever smokers. However, because of the cross-sectional design of the study, researchers could not determine whether the association was temporally correct (i.e., that smoking came before infection). Additionally, smoking may affect the size of the TST and thus the chance for a positive finding, and cross-sectional data are subject to differential survival from higher losses of heavier smokers or of persons with more severe primary infection. Hussain and colleagues (2003) performed a cross-sectional study of prisoners in Pakistan, a region with a moderately high burden of TB that is not influenced substantially by HIV infection. They found an increased risk for latent TB infection among current smokers, with the OR rising from 2.6 for those who smoked 1–5 cigarettes per day to 3.2 for those who smoked more than 10 cigarettes per day.
In a study of Vietnamese immigrants, Plant and colleagues (2002) detected an increased risk of latent TB infection among those who were ever smokers. The strongest ORs were reported among people with a TST induration cutoff of 5 millimeters (mm) (OR = 2.31; 95% CI, 1.58–3.38). The ORs were lower for those with cutoffs of 10 mm (OR = 1.53; 95% CI, 1.13–2.09) or 15 mm (OR = 1.37; 95% CI, 0.95–1.97), which are categories less likely to be contaminated with infections from non-TB mycobacteria. The researchers concluded that smoking increases the risk of mycobacterial infections, of which TB is likely to be the primary contributor. Although smaller tuberculin reactions may be associated with non-TB mycobacteria, smokers may have smaller reactions because of the effects of smoking on cell-mediated immunity. The study reported a 3–5% increase in risk of TB infection per year of smoking exposure.
The majority of studies that have addressed smoking and latent TB infection have not had smoking as the primary exposure of interest but do provide relevant evidence. For example, Kuemmerer and Comstock (1967) noted that children who were living in households where both parents smoked were twice as likely to be infected latently with TB as those who were living in a household with one or no parent smoking. Although the study found that crowding and prior household exposure to TB increased the risk of infection, it did not adjust for these potential confounding factors. In India, Singh and colleagues (2005) investigated the prevalence of TB infection among children of adults with pulmonary TB. For such children, it is well-accepted that a positive TST is the result of recent exposure to TB, and thus exposure to cigarette smoke was likely at the time of acquisition of TB infection. Children exposed to a household TB patient who smoked were almost three times as likely as their counterparts to be infected with TB. Children of adults who smoked and were sputum positive for TB were more likely to be infected than children of adults who did not smoke but were sputum positive for TB, suggesting that smoking raises the risk of TB infection beyond the strong risk of exposure to a highly infectious TB case. A study in South Africa by den Boon and colleagues (2007) suggests that smoking adds to the infectiousness of persons with TB. This study found more than a fourfold increased risk (OR = 4.60; 95% CI, 1.29–16.45) of latent TB infection among children living with an active TB case who were exposed to secondhand smoke (vs. no such exposure). In households without a current TB case, passive smokers were at a moderately increased risk of latent TB infection, but the risk decreased and was not statistically significant after adjusting for other factors.
Nisar and colleagues (1993), who investigated the potential increased risk of latent TB infection among nursing home residents in the United Kingdom, found evidence that smoking increases the prevalence of TB infection in elderly persons and may explain higher rates of positive TB cases in men compared with women. Current smokers had greater TB infection rates than former smokers, who had greater rates than never smokers. The study also found an association between increasing pack-years and increased prevalence of latent TB infection. Length of stay, however, was not associated with increased prevalence of latent TB infection.
Several studies have investigated risk factors for latent TB infection among people in prisons and homeless shelters. In their study of prisoners in Pakistan, Hussain and colleagues (2003) reported weak evidence of a dose-response relationship—with ORs of 2.6, 2.8, and 3.2 for latent infection among current smokers of 1–5, 6–10, and more than 10 cigarettes smoked per day, respectively. Among prisoners in Lebanon, a study by Adib and colleagues (1999) found a modestly increased risk of latent TB infection (OR = 1.2; 95% CI, 1.1–1.3) among current smokers but no indication of a dose-response relationship with amount smoked. In their study of South Carolina inmates, Anderson and colleagues (1997) reported an almost twofold increased risk of latent TB infection (OR = 1.78; 95% CI, 0.98–3.21) among current smokers (vs. never and former smokers). Similarly, in a homeless population with a 75% rate of latent TB infection in Barcelona, Spain, Solsona and colleagues (2001) found an increased risk (OR = 1.72; 95% CI, 1.02–2.86) of infection among current smokers. In a study of migrant workers in California, McCurdy and colleagues (1997) found a threefold increased risk of latent TB infection among former smokers (OR = 3.11; 95% CI, 1.20–8.09) but less than a twofold increase (OR = 1.87; 95% CI, 0.73–4.80) among current smokers.
Tobacco and TB Disease
This section reviews studies of evidence on the association between smoking and clinical TB, with a focus on prospective cohort studies (Table 7.6S).
In studies in Hong Kong, Leung and colleagues (2003, 2004, 2007) investigated the association between smoking and TB disease in younger (≤64 years of age) and older (>64 years of age) TB patients and among people with silicosis (a population at high risk for TB). In most of the populations, smokers had an approximately twofold increased risk for TB, and although alcohol use was a strong contributor to TB risk in each study, smoking had a strong, independent effect after controlling for alcohol consumption. Although former smokers had a lower risk for TB than current smokers, time elapsed since quitting did not affect risk for TB (Leung et al. 2007).
A few studies have specifically addressed the change in risk for TB upon quitting smoking. Based on a follow-up of the British Doctors' Study, Doll and colleagues (1994) reported a 2.8 mortality ratio among current smokers compared with nonsmokers, and a 2.0 mortality ratio among former smokers compared with nonsmokers (Table 7.8S). Prospective cohort studies with large populations and many years of follow-up offer the strongest evidence of a link between smoking and TB. In a cohort based in Taiwan's NHIS, Lin and colleagues (2009a) found a twofold increased risk for TB among current smokers compared with never smokers after controlling for several risk factors. Risk was only slightly diminished for ever smokers. Alcohol use was controlled for in the analysis and reported to be a much stronger risk factor for TB than smoking, although smoking remained associated with TB after controlling for alcohol consumption. In Taiwan, the OR for TB among elderly smokers was 0.78, versus 2.87 for the comparable group in Hong Kong (Leung et al. 2004). This risk difference cannot be attributed to smoking intensity, because the elderly in Taiwan smoked 15 cigarettes per day compared with 11 by those in Hong Kong.
Jee and colleagues (2009)—who conducted a cohort study in Korea using the Korean Cancer Prevention Study of more than 1.3 million middle-class, primarily middle-aged men and women—found increased risk for incident TB disease among male current smokers but not among their female counterparts. The analysis considered alcohol use as a potential confounder and found a dose-response relationship between use of alcohol and amount smoked. Although SES was not included in the Korean analysis, little variability was expected within this relatively homogenous and middle-class population.
Several studies, mostly case-control in design, conducted in India found a strong risk for TB among smokers. When evaluating studies in India and some other countries, the types of products smoked need specific consideration because individuals may smoke cigarettes or bidis, the latter the most common type of smoked tobacco in India, or both. With TB mortality as the outcome of interest, Gupta and colleagues (2005) found that smoking bidis was not less hazardous than smoking cigarettes and that the duration of bidi smoking conveyed greater risk for TB mortality than did the number of bidis smoked per day (Table 7.8S). After adjusting for several sociodemographic factors in a population of primarily low and middle socio-economic classes, Prasad and colleagues (2009) reported a fourfold increased risk for TB among current smokers in a case-control study. The study also found a strong dose-response relationship between increasing pack-years and duration of smoking on risk for TB. However, the analysis found that the number of cigarettes or bidis smoked per day did not affect risk for TB. The authors concluded that the effects of smoking for a prolonged period of time are more important to the development of TB disease than a large number of cigarettes smoked per day.
The effect of passive smoking on risk for TB disease has been reported in several settings. Studies among children are informative because both TB infection and TB disease in children are considered to have been recently acquired. Altet and colleagues (1996), who conducted a case-control study that investigated the effect of passive smoking on the development of TB disease among recently infected children of active smokers, found that exposure to passive smoke increased risk for TB disease fivefold, with the greatest risk among children younger than 10 years of age. Similarly, Kuemmerer and Comstock (1967) reported an increased risk for TB disease among children exposed to two parents who smoked, compared with one or no parent who did so (Table 7.5S). A strong dose-response relationship was found between risk for TB and an increasing number of cigarettes smoked to which children were exposed per day. This study was one of a few to use a biological marker for measuring tobacco exposure, finding that mean levels of cotinine in the urine were significantly greater among contacts that developed TB disease. Use of a biomarker removes potential bias associated with self-reported exposure to tobacco.
In a study by Alcaide and colleagues (1996) that addressed the combined risk of TB among young adults who were exposed to both active and passive smoking (Table 7.6S), active smokers who were contacts of pulmonary TB cases had a 3.6-fold increased risk of developing TB, but those exposed to both active and passive smoking had an OR of 5.1. In a study in Thailand among children younger than 15 years of age, Tipayamongkholgul and colleagues (2005) found a ninefold increased risk for TB disease with close passive exposure to smoke and no known direct contact with a person with TB. In another study from Thailand, in a comparison with nonsmokers and after adjusting for body mass index, Ariyothai and colleagues (2004) found that passively exposed adult smokers were at increased risk for TB (OR = 2.37; 95% CI, 0.94–6.01). The adjusted risks among current (OR = 2.70; 95% CI, 1.04–6.97) and former smokers (OR = 2.88; 95% CI, 0.85–9.78) did not differ materially. Exposure to secondhand smoke in the outdoors or in an office was a more significant factor than such exposure in the home, but very few study participants reported this kind of exposure in the home. By contrast, in a study in Estonia, Tekkel and colleagues (2002) found that exposure to secondhand smoke in the office was not associated with risk of pulmonary TB, but exposure to smoke in the home was associated with a twofold increased risk.
Several studies have included smoking as a potential risk factor for TB in investigations in which smoking was not the primary exposure of interest. For example, in three West African countries, Lienhardt and colleagues (2005) conducted a case-control study on host-related and environment-related factors for TB. After adjusting for various host and environmental factors, current and former smokers had increased risks for TB, about a doubling and a 50% increase, respectively. The study observed a significant dose-response relationship between incidence of TB and three factors: duration of smoking, alcohol use, and drug use. In a study of a population in King County, Washington, Buskin and colleagues (1994) did not find excess risk for TB among smokers after adjusting for age and alcohol consumption. However, among current smokers, the authors observed a dose-response relationship with number of cigarettes smoked per day and number of years of smoking. In China, as part of a mass routine chest radiograph campaign in Shanghai, Yu and colleagues (1988) found heavy smokers to have a twofold increased risk for pulmonary TB (RR = 2.17; 95% CI, 1.29–3.63), but light and moderate smokers did not have an excess risk. Much earlier, Adelstein and Rimington (1967) conducted a mass chest radiograph survey in East Cheshire, United Kingdom, and found that male smokers had a fivefold increased risk for TB compared with nonsmokers, but a very small number of TB cases limited the analyses.
Tobacco and Recurrent TB Disease
The literature investigating smoking as a risk factor for recurrent TB disease is limited and not all studies differentiate between relapse and disease resulting from reactivation of an exogenous reinfection (Table 7.7S). Thomas and colleagues (2005), who investigated predictors of relapse among pulmonary TB patients who had completed therapy in a Directly Observed Treatment, Short course program in South India, found that smoking was associated with increased risk for relapse (OR = 3.1; 95% CI, 1.6–6.0). The authors found three risk factors to be associated with increased risk of TB recurrence: smoking, drug sensitivity profile, and adherence level to TB therapy. Age, gender, education, alcohol use, and initial weight were not associated with increased risk. In a similar study in Brazil, d'Arc Lyra and colleagues (2008) reported an increased risk for relapse among ever smokers (current smokers and former smokers who had given up smoking less than a year from time of interview) compared with never or former smokers (those who had given up smoking 1 year or more) (OR = 2.34; 95% CI, 1.17–4.68). These studies (Thomas et al. 2005; d'Arc Lyra et al. 2008) adjusted for several socioeconomic factors, but none were associated with increased risk for TB relapse.
In their Hong Kong study of tobacco and TB in the elderly, Leung and colleagues (2004) reported an increased risk (OR = 2.48; 95% CI, 1.04–5.89) of being retreated for TB—in this study assumed to be a relapse—among current smokers compared with never smokers after adjusting for many factors, but not SES. Elsewhere, two studies (Chang et al. 2004; Millet et al. 2009) that did not clearly define smoking and appeared to rely on an “ever” versus “never” classification did not find smoking to increase risk of relapse. In Korea, the cohort study by Jee and colleagues (2009), the largest to date to investigate the risk for recurrence in smokers and nonsmokers, used long-term follow-up of cohort members with past TB to investigate recurrent TB, including both relapse and potentially exogenous reinfection. In the study, men who were current smokers had moderately increased risk for recurrence (hazard ratio [HR] = 1.3; 95% CI, 1.2–1.4), but risk was not significantly increased among women who were current smokers, although the HR was of a similar magnitude as that for men (HR = 1.2; 95% CI, 0.8–1.6). Heavy alcohol consumption was associated with recurrent TB in men in this study (HR = 1.2; 95% CI, 1.0–1.3).
Tobacco and TB Mortality
The literature assessing risk of mortality among smokers with TB is somewhat limited, and most studies have not accounted for potential confounding factors, such as delays in diagnosis, HIV infection, or site of disease (Table 7.8S). When Doll (1999), Doll and Hill (1954, 1956, 1964), and Doll and colleagues (1994) began their study of a cohort of British physicians in 1951, treatment for TB was just beginning, and physicians were at high risk from occupational exposure (from their patients). Pulmonary TB was one of more than 25 diseases found in this long-term study to be linked to cigarette smoking. Mortality rates for TB observed after 5 years of follow-up showed trends similar to those observed more than 40 years later (Doll and Hill 1956; Doll et al. 1994). In both the 1956 and 1994 reports, TB mortality rates were elevated in older men who were smokers, and a dose-response relationship was observed with daily amount smoked, with TB mortality as high as 29/100,000 in 1956 and 20/100,000 in 1994 in the group reporting the highest levels of smoking in 1951.
In China, in a retrospective cohort study that assessed the impact of tobacco on the deaths of 1 million people, male smokers had a moderately increased risk for TB mortality compared with their nonsmoking counterparts (RR = 1.20; SE = 0.04) (Liu et al. 1998). The study found a similar, albeit not significant, risk among female smokers (RR = 1.29; SE = 0.08). Urban dwellers of both genders had a higher risk for TB mortality (RR = 1.42; SE = 0.05 for males and RR = 1.56; SE = 0.09 for females) than did those in rural areas. In Hong Kong, a case-control study found, after adjusting for age and education, a 2.5-fold increased risk for TB mortality among middle-aged men and a nonsignificantly increased risk among similarly aged women (Lam et al. 2001). In this study, a strong dose-response relationship with numbers of cigarettes smoked per day was observed among both middle-aged and elderly men. The study identified very few deaths from TB among women, limiting analyses of smoking and TB. In Korea, the cohort study by Jee and colleagues (2009) found that mortality increased 58% in men (HR = 1.58; 95% CI, 1.27–1.97) and 55% in women (HR = 1.55; 95% CI, 1.00–2.41) who were current smokers. Among former smokers, risk of mortality doubled among women (HR = 2.16; 95% CI, 1.35–3.46).
In India, Gajalakshmi and colleagues (2003) and Gupta and colleagues (2005) used different sources of data to attribute between 140,000 and 149,000 TB deaths per annum to smoking, a number that represents half of annual deaths from TB in India. Later, in a nationally representative study in India, Jha and colleagues (2008) attributed 38% of TB deaths among men to smoking. Compared with never smokers, TB mortality increased between twofold and fourfold among male smokers (Gajalakshmi et al. 2003; Jha et al. 2008) and threefold among female smokers (Jha et al. 2008).
Finally, a study in Africa by Sitas and colleagues (2004) compared deaths from diseases known to be associated with tobacco use with deaths from medical conditions unrelated to tobacco use and found that, among deaths from TB, 28% of those for men and 7% of those for women were attributable to smoking. The authors found that smoking was associated with an increased risk of mortality (OR = 1.61; 95% CI, 1.23–2.11).
Tobacco and Type/Severity of TB Disease
Although the association between tobacco use and TB disease has been widely investigated, available studies have not clarified whether smoking affects risk only for pulmonary or also for extrapulmonary TB. In Hong Kong, Leung and colleagues (2004) reported that male current smokers were three times as likely as male never smokers to develop pulmonary TB but were significantly less likely to develop extrapulmonary TB. Later, Lin and associates (2009b) confirmed these results in a study in Taiwan, finding that nonsmokers had increased risk for extrapulmonary TB. In a study in Nepal, Sreeramareddy and colleagues (2008) found that current smokers were 66% less likely to develop extrapulmonary TB than they were pulmonary TB, and this pattern extended to those who had quit smoking 6 months before TB diagnosis (OR = 0.45; 95% CI, 0.21–1.09 for extrapulmonary vs. pulmonary TB). Researchers in a study from Turkey reported similar findings (OR = 0.54; p = 0.025 for an extrapulmonary site vs. a pulmonary site) (Musellim et al. 2005). The meta-analysis by Lin and colleagues (2007) found a higher risk of TB, both pulmonary and extrapulmonary, in smokers than in nonsmokers. When studies were restricted to those that included only pulmonary TB cases, the association was stronger than was found when studies that included both pulmonary and extrapulmonary TB cases were considered (2.01 vs. 1.49). This difference did not reach statistical significance.
One study addressed smoking and the severity of TB. Among a cohort of more than 13,000 TB patients in Spain, Altet-Gomez and colleagues (2005) reported that smokers were 50% more likely than nonsmokers to develop pulmonary disease and almost twice as likely to have cavitary disease.
Evidence Synthesis
Evidence on the relationship between smoking and TB needs to be assessed within the framework offered by the natural history of the infection. Smoking may have an effect at multiple stages of this natural history (Figure 7.7), and thus the evidence should be considered separately for the risks of TB infection, TB disease, recurrent TB disease, and TB mortality (see Table 7.9 for a summary of evidence from three systematic reviews on the association between smoking and three TB outcomes: infection, active disease, and mortality due to TB [van Zyl Smit et al. 2010]). This framework focuses on two questions: (1) Is the risk of incident TB infection higher in smokers than in nonsmokers, and (2) in persons with TB infection, does the course of TB infection differ between smokers and nonsmokers with regard to risk for TB disease, recurrence, and mortality?
Table 7.9
Meta-analysis of the association between smoking and latent tuberculosis (TB) infection, progression to active disease, and mortality from active TB.
TB Infection
The available studies consistently show that smokers are at a greater risk for TB than nonsmokers (RR = 1.2–2.7), but all but one of the studies are cross-sectional in design, leaving the temporality of causation ambiguous. Only the nested case-control study among TST converters found that smoking occurred prior to incident infection (Anderson et al. 1997). The literature has not consistently demonstrated dose-response relationships between TB infection and the number of cigarettes smoked per day or pack-years. Adjusting for alcohol consumption as a potential confounder reduced the strength of the association between tobacco and latent TB infection in a meta-analysis, but the association remained significant (Lin et al. 2007).
Biologic evidence supports the plausibility of increased risk for TB infection among smokers because tobacco smoke has been shown to cause mechanical disruption of ciliary function, alter mucociliary clearance in the airways (Arcavi and Benowitz 2004), and inhibit macrophage responses, thus increasing the likelihood that M. tuberculosis organisms reach the alveoli where TB infection begins (Altet et al. 1996). Although such factors as age, gender, and SES have also been associated with risk of TB infection, smoking has been shown to be an independent risk factor. Exposure to an infectious TB case is necessary for TB infection to occur, but collective evidence suggests that persons exposed to tobacco smoke, either actively or passively, are at greater risk of becoming infected with TB than those who are not exposed to tobacco smoke.
TB Disease
The evidence reviewed in this section implicates smoking as a cause of TB disease. The infectious organism that causes tuberculosis, M. tuberculosis, is, of course, the necessary cause of TB. However, other agents can increase risk for TB by acting to increase the risk for infection or by increasing the risk for disease in those who are infected. Within the framework for causal inference used in the Surgeon General's reports, such additional risk factors that are neither necessary nor sufficient have been interpreted as causal. For example, the 2004 report identified smoking as a causal risk factor for cervical cancer, while acknowledging the necessary role of human papilloma virus (USDHHS 2004). Cohort studies showed that human papilloma virus-infected women who smoked developed cervical intraepithelial neoplasia at a higher rate than nonsmokers. The evidence on smoking and TB is thus interpreted with recognition of the necessary role of M. tuberculosis.
The association between smoking and TB disease is consistent across studies, and findings from several prospective cohort studies confirm previous findings that were largely based on case-control studies. The association is found consistently across different populations and geographic regions, although the most informative studies have been conducted in Asian countries where smoking is common among men and TB disease remains frequent. Most studies have observed dose-response relationships with indicators of the extent of smoking, strongly suggesting that an increased number of cigarettes smoked per day, increased years of smoking, and earlier age at the start of smoking are all associated with increased risk for TB disease.
In interpreting the results of these studies, the main limitation is the incomplete consideration of some potential confounding factors, leaving the possibility of residual confounding. However, prospective cohort studies carried out by Jee and colleagues (2009) and Lin and associates (2009b) have controlled for age, gender, and alcohol use. Aside from smoking, SES has many correlates that are relevant to risk for TB disease, including nutrition, housing, and other exposures. Although various studies have measured SES in different ways, the association between smoking and risk for TB disease persists after adjusting for SES. The body of evidence is greater for pulmonary TB disease than for extrapulmonary TB (Lin et al. 2007). Finally, although research demonstrates a strong association between smoking and TB disease, it is still not clear whether the association reflects an increased risk of infection or of reactivation to active TB disease.
TB Recurrence
Only a few studies present evidence of an association between smoking and risk for recurrent TB disease (Table 7.7S). Unfortunately, when studying recurrent TB, differentiating between relapse due to treatment failure and reactivation of a subsequent infection with M. tuberculosis can be difficult. Studies with short follow-up of patients who have completed treatment—for example, the one by Thomas and colleagues (2005) in South India—suggest that relapse is more likely to occur among smokers than nonsmokers. Studies with longer follow-up are more likely than those with a short follow-up to include both TB cases from relapse and TB cases developing from exogenous reinfection; the former studies report a consistent twofold to threefold increased risk for recurrent TB associated with smoking. Temporality is inherent when investigating recurrent disease, but a dose-response relationship has not been reported. In the Korean cohort study by Jee and colleagues (2009), smoking increased the risk of recurrence by 30% in men but not significantly so in women, and a dose-response relationship with amount smoked was not observed. Overall, the evidence suggests a heightened risk for recurrent TB among smokers.
TB Mortality
The body of evidence for increased risk of mortality among TB patients who smoke has increased considerably over time. Smokers have a greater risk for TB mortality than nonsmokers because of a worsening of the natural history of TB in smokers. Additionally, the impairment of lung function caused by smoking, including the development of COPD, could increase the risk of death from respiratory failure. The several large case-control studies in India and China that have investigated deaths associated with smoking have consistently identified a strong association between smoking and TB mortality, with high attributable risks for smoking. Although most of the mortality evidence comes from studies in India and China, studies from Korea and South Africa provide similar results, suggesting that smoking increases risk for TB mortality across a range of settings in both low- and high-income countries. Two studies found a strong, positive dose-response relationship between number of cigarettes smoked per day and TB mortality (Liu et al. 1998; Lam et al. 2001), but the Korean study did not (Jee et al. 2009). The potential confounders included most commonly in these mortality analyses were age, gender, and education; three of the analyses controlled for alcohol use. Overall, the studies considered in this review consistently show an association between smoking and TB mortality, but the potential limitation among these studies of possible misclassification (in either direction) of TB deaths must be considered.
Conclusions
- The evidence is sufficient to infer a causal relationship between smoking and an increased risk of Mycobacterium tuberculosis disease.
- The evidence is sufficient to infer a causal relationship between smoking and mortality due to tuberculosis.
- The evidence is suggestive of a causal relationship between smoking and the risk of recurrent tuberculosis disease.
- The evidence is inadequate to infer the presence or absence of a causal relationship between active smoking and the risk of tuberculosis infection.
- The evidence is inadequate to infer the presence or absence of a causal relationship between exposure to secondhand smoke and the risk of tuberculosis infection.
- The evidence is inadequate to infer the presence or absence of a causal relationship between exposure to secondhand smoke and the risk of tuberculosis disease.
Implications
Tobacco smoking contributes to the burden of TB worldwide, potentially increasing risk for TB infection, disease, recurrence, and mortality. In 2008, TB killed 1.3 million people worldwide, and tobacco use may have accounted for more than one-half of those deaths in some regions (WHO 2010). Reduction of the consumption of tobacco at the population level will reduce TB infection, disease, and mortality. In short, tobacco control can contribute to TB control. There is currently a strong interest in determining the impact of smoking cessation for newly diagnosed TB patients, with the hypothesis that TB patients who smoke are at increased risk of failing treatment, sustaining a relapse, and/or dying. Cessation efforts among TB patients offer an opportunity to target a vulnerable population that, as a result of having the disease, may be motivated to quit. The effect of smoking on TB disease represents an important motivation for cessation that can be added to the numerous other risks of smoking. The most effective and efficient cessation strategies need to be determined, but they are likely to differ by the epidemiology and smoking behaviors of the targeted population.
Idiopathic Pulmonary Fibrosis
The diffuse parenchymal lung diseases are a heterogeneous group of disorders of known and unknown causes with distinct clinicopathologic characteristics. Among these diseases, the evidence on risk associated with cigarette smoking has been variable. For example, cigarette smoking is associated with an increased risk for IPF, desquamative interstitial pneumonia, and interstitial lung disease but with a decreased risk for sarcoidosis and hypersensitivity pneumonitis (Travis et al. 2002; USDHHS 2004). The focus of the present review is on IPF, the most common and also the most severe of the idiopathic interstitial pneumonias (Raghu et al. 2011). This topic was reviewed in the 2004 Surgeon General's report, The Health Consequences of Smoking; at that time, the evidence was determined to be “inadequate to infer the presence or absence of a causal relationship between active smoking and IPF” (USDHHS 2004).
The prevalence of IPF is much lower than that of COPD (USDHHS 2004). The prevalence of IPF is estimated at only 2 to 43 cases per 100,000 persons (Coultas et al. 1994; Raghu et al. 2006, 2011), but IPF is likely underdiagnosed as well. These prevalence estimates reflect not only the low incidence of the disease but also the high case-fatality rate.
Description of the Literature Review
A MEDLINE search was conducted to identify new studies on the biological mechanisms of IPF and observational studies published during 2005–2012 in order to update a review on this topic published in 2006 (Taskar and Coultas 2006). The search strategy included using the terms “smoking,” “IPF,” and “pulmonary fibrosis”; in addition, after the MEDLINE search was completed, the bibliographies of relevant articles were reviewed to identify literature not found by the search.
Biological Evidence
Although the biological mechanisms leading to pulmonary fibrosis continue to be an active area of investigation, available evidence suggests that both environmental and genetic factors contribute to the disorder (Garantziotis and Schwartz 2006; King et al. 2011; Chilosi et al. 2012; Faner et al. 2012; Macneal and Schwartz 2012). Additionally, smoking has numerous effects on the immune system that may be relevant (see Chapter 10). The process leading to pulmonary fibrosis is posited to start with alveolar epithelial injury from a number of possible inhaled toxicants, such as cigarette smoke or asbestos fibers. This epithelial micro-injury is followed by a complex process that involves multiple pathways of injury and repair (King et al. 2011). The process appears to be lengthy and leads to gradual fibrosis of the lung with stiffening and impaired gas exchange. Some of these same general mechanisms figure in the pathogenesis of COPD (Chilosi et al. 2012; Faner et al. 2012). Emerging evidence points to genetic factors that may be involved in the pathogenesis of IPF; these genes are relevant to host defenses, cell-cell adhesion, and DNA repair (King et al. 2011).
Epidemiologic Evidence
Epidemiologic evidence on the association between cigarette smoking and IPF has been reviewed previously (USDHHS 2004; Taskar and Coultas 2006) and is updated in this section. The sources of epidemiologic evidence have included the results from both descriptive and analytical studies. The descriptive studies have been comprised of a small case series of patients with IPF, disease registries (Coultas et al. 1994; Gribbin et al. 2006), and large health care administrative databases (Raghu et al. 2006). Of the analytical studies, 10 have been case-controls, 1 of familial idiopathic interstitial pneumonia, and 1 autopsy series (Table 7.10S).
Descriptive Studies
The prevalence, incidence, and mortality rates associated with IPF are consistently higher among men than women and increase markedly with advancing age (Coultas et al. 1994; Gribbin et al. 2006; Raghu et al. 2006). These two patterns are consistent with the higher frequency of smoking among men and with the mechanism of repeated micro-injury to the alveolar epithelium occurring with aging.
Subclinical interstitial lung abnormalities are also found among smokers (Lederer et al. 2009; Katzenstein et al. 2010; Washko et al. 2011; Doyle et al. 2012). Using high-resolution CT scanning, Washko and colleagues (2011) examined the lungs of 2,416 smokers 45 years of age or older who had accumulated at least 10 pack-years of smoking. Of these smokers, 8% had interstitial lung abnormalities associated with subpleural abnormalities. These abnormalities were associated with restrictive physiological impairment and impaired 6-minute walking distance (Doyle et al. 2012). Moreover, interstitial lung abnormalities were associated with older age, current smoking (OR = 1.67; 95% CI, 1.14–1.43), and greater exposure to tobacco smoke (OR = 1.08; 95% CI, 1.01–1.15) for each 10 pack-years of smoking (Washko et al. 2011). In addition, two studies of lung specimens obtained from lobectomies performed for lung cancer showed that interstitial fibrosis is common in smokers (Kawabata et al. 2008; Katzenstein et al. 2010). Airspace enlargement with fibrosis was present in 18% of moderate smokers in one of these studies (Kawabata et al. 2008), and in the other, Katzenstein and colleagues (2010) found interstitial fibrosis in more than 25% of the slides taken from the lobectomy specimens in 60% of smokers.
Analytical Studies
Of the 12 studies reported in Table 7.10S, 5 of them, all published 1990–2005, have been reviewed previously (Taskar and Coultas 2006). Of the 10 case-control studies presented in the table, 6 reported significant associations between ever or former smoking and IPF, with the OR (95% CI) ranging from 1.57 (1.01–2.33) to 5.4 (2.30–12.66). These 6 studies were conducted in five different countries: Japan, Mexico, Sweden, the United Kingdom, and the United States. The risk of current smoking was examined in only 1 study and was found to be nonsignificant (Miyake et al. 2005). The largest studies of environmental and occupational risk factors for IPF conducted in the United States (Baumgartner et al. 1997, 2000) and the United Kingdom (Hubbard et al. 1996) had nearly identical results, with an OR (95% CI) for smoking of 1.59 (1.1–2.4) and 1.57 (1.01–2.43), respectively. In 3 studies, there was evidence for a dose-response effect using pack-years of smoking, but the analyses were limited by small numbers in some categories of dose (Hubbard et al. 1996; Baumgartner et al. 1997; Miyake et al. 2005).
These epidemiologic studies had a number of limitations, including inconsistent adjustment for potential confounders, small samples, and missing data. Adjustment for potential confounders was reported in only 4 of these 10 studies. Moreover, when adjustment for confounders was performed, there was variation in the variables used, which included age, gender, region, family history of IPF, occupational and environmental exposures, and comorbid conditions. Small samples with limited statistical power may explain the lack of significant associations in 2 studies (Scott et al. 1990; Miyake et al. 2005). In the study conducted by Hubbard and colleagues (2008) of the association between IPF and cardiovascular disease, misclassification of missing data on the smoking status from 14% of cases and 16% of controls may have resulted in an underestimation of the risk of smoking.
In addition to the case-control design, the association between cigarette smoking and lung fibrosis has been examined using other study designs, including one on familial interstitial pneumonia (Steele et al. 2005) and in an autopsy sample (Schenker et al. 2009). Steele and colleagues (2005) identified 111 families with familial interstitial pneumonia, defined as two or more cases of probable or definite idiopathic interstitial pneumonia in individuals related within three degrees. Among these families, 309 individuals had definite or probable disease, with 80% classified as IPF, and 360 were unaffected. Overall, ever smoking was associated with an increased risk of familial interstitial pneumonia (OR = 3.6; 95% CI, 1.3–9.8 after adjustment for age and gender). Moreover, the average number of pack-years of smoking was significantly higher among affected family members than among those unaffected (16.6 vs. 6.9).
In their autopsy study, Schenker and colleagues (2009) examined 112 consecutive specimens from Hispanic males and described a range of pathologic abnormalities, including smoking-related small airways disease (54.5%) and interstitial fibrosis (19.1%). After adjustment for age and exposure to mineral dust, pathologic evidence of smoking-related small airways disease was strongly associated with interstitial fibrosis (OR = 5.03; 95% CI, 1.12–22.68).
Evidence Synthesis
Since the publication of the 2004 Surgeon General's report on the health consequences of smoking, there have been advances in our understanding of the biological mechanisms involved in the development of IPF and an increase in the number of epidemiologic investigations on this topic. Major issues considered in the interpretation of epidemiologic evidence include the potential for bias and confounding and a lack of statistical power. Although the case-control design, used for most of the studies reported in Table 7.10S, is subject to potential biases, the consistency of the findings, combined with the use of different control groups, including healthy controls, community controls, and clinic/hospital controls, suggests that bias alone is not a likely explanation for the findings. Control for potential confounding factors was not consistent among the epidemiologic investigations reviewed here, but in most studies when there was adjustment for possible confounders, significant associations between smoking and IPF remained. Additionally, given the paucity of confirmed risk factors for IPF, confounding seems an unlikely explanation for this association.
Plausibility is strong, and a causal association is coherent with the current understanding of the toxic biological effects of cigarette smoke, which causes cellular injury. This injury starts a cascade of genetically determined repair responses that may result in fibrosis among persons with genetically abnormal host defenses or repair mechanisms. Moreover, this sequence of biological events, starting with cellular injury and ending with fibrosis, provides support for the criterion of temporality. Looking across the epidemiologic studies, an association of IPF with smoking was found in different populations of patients and controls from different countries and over different periods of time. Although the overall strength of association between smoking and IPF is relatively small (OR = 1.6) (Taskar and Coultas 2006), misclassification of both diagnosis and exposure may have reduced the magnitude of the association. The limited evidence on an increasing strength of association with a greater number of pack-years of smoking supports the biologic-gradient criterion (i.e., dose-response).
Conclusion
- The evidence is suggestive but not sufficient to infer a causal relationship between cigarette smoking and idiopathic pulmonary fibrosis.
Implications
Further research is needed to address gaps in the current evidence, and to establish sufficient evidence for a causal relationship between cigarette smoking and IPF to be adequately assessed.
Impact of Smokefree Policies on Respiratory Outcomes
The evidence of the impact of smokefree policies within indoor environments on multiple health outcomes is reviewed in this and previous reports. In Chapter 8, “Cardiovascular Diseases” it was concluded that “The evidence is sufficient to infer a causal relationship between the implementation of a smokefree law or policy and a reduction in coronary events among populations under 65 years of age.” Previous Surgeon General's reports have concluded that exposure to secondhand smoke causes cough, phlegm, wheeze, and breathlessness among children; lower respiratory illnesses in infants and children; and the onset of wheeze illnesses and exacerbation of asthma among children and adults.
Despite the limited evidence for causal relationships between exposure to secondhand smoke and the risk for other acute and chronic respiratory diseases in adults, researchers have examined the consequences of the implementation of a smokefree law or policy for the number of hospital admissions for respiratory diseases. Eisner and colleagues (2005, 2009a,b) reported findings suggesting that chronic exposure to secondhand smoke increases the risk of COPD and is associated with exacerbation of respiratory symptoms. Evidence reviewed in the 2010 Surgeon General's report documented the mechanisms by which exposure to the complex chemical mixture of combustion compounds in tobacco smoke causes inflammation and oxidative stress. Flouris and Koutedakis (2011) reported results suggesting that exposure to secondhand smoke can produce adverse inflammatory and respiratory effects within 60 minutes of exposure and that these effects persist for at least 3 hours after the exposure. Earlier work by Flouris and colleagues (2009, 2010) provide additional evidence of the acute and short-term effects of exposure to secondhand smoke on lung functions and immune responses. Additionally, as previous Surgeon General's reports have reviewed (USDHHS 2006), the implementation of smokefree laws improves the respiratory health of bar and restaurant workers (Eisner et al. 1998; Menzies et al. 2006; Ayres et al. 2009; Wilson et al. 2012). Hence, there are biological and observational data suggesting that the implementation of smokefree legislation or policies could result in reduced respiratory symptoms and adverse respiratory events.
A recent review (Tan and Glantz 2012) and other recent papers (Vander Weg et al. 2012; Millett et al. 2013; Sims et al. 2013) found significant declines in hospitalizations for respiratory diseases, following the implementation of a smokefree law or policy. In a meta-analysis of 11 studies of smokefree laws covering workplaces, restaurants, and bars, Tan and Glantz (2012) reported a pooled RR of 0.76 (95% CI, 0.68–0.85) for hospital admissions for respiratory disease following the implementation of a smokefree law or policy, with the strongest effects found for asthma and lung infections (Figure 7.9). The 11 studies evaluate comprehensive smokefree laws covering workplaces, restaurants, and bars in countries (Ireland and Scotland), states (Arizona and Delaware), and the city of Toronto, Canada.

Figure 7.9
Forest plot for hospital admissions for respiratory disease following the implementation of a smokefree law or policy. Source: Adapted from Tan and Glantz 2012 with permission from Wolters Kluwer Health, © 2012. Note: CI = confidence interval; (more...)
Millett and colleagues (2013) found a significant decline in admissions for childhood asthma after the implementation of English smokefree legislation in July 2007 (adjusted risk ratio = 0.91; 95% CI, 0.89–0.93). The effect persisted over the first 3 years after implementation and was observed among children from different age, gender, and SES groups and among those residing in urban and rural locations in England. Sims and colleagues (2013) similarly evaluated the July 2007 English smokefree legislation and found that it was associated with a 4.9% (95% CI, 0.6%–9.0%) decline in emergency admissions for asthma in the adult population. Vander Weg and colleagues (2012) analyzed the patterns of hospital admissions for COPD, among Medicare beneficiaries 65 years of age and older, following the implementation of 938 smokefree laws passed by municipalities, counties, and states between 1991–2008. Adjusting for the trend of an increase in COPD admission rates, this analysis found a significant decline in COPD admission following smoking bans. However, in a smaller study in Rhode Island, Roberts and colleagues (2012) did not observe a decline in asthma admissions following the implementation of their statewide smokefree ordinance.
These results suggest that the relationship between acute and chronic exposure to secondhand smoke and respiratory disease outcomes merits further review and investigation. The lack of assessments of pre- and postexposure in almost all studies has been a limitation.
Evidence Summary
This chapter has reviewed updated evidence on COPD, a disease causally linked to smoking in the 1964 report. Mortality from COPD continues to rise, and smoking remains responsible for the majority of cases. For asthma, another obstructive lung disease, the evidence was found to be sufficient to infer that smoking is a cause of incident asthma in adolescents. The benefits of smoke-free policies have been shown previously for workers with asthma; evidence considered in this report points to a reduction in the admissions for respiratory diseases following implementation of a smokefree policy.
TB was once a leading cause of death in the United States. Now far less frequent in the United States, it remains prominent elsewhere and caused 1.4 million deaths worldwide in 2011 (WHO 2013). Evidence reported over the last decade is sufficient to lead to a conclusion that smoking increases the risk for TB and for dying from TB. For IPF, the evidence was suggestive of a causal association.
Implications
The evidence reviewed in this chapter reaffirms the potential for avoiding a substantial burden of respiratory disease through tobacco control. It reaffirms the possibility of avoiding much of the burden of COPD in the United States and reducing the occurrence of asthma in youth and young adults. Most significantly, the evidence considered here points to an opportunity to reduce the burden of disease and mortality from TB. Smoking has received little attention in relation to TB until recently. Smoking cessation should be integral to the management of the millions of people receiving treatment for this disease worldwide. Few etiological risk factors have been found for IPF; continued research on smoking and IPF is needed, given the potential to prevent another respiratory disease with a high fatality rate.
Chapter Conclusions
Chronic Obstructive Pulmonary Disease
- The evidence is sufficient to infer that smoking is the dominant cause of chronic obstructive pulmonary disease (COPD) in men and women in the United States. Smoking causes all elements of the COPD phenotype, including emphysema and damage to the airways of the lung.
- Chronic obstructive pulmonary disease (COPD) mortality has increased dramatically in men and women since the 1964 Surgeon General's report. The number of women dying from COPD now surpasses the number of men.
- The evidence is suggestive but not sufficient to infer that women are more susceptible to develop severe chronic obstructive pulmonary disease at younger ages.
- The evidence is sufficient to infer that severe α1-antitrypsin deficiency and cutis laxa are genetic causes of chronic obstructive pulmonary disease.
Asthma
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and incidence of asthma in adolescents.
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and exacerbation of asthma among children and adolescents.
- The evidence is suggestive but not sufficient to infer a causal relationship between active smoking and the incidence of asthma in adults.
- The evidence is sufficient to infer a causal relationship between active smoking and exacerbation of asthma in adults.
Tuberculosis
- The evidence is sufficient to infer a causal relationship between smoking and an increased risk of Mycobacterium tuberculosis disease.
- The evidence is sufficient to infer a causal relationship between smoking and mortality due to tuberculosis.
- The evidence is suggestive of a causal relationship between smoking and the risk of recurrent tuberculosis disease.
- The evidence is inadequate to infer the presence or absence of a causal relationship between active smoking and the risk of tuberculosis infection.
- The evidence is inadequate to infer the presence or absence of a causal relationship between exposure to secondhand smoke and the risk of tuberculosis infection.
- The evidence is inadequate to infer the presence or absence of a causal relationship between exposure to secondhand smoke and the risk of tuberculosis disease.
Idiopathic Pulmonary Fibrosis
- The evidence is suggestive but not sufficient to infer a causal relationship between cigarette smoking and idiopathic pulmonary fibrosis.
References
- Adelstein AM, Rimington J. Smoking and pulmonary tuberculosis: an analysis based on a study of volunteers for mass miniature radiography. Tubercle. 1967;48(3):219–26.
- Adib SM, Al-Takash H, Al-Hajj C. Tuberculosis in Lebanese jails: prevalence and risk factors. European Journal of Epidemiology. 1999;15(3):253–60. [PubMed: 10395055]
- Aggarwal AN, Chaudhry K, Chhabra SK, D'souza GA, Gupta D, Jindal SK, Katiyar SK, Kumar R, Shah B, Vijayan VK. Prevalence and risk factors for bronchial asthma in Indian adults: a multicentre study. Indian Journal of Chest Diseases and Allied Sciences. 2006;48(1):13–22. [PubMed: 16482947]
- Akinbami LJ, Liu X. Chronic Obstructive Pulmonary Disease Among Adults Aged 18 and Over in the United States, 1998–2009. Hyattsville (MD): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics; 2011. NCHS Data Brief 63.
- Alcaide J, Altet MN, Plans P, Parron I, Folguera L, Salto E, Dominguez A, Pardell H, Salleras L. Cigarette smoking as a risk factor for tuberculosis in young adults: a case-control study. Tubercle and Lung Disease. 1996;77(2):112–6. [PubMed: 8762844]
- Altet MN, Alcaide J, Plans P, Taberner JL, Salto E, Folguera LI, Salleras L. Passive smoking and risk of pulmonary tuberculosis in children immediately following infection. A case-control study. Tubercle and Lung Disease. 1996;77(6):537–44. [PubMed: 9039447]
- Altet-Gomez MN, Alcaide J, Godoy P, Romero MA, Hernandez del Rey I. Clinical and epidemiological aspects of smoking and tuberculosis: a study of 13,038 cases. International Journal of Tuberculosis and Lung Disease. 2005;9(4):430–6. [PubMed: 15830749]
- American Thoracic Society. Standards for the diagnosis and care of patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1995;152(5 Pt 2):S77–S121. [PubMed: 7582322]
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. American Journal of Respiratory and Critical Care Medicine. 2002;165(2):277–304. [PubMed: 11790668]
- Ammous Z, Hackett NR, Butler MW, Raman T, Dolgalev I, O'Connor TP, Harvey BG, Crystal RG. Variability in small airway epithelial gene expression among normal smokers. Chest. 2008;133(6):1344–53. [PMC free article: PMC3632367] [PubMed: 18339782]
- Anderson RH, Sy FS, Thompson S, Addy C. Cigarette smoking and tuberculin skin test conversion among incarcerated adults. American Journal of Preventive Medicine. 1997;13(3):175–81. [PubMed: 9181204]
- Andre E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, et al. Cigarette smoke-induced neurogenic inflammation is mediated by alpha, beta-unsaturated aldehydes and the TRPA1 receptor in rodents. Journal of Clinical Investigation. 2008;118(7):2574–82. [PMC free article: PMC2430498] [PubMed: 18568077]
- Annesi-Maesano I, Oryszczyn MP, Raherison C, Kopferschmitt C, Pauli G, Taytard A, Tunon de LM, Vervloet D, Charpin D. Increased prevalence of asthma and allied diseases among active adolescent tobacco smokers after controlling for passive smoking exposure. A cause for concern? Clinical and Experimental Allergy. 2004;34(7):1017–23. [PubMed: 15248844]
- Apostol GG, Jacobs DR Jr, Tsai AW, Crow RS, Williams OD, Townsend MC, Beckett WS. Early life factors contribute to the decrease in lung function between ages 18 and 40: the Coronary Artery Risk Development in Young Adults study. American Journal of Respiratory and Critical Care Medicine. 2002;166(2):166–72. [PubMed: 12119228]
- Arcavi L, Benowitz NL. Cigarette smoking and infection. Archives of Internal Medicine. 2004;164(20):2206–16. [PubMed: 15534156]
- Ariyothai N, Podhipak A, Akarasewi P, Tornee S, Smithtikarn S, Thongprathum P. Cigarette smoking and its relation to pulmonary tuberculosis in adults. Southeast Asian Journal of Tropical Medicine and Public Health. 2004;35(1):219–27. [PubMed: 15272772]
- Ashraf H, Lo P, Shaker SB, de Bruijne M, Dirksen A, Tonnesen P, Dahlback M, Pedersen JH. Short-term effect of changes in smoking behaviour on emphysema quantification by CT. Thorax. 2011;66(1):55–60. [PubMed: 20978026]
- Austin JB, Selvaraj S, Godden D, Russell G. Deprivation, smoking, and quality of life in asthma. Archives of Disease in Childhood. 2005;90(3):253–7. [PMC free article: PMC1720293] [PubMed: 15723909]
- Avila L, Soto-Martinez ME, Soto-Quiros ME, Celedon JC. Asthma, current wheezing, and tobacco use among adolescents and young adults in Costa Rica. Journal of Asthma. 2005;42(7):543–7. [PubMed: 16169786]
- Ayres JG, Semple S, MacCalman L, Dempsey S, Hilton S, Hurley JF, Miller BG, Naji A, Petticrew M. Bar workers' health and environmental tobacco smoke exposure (BHETSE): symptomatic improvement in bar staff following smoke-free legislation in Scotland. Occupational and Environmental Medicine. 2009;66(5):339–46. [PubMed: 19208693]
- Bacopoulou F, Veltsista A, Vassi I, Gika A, Lekea V, Priftis K, Bakoula C. Can we be optimistic about asthma in childhood? A Greek cohort study. Journal of Asthma. 2009;46(2):171–4. [PubMed: 19253125]
- Baena-Cagnani CE, Gomez RM, Baena-Cagnani R, Canonica GW. Impact of environmental tobacco smoke and active tobacco smoking on the development and outcomes of asthma and rhinitis. Current Opinion in Allergy and Clinical Immunology. 2009;9(2):136–40. [PubMed: 19307883]
- Bankier AA, Madani A, Gevenois PA. CT quantification of pulmonary emphysema: assessment of lung structure and function. Critical Reviews in Computed Tomography. 2002;43(6):399–417. [PubMed: 12521149]
- Barr RG, Bluemke DA, Ahmed FS, Carr JJ, Enright PL, Hoffman EA, Jiang R, Kawut SM, Kronmal RA, Lima JA, et al. Percent emphysema, airflow obstruction, and impaired left ventricular filling. New England Journal of Medicine. 2010;362(3):217–27. [PMC free article: PMC2887729] [PubMed: 20089972]
- Bates MN, Khalakdina A, Pai M, Chang L, Lessa F, Smith KR. Risk of tuberculosis from exposure to tobacco smoke: a systematic review and meta-analysis. Archives of Internal Medicine. 2007;167(4):335–42. [PubMed: 17325294]
- Baumgartner KB, Samet JM, Coultas DB, Stidley CA, Hunt WC, Colby TV, Waldron JA. Occupational and environmental risk factors for idiopathic pulmonary fibrosis: a multicenter case-control study. Collaborating centers. American Journal of Epidemiology. 2000;152(4):307–15. [PubMed: 10968375]
- Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 1997;155(1):242–8. [PubMed: 9001319]
- Becklake MR, Lalloo U. The ‘healthy smoker’: a phenomenon of health selection? Respiration. 1990;57(3):137–44. [PubMed: 2274712]
- Bellia V, Pedone C, Catalano F, Zito A, Davi E, Palange S, Forastiere F, Incalzi RA. Asthma in the elderly: mortality rate and associated risk factors for mortality. Chest. 2007;132(4):1175–82. [PubMed: 17890479]
- Bessac BF, Sivula M, von Hehn CA, Escalera J, Cohn L, Jordt SE. TRPA1 is a major oxidant sensor in murine airway sensory neurons. Journal of Clinical Investigation. 2008;118(5):1899–910. [PMC free article: PMC2289796] [PubMed: 18398506]
- Bhattacharya S, Srisuma S, DeMeo DL, Shapiro SD, Bueno R, Silverman EK, Reilly JJ, Mariani TJ. Molecular biomarkers for quantitative and discrete COPD phenotypes. American Journal of Respiratory Cell and Molecular Biology. 2009;40(3):359–67. [PMC free article: PMC2645534] [PubMed: 18849563]
- Boulet LP, FitzGerald JM, McIvor RA, Zimmerman S, Chapman KR. Influence of current or former smoking on asthma management and control. Canadian Respiratory Journal. 2008;15(5):275–9. [PMC free article: PMC2679551] [PubMed: 18716691]
- Boulet LP, Lemiere C, Archambault F, Carrier G, Descary MC, Deschesnes F. Smoking and asthma: clinical and radiologic features, lung function, and airway inflammation. Chest. 2006;129(3):661–8. [PubMed: 16537865]
- Broide D. New perspectives on mechanisms underlying chronic allergic inflammation and asthma in 2007. Journal of Allergy and Clinical Immunology. 2008;122(3):475–80. [PubMed: 18694589]
- Brown KE, Campbell AH. Tobacco, alcohol and tuberculosis. British Journal of Diseases of the Chest. 1961;55(3):150–8.
- Buskin SE, Gale JL, Weiss NS, Nolan CM. Tuberculosis risk factors in adults in King County, Washington, 1988 through 1990. American Journal of Public Health. 1994;84(11):1750–6. [PMC free article: PMC1615189] [PubMed: 7977912]
- Butland BK, Strachan DP. Asthma onset and relapse in adult life: the British 1958 birth cohort study. Annals of Allergy, Asthma, and Immunology. 2007;98(4):337–43. [PubMed: 17458429]
- Caceres AI, Brackmann M, Elia MD, Bessac BF, del Camino D, D'Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, et al. A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(22):9099–104. [PMC free article: PMC2684498] [PubMed: 19458046]
- Cantral DE, Sisson JH, Veys T, Rennard SI, Spurzem JR. Effects of cigarette smoke extract on bovine bronchial epithelial cell attachment and migration. American Journal of Physiology. 1995;268(5 Pt 1):L723–L728. [PubMed: 7762674]
- Carnevali S, Nakamura Y, Mio T, Liu X, Takigawa K, Romberger DJ, Spurzem JR, Rennard SI. Cigarette smoke extract inhibits fibroblast-mediated collagen gel contraction. American Journal of Physiology. 1998;274(4 Pt 1):L591–L598. [PubMed: 9575878]
- Carter S, Percival T, Paterson J, Williams M. Maternal smoking: risks related to maternal asthma and reduced birth weight in a Pacific Island birth cohort in New Zealand. New Zealand Medical Journal. 2006;119(1238):U2081. [PubMed: 16868578]
- Castaldi PJ, Cho MH, Cohn M, Langerman F, Moran S, Tarragona N, Moukhachen H, Venugopal R, Hasimja D, Kao E, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Human Molecular Genetics. 2010;19(3):526–34. [PMC free article: PMC2798725] [PubMed: 19933216]
- Centers for Disease Control and Prevention. Prevalence of tuberculous infection among U.S. residents of Cuban descent—Dade County, Florida, 1982–84. Morbidity and Mortality Weekly Report. 1992;41(5):74–5. 81. [PubMed: 1732714]
- Centers for Disease Control and Prevention. Chronic obstructive pulmonary disease among adults—United States, 2011. Morbidity and Mortality Weekly Report. 2012;61(46):938–43. [PubMed: 23169314]
- Centers for Disease Control and Prevention. Current cigarette smoking among adults—United States, 2011. Morbidity and Mortality Weekly Report. 2012;61(44):889–94. [PubMed: 23134971]
- Chalmers GW, Macleod KJ, Little SA, Thomson LJ, McSharry CP, Thomson NC. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax. 2002;57(3):226–30. [PMC free article: PMC1746270] [PubMed: 11867826]
- Chang KC, Leung CC, Tam CM. Tuberculosis risk factors in a silicotic cohort in Hong Kong. International Journal of Tuberculosis and Lung Disease. 2001;5(2):177–84. [PubMed: 11258512]
- Chang KC, Leung CC, Tam CM. Risk factors for defaulting from anti-tuberculosis treatment under directly observed treatment in Hong Kong. International Journal of Tuberculosis and Lung Disease. 2004;8(12):1492–8. [PubMed: 15636497]
- Chan-Yeung M, Zhan LX, Tu DH, Li B, He GX, Kauppinen R, Nieminen M, Enarson DA. The prevalence of asthma and asthma-like symptoms among adults in rural Beijing, China. European Respiratory Journal. 2002;19(5):853–8. [PubMed: 12030724]
- Chapman JS, Dyerly MD. Social and other factors in intrafamilial transmission of tuberculosis. American Review of Respiratory Disease. 1964;90:48–60. [PubMed: 14178626]
- Chaudhuri R, Livingston E, McMahon AD, Lafferty J, Fraser I, Spears M, McSharry CP, Thomson NC. Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. American Journal of Respiratory and Critical Care Medicine. 2006;174(2):127–33. [PubMed: 16645173]
- Chaudhuri R, Livingston E, McMahon AD, Thomson L, Borland W, Thomson NC. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. American Journal of Respiratory and Critical Care Medicine. 2003;168(11):1308–11. [PubMed: 12893649]
- Chaudhuri R, McSharry C, McCoard A, Livingston E, Hothersall E, Spears M, Lafferty J, Thomson NC. Role of symptoms and lung function in determining asthma control in smokers with asthma. Allergy. 2008;63(1):132–5. [PubMed: 18053022]
- Chiang CY, Slama K, Enarson DA. Associations between tobacco and tuberculosis. International Journal of Tuberculosis and Lung Disease. 2007;11(3):258–62. [PubMed: 17352089]
- Chilosi M, Poletti V, Rossi A. The pathogenesis of COPD and IPF: distinct horns of the same devil? Respiratory Research. 2012;13:3. [PMC free article: PMC3282644] [PubMed: 22235752]
- Cho MH, Boutaoui N, Klanderman BJ, Sylvia JS, Ziniti JP, Hersh CP, DeMeo DL, Hunninghake GM, Litonjua A, Sparrow D, et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nature Genetics. 2010;42:200–2. [PMC free article: PMC2828499] [PubMed: 20173748]
- Cho MH, Castaldi PJ, Wan ES, Siedlinski M, Hersh CP, DeMeo DL, Himes BE, Sylvia JS, Klanderman BJ, Ziniti JP, et al. A genome-wide association study of COPD identifies a susceptibility locus on chromosome 19q13. Human Molecular Genetics. 2012;21(4):947–57. [PMC free article: PMC3298111] [PubMed: 22080838]
- Cho MH, Ciulla DM, Klanderman BJ, Hersh CP, Litonjua AA, Sparrow D, Raby BA, Silverman EK. Analysis of exonic elastin variants in severe, early-onset chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology. 2009;40(6):751–5. [PMC free article: PMC2689920] [PubMed: 19029017]
- ClinicalTrials.gov. Examining the Genetic Factors That May Cause Chronic Obstructive Pulmonary Disease (COPD) (COPDGene). 2013. [February 11, 2013]. < http:
//clinicaltrials .gov/show/NCT00608764>. - Comstock GW. Frost revisited: the modern epidemiology of tuberculosis. American Journal of Epidemiology. 1975;101(5):363–82. [PubMed: 1093397]
- Comstock GW, Livesay VT, Woolpert SF. The prognosis of a positive tuberculin reaction in childhood and adolescence. American Journal of Epidemiology. 1974;99(2):131–8. [PubMed: 4810628]
- Corbett E, Glaisyer H, Chan C, Madden B, Khaghani A, Yacoub M. Congenital cutis laxa with a dominant inheritance and early onset emphysema. Thorax. 1994;49(8):836–7. [PMC free article: PMC475135] [PubMed: 8091333]
- Coultas DB, Zumwalt RE, Black WC, Sobonya RE. The epidemiology of interstitial lung diseases. American Journal of Respiratory and Critical Care Medicine. 1994;150(4):967–72. [PubMed: 7921471]
- Coxson HO. Quantitative computed tomography assessment of airway wall dimensions: current status and potential applications for phenotyping chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society. 2008;5(9):940–5. [PMC free article: PMC2720108] [PubMed: 19056721]
- Crampin AC, Glynn JR, Floyd S, Malema SS, Mwinuka VK, Ngwira BM, Mwaungulu FD, Warndorff DK, Fine PE. Tuberculosis and gender: exploring the patterns in a case control study in Malawi. International Journal of Tuberculosis and Lung Disease. 2004;8(2):194–203. [PubMed: 15139448]
- Dance A. Health impact: breathless. Nature. 2012;489(7417):S2–S3. [PubMed: 23013713]
- d'Arc Lyra BJ, de Fátima Pessoa Militão de Albuquerque M, de Alencar Ximenes RA, Rodrigues LC. Smoking increases the risk of relapse after successful tuberculosis treatment. International Journal of Epidemiology. 2008;37(4):841–51. [PMC free article: PMC2483312] [PubMed: 18556729]
- Davies PD, Yew WW, Ganguly D, Davidow AL, Reichman LB, Dheda K, Rook GA. Smoking and tuberculosis: the epidemiological association and immunopathogenesis. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2006;100(4):291–8. [PubMed: 16325875]
- de Marco R, Marcon A, Jarvis D, Accordini S, Almar E, Bugiani M, Carolei A, Cazzoletti L, Corsico A, Gislason D, et al. Prognostic factors of asthma severity: a 9-year international prospective cohort study. Journal of Allergy and Clinical Immunology. 2006;117(6):1249–56. [PubMed: 16750983]
- de Vries MP, van den Bemt L, Lince S, Muris JW, Thoonen BP, van Schayck CP. Factors associated with asthma control. Journal of Asthma. 2005;42(8):659–65. [PubMed: 16266957]
- Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet. 2013;1(1):73–83. [PubMed: 24321806]
- DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A, Bueno R, Pillai SG, Lomas DA, Sparrow D, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. American Journal of Human Genetics. 2009;85(4):493–502. [PMC free article: PMC2756547] [PubMed: 19800047]
- DeMeo DL, Mariani TJ, Lange C, Srisuma S, Litonjua AA, Celedon JC, Lake SL, Reilly JJ, Chapman HA, Mecham BH, et al. The SERPINE2 gene is associated with chronic obstructive pulmonary disease. American Journal of Human Genetics. 2006;78(2):253–64. [PMC free article: PMC1380249] [PubMed: 16358219]
- den Boon S, van Lill SW, Borgdorff MW, Verver S, Bateman ED, Lombard CJ, Enarson DA, Beyers N. Association between smoking and tuberculosis infection: a population survey in a high tuberculosis incidence area. Thorax. 2005;60(7):555–7. [PMC free article: PMC1747449] [PubMed: 15994262]
- den Boon S, Verver S, Marais BJ, Enarson DA, Lombard CJ, Bateman ED, Irusen E, Jithoo A, Gie RP, Borgdorff MW, et al. Association between passive smoking and infection with Mycobacterium tuberculosis in children. Pediatrics. 2007;119(4):734–9. [PubMed: 17403844]
- Dickens JA, Miller BE, Edwards LD, Silverman EK, Lomas DA, Tal-Singer R. COPD association and repeatability of blood biomarkers in the ECLIPSE cohort. Respiratory Research. 2011;12:146. [PMC free article: PMC3247194] [PubMed: 22054035]
- Diette GB, Krishnan JA, Dominici F, Haponik E, Skinner EA, Steinwachs D, Wu AW. Asthma in older patients: factors associated with hospitalization. Archives of Internal Medicine. 2002;162(10):1123–32. [PubMed: 12020182]
- Dirksen A. Monitoring the progress of emphysema by repeat computed tomography scans with focus on noise reduction. Proceedings of the American Thoracic Society. 2008;5(9):925–8. [PubMed: 19056718]
- Doll R. Risk from tobacco and potentials for health gain. International Journal of Tuberculosis and Lung Disease. 1999;3(2):90–9. [PubMed: 10091873]
- Doll R, Hill AB. The mortality of doctors in relation to their smoking habits; a preliminary report. British Medical Journal. 1954;1(4877):1451–5. [PMC free article: PMC2085438] [PubMed: 13160495]
- Doll R, Hill AB. Lung cancer and other causes of death in relation to smoking; a second report on the mortality of British doctors. British Medical Journal. 1956;2(5001):1071–81. [PMC free article: PMC2035864] [PubMed: 13364389]
- Doll R, Hill AB. Mortality in relation to smoking: ten years' observations of British doctors. British Medical Journal. 1964;1(5396):1460–7. [PMC free article: PMC1814697] [PubMed: 14132080]
- Doll R, Peto R, Wheatley K, Gray R, Sutherland I. Mortality in relation to smoking: 40 years' observations on male British doctors. British Medical Journal. 1994;309(6959):901–11. [PMC free article: PMC2541142] [PubMed: 7755693]
- Dove MS, Dockery DW, Mittleman MA, Schwartz J, Sullivan EM, Keithly L, Land T. The impact of Massachusetts' smoke-free workplace laws on acute myocardial infarction deaths. American Journal of Public Health. 2010;100(11):2206–12. [PMC free article: PMC2951939] [PubMed: 20864706]
- Doyle TJ, Washko GR, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, Divo MJ, Celli BR, Sciurba FC, Silverman EK, et al. Interstitial lung abnormalities and reduced exercise capacity. American Journal of Respiratory and Critical Care Medicine. 2012;185(7):756–62. [PMC free article: PMC3326424] [PubMed: 22268134]
- Eagan TM, Bakke PS, Eide GE, Gulsvik A. Incidence of asthma and respiratory symptoms by sex, age and smoking in a community study. European Respiratory Journal. 2002;19(4):599–605. [PubMed: 11998986]
- Eda S, Kubo K, Fujimoto K, Matsuzawa Y, Sekiguchi M, Sakai F. The relations between expiratory chest CT using helical CT and pulmonary function tests in emphysema. American Journal of Respiratory and Critical Care Medicine. 1997;155(4):1290–4. [PubMed: 9105069]
- Eisner MD, Balmes J, Katz PP, Trupin L, Yelin EH, Blanc PD. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environmental Health. 2005;4(1):7. [PMC free article: PMC1145187] [PubMed: 15890079]
- Eisner MD, Iribarren C. The influence of cigarette smoking on adult asthma outcomes. Nicotine & Tobacco Research. 2007;9(1):53–6. [PubMed: 17365736]
- Eisner MD, Iribarren C, Yelin EH, Sidney S, Katz PP, Sanchez G, Blanc PD. The impact of SHS exposure on health status and exacerbations among patients with COPD. International Journal of Chronic Obstructive Pulmonary Disease. 2009;4:169–76. [PMC free article: PMC2685143] [PubMed: 19516915]
- Eisner MD, Jacob P 3rd, Benowitz NL, Balmes J, Blanc PD. Longer term exposure to secondhand smoke and health outcomes in COPD: impact of urine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Nicotine & Tobacco Research. 2009;11(8):945–53. [PMC free article: PMC2711983] [PubMed: 19587064]
- Eisner MD, Smith AK, Blanc PD. Bartenders' respiratory health after establishment of smoke-free bars and taverns. JAMA: the Journal of the American Medical Association. 1998;280(22):1909–14. [PubMed: 9851475]
- Eisner MD, Yelin EH, Katz PP, Lactao G, Iribarren C, Blanc PD. Risk factors for work disability in severe adult asthma. American Journal of Medicine. 2006;119(10):884–91. [PubMed: 17000221]
- Enomoto T, Usuki J, Azuma A, Nakagawa T, Kudoh S. Diabetes mellitus may increase risk for idiopathic pulmonary fibrosis. Chest. 2003;123(6):2007–11. [PubMed: 12796182]
- Eriksson S. Studies in alpha 1-antitrypsin deficiency. Acta Medica Scandinavica. Supplementum. 1965;432:1–85. [PubMed: 4160491]
- Faner R, Rojas M, Macnee W, Agusti A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2012;186(4):306–13. [PubMed: 22582162]
- Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, Zhang Y, Sciurba FC, Duncan SR. Autoantibodies in patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2008;177(2):156–63. [PMC free article: PMC2204079] [PubMed: 17975205]
- Feinleib M, Rosenberg HM, Collins JG, Delozier JE, Pokras R, Chevarley FM. Trends in COPD morbidity and mortality in the United States. American Review of Respiratory Disease. 1989;140(3 Pt 2):S9–S18. [PubMed: 2782766]
- Fernandez-Benitez M, Anton J, Guillen GF. Risk factors associated to the prevalence of asthma in adolescence. Allergologia Immunopathologia (Madr). 2007;35(5):193–6. [PubMed: 17923073]
- Ferris BG Jr., Anderson DO. The prevalence of chronic respiratory disease in a New Hampshire town. American Review of Respiratory Disease. 1962;86:165–77. [PubMed: 13892585]
- Fletcher CM. Chronic bronchitis. Its prevalence, nature, and pathogenesis. American Review of Respiratory Disease. 1959;80:483–94. [PubMed: 13823476]
- Fletcher C, Peto R. The natural history of chronic airflow obstruction. British Medical Journal. 1977;1(6077):1645–8. [PMC free article: PMC1607732] [PubMed: 871704]
- Fletcher CM, Gilson JG, Hugh-Jones P, Scadding JG. Terminology, definitions, and classification of chronic pulmonary emphysema and related conditions. A report of the conclusions of a Ciba guest symposium. Thorax. 1959;14:286–99.
- Flouris AD, Koutedakis Y. Immediate and short-term consequences of secondhand smoke exposure on the respiratory system. Current Opinion in Pulmonary Medicine. 2011;17(2):110–5. [PubMed: 21178628]
- Flouris AD, Metsios GS, Carrillo AE, Jamurtas AZ, Gourgoulianis K, Kiropoulos T, Tzatzarakis MN, Tsatsakis AM, Koutedakis Y. Acute and short-term effects of secondhand smoke on lung function and cytokine production. American Journal of Respiratory and Critical Care Medicine. 2009;179(11):1029–33. [PubMed: 19264972]
- Flouris AD, Metsios GS, Jamurtas AZ, Koutedakis Y. Cardiorespiratory and immune response to physical activity following exposure to a typical smoking environment. Heart. 2010;96(11):860–4. [PubMed: 20478865]
- Ford ES, Croft JB, Mannino DM, Wheaton AG, Zhang X, Giles WH. COPD surveillance—United States, 1999–2011. Chest. 2013;144(1):284–305. [PMC free article: PMC3707177] [PubMed: 23619732]
- Ford ES, Mannino DM, Redd SC, Moriarty DG, Mokdad AH. Determinants of quality of life among people with asthma: findings from the Behavioral Risk Factor Surveillance System. Journal of Asthma. 2004;41(3):327–36. [PubMed: 15260466]
- Ford ES, Mannino DM, Zhao G, Li C, Croft JB. Changes in mortality among U.S. adults with COPD in two national cohorts recruited from 1971–1975 and 1988–1994. Chest. 2012;141(1):101–10. [PubMed: 21700689]
- Foreman MG, Zhang L, Murphy J, Hansel NN, Make B, Hokanson JE, Washko G, Regan EA, Crapo JD, Silver-man EK, et al. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPDGene Study. American Journal of Respiratory and Critical Care Medicine. 2011;184(4):414–20. [PMC free article: PMC3175544] [PubMed: 21562134]
- Frank P, Morris J, Hazell M, Linehan M, Frank T. Smoking, respiratory symptoms and likely asthma in young people: evidence from postal questionnaire surveys in the Wythenshawe Community Asthma Project (WYCAP). BMC Pulmonary Medicine. 2006;6:10. [PMC free article: PMC1489948] [PubMed: 16716223]
- Gaga M, Papageorgiou N, Yiourgioti G, Karydi P, Liapikou A, Bitsakou H, Zervas E, Koulouris NG, Holgate ST. Risk factors and characteristics associated with severe and difficult to treat asthma phenotype: an analysis of the ENFUMOSA group of patients based on the ECRHS questionnaire. Clinical and Experimental Allergy. 2005;35(7):954–9. [PubMed: 16008684]
- Gajalakshmi V, Peto R. Smoking, drinking and incident tuberculosis in rural India: population-based case-control study. International Journal of Epidemiology. 2009;38(4):1018–25. [PubMed: 19498083]
- Gajalakshmi V, Peto R, Kanaka TS, Jha P. Smoking and mortality from tuberculosis and other diseases in India: retrospective study of 43000 adult male deaths and 35000 controls. Lancet. 2003;362(9383):507–15. [PubMed: 12932381]
- Gan WQ, Man SF, Postma DS, Camp P, Sin DD. Female smokers beyond the perimenopausal period are at increased risk of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Respiratory Research. 2006;7:52. [PMC free article: PMC1435894] [PubMed: 16571126]
- Garantziotis S, Schwartz DA. Host-environment interactions in pulmonary fibrosis. Seminars in Respiratory and Critical Care Medicine. 2006;27(6):574–80. [PubMed: 17195134]
- Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, Navarro C, Perez-Padilla R, Vargas MH, Loyd JE, Selman M. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respiratory Medicine. 2011;105(12):1902–7. [PubMed: 21917441]
- Genuneit J, Weinmayr G, Radon K, Dressel H, Windstetter D, Rzehak P, Vogelberg C, Leupold W, Nowak D, von Mutius E, et al. Smoking and the incidence of asthma during adolescence: results of a large cohort study in Germany. Thorax. 2006;61(7):572–8. [PMC free article: PMC2104663] [PubMed: 16537668]
- Gevenois PA, de Maertelaer V, De Vuyst P, Zanen J, Yernault JC. Comparison of computed density and macroscopic morphometry in pulmonary emphysema. American Journal of Respiratory and Critical Care Medicine. 1995;152(2):653–7. [PubMed: 7633722]
- Gevenois PA, De Vuyst P, de Maertelaer V, Zanen J, Jacobovitz D, Cosio MG, Yernault JC. Comparison of computed density and microscopic morphometry in pulmonary emphysema. American Journal of Respiratory and Critical Care Medicine. 1996;154:187–92. [PubMed: 8680679]
- Gilliland FD, Islam T, Berhane K, Gauderman WJ, McConnell R, Avol E, Peters JM. Regular smoking and asthma incidence in adolescents. American Journal of Respiratory and Critical Care Medicine. 2006;174(10):1094–100. [PMC free article: PMC2648110] [PubMed: 16973983]
- Global Initiative for Chronic Obstructive Lung Disease. Global Strategy for the Diagnosis, Management and Prevention of COPD. Feb, 2013. [October 29, 2013]. < http://www
.goldcopd.org /uploads/users/files /GOLD_Report_2013_Feb20.pdf>. - Global Initiative for Chronic Obstructive Pulmonary Disease. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease. 2011. [August 6, 2013]. < http://www
.goldcopd.org /uploads/users/files /GOLD_Report_2011Dec30.pdf>. [PubMed: 17507545] - Golpon HA, Coldren CD, Zamora MR, Cosgrove GP, Moore MD, Tuder RM, Geraci MW, Voelkel NF. Emphysema lung tissue gene expression profiling. American Journal of Respiratory Cell and Molecular Biology. 2004;31(6):595–600. [PubMed: 15284076]
- Golub JE, Coberly J, Chaisson RE. Tuberculosis. In: Nelson KE, Masters Williams C, editors. Infectious Disease Epidemiology: Theory and Practice. 3rd ed. Burlington (MA): Jones and Bartlett Learning; 2013.
- Gomez M, Vollmer WM, Caceres ME, Jossen R, Baena-Cagnani CE. Adolescent smokers are at greater risk for current asthma and rhinitis. International Journal of Tuberculosis and Lung Disease. 2009;13(8):1023–8. [PubMed: 19723384]
- Gorbunova V, Jacobs SS, Lo P, Dirksen A, Nielsen M, Bab-Hadiashar A, de Bruijne M. Early detection of emphysema progression. Medical image computing and computer-assisted intervention: MICCAI International Conference on Medical Image Computing and Computer-Assisted Intervention. 2010;13(Pt 2):193–200. [PubMed: 20879315]
- Gribbin J, Hubbard RB, Le Jeune I, Smith CJ, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61(11):980–5. [PMC free article: PMC2121155] [PubMed: 16844727]
- Gross P, Pfitzer EA, Tolker E, Babyak MA, Kaschak M. Experimental Emphysema: Its Production with Papain in Normal and Silicotic Rats. Archives of Environmental Health. 1965;11:50–8. [PubMed: 14312390]
- Grumelli S, Corry D, Song L, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis DE, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Medicine. 2004;1(1):e8. [PMC free article: PMC523885] [PubMed: 15526056]
- Gupta BN, Mathur N, Mahendra PN, Srivastava AK, Swaroop V. Smoking increases risk of pulmonary tuberculosis. Energy Environment Monitor. 1997;13(1):61–7.
- Gupta PC, Pednekar MS, Parkin DM, Sankaranarayanan R. Tobacco associated mortality in Mumbai (Bombay) India. Results of the Bombay Cohort Study. International Journal of Epidemiology. 2005;34(6):1395–402. [PubMed: 16249218]
- Gupta RK, Gupta A, Jamwal DS, Suri SP. A socio-epidemiological study of tuberculosis in a rural area. JK Science. 2002;4(3):119–22.
- Gustafson T, Dahlman-Hoglund A, Nilsson K, Strom K, Tornling G, Toren K. Occupational exposure and severe pulmonary fibrosis. Respiratory Medicine. 2007;101(10):2207–12. [PubMed: 17628464]
- Gwynn RC. Risk factors for asthma in US adults: results from the 2000 Behavioral Risk Factor Surveillance System. Journal of Asthma. 2004;41(1):91–8. [PubMed: 15046383]
- Han MK, Kazerooni EA, Lynch DA, Liu LX, Murray S, Curtis JL, Criner GJ, Kim V, Bowler RP, Hanania NA, et al. Chronic obstructive pulmonary disease exacerbations in the COPDGene Study: associated radiologic phenotypes. Radiology. 2011;261(1):274–82. [PMC free article: PMC3184233] [PubMed: 21788524]
- Hancock DB, Artigas MS, Gharib SA, Henry A, Manichaikul A, Ramasamy A, Loth DW, Imboden M, Koch B, McArdle WL, et al. Genome-wide joint meta-analysis of SNP and SNP-by-smoking interaction identifies novel loci for pulmonary function. PLoS genetics. 2012;8(12):e1003098. [PMC free article: PMC3527213] [PubMed: 23284291]
- Hancock DB, Eijgelsheim M, Wilk JB, Gharib SA, Loehr LR, Marciante KD, Franceschini N, van Durme YM, Chen TH, Barr RG, et al. Meta-analyses of genomewide association studies identify multiple loci associated with pulmonary function. Nature Genetics. 2010;42(1):45–52. [PMC free article: PMC2832852] [PubMed: 20010835]
- Hansel NN, Ruczinski I, Rafaels N, Sin DD, Daley D, Malinina A, Huang L, Sandford A, Murray T, Kim Y, et al. Genome-wide study identifies two loci associated with lung function decline in mild to moderate COPD. Human Genetics. 2013;132(1):79–90. [PMC free article: PMC3536920] [PubMed: 22986903]
- Harling G, Ehrlich R, Myer L. The social epidemiology of tuberculosis in South Africa: a multilevel analysis. Social Science and Medicine. 2008;66(2):492–505. [PubMed: 17920743]
- Hasegawa M, Nasuhara Y, Onodera Y, Makita H, Nagai K, Fuke S, Ito Y, Betsuyaku T, Nishimura M. Airflow limitation and airway dimensions in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2006;173(12):1309–15. [PubMed: 16556695]
- Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–4. [PubMed: 9302297]
- Hedlund U, Eriksson K, Ronmark E. Socio-economic status is related to incidence of asthma and respiratory symptoms in adults. European Respiratory Journal. 2006;28(2):303–10. [PubMed: 16540503]
- Herman PM, Walsh ME. Hospital admissions for acute myocardial infarction, angina, stroke, and asthma after implementation of Arizona's comprehensive statewide smoking ban. American Journal of Public Health. 2011;101(3):491–6. [PMC free article: PMC3036684] [PubMed: 20466955]
- Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. Chronic obstructive pulmonary disease in alpha1-antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax. 2004;59(10):843–9. [PMC free article: PMC1746834] [PubMed: 15454649]
- Hersh CP, Pillai SG, Zhu G, Lomas DA, Bakke P, Gulsvik A, DeMeo DL, Klanderman BJ, Lazarus R, Litonjua AA, et al. Multistudy fine mapping of chromosome 2q identifies XRCC5 as a chronic obstructive pulmonary disease susceptibility gene. American Journal of Respiratory and Critical Care Medicine. 2010;182(5):605–13. [PMC free article: PMC2937234] [PubMed: 20463177]
- Hersh CP, Silverman EK, Gascon J, Bhattacharya S, Klanderman BJ, Litonjua AA, Lefebvre V, Sparrow D, Reilly JJ, Anderson WH, et al. SOX5 is a candidate gene for chronic obstructive pulmonary disease susceptibility and is necessary for lung development. American Journal of Respiratory and Critical Care Medicine. 2011;183(11):1482–9. [PMC free article: PMC3137139] [PubMed: 21330457]
- Higgins ITT. Epidemiology of Chronic Respiratory Disease: a Literature Review. Washington: Environmental Protection Agency, Office of Research and Development; 1974. Environmental Health Effects Research Series. Publication No. EPA-650/1-74-007.
- Hnizdo E, Murray J. Risk of pulmonary tuberculosis relative to silicosis and exposure to silica dust in South African gold miners. Occupational and Environmental Medicine. 1998;55(7):496–502. [PMC free article: PMC1757613] [PubMed: 9816385]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. New England Journal of Medicine. 2004;350(26):2645–53. [PubMed: 15215480]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. Journal of Clinical Investigation. 2006;116(3):753–9. [PMC free article: PMC1361346] [PubMed: 16470245]
- Huang JT, Chaudhuri R, Albarbarawi O, Barton A, Grierson C, Rauchhaus P, Weir CJ, Messow M, Stevens N, McSharry C, et al. Clinical validity of plasma and urinary desmosine as biomarkers for chronic obstructive pulmonary disease. Thorax. 2012;67(6):502–8. [PMC free article: PMC3358730] [PubMed: 22250098]
- Hubbard R, Lewis S, Richards K, Johnston I, Britton J. Occupational exposure to metal or wood dust and aetiology of cryptogenic fibrosing alveolitis. Lancet. 1996;347(8997):284–9. [PubMed: 8569361]
- Hubbard RB, Smith C, Le Jeune I, Gribbin J, Fogarty AW. The association between idiopathic pulmonary fibrosis and vascular disease: a population-based study. American Journal of Respiratory and Critical Care Medicine. 2008;178(12):1257–61. [PubMed: 18755924]
- Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, Stidley C, Melen E, Soderhall C, Hallberg J, et al. MMP12, lung function, and COPD in high-risk populations. New England Journal of Medicine. 2009;361(27):2599–608. [PMC free article: PMC2904064] [PubMed: 20018959]
- Hussain H, Akhtar S, Nanan D. Prevalence of and risk factors associated with Mycobacterium tuberculosis infection in prisoners, North West Frontier Province, Pakistan. International Journal of Epidemiology. 2003;32(5):794–9. [PubMed: 14559752]
- Ikaheimo P, Hartikainen S, Tuuponen T, Hakko H, Kiuttu J, Klaukka T. What lies behind relief and worsening of asthma symptoms? A register-based study of adults with asthma and other chronic obstructive pulmonary diseases in Finland. Primary Care Respiratory Journal: Journal of the General Practice Airways Group. 2006;15(5):278–85. [PMC free article: PMC6730825] [PubMed: 16979379]
- Imboden M, Bouzigon E, Curjuric I, Ramasamy A, Kumar A, Hancock DB, Wilk JB, Vonk JM, Thun GA, Siroux V, et al. Genome-wide association study of lung function decline in adults with and without asthma. Journal of Allergy and Clinical Immunology. 2012;129(5):1218–28. [PMC free article: PMC3340499] [PubMed: 22424883]
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. Journal of Experimental Medicine. 2006;203(1):7–13. [PMC free article: PMC2118081] [PubMed: 16380507]
- Iwai K, Mori T, Yamada N, Yamaguchi M, Hosoda Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. American Journal of Respiratory and Critical Care Medicine. 1994;150(3):670–5. [PubMed: 8087336]
- James AL, Palmer LJ, Kicic E, Maxwell PS, Lagan SE, Ryan GF, Musk AW. Decline in lung function in the Busselton Health Study: the effects of asthma and cigarette smoking. American Journal of Respiratory and Critical Care Medicine. 2005;171(2):109–14. [PubMed: 15486340]
- Jang AS, Park JS, Lee JH, Park SW, Kim DJ, Uh ST, Kim YH, Park CS. The impact of smoking on clinical and therapeutic effects in asthmatics. Journal of Korean Medical Science. 2009;24(2):209–14. [PMC free article: PMC2672118] [PubMed: 19399260]
- Janoff A. Elastases and emphysema. Current assessment of the protease-antiprotease hypothesis. American Review of Respiratory Disease. 1985;132(2):417–33. [PubMed: 3896082]
- Janoff A, Sloan B, Weinbaum G, Damiano V, Sandhaus RA, Elias J, Kimbel P. Experimental emphysema induced with purified human neutrophil elastase: tissue localization of the instilled protease. American Review of Respiratory Disease. 1977;115(3):461–78. [PubMed: 842956]
- Jee SH, Golub JE, Jo J, Park IS, Ohrr H, Samet JM. Smoking and risk of tuberculosis incidence, mortality, and recurrence in South Korean men and women. American Journal of Epidemiology. 2009;170(12):1478–85. [PMC free article: PMC2800271] [PubMed: 19917554]
- Jentoft HF, Omenaas E, Eide GE, Gulsvik A. Tuberculin reactivity: prevalence and predictors in BCG-vaccinated young Norwegian adults. Respiratory Medicine. 2002;96(12):1033–9. [PubMed: 12477220]
- Jha P, Jacob B, Gajalakshmi V, Gupta PC, Dhingra N, Kumar R, Sinha DN, Dikshit RP, Parida DK, Kamadod R, et al. A nationally representative case-control study of smoking and death in India. New England Journal of Medicine. 2008;358(11):1137–47. [PubMed: 18272886]
- Jick SS, Lieberman ES, Rahman MU, Choi HK. Glucocorticoid use, other associated factors, and the risk of tuberculosis. Arthritis and Rheumatism. 2006;55(1):19–26. [PubMed: 16463407]
- Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. Journal of Clinical Investigation. 2000;106(11):1311–9. [PMC free article: PMC387249] [PubMed: 11104784]
- Katzenstein AL, Mukhopadhyay S, Zanardi C, Dexter E. Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Human Pathology. 2010;41(3):316–25. [PubMed: 20004953]
- Kawabata Y, Hoshi E, Murai K, Ikeya T, Takahashi N, Saitou Y, Kurashima K, Ubukata M, Takayanagi N, Sugita H, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: a semiquantitative study with correlation to postoperative course. Histopathology. 2008;53(6):707–14. [PubMed: 19102010]
- Kelleher CM, Silverman EK, Broekelmann T, Litonjua AA, Hernandez M, Sylvia JS, Stoler J, Reilly JJ, Chapman HA, Speizer FE, et al. A functional mutation in the terminal exon of elastin in severe, early-onset chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology. 2005;33(4):355–62. [PMC free article: PMC2715343] [PubMed: 16081882]
- Kent BD, Sulaiman I, Nicholson TT, Lane SJ, Moloney ED. Acute pulmonary admissions following implementation of a national workplace smoking ban. Chest. 2012;142(3):673–9. [PubMed: 22383660]
- Kim TB, Park CS, Bae YJ, Cho YS, Moon HB. Factors associated with severity and exacerbation of asthma: a baseline analysis of the cohort for reality and evolution of adult asthma in Korea (COREA). Annals of Allergy, Asthma, and Immunology. 2009;103(4):311–7. [PubMed: 19852195]
- Kim WJ, Silverman EK, Hoffman E, Criner GJ, Mosenifar Z, Sciurba FC, Make BJ, Carey V, Estepar RS, Diaz A, et al. CT metrics of airway disease and emphysema in severe COPD. Chest. 2009;136(2):396–404. [PMC free article: PMC2733947] [PubMed: 19411295]
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378(9807):1949–61. [PubMed: 21719092]
- Kohri K, Ueki IF, Nadel JA. Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activation. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2002;283(3):L531–L40. [PubMed: 12169572]
- Kolappan C, Gopi PG. Tobacco smoking and pulmonary tuberculosis. Thorax. 2002;57(11):964–6. [PMC free article: PMC1746222] [PubMed: 12403879]
- Kolappan C, Subramani R. Association between biomass fuel and pulmonary tuberculosis: a nested case-control study. Thorax. 2009;64(8):705–8. [PubMed: 19359267]
- Kong X, Cho MH, Anderson W, Coxson HO, Muller N, Washko G, Hoffman EA, Bakke P, Gulsvik A, Lomas DA, et al. Genome-wide association study identifies BICD1 as a susceptibility gene for emphysema. American Journal of Respiratory and Critical Care Medicine. 2011;183(1):43–9. [PMC free article: PMC3040393] [PubMed: 20709820]
- Kotton DN, Fabian AJ, Mulligan RC. Failure of bone marrow to reconstitute lung epithelium. American Journal of Respiratory Cell and Molecular Biology. 2005;33(4):328–34. [PMC free article: PMC2715341] [PubMed: 15961722]
- Kubo K, Eda S, Yamamoto H, Fujimoto K, Matsuzawa Y, Maruyama Y, Hasegawa M, Sone S, Sakai F. Expiratory and inspiratory chest computed tomography and pulmonary function tests in cigarette smokers. European Respiratory Journal. 1999;13(2):252–6. [PubMed: 10065664]
- Kuemmerer JM, Comstock GW. Sociologic concomitants of tuberculin sensitivity. American Review of Respiratory Disease. 1967;96(5):885–92. [PubMed: 6059198]
- Laforest L, Van Ganse E, Devouassoux G, Bousquet J, Chretin S, Bauguil G, Pacheco Y, Chamba G. Influence of patients' characteristics and disease management on asthma control. Journal of Allergy and Clinical Immunology. 2006;117(6):1404–10. [PubMed: 16751004]
- Lam TH, Ho SY, Hedley AJ, Mak KH, Peto R. Mortality and smoking in Hong Kong: case-control study of all adult deaths in 1998. British Medical Journal. 2001;323(7309):361. [PMC free article: PMC37393] [PubMed: 11509422]
- Lange P, Parner J, Vestbo J, Schnohr P, Jensen G. A 15-year follow-up study of ventilatory function in adults with asthma. New England Journal of Medicine. 1998;339(17):1194–200. [PubMed: 9780339]
- Laurell CB, Eriksson S. The electrophoretic alpha-1-globulin pattern of serum in alpha-1-antitrypsin deficiency. Scandinavian Journal of Clinical and Laboratory Investigation. 1963;15:132–40. [PubMed: 23527532]
- Laurent P, Janoff A, Kagan HM. Cigarette smoke blocks cross-linking of elastin in vitro. American Review of Respiratory Disease. 1983;127(2):189–92. [PubMed: 6830036]
- Lazarus SC, Chinchilli VM, Rollings NJ, Boushey HA, Cherniack R, Craig TJ, Deykin A, DiMango E, Fish JE, Ford JG, et al. Smoking affects response to inhaled corticosteroids or leukotriene receptor antagonists in asthma. American Journal of Respiratory and Critical Care Medicine. 2007;175(8):783–90. [PMC free article: PMC1899291] [PubMed: 17204725]
- Lederer DJ, Enright PL, Kawut SM, Hoffman EA, Hunninghake G, van Beek EJ, Austin JH, Jiang R, Lovasi GS, Barr RG. Cigarette smoking is associated with subclinical parenchymal lung disease: the Multi-Ethnic Study of Atherosclerosis (MESA)-lung study. American Journal of Respiratory and Critical Care Medicine. 2009;180(5):407–14. [PMC free article: PMC2742759] [PubMed: 19542480]
- Lee SH, Goswami S, Grudo A, Song LZ, Bandi V, Goodnight-White S, Green L, Hacken-Bitar J, Huh J, Bakaeen F, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nature Medicine. 2007;13(5):567–9. [PubMed: 17450149]
- Leung CC, Li T, Lam TH, Yew WW, Law WS, Tam CM, Chan WM, Chan CK, Ho KS, Chang KC. Smoking and tuberculosis among the elderly in Hong Kong. American Journal of Respiratory and Critical Care Medicine. 2004;170(9):1027–33. [PubMed: 15282201]
- Leung CC, Yew WW, Chan CK, Tam CM, Lam CW, Chang KC, Chau CH, Lau KS, Law WS. Smoking and tuberculosis in Hong Kong. International Journal of Tuberculosis and Lung Disease. 2003;7(10):980–6. [PubMed: 14552569]
- Leung CC, Yew WW, Law WS, Tam CM, Leung M, Chung YW, Cheung KW, Chan KW, Fu F. Smoking and tuberculosis among silicotic patients. European Respiratory Journal. 2007;29(4):745–50. [PubMed: 17182648]
- Levine SJ, Wenzel SE. Narrative review: the role of Th2 immune pathway modulation in the treatment of severe asthma and its phenotypes. Annals of Internal Medicine. 2010;152(4):232–7. [PMC free article: PMC2846792] [PubMed: 20157138]
- Lewis JG, Chamberlain DA. Alcohol consumption and smoking habits in male patients with pulmonary tuberculosis. British Journal of Preventive and Social Medicine. 1963;17:149–52. [PMC free article: PMC1058910] [PubMed: 14044850]
- Lewitter FI, Tager IB, McGue M, Tishler PV, Speizer FE. Genetic and environmental determinants of level of pulmonary function. American Journal of Epidemiology. 1984;120:518–29. [PubMed: 6475921]
- Lienhardt C, Fielding K, Sillah JS, Bah B, Gustafson P, Warndorff D, Palayew M, Lisse I, Donkor S, Diallo S, et al. Investigation of the risk factors for tuberculosis: a case-control study in three countries in West Africa. International Journal of Epidemiology. 2005;34(4):914–23. [PubMed: 15914505]
- Lin HH, Ezzati M, Chang HY, Murray M. Association between tobacco smoking and active tuberculosis in Taiwan: prospective cohort study. American Journal of Respiratory and Critical Care Medicine. 2009;180(5):475–80. [PubMed: 19542475]
- Lin HH, Ezzati M, Murray M. Tobacco smoke, indoor air pollution and tuberculosis: a systematic review and meta-analysis. PLoS Medicine. 2007;4(1):e20. [PMC free article: PMC1769410] [PubMed: 17227135]
- Lin JN, Lai CH, Chen YH, Lee SS, Tsai SS, Huang CK, Chung HC, Liang SH, Lin HH. Risk factors for extra-pulmonary tuberculosis compared to pulmonary tuberculosis. International Journal of Tuberculosis and Lung Disease. 2009;13(5):620–5. [PubMed: 19383196]
- Lin YS, Hsu CC, Bien MY, Hsu HC, Weng HT, Kou YR. Activations of TRPA1 and P2X receptors are important in ROS-mediated stimulation of capsaicin-sensitive lung vagal afferents by cigarette smoke in rats. Journal of Applied Physiology. 2010;108(5):1293–303. [PubMed: 20167675]
- Liu BQ, Peto R, Chen ZM, Boreham J, Wu YP, Li JY, Campbell TC, Chen JS. Emerging tobacco hazards in China: 1. Retrospective proportional mortality study of one million deaths. British Medical Journal. 1998;317(7170):1411–22. [PMC free article: PMC28719] [PubMed: 9822393]
- Loeys B, Van Maldergem L, Mortier G, Coucke P, Gerniers S, Naeyaert JM, De Paepe A. Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Human Molecular Genetics. 2002;11(18):2113–8. [PubMed: 12189163]
- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357(6379):605–7. [PubMed: 1608473]
- Lomas DA, Silverman EK, Edwards LD, Locantore NW, Miller BE, Horstman DH, Tal-Singer R. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. European Respiratory Journal. 2009;34(1):95–102. [PubMed: 19164344]
- Lomas DA, Silverman EK, Edwards LD, Miller BE, Coxson HO, Tal-Singer R. Evaluation of serum CC-16 as a biomarker for COPD in the ECLIPSE cohort. Thorax. 2008;63(12):1058–63. [PubMed: 18757456]
- Lonnroth K, Castro KG, Chakaya JM, Chauhan LS, Floyd K, Glaziou P, Raviglione MC. Tuberculosis control and elimination 2010-50: cure, care, and social development. Lancet. 2010;375(9728):1814–29. [PubMed: 20488524]
- Lonnroth K, Williams BG, Stadlin S, Jaramillo E, Dye C. Alcohol use as a risk factor for tuberculosis—a systematic review. BMC Public Health. 2008;8:289. [PMC free article: PMC2533327] [PubMed: 18702821]
- Lowe CR. An association between smoking and respiratory tuberculosis. British Medical Journal. 1956;2(5001):1081–6. [PMC free article: PMC2035839] [PubMed: 13364390]
- Luisetti M, Ma S, Iadarola P, Stone PJ, Viglio S, Casado B, Lin YY, Snider GL, Turino GM. Desmosine as a biomarker of elastin degradation in COPD: current status and future directions. European Respiratory Journal. 2008;32(5):1146–57. [PubMed: 18978133]
- Ma S, Lieberman S, Turino GM, Lin YY. The detection and quantitation of free desmosine and isodesmosine in human urine and their peptide-bound forms in sputum. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(22):12941–3. [PMC free article: PMC240723] [PubMed: 14563926]
- Mackay D, Haw S, Ayres JG, Fischbacher C, Pell JP. Smoke-free legislation and hospitalizations for childhood asthma. New England Journal of Medicine. 2010;363(12):1139–45. [PubMed: 20843248]
- Macneal K, Schwartz DA. The genetic and environmental causes of pulmonary fibrosis. Proceedings of the American Thoracic Society. 2012;9(3):120–5. [PubMed: 22802285]
- Madani A, Van Muylem A, de Maertelaer V, Zanen J, Gevenois PA. Pulmonary emphysema: size distribution of emphysematous spaces on multidetector CT images—comparison with macroscopic and microscopic morphometry. Radiology. 2008;248(3):1036–41. [PubMed: 18710992]
- Madani A, Zanen J, de Maertelaer V, Gevenois PA. Pulmonary emphysema: objective quantification at multi-detector row CT—comparison with macroscopic and microscopic morphometry. Radiology. 2006;238(3):1036–43. [PubMed: 16424242]
- Maeno T, Houghton AM, Quintero PA, Grumelli S, Owen CA, Shapiro SD. CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in mice. Journal of Immunology. 2007;178(12):8090–6. [PubMed: 17548647]
- Mahadeva R, Atkinson C, Li Z, Stewart S, Janciauskiene S, Kelley DG, Parmar J, Pitman R, Shapiro SD, Lomas DA. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. American Journal of Pathology. 2005;166(2):377–86. [PMC free article: PMC3278851] [PubMed: 15681822]
- Mallol J, Castro-Rodriguez JA, Cortez E. Effects of active tobacco smoking on the prevalence of asthma-like symptoms in adolescents. International Journal of Chronic Obstructive Pulmonary Disease. 2007;2(1):65–9. [PMC free article: PMC2692110] [PubMed: 18044067]
- Mannino DM, Gagnon RC, Petty TL, Lydick E. Obstructive lung disease and low lung function in adults in the United States: data from the National Health and Nutrition Examination Survey, 1988–1994. Archives of Internal Medicine. 2000;160(11):1683–9. [PubMed: 10847262]
- Mannino DM, Homa DM, Akinbami LJ, Ford ES, Redd SC. Chronic obstructive pulmonary disease surveillance—United States, 1971–2000. Morbidity and Mortality Weekly Report: Surveillance Summaries. 2002;51(SS-06):1–16. [PubMed: 12198919]
- Martinez FJ, Curtis JL, Sciurba F, Mumford J, Giardino ND, Weinmann G, Kazerooni E, Murray S, Criner GJ, Sin DD, et al. Sex differences in severe pulmonary emphysema. American Journal of Respiratory and Critical Care Medicine. 2007;176(3):243–52. [PMC free article: PMC1994221] [PubMed: 17431226]
- Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nature Medicine. 1997;3(6):675–7. [PubMed: 9176496]
- Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Medicine. 2006;3(11):e442. [PMC free article: PMC1664601] [PubMed: 17132052]
- Matsui EC, Hansel NN, McCormack MC, Rusher R, Breysse PN, Diette GB. Asthma in the inner city and the indoor environment. Immunology and Allergy Clinics of North America. 2008;28(3):665–86. [PMC free article: PMC5516633] [PubMed: 18572113]
- Matsuoka S, Kurihara Y, Yagihashi K, Hoshino M, Watanabe N, Nakajima Y. Quantitative assessment of air trapping in chronic obstructive pulmonary disease using inspiratory and expiratory volumetric MDCT. American Journal of Roentgenology. 2008;190(3):762–9. [PubMed: 18287450]
- Matsuoka S, Kurihara Y, Yagihashi K, Nakajima Y. Morphological progression of emphysema on thin-section CT: Analysis of longitudinal change in the number and size of low-attenuation clusters. Journal of Computer Assisted Tomography. 2006;30(4):669–74. [PubMed: 16845301]
- Matsuoka S, Kurihara Y, Yagihashi K, Nakajima Y. Quantitative assessment of peripheral airway obstruction on paired expiratory/inspiratory thin-section computed tomography in chronic obstructive pulmonary disease with emphysema. Journal of Computer Assisted Tomography. 2007;31(3):384–9. [PubMed: 17538284]
- Matsuoka S, Washko GR, Dransfield MT, Yamashiro T, San Jose Estepar R, Diaz A, Silverman EK, Patz S, Hatabu H. Quantitative CT measurement of cross-sectional area of small pulmonary vessel in COPD: correlations with emphysema and airflow limitation. Academic Radiology. 2010;17(1):93–9. [PMC free article: PMC2790546] [PubMed: 19796970]
- McCurdy SA, Arretz DS, Bates RO. Tuberculin reactivity among California Hispanic migrant farm workers. American Journal of Industrial Medicine. 1997;32(6):600–5. [PubMed: 9358916]
- McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. New England Journal of Medicine. 2011;365(17):1567–75. [PMC free article: PMC3238466] [PubMed: 22029978]
- Mecham RP, Broekelmann T, Davis EC, Gibson MA, Brown-Augsburger P. Elastic fibre assembly: macromolecular interactions. Ciba Foundation Symposium. 1995;192:172–81. discussion 81–4. [PubMed: 8575256]
- Meng YY, Wilhelm M, Rull RP, English P, Nathan S, Ritz B. Are frequent asthma symptoms among low-income individuals related to heavy traffic near homes, vulnerabilities, or both? Annals of Epidemiology. 2008;18(5):343–50. [PubMed: 18433665]
- Menzies D, Nair A, Williamson PA, Schembri S, Al-Khairalla MZ, Barnes M, Fardon TC, McFarlane L, Magee GJ, Lipworth BJ. Respiratory symptoms, pulmonary function, and markers of inflammation among bar workers before and after a legislative ban on smoking in public places. JAMA: the Journal of the American Medical Association. 2006;296(14):1742–8. [PubMed: 17032987]
- Merchant RK, Schwartz DA, Helmers RA, Dayton CS, Hunninghake GW. Bronchoalveolar lavage cellularity. The distribution in normal volunteers. American Review of Respiratory Disease. 1992;146(2):448–53. [PubMed: 1489138]
- Miguez-Burbano MJ, Burbano X, Ashkin D, Pitchenik A, Allan R, Pineda L, Rodriguez N, Shor-Posner G. Impact of tobacco use on the development of opportunistic respiratory infections in HIV seropositive patients on antiretroviral therapy. Addiction Biology. 2003;8(1):39–43. [PubMed: 12745414]
- Millet JP, Orcau A, de Olalla PG, Casals M, Rius C, Cayla JA. Tuberculosis recurrence and its associated risk factors among successfully treated patients. Journal of Epidemiology and Community Health. 2009;63(10):799–804. [PubMed: 19179367]
- Millett C, Lee JT, Laverty AA, Glantz SA, Majeed A. Hospital admissions for childhood asthma after smoke-free legislation in England. Pediatrics. 2013;131(2):e495–501. [PMC free article: PMC4528337] [PubMed: 23339216]
- Miyake Y, Sasaki S, Yokoyama T, Chida K, Azuma A, Suda T, Kudoh S, Sakamoto N, Okamoto K, Kobashi G, et al. Occupational and environmental factors and idiopathic pulmonary fibrosis in Japan. Annals of Occupational Hygiene. 2005;49(3):259–65. [PubMed: 15640309]
- Moorman JE, Rudd RA, Johnson CA, King M, Minor P, Bailey C, Scalia MR, Akinbami LJ. National surveillance for asthma—United States, 1980–2004. Morbidity and Mortality Weekly Report. 2007;56(SS-8):1–54. [PubMed: 17947969]
- Moraros J, Bird Y, Chen S, Buckingham R, Meltzer RS, Prapasiri S, Solis LH. The impact of the 2002 Delaware smoking ordinance on heart attack and asthma. International Journal of Environmental Research and Public Health. 2010;7(12):4169–78. [PMC free article: PMC3037047] [PubMed: 21318001]
- Musellim B, Erturan S, Sonmez DE, Ongen G. Comparison of extra-pulmonary and pulmonary tuberculosis cases: factors influencing the site of reactivation. International Journal of Tuberculosis and Lung Disease. 2005;9(11):1220–3. [PubMed: 16333928]
- Nadel JA. Role of neutrophil elastase in hypersecretion during COPD exacerbations, and proposed therapies. Chest. 2000;117(5 Suppl 2):386S–389S. [PubMed: 10843982]
- Naiman A, Glazier RH, Moineddin R. Association of anti-smoking legislation with rates of hospital admission for cardiovascular and respiratory conditions. Canadian Medical Association Journal. 2010;182(8):761–7. [PMC free article: PMC2871198] [PubMed: 20385737]
- Nakamura K, Nagata C, Fujii K, Kawachi T, Takatsuka N, Oba S, Shimizu H. Cigarette smoking and the adult onset of bronchial asthma in Japanese men and women. Annals of Allergy, Asthma, and Immunology. 2009;102(4):288–93. [PubMed: 19441599]
- Nakamura Y, Miyata M, Ohba T, Ando T, Hatsushika K, Suenaga F, Shimokawa N, Ohnuma Y, Katoh R, Ogawa H, et al. Cigarette smoke extract induces thymic stromal lymphopoietin expression, leading to T(H)2-type immune responses and airway inflammation. Journal of Allergy and Clinical Immunology. 2008;122(6):1208–14. [PubMed: 18926564]
- Nakamura Y, Romberger DJ, Tate L, Ertl RF, Kawamoto M, Adachi Y, Mio T, Sisson JH, Spurzem JR, Rennard SI. Cigarette smoke inhibits lung fibroblast proliferation and chemotaxis. American Journal of Respiratory and Critical Care Medicine. 1995;151(5):1497–503. [PubMed: 7735606]
- Nakano Y, Wong JC, de Jong PA, Buzatu L, Nagao T, Coxson HO, Elliott WM, Hogg JC, Pare PD. The prediction of small airway dimensions using computed tomography. American Journal of Respiratory and Critical Care Medicine. 2005;171(2):142–6. [PubMed: 15516531]
- National Asthma Education and Prevention Program. Expert Panel report 3 (EPR-3): guidelines for the diagnosis and management of asthma-summary report 2007. Journal of Allergy and Clinical Immunology. 2007;120(5 Suppl):S94–S138. [PubMed: 17983880]
- National Center for Health Statistics. Deaths, percent of total deaths, and death rates for the 15 leading causes of death: United States and each state, 2010. Nov 21, 2012. [July 16, 2013]. < http://www
.cdc.gov/nchs /data/dvs/LCWK9_2010.pdf>. - National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda (MD): U.S. Department of Health and Human Services, National Institutes of Health, National Heart, Lung, and Blood Institute, National Asthma Education and Prevention Program, 2007; NIH Publication No. 07-4051.
- Navon L, Fiore B, Anderson H. Asthma and tobacco: double trouble for Wisconsin adolescents. WMJ. 2005;104(7):47–53. [PubMed: 16294600]
- Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. New England Journal of Medicine. 1974;291(15):755–8. [PubMed: 4414996]
- Ning W, Li CJ, Kaminski N, Feghali-Bostwick CA, Alber SM, Di YP, Otterbein SL, Song R, Hayashi S, Zhou Z, et al. Comprehensive gene expression profiles reveal pathways related to the pathogenesis of chronic obstructive pulmonary disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(41):14895–900. [PMC free article: PMC522001] [PubMed: 15469929]
- Nisar M, Williams CS, Ashby D, Davies PD. Tuberculin testing in residential homes for the elderly. Thorax. 1993;48(12):1257–60. [PMC free article: PMC464987] [PubMed: 8303634]
- Nishimura M, Makita H, Nagai K, Konno S, Nasuhara Y, Hasegawa M, Shimizu K, Betsuyaku T, Ito YM, Fuke S, et al. Annual change in pulmonary function and clinical phenotype in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2012;185(1):44–52. [PubMed: 22016444]
- Nouri-Shirazi M, Guinet E. A possible mechanism linking cigarette smoke to higher incidence of respiratory infection and asthma. Immunology Letters. 2006;103(2):167–76. [PubMed: 16325267]
- Omachi TA, Iribarren C, Sarkar U, Tolstykh I, Yelin EH, Katz PP, Blanc PD, Eisner MD. Risk factors for death in adults with severe asthma. Annals of Allergy, Asthma, and Immunology. 2008;101(2):130–6. [PMC free article: PMC2767233] [PubMed: 18727467]
- O'Reilly P, Jackson PL, Noerager B, Parker S, Dransfield M, Gaggar A, Blalock JE. N-alpha-PGP and PGP, potential biomarkers and therapeutic targets for COPD. Respiratory Research. 2009;10:38. [PMC free article: PMC2693492] [PubMed: 19450278]
- Orie NGM, Sluiter HJ, de Vries K, Tammeling GJ, Witkop J. The host factor in bronchitis. In: Orie NGM, Sluiter HJ, editors. Bronchitis: An International Symposium, 27–29 April 1960. Assen (The Netherlands): Royal Van Gorcum; 1961. pp. 43–59.
- Orlandi I, Moroni C, Camiciottoli G, Bartolucci M, Pistolesi M, Villari N, Mascalchi M. Chronic obstructive pulmonary disease: thin-section CT measurement of airway wall thickness and lung attenuation. Radiology. 2005;234(2):604–10. [PubMed: 15671010]
- Osman M, Cantor JO, Roffman S, Keller S, Turino GM, Mandl I. Cigarette smoke impairs elastin resynthesis in lungs of hamsters with elastase-induced emphysema. American Review of Respiratory Disease. 1985;132(3):640–3. [PubMed: 3929658]
- Pai M, Mohan A, Dheda K, Leung CC, Yew WW, Christopher DJ, Sharma SK. Lethal interaction: the colliding epidemics of tobacco and tuberculosis. Expert Review of Anti-Infective Therapy. 2007;5(3):385–91. [PubMed: 17547503]
- Palmer LJ, Celedon JC, Chapman HA, Speizer FE, Weiss ST, Silverman EK. Genome-wide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary disease. Human Molecular Genetics. 2003;12(10):1199–210. [PubMed: 12719384]
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha1-antitrypsin deficiency influences lung function impairment. American Journal of Respiratory and Critical Care Medicine. 2004;170(11):1172–8. [PubMed: 15306534]
- Parrish NM, Dick JD, Bishai WR. Mechanisms of latency in Mycobacterium tuberculosis. Trends in Microbiology. 1998;6(3):107–12. [PubMed: 9582936]
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. American Journal of Respiratory and Critical Care Medicine. 2001;163(5):1256–76. [PubMed: 11316667]
- Pednekar MS, Gupta PC. Prospective study of smoking and tuberculosis in India. Preventive Medicine. 2007;44(6):496–8. [PubMed: 17391745]
- Perez-Padilla R, Perez-Guzman C, Baez-Saldana R, Torres-Cruz A. Cooking with biomass stoves and tuberculosis: a case control study. International Journal of Tuberculosis and Lung Disease. 2001;5(5):441–7. [PubMed: 11336275]
- Peters AT, Klemens JC, Haselkorn T, Weiss ST, Grammer LC, Lee JH, Chen H. Insurance status and asthma-related health care utilization in patients with severe asthma. Annals of Allergy, Asthma, and Immunology. 2008;100(4):301–7. [PubMed: 18450113]
- Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nature Medicine. 2005;11(5):491–8. [PMC free article: PMC1352344] [PubMed: 15852018]
- Piipari R, Jaakkola JJ, Jaakkola N, Jaakkola MS. Smoking and asthma in adults. European Respiratory Journal. 2004;24(5):734–9. [PubMed: 15516665]
- Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, Feng S, Hersh CP, Bakke P, Gulsvik A, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genetics. 2009;5(3):e1000421. [PMC free article: PMC2650282] [PubMed: 19300482]
- Pillai SG, Kong X, Edwards LD, Cho MH, Anderson WH, Coxson HO, Lomas DA, Silverman EK. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2010;182(12):1498–505. [PMC free article: PMC3029936] [PubMed: 20656943]
- Plant AJ, Watkins RE, Gushulak B, O'Rourke T, Jones W, Streeton J, Sang D. Predictors of tuberculin reactivity among prospective Vietnamese migrants: the effect of smoking. Epidemiology and Infection. 2002;128(1):37–45. [PMC free article: PMC2869793] [PubMed: 11895089]
- Polosa R, Knoke JD, Russo C, Piccillo G, Caponnetto P, Sarva M, Proietti L, Al-Delaimy WK. Cigarette smoking is associated with a greater risk of incident asthma in allergic rhinitis. Journal of Allergy and Clinical Immunology. 2008;121(6):1428–34. [PubMed: 18436295]
- Prasad R, Garg R, Singhal S, Dawar R, Agarwal GG. A case-control study of tobacco smoking and tuberculosis in India. Annals of Thoracic Medicine. 2009;4(4):208–10. [PMC free article: PMC2801046] [PubMed: 19881167]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. American Journal of Respiratory and Critical Care Medicine. 2011;183(6):788–824. [PMC free article: PMC5450933] [PubMed: 21471066]
- Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. American Journal of Respiratory and Critical Care Medicine. 2006;174(7):810–6. [PubMed: 16809633]
- Raherison C, Baldi I, Tunon-De-Lara JM, Taytard A, Annesi-Maesano I. Asthma phenotypes according to the timing of smoking onset in young adults. International Journal of Tuberculosis and Lung Disease. 2003;7(1):84–92. [PubMed: 12701840]
- Rahimi-Rad MH, Gaderi-Pakdel F, Salari-Lak S. Smoking and asthma in 20-44-year-old adults in Urmia, Islamic Republic of Iran. Eastern Mediterranean Health Journal. 2008;14(1):6–16. [PubMed: 18557447]
- Ramin B, Kam D, Feleke B, Jacob B, Jha P. Smoking, HIV and non-fatal tuberculosis in an urban African population. International Journal of Tuberculosis and Lung Disease. 2008;12(6):695–7. [PubMed: 18492341]
- Rayens MK, Burkhart PV, Zhang M, Lee S, Moser DK, Mannino D, Hahn EJ. Reduction in asthma-related emergency department visits after implementation of a smoke-free law. Journal of Allergy and Clinical Immunology. 2008;122(3):537–41. e3. [PubMed: 18692884]
- Redline S, Tishler PV, Rosner B, Lewitter FI, Vandenburgh M, Weiss ST, Speizer FE. Genotypic and phenotypic similarities in pulmonary function among family members of adult monozygotic and dizygotic twins. American Journal of Epidemiology. 1989;129(4):827–36. [PubMed: 2923128]
- Rehm J, Samokhvalov AV, Neuman MG, Room R, Parry C, Lonnroth K, Patra J, Poznyak V, Popova S. The association between alcohol use, alcohol use disorders and tuberculosis (TB). A systematic review. BMC Public Health. 2009;9:450. [PMC free article: PMC2796667] [PubMed: 19961618]
- Reid DD, Anderson DO, Ferris BG, Fletcher CM. An Anglo-American comparison of the prevalence of bronchitis. British Medical Journal. 1964;2(5423):1487–91. [PMC free article: PMC1817179] [PubMed: 14214183]
- Repapi E, Sayers I, Wain LV, Burton PR, Johnson T, Obeidat M, Zhao JH, Ramasamy A, Zhai G, Vitart V, et al. Genome-wide association study identifies five loci associated with lung function. Nature Genetics. 2010;42(1):36–44. [PMC free article: PMC2862965] [PubMed: 20010834]
- Revel MP, Faivre JB, Remy-Jardin M, Deken V, Duhamel A, Marquette CH, Tacelli N, Bakai AM, Remy J. Automated lobar quantification of emphysema in patients with severe COPD. European Radiology. 2008;18(12):2723–30. [PubMed: 18604539]
- Rieder HL. Opportunity for exposure and risk of infection: the fuel for the tuberculosis pandemic. Infection. 1995;23(1):1–3. [PubMed: 7744486]
- Robays LJ, Lanckacker EA, Moerloose KB, Maes T, Bracke KR, Brusselle GG, Joos GF, Vermaelen KY. Concomitant inhalation of cigarette smoke and aerosolized protein activates airway dendritic cells and induces allergic airway inflammation in a TLR-independent way. Journal of Immunology. 2009;183(4):2758–66. [PubMed: 19635922]
- Roberts C, Davis PJ, Taylor KE, Pearlman DN. The impact of Rhode Island's statewide smoke-free ordinance on hospital admissions and costs for acute myocardial infarction and asthma. Medicine and Health, Rhode Island. 2012;95(1):23–5. [PubMed: 22439460]
- Ronmark E, Lindberg A, Watson L, Lundback B. Outcome and severity of adult onset asthma—report from the obstructive lung disease in northern Sweden studies (OLIN). Respiratory Medicine. 2007;101(11):2370–7. [PubMed: 17689949]
- Rose D, Mannino DM, Leaderer BP. Asthma prevalence among US adults, 1998–2000: role of Puerto Rican ethnicity and behavioral and geographic factors. American Journal of Public Health. 2006;96(5):880–8. [PMC free article: PMC1470587] [PubMed: 16571713]
- Rosenberg SR, Kalhan R. Biomarkers in chronic obstructive pulmonary disease. Translational Research. 2012;159(4):228–37. [PubMed: 22424427]
- Roth MD, Connett JE, D'Armiento JM, Foronjy RF, Friedman PJ, Goldin JG, Louis TA, Mao JT, Muindi JR, O'Connor GT, et al. Feasibility of retinoids for the treatment of emphysema study. Chest. 2006;130(5):1334–45. [PubMed: 17099008]
- Schenker MB, Pinkerton KE, Mitchell D, Vallyathan V, Elvine-Kreis B, Green FH. Pneumoconiosis from agricultural dust exposure among young California farmworkers. Environmental Health Perspectives. 2009;117(6):988–94. [PMC free article: PMC2702418] [PubMed: 19590695]
- Scott J, Johnston I, Britton J. What causes cryptogenic fibrosing alveolitis? A case-control study of environmental exposure to dust. British Medical Journal. 1990;301(6759):1015–7. [PMC free article: PMC1664043] [PubMed: 2249047]
- Seabra B, Guimaraes M, Carvalho A, Duarte R. The smoking rate and its repercussions on an asthmatic population sample. Revista Portuguesa de Pneumologia. 2008;14(5):617–24. [PubMed: 18781263]
- Sears MR, Greene JM, Willan AR, Wiecek EM, Taylor DR, Flannery EM, Cowan JO, Herbison GP, Silva PA, Poulton R. A longitudinal, population-based, cohort study of childhood asthma followed to adulthood. New England Journal of Medicine. 2003;349(15):1414–22. [PubMed: 14534334]
- Senior RM, Griffin GL, Mecham RP, Wrenn DS, Prasad KU, Urry DW. Val-Gly-Val-Ala-Pro-Gly, a repeating peptide in elastin, is chemotactic for fibroblasts and monocytes. Journal of Cell Biology. 1984;99(3):870–4. [PMC free article: PMC2113419] [PubMed: 6547961]
- Senior RM, Tegner H, Kuhn C, Ohlsson K, Starcher BC, Pierce JA. The induction of pulmonary emphysema with human leukocyte elastase. American Review of Respiratory Disease. 1977;116(3):469–75. [PubMed: 900634]
- Shah JR, Warawadekar MS, Deshmukh PA, Phutane PN. Institutional survey of pulmonary tuberculosis with special reference to smoking habits. Indian Journal of Medical Sciences. 1959;13(5):381–92. [PubMed: 13672620]
- Shaker SB, Dirksen A, Laursen LC, Maltbaek N, Christensen L, Sander U, Seersholm N, Skovgaard LT, Nielsen L, Kok-Jensen A. Short-term reproducibility of computed tomography-based lung density measurements in alpha-1 antitrypsin deficiency and smokers with emphysema. Acta Radiologica. 2004;45(4):424–30. [PubMed: 15323395]
- Shapiro SD, Goldstein NM, Houghton AM, Kobayashi DK, Kelley D, Belaaouaj A. Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. American Journal of Pathology. 2003;163(6):2329–35. [PMC free article: PMC1892384] [PubMed: 14633606]
- Shavit O, Swern A, Dong Q, Newcomb K, Sazonov KV, Taylor SD. Impact of smoking on asthma symptoms, healthcare resource use, and quality of life outcomes in adults with persistent asthma. Quality of Life Research. 2007;16(10):1555–65. [PubMed: 17917792]
- Shetty N, Shemko M, Vaz M, D'souza G. An epidemiological evaluation of risk factors for tuberculosis in South India: a matched case control study. International Journal of Tuberculosis and Lung Disease. 2006;10(1):80–6. [PubMed: 16466042]
- Shifren A, Mecham RP. The stumbling block in lung repair of emphysema: elastic fiber assembly. Proceedings of the American Thoracic Society. 2006;3(5):428–33. [PMC free article: PMC2658707] [PubMed: 16799087]
- Short JJ, Johnson HJ, Ley H. The Effects of Tobacco Smoking on Health—Study of 2,031 Medical Records. 1939. Research Collection. Bates No. 4102. < http://legacy
.library .ucsf.edu/tid/vrs66b00>. - Silverman EK. Perspective: how can genetics help? Nature. 2012;489(7417):S7. [PubMed: 23013715]
- Silverman EK, Chapman HA, Drazen JM, Weiss ST, Rosner B, Campbell EJ, O'Donnell WJ, Reilly JJ, Ginns L, Mentzer S, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease: risk to relatives for airflow obstruction and chronic bronchitis. American Journal of Respiratory and Critical Care Medicine. 1998;157:1770–8. [PubMed: 9620904]
- Silverman EK, Miletich JP, Pierce JA, Sherman LA, Endicott SK, Broze GJ Jr, Campbell EJ. Alpha-1-antitrypsin deficiency: high prevalence in the St. Louis area determined by direct population screening. American Review of Respiratory Disease. 1989;140:961–6. [PubMed: 2679271]
- Silverman EK, Mosley JD, Palmer LJ, Barth M, Senter JM, Brown A, Drazen JM, Kwiatkowski DJ, Chapman HA, Campbell EJ, et al. Genome-wide linkage analysis of severe, early-onset chronic obstructive pulmonary disease: Airflow obstruction and chronic bronchitis phenotypes. Human Molecular Genetics. 2002;11(6):623–32. [PubMed: 11912177]
- Silverman EK, Palmer LJ, Mosley JD, Barth M, Senter JM, Brown A, Drazen JM, Kwiatkowski DJ, Chapman HA, Campbell EJ, et al. Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary disease. American Journal of Human Genetics. 2002;70(5):1229–39. [PMC free article: PMC447597] [PubMed: 11914989]
- Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. New England Journal of Medicine. 2009;360(26):2749–57. [PubMed: 19553648]
- Simon SA, Liedtke W. How irritating: the role of TRPA1 in sensing cigarette smoke and aerogenic oxidants in the airways. Journal of Clinical Investigation. 2008;118(7):2383–6. [PMC free article: PMC2430501] [PubMed: 18568080]
- Sims M, Maxwell R, Gilmore A. Short-term impact of the smokefree legislation in England on emergency hospital admissions for asthma among adults: a population-based study. Thorax. 2013;68(7):619–24. [PubMed: 23589509]
- Sin DD, Miller BE, Duvoix A, Man SF, Zhang X, Silverman EK, Connett JE, Anthonisen NA, Wise RA, Tashkin D, et al. Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2011;183(9):1187–92. [PMC free article: PMC3114051] [PubMed: 21216880]
- Singh M, Mynak ML, Kumar L, Mathew JL, Jindal SK. Prevalence and risk factors for transmission of infection among children in household contact with adults having pulmonary tuberculosis. Archives of Disease in Childhood. 2005;90(6):624–8. [PMC free article: PMC1720464] [PubMed: 15908630]
- Sitas F, Urban M, Bradshaw D, Kielkowski D, Bah S, Peto R. Tobacco attributable deaths in South Africa. Tobacco Control. 2004;13(4):396–9. [PMC free article: PMC1747967] [PubMed: 15564624]
- Slama K, Chiang CY, Enarson DA, Hassmiller K, Fanning A, Gupta P, Ray C. Tobacco and tuberculosis: a qualitative systematic review and meta-analysis. International Journal of Tuberculosis and Lung Disease. 2007;11(10):1049–61. [PubMed: 17945060]
- Snider GL. Emphysema: the first two centuries—and beyond. A historical overview, with suggestions for future research: part 1. American Review of Respiratory Disease. 1992;146(5 Pt 1):1334–44. [PubMed: 1443893]
- Snider GL. Emphysema: the first two centuries—and beyond. A historical overview, with suggestions for future research: part 2. American Review of Respiratory Disease. 1992;146(6):1615–22. [PubMed: 1456587]
- Snider GL, Lucey EC, Christensen TG, Stone PJ, Calore JD, Catanese A, Franzblau C. Emphysema and bronchial secretory cell metaplasia induced in hamsters by human neutrophil products. American Review of Respiratory Disease. 1984;129(1):155–60. [PubMed: 6561016]
- Soler Artigas M, Wain LV, Repapi E, Obeidat M, Sayers I, Burton PR, Johnson T, Zhao JH, Albrecht E, Dominiczak AF, et al. Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. American Journal of Respiratory and Critical Care Medicine. 2011;184(7):786–95. [PMC free article: PMC3398416] [PubMed: 21965014]
- Solsona J, Caylá JA, Nadal J, Bedia M, Mata C, Brau J, Maldonado J, Milá C, Alcaide J, Altet N, et al. Screening for tuberculosis upon admission to shelters and free-meal services. European Journal of Epidemiology. 2001;17(2):123–8. [PubMed: 11599684]
- Sorheim IC, Bakke P, Gulsvik A, Pillai SG, Johannessen A, Gaarder PI, Campbell EJ, Agusti A, Calverley PM, Donner CF, et al. Alpha(1)-antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohorts. Chest. 2010;138(5):1125–32. [PMC free article: PMC2972629] [PubMed: 20595457]
- Sorheim IC, Johannessen A, Gulsvik A, Bakke PS, Silverman EK, DeMeo DL. Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax. 2010;65(6):480–5. [PMC free article: PMC8191512] [PubMed: 20522842]
- Spira A, Beane J, Pinto-Plata V, Kadar A, Liu G, Shah V, Celli B, Brody JS. Gene expression profiling of human lung tissue from smokers with severe emphysema. American Journal of Respiratory Cell and Molecular Biology. 2004;31(6):601–10. [PubMed: 15374838]
- Spira A, Beane J, Shah V, Liu G, Schembri F, Yang X, Palma J, Brody JS. Effects of cigarette smoke on the human airway epithelial cell transcriptome. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(27):10143–8. [PMC free article: PMC454179] [PubMed: 15210990]
- Sreeramareddy CT, Panduru KV, Verma SC, Joshi HS, Bates MN. Comparison of pulmonary and extrapulmonary tuberculosis in Nepal- a hospital-based retrospective study. BMC Infectious Diseases. 2008;8:8. [PMC free article: PMC2245948] [PubMed: 18218115]
- Stallberg B, Lisspers K, Hasselgren M, Johansson G, Svardsudd K. Factors related to the level of severity of asthma in primary care. Respiratory Medicine. 2007;101(10):2076–83. [PubMed: 17628463]
- Stampfli MR, Anderson GP. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nature Reviews Immunology. 2009;9(5):377–84. [PubMed: 19330016]
- Steele MP, Speer MC, Loyd JE, Brown KK, Herron A, Slifer SH, Burch LH, Wahidi MM, Phillips JA 3rd, Sporn TA, et al. Clinical and pathologic features of familial interstitial pneumonia. American Journal of Respiratory and Critical Care Medicine. 2005;172(9):1146–52. [PMC free article: PMC2718398] [PubMed: 16109978]
- Stoel BC, Putter H, Bakker ME, Dirksen A, Stockley RA, Piitulainen E, Russi EW, Parr D, Shaker SB, Reiber JH, et al. Volume correction in computed tomography densitometry for follow-up studies on pulmonary emphysema. Proceedings of the American Thoracic Society. 2008;5(9):919–24. [PubMed: 19056717]
- Stolk J, Stockley RA, Stoel BC, Cooper BG, Piitulainen E, Seersholm N, Chapman KR, Burdon JG, Decramer M, Abboud RT, et al. Randomised controlled trial for emphysema with a selective agonist of the gamma-type retinoic acid receptor. European Respiratory Journal. 2012;40(2):306–12. [PubMed: 22282548]
- Stone PJ, Gottlieb DJ, O'Connor GT, Ciccolella DE, Breuer R, Bryan-Rhadfi J, Shaw HA, Franzblau C, Snider GL. Elastin and collagen degradation products in urine of smokers with and without chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1995;151:952–9. [PubMed: 7697272]
- Strine TW, Balluz LS, Ford ES. The associations between smoking, physical inactivity, obesity, and asthma severity in the general US population. Journal of Asthma. 2007;44(8):651–8. [PubMed: 17943577]
- Stuart-Harris CH. The epidemiology and evolution of chronic bronchitis. British Journal of Tuberculosis and Diseases of the Chest. 1954;48(3):169–78. [PubMed: 13182194]
- Sturm JJ, Yeatts K, Loomis D. Effects of tobacco smoke exposure on asthma prevalence and medical care use in North Carolina middle school children. American Journal of Public Health. 2004;94(2):308–13. [PMC free article: PMC1448248] [PubMed: 14759947]
- Suzuki K, Tanaka H, Kaneko S, Nishi M, Teramoto S, Itoh S, Abe S. Respiratory symptoms and cigarette smoking in 3,197 pulmonologist-based asthmatic patients with a highly prevalent use of inhaled corticosteroid. Journal of Asthma. 2003;40(3):243–50. [PubMed: 12807167]
- Tan CE, Glantz SA. Association between smoke-free legislation and hospitalizations for cardiac, cerebrovascular, and respiratory diseases: a meta-analysis. Circulation. 2012;126(18):2177–83. [PMC free article: PMC3501404] [PubMed: 23109514]
- Taskar VS, Coultas DB. Is idiopathic pulmonary fibrosis an environmental disease? Proceedings of the American Thoracic Society. 2006;3(4):293–8. [PubMed: 16738192]
- Tekkel M, Rahu M, Loit HM, Baburin A. Risk factors for pulmonary tuberculosis in Estonia. International Journal of Tuberculosis and Lung Disease. 2002;6(10):887–94. [PubMed: 12365575]
- Thomas A, Gopi PG, Santha T, Chandrasekaran V, Subramani R, Selvakumar N, Eusuff SI, Sadacharam K, Narayanan PR. Predictors of relapse among pulmonary tuberculosis patients treated in a DOTS programme in South India. International Journal of Tuberculosis and Lung Disease. 2005;9(5):556–61. [PubMed: 15875929]
- Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, Sulem P, Rafnar T, Esko T, Walter S, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nature Genetics. 2010;42(5):448–53. [PMC free article: PMC3080600] [PubMed: 20418888]
- Thun MJ, Carter BD, Feskanich D, Freedman ND, Prentice R, Lopez AD, Hartge P, Gapstur SM. 50-year trends in smoking-related mortality in the United States. New England Journal of Medicine. 2013;368(4):351–64. [PMC free article: PMC3632080] [PubMed: 23343064]
- Thurlbeck WM, Ryder RC, Sternby N. A comparative study of the severity of emphysema in necropsy populations in three different countries. American Review of Respiratory Disease. 1974;109(2):239–48. [PubMed: 4811778]
- Tipayamongkholgul M, Podhipak A, Chearskul S, Sunakorn P. Factors associated with the development of tuberculosis in BCG immunized children. Southeast Asian Journal of Tropical Medicine and Public Health. 2005;36(1):145–50. [PubMed: 15906658]
- Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nature Genetics. 2010;42(5):441–7. [PMC free article: PMC2914600] [PubMed: 20418890]
- Tocque K, Bellis MA, Beeching NJ, Syed Q, Remmington T, Davies PD. A case-control study of lifestyle risk factors associated with tuberculosis in Liverpool, North-West England. European Respiratory Journal. 2001;18(6):959–64. [PubMed: 11829102]
- Tomlinson JE, McMahon AD, Chaudhuri R, Thompson JM, Wood SF, Thomson NC. Efficacy of low and high dose inhaled corticosteroid in smokers versus non-smokers with mild asthma. Thorax. 2005;60(4):282–7. [PMC free article: PMC1747368] [PubMed: 15790982]
- Toren K, Olin AC, Hellgren J, Hermansson BA. Rhinitis increase the risk for adult-onset asthma—a Swedish population-based case-control study (MAP-study). Respiratory Medicine. 2002;96(8):635–41. [PubMed: 12195846]
- Turino GM, Barker AF, Brantly ML, Cohen AB, Connelly RP, Crystal RG, Eden E, Schluchter MD, Stoller JK. Clinical features of individuals with PI*SZ phenotype of alpha-1-antitrypsin deficiency. American Journal of Respiratory and Critical Care Medicine. 1996;154:1718–25. [PubMed: 8970361]
- Tutor CG, Campbell JE. Association of obesity and smoking with chronic diseases among Oklahoma adults. Journal—Oklahoma State Medical Association. 2004;97(10):443–7. [PubMed: 15552241]
- U.S. Department of Health and Human Services. The Health Consequences of Smoking for Women. A Report of the Surgeon General. Washington: U.S. Department of Health and Human Services, Public Health Service, Office of the Assistant Secretary for Health, Office on Smoking and Health; 1980.
- U.S. Department of Health and Human Services. The Health Consequences of Smoking: Chronic Obstructive Lung Disease. A Report of the Surgeon General. Rockville (MD): U.S. Department of Health and Human Services, Public Health Service Office on Smoking and Health; 1984. DHHS Publication No. (PHS) 84-50205.
- U.S. Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General. Atlanta (GA): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004.
- U.S. Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. Atlanta (GA): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2006. [PubMed: 20669524]
- U.S. Department of Health and Human Services. How Tobacco Smoke Causes Disease—The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta (GA): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2010. [PubMed: 21452462]
- U.S. Department of Health, Education, and Welfare. Smoking and Health: Report of the Advisory Committee to the Surgeon General of the Public Health Service. Washington: U.S. Department of Health, Education, and Welfare, Public Health Service, Center for Disease Control; 1964. PHS Publication No. 1103.
- Van de Ven MO, Engels RC, Kerstjens HA, Van den Eijnden RJ. Bidirectionality in the relationship between asthma and smoking in adolescents: a population-based cohort study. Journal of Adolescent Health. 2007;41(5):444–54. [PubMed: 17950164]
- van Durme YM, Eijgelsheim M, Joos GF, Hofman A, Uitterlinden AG, Brusselle GG, Stricker BH. Hedgehog-interacting protein is a COPD susceptibility gene: the Rotterdam Study. European Respiratory Journal. 2010;36(1):89–95. [PubMed: 19996190]
- van Durme YM, Verhamme KM, Aarnoudse AJ, Van Pottelberge GR, Hofman A, Witteman JC, Joos GF, Brusselle GG, Stricker BH. C-reactive protein levels, haplotypes, and the risk of incident chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 2009;179(5):375–82. [PubMed: 19096002]
- Van Hove CL, Moerloose K, Maes T, Joos GF, Tournoy KG. Cigarette smoke enhances Th-2 driven airway inflammation and delays inhalational tolerance. Respiratory Research. 2008;9:42. [PMC free article: PMC2408577] [PubMed: 18489797]
- van Zyl Smit RN, Pai M, Yew WW, Leung CC, Zumla A, Bateman ED, Dheda K. Global lung health: the colliding epidemics of tuberculosis, tobacco smoking, HIV and COPD. European Respiratory Journal. 2010;35(1):27–33. [PMC free article: PMC5454527] [PubMed: 20044459]
- Vander Weg MW, Rosenthal GE, Vaughan Sarrazin M. Smoking bans linked to lower hospitalizations for heart attacks and lung disease among Medicare beneficiaries. Health Affairs. 2012;31(12):2699–707. [PubMed: 23213154]
- Vestbo J, Edwards LD, Scanlon PD, Yates JC, Agusti A, Bakke P, Calverley PM, Celli B, Coxson HO, Crim C, et al. Changes in forced expiratory volume in 1 second over time in COPD. New England Journal of Medicine. 2011;365(13):1184–92. [PubMed: 21991892]
- Vogelberg C, Hirsch T, Radon K, Dressel H, Windstetter D, Weinmayr G, Weiland SK, von Mutius E, Nowak D, Leupold W. Leisure time activity and new onset of wheezing during adolescence. European Respiratory Journal. 2007;30(4):672–6. [PubMed: 17596269]
- Wang H, Liu X, Umino T, Skold CM, Zhu Y, Kohyama T, Spurzem JR, Romberger DJ, Rennard SI. Cigarette smoke inhibits human bronchial epithelial cell repair processes. American Journal of Respiratory Cell and Molecular Biology. 2001;25(6):772–9. [PubMed: 11726404]
- Wang IM, Stepaniants S, Boie Y, Mortimer JR, Kennedy B, Elliott M, Hayashi S, Loy L, Coulter S, Cervino S, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. American Journal of Respiratory and Critical Care Medicine. 2008;177(4):402–11. [PubMed: 17975202]
- Wang Z, Zheng T, Zhu Z, Homer RJ, Riese RJ, Chapman HA Jr, Shapiro SD, Elias JA. Interferon gamma induction of pulmonary emphysema in the adult murine lung. Journal of Experimental Medicine. 2000;192(11):1587–600. [PMC free article: PMC2193095] [PubMed: 11104801]
- Washko GR, Dransfield MT, Estepar RS, Diaz A, Matsuoka S, Yamashiro T, Hatabu H, Silverman EK, Bailey WC, Reilly JJ. Airway wall attenuation: a biomarker of airway disease in subjects with COPD. Journal of Applied Physiology. 2009;107(1):185–91. [PMC free article: PMC2711787] [PubMed: 19407254]
- Washko GR, Hunninghake GM, Fernandez IE, Nishino M, Okajima Y, Yamashiro T, Ross JC, Estepar RS, Lynch DA, Brehm JM, et al. Lung volumes and emphysema in smokers with interstitial lung abnormalities. New England Journal of Medicine. 2011;364(10):897–906. [PMC free article: PMC3074462] [PubMed: 21388308]
- Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nature Medicine. 2006;12(3):317–23. [PubMed: 16474398]
- Wells JM, Washko GR, Han MK, Abbas N, Nath H, Mamary AJ, Regan E, Bailey WC, Martinez FJ, Westfall E, et al. Pulmonary arterial enlargement and acute exacerbations of COPD. New England Journal of Medicine. 2012;367(10):913–21. [PMC free article: PMC3690810] [PubMed: 22938715]
- Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, Borecki IB, Silverman EK, Weiss ST, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5(3):e1000429. [PMC free article: PMC2652834] [PubMed: 19300500]
- Wilk JB, Shrine NR, Loehr LR, Zhao JH, Manichaikul A, Lopez LM, Smith AV, Heckbert SR, Smolonska J, Tang W, et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. American Journal of Respiratory and Critical Care Medicine. 2012;186(7):622–32. [PMC free article: PMC3480517] [PubMed: 22837378]
- Wilson T, Shamo F, Boynton K, Kiley J. The impact of Michigan's Dr. Ron Davis smoke-free air law on levels of cotinine, tobacco-specific lung carcinogen and severity of self-reported respiratory symptoms among non-smoking bar employees. Tobacco Control. 2012;21(6):593–5. [PubMed: 22705599]
- World Health Organization. Global Tuberculosis Control—Surveillance, Planning, Financing. Geneva (Switzerland): World Health Organization; 2008.
- World Health Organization. Global Tuberculosis Control: WHO Report 2010. Geneva (Switzerland): World Health Organization; 2010.
- World Health Organization. Tuberculosis: fact sheet no. 104. 2013. [April 29, 2013]. < http://www
.who.int/mediacentre /factsheets/fs104/en/>. - World Health Organization and International Union Against Tuberculosis and Lung Disease. A WHO/The Union Monograph on TB and Tobacco Control: Joining Efforts to Control Two Related Global Epidemics. Geneva (Switzerland): World Health Organization; 2007.
- Wright JL. Emphysema: concepts under change—a pathologist's perspective. Modern Pathology. 1995;8(8):873–80. [PubMed: 8552579]
- Wright JL, Churg A. Smoke-induced emphysema in guinea pigs is associated with morphometric evidence of collagen breakdown and repair. American Journal of Physiology. 1995;268(1 Pt 1):L17–L20. [PubMed: 7840223]
- Yarnell JW, Stevenson MR, MacMahon J, Shields M, McCrum EE, Patterson CC, Evans AE, Manning PJ, Clancy L. Smoking, atopy and certain furry pets are major determinants of respiratory symptoms in children: the International Study of Asthma and Allergies in Childhood Study (Ireland). Clinical and Experimental Allergy. 2003;33(1):96–100. [PubMed: 12534556]
- Yoon HI, Sin DD. Biomarkers of therapeutic response in patients with chronic obstructive pulmonary disease: a critical review of the literature. Drugs. 2011;71(14):1821–37. [PubMed: 21942975]
- Yoshida T, Mett I, Bhunia AK, Bowman J, Perez M, Zhang L, Gandjeva A, Zhen L, Chukwueke U, Mao T, et al. Rtp801, a suppressor of mTOR signaling, is an essential mediator of cigarette smoke-induced pulmonary injury and emphysema. Nature Medicine. 2010;16(7):767–73. [PMC free article: PMC3956129] [PubMed: 20473305]
- Young RP, Hopkins RJ, Hay BA, Whittington CF, Epton MJ, Gamble GD. FAM13A locus in COPD is independently associated with lung cancer—evidence of a molecular genetic link between COPD and lung cancer. Application of Clinical Genetics. 2010;2011:1–10. [PMC free article: PMC3681173] [PubMed: 23776362]
- Young RP, Whittington CF, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Chromosome 4q31 locus in COPD is also associated with lung cancer. European Respiratory Journal. 2010;36(6):1375–82. [PubMed: 21119205]
- Yu GP, Hsieh CC, Peng J. Risk factors associated with the prevalence of pulmonary tuberculosis among sanitary workers in Shanghai. Tubercle. 1988;69(2):105–12. [PubMed: 3188230]
- Zeskind JE, Lenburg ME, Spira A. Translating the COPD transcriptome: insights into pathogenesis and tools for clinical management. Proceedings of the American Thoracic Society. 2008;5(8):834–41. [PMC free article: PMC2645236] [PubMed: 19017738]
- Zhang LX, Enarson DA, He GX, Li B, Chan-Yeung M. Occupational and environmental risk factors for respiratory symptoms in rural Beijing, China. European Respiratory Journal. 2002;20(6):1525–31. [PubMed: 12503714]
- Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ Jr, Chapman HA Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. Journal of Clinical Investigation. 2000;106(9):1081–93. [PMC free article: PMC301418] [PubMed: 11067861]
Footnotes
- 1
Pack-years = the number of years of smoking multiplied by the number of packs of cigarettes smoked per day.
- Respiratory Diseases - The Health Consequences of Smoking—50 Years of ProgressRespiratory Diseases - The Health Consequences of Smoking—50 Years of Progress
- TPT1 tumor protein, translationally-controlled 1 [Homo sapiens]TPT1 tumor protein, translationally-controlled 1 [Homo sapiens]Gene ID:7178Gene
- 7178[uid] AND (alive[prop]) (1)Gene
- POLR2E RNA polymerase II, I and III subunit E [Homo sapiens]POLR2E RNA polymerase II, I and III subunit E [Homo sapiens]Gene ID:5434Gene
- 5434[uid] AND (alive[prop]) (1)Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...