Affected Females
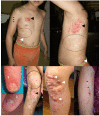
Ectodermal manifestations. The most characteristic features of PORCN-related developmental disorders are the skin manifestations (see ). The cutaneous findings typically follow the lines of Blaschko and include patchy areas of skin aplasia/hypoplasia, skin hypo- and/or hyperpigmentation, and nodular fat herniation. The lines of Blaschko represent cell migration pathways that are linear on the limbs and circumferential on the trunk. The lines are classically described as V shaped overlying the upper spine, S shaped on the abdomen, and an inverted-U shape from the breast area to the upper arms. These findings are typically evident at birth, but the distribution and severity may change over time. Unilateral involvement has also been reported [Bree et al 2016].
Other integumentary system abnormalities include wiry hair, sparse hair, patchy alopecia of the scalp, and abnormal nails. The nails can be absent (anonychia), small (micronychia), hypoplastic, or dysplastic, often with longitudinal ridging, splitting, or V-shaped nicking [Bree et al 2016].
Papillomatosis. Papillomas and telangiectasias are typically not present at birth but develop with age.
Verrucous papillomas are found in the oral mucosa of the mouth, nose, pharynx, larynx, trachea, and esophagus. Large papillomas of the larynx can obstruct breathing during anesthesia or can cause obstructive sleep apnea. Papillomas in the esophagus or larynx can also cause or contribute to severe gastroesophageal reflux disease (GERD).
The vaginal or rectal mucosa are also common sites for papillomas, where they can be confused with genital warts.
Dental abnormalities and eye findings, both of which result from abnormalities in ectodermal appendage development, are described separately below.
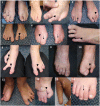
Limb and skeletal manifestations. Most individuals with a PORCN-related developmental disorder have limb malformations noted at birth, including syndactyly, oligodactyly, and split-hand/foot malformation or ectrodactyly (see and ). These malformations, which do not change over time, may impair function.
Additionally, reduction defects of the long bones ranging from leg length discrepancies to transverse defects of the distal radius/ulna or tibia/fibula are commonly seen.
Less common limb malformations that may be present at birth and impair function include camptodactyly (contraction deformities of the digits) and brachydactyly (shortening of the digits).
Costovertebral segmentation abnormalities including fused ribs, bifid ribs, hemivertebrae, and butterfly vertebrae are present at birth but are often not evident on physical examination and may only be seen on x-ray of the chest and/or spine. Although these malformations do not typically cause problems in infancy or early childhood, they may cause scoliosis as the child grows. Kyphosis or kyphoscoliosis is seen in approximately 10% of affected individuals [Smith & Hunt 2016]. More often, these segmentation abnormalities do not cause health issues.
Diastasis pubis, an abnormal separation of the symphysis pubis, may be an incidental finding or may present in adolescence or adulthood with pain. The gap between the pubic bones in the average non-pregnant adult is 4-5 mm. An abnormal gap is considered to be 1 cm or more, sometimes with the two bones being slightly out of alignment. In some individuals, diastasis pubis may cause pain with walking or in the symphysis pubis, legs, groin, and lower abdomen.
Fibrous dysplasia of bone (i.e., replacement of medullary bone with trabeculae of woven bone containing fluid-filled cysts embedded in a fibrous matrix) may affect any bone at any time. On x-ray the bone appears radiolucent, with what is classically described as a "ground-glass" appearance. Fibrous dysplasia may be asymptomatic or become evident when it is the site of a pathologic fracture.
Giant cell-like tumors of long bones, reported on occasion, may develop in childhood, adolescence, or adulthood. They typically become evident when a pathologic bone fracture occurs at the site of the lesion [Selzer et al 1974, Joannides et al 1983, Tanaka et al 1990]. In the small number of reports to date, none of these tumors has been malignant.
Osteopathia striata, a striated appearance of the bones evident on plain x-rays, is common and may be seen in childhood, adolescence, and adulthood. It is currently unclear if individuals with this finding are at increased risk for general osteoporosis. Of note, a spontaneous patella fracture related to osteoporosis in an individual with a PORCN-related developmental disorder has been reported [Altschuler et al 2012].
Eye findings. Developmental abnormalities of the eyes are common and are evident at birth; PORCN plays an important role in the development of the optic cup [Fuhrmann et al 2022]. Depending on the severity of the manifestations, vision can range from 20/20 to no light perception. Reported eye abnormalities include anophthalmia/microphthalmia; microcornea; iris, chorioretinal, and eyelid colobomas; lacrimal duct abnormalities; and cataracts (cortical and subcapsular) [Gisseman & Herce 2016].
Strabismus and/or nystagmus can be observed when visual impairment in infancy is significant.
Craniofacial findings. Facial features are variable and include facial asymmetry, notched alae nasi, pointed chin, and small, underfolded pinnae. These facial characteristics are not typically evident at birth but develop with time (see ) [Bostwick et al 2016].
Note facial features of pointed chin and small right ear.
Cleft lip and palate can be present and may lead to difficulty with feeding. More severe facial clefting can cause feeding, breathing, and vision problems, as well as significant cosmetic concerns [Wright et al 2016].
Oral and dental findings. Oral manifestations are seen in more than half of affected individuals and include both soft tissue and hard tissue abnormalities.
Enamel hypoplasia that predisposes to dental caries is the most common problem. Other findings include: hypodontia, oligodontia, supernumerary teeth, and dental crowding leading to malocclusion of both primary and secondary dentition; vertical grooving of the teeth; microdontia (small teeth); taurodontia (prism-shaped molars); fused teeth; and abnormal root morphology [Balmer et al 2004, Tejani et al 2005, Murakami et al 2011]. Affected individuals may also have problems with the eruption and position of teeth.
Soft tissue abnormalities include generalized gingivitis and intraoral lipomas and papillomas [Wright et al 2016].
Gastrointestinal and nutrition. Findings include poor weight gain (77% of individuals), short stature (65%), oral motor dysfunction (41%),GERD (24%), gastroparesis (35%), and constipation (35%) [Motil et al 2016, Hsu et al 2019]. Food allergies – primarily to milk, soy, and shellfish – are present in 12% of affected individuals.
Other developmental abnormalities of the digestive system are rare but may have severe consequences; they include abdominal wall defects and diaphragmatic hernia (see Congenital Diaphragmatic Hernia Overview).
Severe GERD has been reported in infancy and childhood, leading to feeding difficulties with frequent vomiting and/or discomfort/distress. GERD likely results from esophageal papillomas [Brinson et al 1987].
Renal and urogenital. Genital labial hypoplasia is present in most females [Adeyemi-Fowode et al 2016]. Occasional affected individuals with müllerian anomalies, including bicornuate uterus, have been described [Reddy & Laufer 2009, Lopez-Porras et al 2011]. Renal structural abnormalities are uncommon.
Cognitive and psychological. Development and intellectual ability are normal in most individuals. Intellectual impairment (15%-20% of individuals), behavioral issues (~20%), emotional lability (40%-50%), and withdrawn behavior (65%) have been reported [Deidrick et al 2016]. In those with emotional, behavioral, adaptive, and intellectual impairment, the spectrum of severity varies widely.
Structural brain abnormalities and spina bifida [Goltz et al 1970, Almeida et al 1988] have been reported but are uncommon.
Epilepsy has been reported [Kanemura et al 2011].
Other. Mixed conductive and sensorineural hearing loss has been reported on occasion.
An adult female with multiple cutaneous basal cell carcinomas has been reported. Whether basal cell carcinoma is more prevalent in individuals with a PORCN-related developmental disorder is currently unknown, but heightened surveillance and appropriate treatment for such lesions may be indicated [Patrizi et al 2012].
When the first affected female in the family has milder manifestations than affected females in subsequent generations [Heinz et al 2019], it is most likely that she has either mosaicism for the PORCN pathogenic variant or skewing of X-chromosome inactivation. Alternative explanations could be reduced reproductive fitness in severely affected females, such that only mildly affected females reproduce.