Clinical Description
Laing distal myopathy is characterized by muscle weakness and atrophy beginning in the lower legs [Lamont et al 2006]. Onset is often before age five years. In a few children, onset has been so early as to delay walking. In two families, weakness was not recognized until the teenage years [Zimprich et al 2000, Hedera et al 2003, Lamont et al 2006]. In one family with 20 affected members, onset of lower-limb weakness occurred between early childhood and the fourth decade [Lamont et al 2014]. Onset as late as the sixth decade has been described [Hara et al 2019].
More than 200 individuals have been identified with a pathogenic variant in MYH7 associated with Laing distal myopathy. The following description of the phenotypic features associated with this condition is based on the reports of Lamont et al [2014], Fiorillo et al [2016], and Dabaj et al [2018].
Table 2.
Laing Distal Myopathy: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| Lower leg muscle weakness & atrophy | 100% | |
| Finger extensor weakness | 100% | Variable time of onset |
| Mild facial weakness | 80% | |
| Neck flexor weakness | 100% | |
| Proximal muscle weakness | 100% | |
| Spinal manifestations | ~30% | |
| Cardiac problems | 30% | |
Lower leg weakness follows a typical sequence: initially dorsiflexion of the ankle and great toe is affected and leads to a high-stepping gait, dropped big toe, and secondary tightening of the Achilles tendon (see ).
Early development of anterior compartment weakness has led to marked tightening of the Achilles tendon bilaterally, with the affected individual unable to place his heels on the ground.
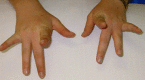
Weakness of finger extensors develops between months and several decades after the onset of leg weakness [Lamont et al 2014]. The third and fourth fingers appear to be more severely affected than the other fingers (see ), although any of the fingers can be affected. The thumb is spared. Weakness of the finger extensors is often accompanied by a postural and action tremor of the hands.
Individual with Laing distal myopathy attempting to extend her second to fifth fingers. Note marked weakness of third- and fourth-finger extension.
Mild facial weakness is often present, leading to inability to bury the eyelashes completely when closing the eyes tightly, and inability to keep the lips pursed against resistance. One affected individual has a mild Bell's phenomenon.
Weakness of neck flexion, seen in all affected individuals, is usually early in onset, though weakness of neck flexion did not manifest in one family until the sixth decade. In most affected individuals and sites, the weakness is symmetric.
Proximal weakness. After distal weakness has been present for more than ten years, mild proximal weakness occurs, with a slight Trendelenburg gait and mild scapular winging (see ). Axial musculature may be mildly weak as well (manifesting as, e.g., inability to do a sit-up).
Mild scapular winging and weakness develops later.
Progression is usually extremely slow; however, in one person the weakness became generalized and a wheelchair was required for mobility by age 15 years [Lamont et al 2014].
Spinal manifestations, which can include kyphoscoliosis, spinal rigidity, and spinal extensor muscle contractures, occur in one third of individuals and can vary within a family [Feinstein-Linial et al 2016, Fiorillo et al 2016]. Severe axial involvement with scoliosis, cervical hyperextension, and bent spine has been described [Dabaj et al 2018].
Cardiac problems are common. In their review of 88 affected individuals from 22 families, Lamont et al [2014] reported cardiac involvement ranging from hypertrophic cardiomyopathy with onset from birth to the third decade of life, to dilated cardiomyopathy with onset from birth to the second decade of life. In an earlier report, a father and son in one family developed a dilated cardiomyopathy for which no other cause was found [Hedera et al 2003].
Respiratory issues, present in approximately 40% of individuals in the form of reduced forced vital capacity, are not usually life threatening [Lamont et al 2014]. Sleep apnea or sleep-related respiratory insufficiency may develop [Yu et al 2020].
CNS involvement with white matter lesions and epilepsy has been described in a single family including three of 14 family members over three generations [Lefter et al 2015].
Muscle pathology in Laing distal myopathy is highly variable [Lamont et al 2006, Lamont et al 2014].
Penetrance
Penetrance appears to be at least 85%.
Muelas et al [2010] reported a large Spanish family in which the age of onset ranged from birth to the sixth decade; 15% of family members with the pathogenic variant were reported to be asymptomatic. (Note, however, that individual ages at the time of reporting were not clearly stated.)
In one apparent instance of a de novo pathogenic variant, the supposedly unaffected father was found to have somatic mosaicism; however, when examined, he did have mild weakness [Lamont et al 2014].
Prevalence
The prevalence of Laing distal myopathy is unknown. It is thought to be the most common distal myopathy worldwide [B Udd, personal communication], accounting for approximately 50% of early-onset distal myopathy [Author, personal observation]. The frequency of de novo pathogenic variants would also suggest a relatively high prevalence.
Laing distal myopathy has been reported in most populations [Park et al 2013, Lamont et al 2014, Hara et al 2019] and does not appear to be more prevalent in any specific populations [Author, personal observation].