Clinical Description
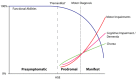
During the prodromal phase of Huntington disease (HD) individuals may have subtle changes in motor skills, cognition, and personality (see ) [Tabrizi et al 2013, Ross et al 2014, Liu et al 2015]. These subtle changes can occur as early as 15-20 years prior to the clinical onset of manifest HD.
Natural history of Huntington disease (HD). Presymptomatic individuals are free from signs and symptoms of HD. During the prodromal phase, subtle signs and symptoms may be present prior to the diagnosis of HD, which is usually based on motor symptoms. (more...)
Reilmann et al [2014] present guidelines for the diagnostic criteria of presymptomatic, prodromal, and manifest HD; see Table 2. This table can be used to place individuals into different diagnostic categories, which may have clinical management implications over time. For example, awareness of presymptomatic and prodromal HD may allow for preventive (rather than symptomatic) therapies. Note the clear differentiation of genetically confirmed HD in the classification system.
Table 2.
Categories of Huntington Disease Diagnosis
View in own window
| HD Classification | HD Signs/Symptoms |
|---|
| Genetically Confirmed | NOT Genetically Confirmed |
|---|
| Presymptomatic HD: HD, genetically confirmed, presymptomatic | Clinically at risk for HD: HD, not genetically confirmed, clinically at risk | No clinical motor signs/symptoms (motor DCL = 0 or 1) No cognitive signs/symptoms May have changes in imaging, quantitative motor assessments, or other biomarkers No symptomatic treatment indicated Disease-modifying treatment when safe & available
|
| Prodromal HD: HD, genetically confirmed, prodromal | Clinically prodromal HD: HD, not genetically confirmed, clinically prodromal | Subtle motor signs (usually motor DCL = 2) AND/OR subtle cognitive signs or symptoms Minor decline from individual premorbid level of function possibly detectable, but not required & not detectable on TFC Apathy or depression or other behavioral changes judged related to HD may be present. Usually changes in imaging & quantitative motor assessments May/may not require symptomatic treatment (e.g., for depression) Disease-modifying treatment appropriate
|
| Manifest
HD: HD, genetically confirmed, manifest | Clinically manifest HD: HD, not genetically confirmed, clinically manifest 1 | Presence of clinical motor &/or cognitive signs & symptoms that have an impact on life, with: Functional changes (e.g., ↓ TFC); Motor DCL = 3 or 4 (or motor DCL of 2 if cognitive changes significant AND evidence of progression)
Symptomatic & disease-modifying treatment appropriate
|
DCL = diagnostic confidence level (from the UHDRS rating scale); HD = Huntington disease; TFC = total functional capacity
- 1.
Requires motor DCL = 4, plus cognitive changes
The mean age of onset for HD is approximately 45 years [Bates et al 2015]. About two thirds of affected individuals first present with neurologic manifestations; others present with psychiatric changes. In the early stages following diagnosis, manifestations include subtle changes in eye movements, coordination, minor involuntary movements, difficulty in mental planning, and often a depressed or irritable mood (see Table 3). Affected individuals are usually able to perform most of their ordinary activities and to continue working [Ross et al 2014, Bates et al 2015].
In approximately 25% of individuals with HD, the onset is delayed until after age 50 years, with some after age 70 years. These individuals have chorea, gait disturbances, and dysphagia, but a more prolonged and benign course than the typical individual.
In the next stage, chorea becomes more prominent, voluntary activity becomes increasingly difficult, and dysarthria and dysphagia worsen. Most individuals are forced to give up their employment and depend increasingly on others for help, although they are still able to maintain a considerable degree of personal independence. The impairment is usually considerable, sometimes with intermittent outbursts of aggressive behaviors and social disinhibition.
In late stages of HD, motor disability becomes severe and the individual is often totally dependent, mute, and incontinent. The median survival time after onset is 15 to 18 years (range: 5 to >25 years). The average age at death is 54 to 55 years [Bates et al 2015].
Table 3.
Onset of Clinical Signs and Symptoms in HD
View in own window
| Clinical Signs in HD |
|---|
|
Early
| Clumsiness Agitation Irritability Apathy Anxiety Disinhibition Delusions Hallucinations Abnormal eye movements Depression Olfactory dysfunction
|
|
Middle
| Dystonia Involuntary movements Trouble w/balance & walking Chorea, twisting & writhing motions, jerks, staggering, swaying, disjointed gait (can seem like intoxication) Trouble w/activities that require manual dexterity Slow voluntary movements; difficulty initiating movement Inability to control speed & force of movement Slow reaction time General weakness Weight loss Speech difficulties Stubbornness
|
|
Late
| Rigidity Bradykinesia (difficulty initiating & continuing movements) Severe chorea (less common) Significant weight loss Inability to walk Inability to speak Swallowing difficulties; danger of choking Inability to care for oneself
|
Abnormalities of movement. Disturbances of both involuntary and voluntary movements occur in individuals with HD. Chorea, an involuntary movement disorder consisting of nonrepetitive, nonperiodic jerking of limbs, face, or trunk, is the major sign of the disease. Chorea is present in more than 90% of individuals, and typically increases in severity during the first ten years. The choreic movements are continuously present during waking hours, cannot be suppressed voluntarily, and are worsened by stress.
With advancing disease duration, other involuntary movements such as bradykinesia, rigidity, and dystonia occur. Impairment in voluntary motor function is an early sign. Affected individuals and their families describe clumsiness in common daily activities. Motor speed, fine motor control, and gait are affected. Oculomotor disturbances occur early and worsen progressively. Difficulty in initiating ocular saccades, slow and hypometric saccades, and problems in gaze fixation may be seen in up to 75% of symptomatic individuals [Blekher et al 2006, Golding et al 2006]. Dysarthria occurs early and is common. Dysphagia occurs in the late stages. Hyperreflexia occurs early in 90% of individuals, while clonus and extensor plantar responses occur late and less frequently.
Abnormalities of cognition. A global and progressive decline in cognitive capabilities occurs in all individuals with HD. Cognitive changes include forgetfulness, slowness of thought processes, impaired visuospatial abilities, and impaired ability to manipulate acquired knowledge. Several studies have identified subtle but definite cognitive deficits prior to the onset of motor symptoms [Bourne et al 2006, Montoya et al 2006, Paulsen et al 2008, Tabrizi et al 2009, Rupp et al 2010]. The initial changes often involve loss of mental flexibility and impairment of executive functions such as mental planning and organization of sequential activities.
Memory deficits with greater impairment for retrieval of information occur early, but verbal cues, priming, and sufficient time may lead to partial or correct recall. Early in the disease the memory deficits in HD are usually much less severe than in Alzheimer disease.
The overall cognitive and behavioral syndrome in individuals with HD is more similar to frontotemporal dementia than to Alzheimer disease. Attention and concentration are involved early [Peinemann et al 2005], resulting in easy distractibility. Language functions are relatively preserved, but a diminished level of syntactic complexity, cortical speech abnormalities, paraphasic errors, and word-finding difficulties are common in late stages.
Neuropsychologic testing reveals impaired visuospatial abilities, particularly in late stages of the disease. Lack of awareness, especially of one's own disabilities, is common [Ho et al 2006, Bates et al 2015].
Psychiatric disturbances. Individuals with HD develop significant personality changes, affective psychosis, or schizophrenic psychosis [Rosenblatt 2007]. Prior to onset of HD, they tend to score high on measures of depression, hostility, obsessive-compulsiveness, anxiety, and psychoticism [Duff et al 2007]. Unlike the progressive cognitive and motor disturbances, the psychiatric changes tend not to progress with disease severity [Epping et al 2016]. Behavioral disturbances such as intermittent explosiveness, apathy, aggression, alcohol abuse, sexual dysfunction and deviations, and increased appetite are frequent. Delusions, often paranoid, are common. Hallucinations are less common.
Depression and suicide risk. The incidence of depression in preclinical and symptomatic individuals is more than twice that in the general population [Paulsen et al 2005b, Marshall et al 2007]. The etiology of depression in HD is unclear; it may be a pathologic rather than a psychological consequence of having the disease [Slaughter et al 2001, Pouladi et al 2009]. Suicide and suicide ideation are common in persons with HD, but the incidence rate changes with disease course and predictive testing results [Larsson et al 2006, Robins Wahlin 2007, van Duijn et al 2018]. The critical periods for suicide risk were found to be just prior to receiving a diagnosis and later, when affected individuals experience a loss of independence [Baliko et al 2004, Paulsen et al 2005a, Eddy et al 2016].
Other. Persons with HD tend to have a lower body mass index than controls [Pratley et al 2000, Stoy & McKay 2000, Djoussé et al 2002, Robbins et al 2006], which may be related to altered metabolism [Duan et al 2014] and could represent a biomarker of clinical progression [van der Burg et al 2017]. Individuals with HD also demonstrate disturbed cholesterol metabolism [Valenza & Cattaneo 2006, Wang et al 2014]. It is also common for persons with HD to demonstrate increased appetite and energy expenditure [Pratley et al 2000, Trejo et al 2004, Gaba et al 2005].
Sleep and circadian rhythms are disrupted in individuals with HD [Goodman & Barker 2010, Morton 2013], possibly as a result of hypothalamic dysfunction [Petersén & Björkqvist 2006] and/or alterations in melatonin secretion [Kalliolia et al 2014]. Insomnia and daytime somnolence may also be present, although this is more commonly due to psychiatric changes, depression, or chorea [Videnovic et al 2009].
Neuropathology. The primary neuropathologic feature of HD is degeneration of neurons in the caudate and putamen as well as the cerebral cortex [Waldvogel et al 2015]. The preferential degeneration of enkephalin-containing, medium spiny neurons of the indirect pathway of movement control in the basal ganglia provides the neurobiologic basis for chorea [Galvan et al 2012]. The additional loss of substance P-containing medium spiny neurons of the direct pathway results in akinesia and dystonia [Galvan et al 2012]. There is also evidence for loss of neurons in the globus pallidus, subthalamic nucleus, thalamus, hypothalamus, substantia nigra, and hippocampus [Vonsattel et al 1985, Vonsattel & DiFiglia 1998, Heinsen et al 1999, Petersén et al 2005, Guo et al 2012, Domínguez et al 2013, Singh-Bains et al 2016]. Region-specific patterns of neuronal loss in the basal ganglia and cortex may underlie the most evident symptoms in affected individuals and could contribute to the phenotypic variability among individuals [Thu et al 2010, Hadzi et al 2012, Kim et al 2014, Waldvogel et al 2015, Mehrabi et al 2016]. Pathology is also observed in peripheral tissues [Björkqvist et al 2008, van der Burg et al 2009].
Intraneuronal inclusions containing huntingtin, the protein expressed from HTT, are also a prominent neuropathologic feature of the disease. However, the expression of the huntingtin protein and the pattern and timing of huntingtin-containing inclusions in the brain do not correlate with the selective degeneration of the disease and are not believed to be primary determinants of pathology [Kuemmerle et al 1999, Michalik & Van Broeckhoven 2003, Arrasate et al 2004, Slow et al 2005, Slow et al 2006].
Neuroimaging. Imaging studies provide additional support for the clinical diagnosis of HD and are valuable tools for studying progression of the disease [Biglan et al 2009, Paulsen 2009, Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]. In addition to significant striatal atrophy in symptomatic individuals, regional and whole-brain gray and white matter changes have been detected [Majid et al 2011, Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]. Furthermore, MRI studies have revealed progressive gray and white matter atrophy many years prior to predicted disease onset [Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]. Numerous studies in recent years have used neuroimaging to elucidate the clinical progression of HD, with the specific interest of using these objective measures in clinical trials for testing efficacy of experimental therapeutics [Tabrizi et al 2012, Tabrizi et al 2013].
Juvenile HD is defined by the onset of symptoms before age 20 years and accounts for 5%-10% of individuals with HD [Gonzalez-Alegre & Afifi 2006, Quarrell et al 2013]. The motor, cognitive, and psychiatric disturbances observed in adult HD are also observed in juvenile HD, but the clinical presentation of these disturbances is different. Severe mental deterioration, prominent motor and cerebellar symptoms, speech and language delay, and rapid decline are also characteristic of juvenile HD [Nance & Myers 2001, Gonzalez-Alegre & Afifi 2006, Squitieri et al 2006, Yoon et al 2006]. Epileptic seizures, unique to the youngest-onset group, are present in 30%-50% of those with onset of HD before age ten years [Gonzalez-Alegre & Afifi 2006].
In teenagers, symptoms are more similar to adult HD, in which chorea and severe behavioral disturbances are common initial manifestations [Nance & Myers 2001].
Intermediate alleles (IA). An individual with a CAG repeat in the 27-35 range is not believed to be at risk of developing HD but, because of instability in the CAG tract, may be at risk of having a child with an allele in the pathogenic CAG range [Semaka et al 2006, Kay et al 2018]. Limited data suggest that individuals with IAs may exhibit behavioral changes as well as motor and cognitive impairments, although more research is required in this regard [Killoran et al 2013, Cubo et al 2016].