Summary
Clinical characteristics.
Hypophosphatasia is characterized by defective mineralization of growing or remodeling bone, with or without root-intact tooth loss, in the presence of low activity of serum and bone alkaline phosphatase. Clinical features range from stillbirth without mineralized bone at the severe end to pathologic fractures of the lower extremities in later adulthood at the mild end. While the disease spectrum is a continuum, seven clinical forms of hypophosphatasia are usually recognized based on age at diagnosis and severity of features:
Perinatal (severe): Characterized by pulmonary insufficiency and hypercalcemia
Perinatal (benign): Prenatal skeletal manifestations that slowly resolve into one of the milder forms
Infantile: Onset between birth and age six months of clinical features of rickets without elevated serum alkaline phosphatase activity
Severe childhood (juvenile): Variable presenting features progressing to rickets
Mild childhood: Low bone mineral density for age, increased risk of fracture, and premature loss of primary teeth with intact roots
Adult: Characterized by stress fractures and pseudofractures of the lower extremities in middle age, sometimes associated with early loss of adult dentition
Odontohypophosphatasia: Characterized by premature exfoliation of primary teeth and/or severe dental caries without skeletal manifestations
Management.
Targeted therapy: Asfotase alfa (Strensiq®) enzyme replacement therapy (ERT) has been shown to improve pulmonary function, calcium homeostasis / bone health, and survival in individuals with the infantile and early childhood (juvenile) type of hypophosphatasia. There is growing experience with ERT in individuals with the perinatal (severe) type and emerging experience with ERT in treating osteomalacia in adults.
Supportive care: For the perinatal (severe) type: expectant management and family support; respiratory support; management of calcium homeostasis and bone health per endocrinologist and orthopedist; pain management; neurosurgical management of craniosynostosis; management of kidney disease per nephrologist; dental care. For the infantile and early childhood (juvenile) types: respiratory support; management of calcium homeostasis and bone health per endocrinologist and orthopedist; pain management; treatment of seizures with vitamin B6; neurosurgical management of craniosynostosis; management of kidney disease per nephrologist; dental care. For all other types: dental care starting at age one year; nonsteroidal anti-inflammatory drugs for osteoarthritis, bone pain, and osteomalacia; internal fixation for pseudofractures and stress fractures. In adult hypophosphatasia, there is limited experience in treating osteomalacia with teriparatide.
Surveillance: Monitor calcium homeostasis and bone health per endocrinologist, nephrologist, and orthopedist; physical medicine and rehabilitation, physical therapy, and occupational therapy evaluations as needed; monitor children with infantile type for increased intracranial pressure secondary to craniosynostosis; nephrology evaluations as needed for kidney disease; neurology evaluations as needed for seizures; dental visits twice yearly starting at age one year.
Agents/circumstances to avoid: Bisphosphonates and excess vitamin D; teriparatide is contraindicated in children.
Pregnancy management: The use of asfotase alfa (Strensiq®) ERT during human pregnancy has not been extensively studied; therefore, any potential risk to the fetus of a pregnant woman taking this therapy during pregnancy is unknown.
Genetic counseling.
Perinatal and infantile hypophosphatasia are typically inherited in an autosomal recessive manner. The milder forms, especially adult and odontohypophosphatasia, may be inherited in an autosomal recessive or autosomal dominant manner depending on the effect that the ALPL pathogenic variant has on TNSALP (alkaline phosphatase, tissue-nonspecific isozyme) activity.
Autosomal recessive hypophosphatasia: If both parents are known to be heterozygous for an ALPL pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting biallelic pathogenic variants and being affected, a 50% chance of being heterozygous, and a 25% chance of inheriting neither of the familial pathogenic variants. Depending on the ALPL pathogenic variant, heterozygous sibs may be either clinically asymptomatic (manifesting only biochemical abnormality) or have milder clinical symptoms than the proband.
Autosomal dominant hypophosphatasia: All individuals reported to date with hypophosphatasia caused by a heterozygous ALPL variant with a dominant-negative effect inherited the ALPL pathogenic variant from a parent (who may or may not have clinical manifestations of hypophosphatasia). Unless an individual with autosomal dominant hypophosphatasia has children with an individual who has a heterozygous or biallelic ALPL pathogenic variant(s), offspring have a 50% chance of inheriting the ALPL pathogenic variant.
Once the ALPL pathogenic variant(s) have been identified in an affected family member, heterozygote testing for at-risk relatives, prenatal testing, and preimplantation genetic testing for hypophosphatasia are possible. Recurrence of perinatal and infantile hypophosphatasia may reliably be identified by prenatal ultrasound examination.
Diagnosis
No consensus clinical diagnostic criteria for hypophosphatasia have been published.
Suggestive Findings
Hypophosphatasia should be suspected in probands with the following clinical, laboratory, and radiographic features.
Clinical features
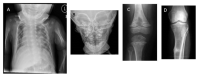
Clinical features of infantile rickets: growth failure, craniotabes, craniosynostosis, blue sclerae, flail chest, costochondral enlargement ("rachitic rosary"), scoliosis, thickening of wrists, knees, and ankles, bowing of legs, lax ligaments, and hypotonia
Premature loss of deciduous teeth beginning with the incisors. Unusually and characteristically, the dental root remains attached to the lost tooth. Dental caries and early loss or extraction of adult teeth is also seen (see ).
Vitamin B6 (pyridoxine)-responsive seizures
Bone pain
Lost incisors with and without hypophosphatasia A. Hypophosphatasia: root intact
Laboratory features
Hypercalciuria particularly during the first year of life with or without hypercalcemia
Typically normal serum calcium and ionized calcium. Note: May be elevated, particularly in the first year of life.
Typically normal serum and urine inorganic phosphate. Note: May be elevated.
Normal serum vitamin D (25-hydroxy and 1,25-dihydroxy) and parathyroid hormone
Elevated plasma vitamin B6 without oral supplementation
Elevated serum pyridoxal 5'-phosphate (PLP), a biologically active metabolite of vitamin B6. Note: (1) Reference laboratories may measure PLP and report as "vitamin B6." (2) Use of multivitamin or calcium supplements containing vitamin B6 within a week of assaying serum PLP may lead to false positive results.
Elevated urine phosphoethanolamine (PEA) and proline on urine amino acid chromatogram. Note: (1) Urine PEA may be elevated with other metabolic bone diseases. (2) Urine PEA may be normal in affected individuals and can be elevated in asymptomatic heterozygotes.
Elevated urine inorganic pyrophosphate (PPi). Note: (1) Assay is not available in North American clinical laboratories. (2) Asymptomatic heterozygotes can have elevated urine PPi.
Reduced serum unfractionated alkaline phosphatase (ALP) activity. Note: (1) Transient increases in serum ALP activity can occur during pregnancy, with liver disease, and after acute fracture or surgery. Thus, serial measurements may be necessary in toddlers with unexplained fractures. Quantitation of the activity of the bone isoform of ALP in serum may be necessary in the setting of liver disease. The bone isoform is heat labile; the liver isoform is heat stable. (2) Asymptomatic heterozygotes can have reduced serum ALP activity.
Radiographic features
Prenatal long bone bowing with osteochondral spurs
Infantile rickets: undermineralized bones, widened-appearing sutures, brachycephaly, rachitic costochondral rib changes (see ), flared metaphyses, poorly ossified epiphyses, and bowed long bones
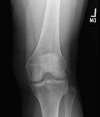
Focal bony defects of the metaphyses resembling radiolucent "tongues" (see ) are fairly specific for childhood hypophosphatasia.
Defective mineralization of growing/remodeling bone and/or teeth. Bone mineral content increases with age, and there may be improved mineralization during adolescence with decreased mineralization in middle age.
Alveolar bone loss resulting in premature loss of deciduous teeth typically involving the anterior mandible, with the central incisors lost first. However, any tooth may be affected (see ).
Pathologic fractures. Growing children may have a predilection to metaphyseal fractures; however, epiphyseal and diaphyseal fractures are also seen. In adults, metatarsal stress fractures and femoral pseudofractures prevail.
Osteomalacia with lateral pseudofractures ("Looser zones") in adult hypophosphatasia (see )
Radiographic signs of hypophosphatasia A. Rachitic rib changes, flail chest, and metaphyseal dysplasia (proximal humerus) in infantile hypophosphatasia
Establishing the Diagnosis
The clinical diagnosis of hypophosphatasia can be established in a proband with suggestive clinical, radiographic, and laboratory features by identification of reduced serum unfractionated ALP activity.
The molecular diagnosis
can be established in a proband with suggestive findings by identification of ONE of the following on molecular genetic testing (see Table 1):
Note: (1) Individuals with a heterozygous loss-of-function ALPL variant can have mild features of adult hypophosphatasia [Mornet et al 2021] (see Clinical Description, heterozygous loss-of-function variants). (2) Identification of a biallelic or heterozygous ALPL variant(s) of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other skeletal dysplasias are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of ALPL is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A multigene panel that includes ALPL and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other skeletal dysplasias, comprehensive
genomic testing (which does not require the clinician to determine which gene is likely involved) is likely the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hypophosphatasia
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
ALPL
| Sequence analysis 3 | ~95% 4 |
| Gene-targeted deletion/duplication analysis 5 | <5% 6 |
| Unknown 7 | NA | <1% |
- 1.
- 2.
- 3.
- 4.
In individuals with severe (perinatal and infantile) hypophosphatasia, biallelic ALPL pathogenic variants are identified in approximately 95% of individuals of European ancestry. In other clinical phenotypes, the proportion of pathogenic variants detected is difficult to estimate.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
- 7.
Anecdotal reports of individuals with clinical and biochemical features of adult hypophosphatasia with no detected ALPL pathogenic variant(s) suggest a potential second locus, not yet identified.
Clinical Characteristics
Clinical Description
Hypophosphatasia is characterized by defective mineralization of bone and/or teeth and reduced serum alkaline phosphatase (ALP). The phenotypic spectrum ranges from stillbirth without mineralized bone at the severe end to pathologic stress fractures of the lower extremities in older adults at the mild end (Table 2). Intrafamilial clinical variability is common, particularly when some affected family members have a heterozygous ALPL pathogenic variant and other affected family members have biallelic pathogenic variants. Sibs with compound heterozygous variants tend to display less clinical variability at the severe end of the spectrum and more variability at the milder end of the spectrum.
Table 2.
Select Clinical, Radiographic, and Laboratory Features of Hypophosphatasia by Type
View in own window
| Type | MOI | Cardinal Features | Additional Features |
|---|
Perinatal
(severe)
| AR | Hypomineralization Osteochondral spurs
| Long bone bowing Pretibial dimpling
|
Perinatal
(benign)
| AR/AD | Long bone bowing Benign postnatal course
| |
|
Infantile
| Mostly AR |
| Additional clinical & radiographic features of infantile rickets 1, 2 Alveolar bone loss (anterior mandible) Premature loss of deciduous teeth
|
Severe
childhood
(juvenile)
| AR/AD |
| Premature loss of deciduous teeth (incisors) |
Mild
childhood
| AR/AD | ↑ fractures | Premature loss of deciduous teeth (incisors) |
|
Adult
| AR/AD |
|
|
Odontohypo-
phosphatasia
| AR/AD | Alveolar bone loss | Exfoliation (incisors) Dental caries
|
- 1.
Clinical features of infantile rickets: growth failure, craniotabes, blue sclerae, scoliosis, thickening of wrists and ankles, bowing of lower extremities, lax ligaments, and hypotonia
- 2.
Radiographic features of infantile rickets: widened-appearing sutures, brachycephaly, flail chest, flared metaphyses, poorly ossified epiphyses, and bowed long bones in the lower extremities
Perinatal (severe) hypophosphatasia is typically identified by prenatal ultrasound examination. Pregnancies may end in stillbirth. Small thoracic cavity and short, bowed limbs are seen in both stillborn and live-born infants. A flail chest may be present (see ). Infants with perinatal hypophosphatasia may experience pulmonary insufficiency; restrictive lung disease is the most frequent cause of death. Hypercalcemia is common and may be associated with apnea or seizures. In those treated with asfotase alfa enzyme replacement therapy (ERT), a new phenotype of "treated perinatal and infantile hypophosphatasia" is emerging. However, even when the diagnosis is made expediently, unfavorable outcomes with ERT are possible [Duffus et al 2018]. Infants with perinatal (severe) hypophosphatasia started on ERT between age one day and age 78 months showed improvement in pulmonary function and survival. The effect of ERT on fractures remains unclear [Whyte et al 2019]. In the past, individuals with severe phenotypes died before dental eruption; emerging data suggest the possibility of dental features in infants treated with ERT.
Perinatal (benign) hypophosphatasia is typically identified by prenatal ultrasound examination showing short and bowed long bones but normal or slightly decreased mineralization. Postnatally, skeletal manifestations slowly resolve with a less severe hypophosphatasia phenotype [Wenkert et al 2011].
Infantile hypophosphatasia. There may be no clinical features apparent at birth. Clinical signs may be recognized between birth and age six months and resemble rickets (see ). Clinical severity depends on the degree of pulmonary insufficiency; the infantile phenotype has high mortality. Prior to the availability of ERT, 50% of individuals succumbed to respiratory failure caused by undermineralization of the ribs. Other complications include hypercalcemia, irritability, poor feeding, failure to thrive, hypotonia, and more rarely vitamin B6-responsive seizures (see Management). Open fontanels and wide sutures may be deceptive, in that the hypomineralized bone causing this radiographic appearance is prone to premature fusion. Craniosynostosis and intracranial hypertension are potential complications. Older children may have kidney damage. Clinical trials with ERT have shown improvement in developmental milestones and pulmonary function (see ) [Whyte et al 2019].
Radiograph of treated hypophosphatasia. Individual from Figure 2A after 12 months of asfotase alfa enzyme replacement therapy. Note tracheostomy tube, placed for laryngomalacia and bronchomalacia, features of the treated disease. Rachitic rib and metaphyseal (more...)
Severe childhood (juvenile) hypophosphatasia displays wide variability in initial clinical presentation but often progresses to rickets. More severely affected toddlers have short stature and delay in walking, developing a waddling myopathic gait. Bone and joint pain are typical. Diaphyseal and metaphyseal fractures may occur. Gait, six-minute walk test, and step length improved in individuals treated with ERT. To date, data are insufficient to assess the effect of ERT on fractures in juvenile hypophosphatasia [Whyte et al 2016].
Mild childhood hypophosphatasia is characterized by low bone mineral density for age with unexplained fractures. Children may have premature loss of deciduous teeth (prior to age 5 years), usually beginning with incisors, with the dental root characteristically remaining attached to the lost tooth. Bone and joint pain are atypical.
Adult hypophosphatasia is sometimes associated with a history of transient rickets in childhood and/or premature loss of deciduous teeth. Early loss of adult dentition is common. Other dental problems in adolescents and adults with hypophosphatasia are more poorly characterized, although enamel hypoplasia and tooth mobility have been described. Adult hypophosphatasia is usually recognized in middle age, the cardinal features being stress fractures and pseudofractures of the lower extremities. Foot pain and slow-to-heal stress fractures of the metatarsals are common. Thigh and hip pain may reflect pseudofractures ("Looser zones") in the lateral cortex of the femoral diaphysis (see ). Chondrocalcinosis and osteoarthropathy may develop with age (see ). Osteomalacia distinguishes adult hypophosphatasia from odontohypophosphatasia.
Radiograph of treated adult hypophosphatasia: linear sclerosis in remodeling distal femur and proximal tibia, osteophytes mid-proximal tibia, and chondrocalcinosis medial lateral compartment
Odontohypophosphatasia can be seen as an isolated finding without additional abnormalities of the skeletal system or can be variably seen in the above forms of hypophosphatasia. Caution should be exercised in citing extradental manifestations of other forms of hypophosphatasia in individuals with odontohypophosphatasia, in that such features may be common and multifactorial (e.g., low bone density for age). Premature exfoliation of primary teeth and/or severe dental caries may be seen, with the incisors most frequently lost.
Phenotype in those with heterozygous loss-of-function variants. Heterozygous loss-of-function ALPL variants have been identified in adults with osteoporosis, musculoskeletal pain, and an increased risk of fractures [Mornet et al 2021]. These individuals are ascertained by low serum ALP and tend to have additional biochemical evidence of hypophosphatasia (elevated serum pyridoxal 5'-phosphate [PLP] or urine phosphoethanolamine [PEA]). Those ascertained as an incidental finding on molecular testing have lower ALP activity but may not display additional biochemical evidence. In this latter circumstance, elevated serum PLP or urine PEA may predict disease potential.
Histopathology
Bone histology reveals rachitic abnormalities of the growth plate. Histochemical testing of osteoclasts reveals lack of membrane-associated ALP activity. Osteoclasts and osteoblasts otherwise appear normal.
Tooth histology reveals a decrease in cementum, which varies with the severity of the disease.
Nomenclature
Hypophosphatasia takes its name from low activity of the enzyme ALP, rather than reflecting serum concentration of phosphorus.
In classifications of genetic conditions, hypophosphatasia may be considered a metabolic bone disease, a skeletal dysplasia, a metaphyseal dysplasia, a dental disorder, or a disorder of membrane-bound ectoenzyme activity in the extracellular matrix.
Prevalence
Based on pediatric hospital records in Ontario, Canada, the birth prevalence of (autosomal recessive) perinatal and infantile hypophosphatasia was estimated at 1:100,000 [Fraser 1957]. Applying the Hardy-Weinberg equation to this estimate, the frequency of heterozygotes for ALPL pathogenic variants in Ontario, Canada, is about 1:150.
In the Canadian Mennonite population, the prevalence of the perinatal (severe) form is 1:2,500 (carrier frequency 1:25) due to founder the variant p.Gly334Asp [Triggs-Raine et al 2016].
On the basis of molecular diagnosis in France and elsewhere in Europe, the prevalence of severe forms has been estimated at 1:300,000. For mild forms (perinatal benign, mild childhood, adult, and odontohypophosphatasia), the prevalence is expected to be as high as 1:6,300 [Mornet et al 2011] because heterozygotes may express the disease with low selective pressure. Applying the Hardy-Weinberg equation to this estimate for severe forms, the frequency of heterozygotes for ALPL pathogenic variants in France is about 1:275.
In Japan, the birth prevalence of severe hypophosphatasia may be estimated at 1:150,000 on the basis on the frequency of individuals homozygous for the pathogenic variant c.1559delT (1:900,000 [Watanabe et al 2011]) and on the proportion of this pathogenic variant in affected individuals of Japanese ancestry (45.4% [Michigami et al 2020]).
In China, some pathogenic variants have been reported [Wei et al 2010, Zhang et al 2012, Yang et al 2013] but the birth prevalence is unknown.
In Africa, no individuals with hypophosphatasia have been reported in the medical literature outside of North Africa and South Africa; however, clinical ascertainment bias is significant. African American individuals with hypophosphatasia are rare; it is assumed that pathogenic variants in this population represent European admixture.
Differential Diagnosis
The differential diagnosis of hypophosphatasia depends on the age at which the diagnosis is considered. Clinical features that help differentiate hypophosphatasia from other conditions include bone hypomineralization prenatally and immediately postnatally; elevated serum concentrations of calcium and phosphorus postnatally; and persistently low serum alkaline phosphatase (ALP) enzyme activity.
In Utero
Early prenatal ultrasound examination may lead to a consideration of osteogenesis imperfecta (OI) type II, campomelic dysplasia, and chondrodysplasias with defects in bone mineralization, as well as hypophosphatasia. Experienced sonographers usually have little difficulty in distinguishing among these disorders. Fetal radiographs are sometimes helpful in recognizing the undermineralization of bone that is more typical of perinatal hypophosphatasia than of the other disorders considered in the differential diagnosis.
At Birth
Outwardly difficult to distinguish, OI type II, thanatophoric dysplasia, campomelic dysplasia, and chondrodysplasias with bone mineralization defects are readily distinguished from hypophosphatasia by radiograph. In individuals in which the diagnosis is in doubt, analysis of serum ALP activity, pyridoxal 5'-phosphate (PLP) or vitamin B6, and urine phosphoethanolamine (PEA) can suggest the diagnosis pending confirmation with molecular genetic testing.
Infancy and Childhood
Irritability, poor feeding, failure to thrive, hypotonia, and seizures place the infantile type in a broad differential diagnosis that includes inborn errors of energy metabolism, organic acidemia, primary and secondary rickets, neglect, and non-accidental trauma. Infantile hypophosphatasia is suspected with low serum ALP enzyme activity, making the argument for routine screening of serum ALP enzyme activity in infants and children with failure to thrive, unexplained seizures, and suspected non-accidental skeletal injury.
Table 3.
Acquired Disorders and Disorders of Unknown Cause in the Differential Diagnosis of Infantile and Childhood-Onset Hypophosphatasia
View in own window
| Disorder | Clinical Features / Comment |
|---|
Intractable
seizures
| May present prior to biochemical or radiographic manifestations of rickets in early hypophosphatasia |
|
Rickets
| The clinical & radiographic features of rickets are present in perinatal & infantile presentations of hypophosphatasia. However, rickets caused by nutritional &/or vitamin D deficiency, vitamin D resistance, or renal osteodystrophy are readily distinguished from hypophosphatasia by lab findings. In these causes of rickets, the following are characteristic:
↑ serum alkaline phosphatase activity Low serum calcium & phosphorus Low serum vitamin D ↑ serum parathyroid hormone
|
Idiopathic
juvenile
osteoporosis
| Typically presents in preadolescents w/fractures & osteoporosis. The fracture susceptibility & osteoporosis usually resolve spontaneously w/puberty. |
Renal
osteodystrophy
| May be confused w/late presentation of the childhood (juvenile) type assoc w/kidney damage; however, characteristic biochemical findings distinguish the disorders. |
Non-accidental
trauma
(child abuse)
| Like OI, medical history, family history, physical exam, routine lab tests, radiographic imaging, & clinical course all contribute to distinguishing hypophosphatasia from child abuse. Multiple fractures are less typical of hypophosphatasia. Family history may be particularly instructive: the perinatal (severe) type is AR, & childhood (juvenile), adult, & odontohypophosphatasia types are AD; all have been reported in a single family ascertained by unexplained fracture in a child. 1 Serial measurement of serum ALP activity is usually sufficient to identify hypophosphatasia in this circumstance. |
Pseudohypo-
phosphatasia
| Characterized by clinical, biochemical, & radiographic findings reminiscent of infantile hypophosphatasia, w/exception that clinical lab assays of serum ALP activity are in normal range. |
Periodontal
disease
| In advanced, Stage V periodontitis, loss of mandibular bone may → tooth loss w/intact root. This is unusual prior to adulthood. |
Table 4.
Hereditary Disorders in the Differential Diagnosis of Infantile and Childhood-Onset Hypophosphatasia
View in own window
| Gene | Disorder | MOI | Clinical Features / Comment |
|---|
COL1A1
COL1A2 1
| Osteogenesis imperfect (OI) (See COL1A1/2 Osteogenesis Imperfecta.) | AD | OI w/deformation (typically type III in infancy or type IV later on) may resemble hypophosphatasia clinically. |
|
DSPP
| Dentinogenesis imperfect (OMIM DSPP Clinical Synopsis) | AD | Whether part of OI or an isolated finding, dentinogenesis imperfecta is distinguishable from dental presentation of hypophosphatasia. |
|
LIFR
| Stuve-Wiedemann syndrome (OMIM 601559) | AR | Presents w/temperature dysregulation, diminished reflexes, & contractures, but severe perinatal presentation shares several features w/hypophosphatasia: respiratory insufficiency, bowing of long bones, metaphyseal dysplasia, low bone density for age, & fracture predilection. |
|
NOTCH2
| Hadju-Cheney syndrome (OMIM 102500) | AD | Characterized by failure to thrive, dysmorphic facial features, early tooth loss, genitourinary anomalies, osteopenia, pathologic fractures, wormian bones, failure of suture ossification, basilar impression, vertebral abnormalities, joint laxity, bowed fibulae, short distal digits, acroosteolysis, & hirsutism |
P4HB
SEC24D
| Cole-Carpenter syndrome (OMIM PS112240) | AD
AR | Characterized by bone deformities, multiple fractures, proptosis, shallow orbits, orbital craniosynostosis, frontal bossing, & hydrocephalus |
|
RUNX2
|
Cleidocranial dysplasia spectrum disorder
| AD | Characterized by late closure of fontanels & cranial sutures, aplastic clavicles, delayed mineralization of the pubic rami, & delayed eruption of deciduous & permanent teeth. Skeletal dysplasia is distinguishable from hypophosphatasia on clinical exam & skeletal survey. Dental dysplasia does not result in early tooth loss, & enamel hypoplasia is readily distinguishable from odontohypophosphatasia. |
Adult and Odontohypophosphatasia
Table 5.
Acquired Disorders and Disorders of Unknown Cause in the Differential Diagnosis of Adult-Onset Hypophosphatasia and Odontohypophosphatasia
View in own window
| Disorder | Clinical Features / Comment |
|---|
| Osteoarthritis
& pseudogout (secondary to calcium pyrophosphate dehydrate deposition) | Both are presentations of adult hypophosphatasia, distinguished from the more common disorders by clinical history & lab findings. |
|
Osteopenia/osteoporosis
| Must be distinguished from adult hypophosphatasia, in that bisphosphonates may be contraindicated (See Management, Agents/Circumstances to Avoid.) |
|
Periodontal disease
| May be difficult to distinguish from hypophosphatasia, in that alveolar bone loss can be seen w/severe gingivitis. However, gingival inflammation is unusual w/odontohypophosphatasia. |
|
Adult pseudohypophosphatasia
| Characterized by clinical, biochemical, & radiographic findings reminiscent of adult hypophosphatasia, w/exception that clinical lab assays of serum ALP activity are in normal range. |
ALP = alkaline phosphatase
Table 6.
Hereditary Disorders in the Differential Diagnosis of Adult-Onset Hypophosphatasia and Odontohypophosphatasia
View in own window
| Gene | Disorder | MOI | Clinical Features / Comment |
|---|
C1R
C1S
COL3A1
| Familial periodontal disease as part of connective tissue disorder (e.g., vascular Ehlers-Danlos syndrome [EDS] or periodontal EDS) | AD
(AR 1) | Periodontal EDS may present w/root-intact tooth loss, the distinction being low serum ALP in odontohypophosphatasia. |
|
CTSC
| Aggressive periodontitis 1 (OMIM 170650) | AR | Familial periodontal disease |
| Papillon-Lefevre syndrome (OMIM 245000) | AR | Rarer disorders assoc w/premature tooth loss & periodontal disease. The periodontal disease is usually earlier in onset & more severe than that seen w/odontohypophosphatasia. Both Papillon-Lefevre syndrome & HMS are usually assoc w/palmar keratosis, further distinguishing them from odontohypophosphatasia. Measurement of serum ALP enzyme activity is reasonable when either disorder is considered. |
| Haim-Munk syndrome (HMS) (OMIM 245010) | AR |
|
DSPP
| Dentinogenesis imperfecta (OMIM DSPP Clinical Synopsis) | AD | Dentinogenesis imperfecta is readily distinguishable from odontohypophosphatasia on biochemical findings. |
|
ELANE
| Familial periodontal disease assoc w/neutropenia (e.g., ELANE-related neutropenia) | AD | ELANE-related neutropenia includes congenital neutropenia & cyclic neutropenia, both of which are primary hematologic disorders characterized by recurrent fever, skin & oropharyngeal inflammation (e.g., mouth ulcers, gingivitis, sinusitis, & pharyngitis), & cervical adenopathy. |
- 1.
Vascular EDS is almost always inherited in an autosomal dominant manner, but rare examples of biallelic inheritance have been reported.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hypophosphatasia, the evaluations summarized in Table 7 and Table 8 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 7.
Recommended Evaluations Following Initial Diagnosis in Infants with Perinatal Hypophosphatasia
View in own window
| System/Concern | Evaluation | Comment |
|---|
Calcium
homeostasis
| Serum calcium, phosphorus, magnesium Referral to endocrinologist for mgmt of bone health
| To identify those at risk of apnea &/or seizures due to hypercalcemia |
Pulmonary
insufficiency
| Clinical assessment of pulmonary function | To assist in prognosis & distinguishing between severe & benign perinatal types |
Orthopedic
manifestations
|
| |
|
Seizures
| Eval by neurologist for suspected seizures | |
|
Craniosynostosis
| Eval by craniofacial specialists &/or neurosurgeon for those w/craniosynostosis | |
|
Renal function
|
| |
Family support
& resources
| Assess need for:
| |
Genetic
counseling
| By genetics professionals 1 | To inform affected persons & families re nature, MOI, & implications of hypophosphatasia to facilitate medical & personal decision making |
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Table 8.
Recommended Evaluations Following Initial Diagnosis in Older Individuals with Hypophosphatasia
View in own window
| System/Concern | Evaluation | Comment |
|---|
Orthopedic
manifestations
|
| If head shape is abnormal, consider 3D CT scan to further evaluate craniosynostosis. |
Calcium
homeostasis
| Serum 25-hydroxy vitamin D, 1,25-dihydroxy vitamin D, & nPTH to assess for confounding comorbidity (e.g., vitamin D deficiency) Referral to endocrinologist for mgmt of bone health
| |
|
Renal function
|
| |
Dental
complications
| Dental eval | By age 1 yr |
Genetic
counseling
| By genetics professionals 1 | To inform affected persons & families re nature, MOI, & implications of hypophosphatasia to facilitate medical & personal decision making |
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
There is no cure for hypophosphatasia. Targeted therapy in the form of enzyme replacement therapy (ERT) is available, and supportive care by specialists is recommended.
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Asfotase alfa (Strensiq®) ERT has been shown to improve pulmonary function, calcium homeostasis / bone health, and survival in individuals with the infantile and early childhood (juvenile) type of hypophosphatasia. There is growing experience with ERT in individuals with the perinatal (severe) type and emerging experience with ERT in treating osteomalacia in adults.
Table 9.
Targeted Treatment of Hypophosphatasia
View in own window
| Targeted Treatment | Dosage | Considerations |
|---|
| Asfotase alfa ERT 1, 2 | Asfotase alfa ERT is given as a subcutaneous injection.
| The most common regimens are a 1 mg/kg injection 6x per week or a 2 mg/kg injection 3x per week. In absence of ERT, calcitonin & steroids could be attempted short term; efficacy is limited. 3
|
ERT = enzyme replacement therapy
- 1.
The treatment duration and long-term effects of ERT with asfotase alfa remain unknown for perinatal and infantile hypophosphatasia. In theory, ERT would be less effective once endochondral bone formation is complete after the epiphyses fuse.
- 2.
Clinical trials in adults are limited to those with documented childhood disease, and in theory the treatment has occurred after endochondral bone formation is complete (remodeling phase). Biochemical and limited functional improvement can be documented, but treatment end points, duration, and long-term effects are unknown for adult hypophosphatasia.
- 3.
Supportive Care
At all ages, supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 10).
Table 10.
Treatment of Manifestations in Individuals with Hypophosphatasia
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
Respiratory
compromise
|
| Comfort care & supportive mgmt of infants w/perinatal (severe) type remains an option for those w/o access to ERT. |
Calcium
homeostasis /
Bone health
| Mgmt per endocrinologist to optimize bone homeostasis & avoid exacerbating treatments | Mgmt of calcium homeostasis can further be complicated by recalcitrant hypercalcemia/ hypercalciuria, & optimal mgmt remains unclear: hypercalcemia/ hypercalciuria is typically resistant to hydration & furosemide treatment, & bisphosphonates would be contraindicated (see Agents/Circumstances to Avoid). |
| Asfotase alfa ERT (See Table 9.) | |
Physical medicine & rehab, PT, & OT to optimize mobility & autonomy Low-impact physical activity & exercise
| Supervision by physician specialist familiar w/hypophosphatasia is suggested. |
| Adults: calcium & vitamin D supplementation may prevent secondary hyperparathyroidism. | This should only be pursued w/close monitoring by physician specialist familiar w/hypophosphatasia. |
|
Fractures
| Mgmt of primary & secondary skeletal manifestations per orthopedist Internal fixation has been suggested as optimal mgmt. Consider foot orthotics for tarsal fractures & pseudofractures in adults.
| Pseudofractures & stress fractures are difficult to manage. |
Bone pain &
osteomalacia
| Adults: Teriparatide may improve pain, mobility, & fracture repair. 1 |
|
|
| Bisphosphonates are contraindicated (see Agents/Circumstances to Avoid). |
|
Osteoarthritis
| May respond to NSAIDs | |
|
Craniosynostosis
| Mgmt per neurosurgeon to monitor & manage complications incl:
| Craniosynostosis in those w/infantile type is variable. |
|
Kidney disease
| Mgmt per nephrologist to monitor calcium homeostasis & assess for nephrocalcinosis | |
|
Seizures & myopathy
|
| PLP is one of the natural substrates of ALP; PLP deficiency in CNS may ↓ seizure threshold by ↓ing neurotransmitter synthesis. |
|
Dental complications
| Pediatric & adult dentistry to preserve primary dentition (to support nutrition) & to preserve or replace secondary dentition. | By age 1 yr |
Family support
& resources
| Psychological support & social work support | The involvement of multiple specialists treating complex interrelated medical issues mandates case mgmt & social work support. |
ALP = alkaline phosphatase; CNS = central nervous system; ERT = enzyme replacement therapy; NSAIDs = nonsteroidal anti-inflammatory drugs; OT = occupational therapy; PLP = pyridoxal phosphate; PT = physical therapy; TNSALP = alkaline phosphatase, tissue-nonspecific isozyme
- 1.
- 2.
Surveillance
Table 11.
Recommended Surveillance for Individuals with Hypophosphatasia
View in own window
| System/Concern | Evaluation | Frequency |
|---|
Calcium
homeostasis /
Bone health
| Endocrinology &/or nephrology follow up | Per endocrinologist or nephrologist |
| Physical medicine & rehab, PT, & OT | As needed |
| Orthopedic follow up | Per orthopedist |
|
Craniosynostosis
| Neurosurgery follow up to monitor for ↑ intracranial pressure secondary to craniosynostosis in infantile type | Per neurosurgeon |
|
Kidney disease
| Nephrology follow up | Per nephrologist |
|
Seizures
| Neurology eval | As needed |
Dental
complications
| Pediatric dental eval | Every 6 mos beginning at age 1 yr |
OT = occupational therapy; PT = physical therapy
Agents/Circumstances to Avoid
Bisphosphonates are relatively contraindicated in hypophosphatasia. Although adverse outcomes have not been identified in children with the infantile type [Deeb et al 2000], theoretic concern has long been raised based on the structure of bisphosphonates. The phosphate motifs in bisphosphonates have a similar conformation to inorganic pyrophosphate (PPi), the natural substrate of TNSALP (alkaline phosphatase, tissue-nonspecific isozyme); thus, treatment with bisphosphonates is thought to be analogous to "adding fuel to the fire." In adults with hypophosphatasia and osteomalacia treated with bisphosphonates, lateral subtrochanteric femoral pseudofractures have been described [Whyte 2009]. As the prevalence of adult hypophosphatasia is not known and many undiagnosed adults undoubtedly are treated with bisphosphonates, the frequency of this unusual complication is not known.
Excess vitamin D can exacerbate hypercalcemia/hypercalciuria in children with infantile hypophosphatasia who have hypercalcemia.
Teriparatide (recombinant human parathyroid hormone fragment, amino acids 1-34) at high doses induces osteosarcoma in rats and may increase the risk of radiation-induced osteosarcoma (a pediatric growth plate tumor) in humans. Thus, it is contraindicated in children with hypophosphatasia.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
The use of asfotase alfa (Strensiq®) ERT during human pregnancy has not been extensively studied; therefore, any potential risk to the fetus of a pregnant woman taking this therapy during pregnancy is unknown.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Osteoblast enhancement by anti-sclerostin antibodies. Teriparatide enhances osteoblast production of TNSALP, and sclerostin inhibits osteoblast differentiation. Anti-sclerostin therapies have emerged for metabolic bone diseases. A specific Phase II clinical open-label trial for eight adults with hypophosphatasia (mean age 47.8 years) using anti-sclerostin monoclonal antibodies (BPS804) showed early improvement in bone density and markers of bone turnover in seven individuals completing the 16-week study period. Hypophosphatasia-specific biomarkers other than serum alkaline phosphatase were not reported, and functional assessments were beyond the scope of a Phase II study [Seefried et al 2017].
Bone marrow transplantation (hematopoietic cell transplantation) was used to treat an eight-month-old girl with severe hypophosphatasia with prolonged, significant clinical and radiologic improvement [Whyte et al 2003]. Seven years after transplantation, she was reported to be active and growing, and to have the clinical phenotype of the childhood (juvenile) form of hypophosphatasia [Cahill et al 2007]. In another trial, both bone marrow and allogenic mesenchymal stem cells were implanted in an eight-month-old infant, resulting in improvement of respiratory conditions [Tadokoro et al 2009]. However, the infant developed therapy-related leukemia [Taketani et al 2013]. Transplantation of ex vivo expanded mesenchymal stem cells for individuals who had previously undergone bone marrow transplantation improved bone mineralization, muscle mass, respiratory function, intellectual development, and survival [Taketani et al 2015].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Perinatal and infantile hypophosphatasia are typically inherited in an autosomal recessive manner.
Milder forms of hypophosphatasia, especially adult and odontohypophosphatasia, may be inherited in an autosomal recessive or autosomal dominant manner depending on the effect of the ALPL pathogenic variant on TNSALP (alkaline phosphatase, tissue-nonspecific isozyme) activity [Mornet et al 2021]. ALPL variants with a dominant-negative effect are associated with autosomal dominant inheritance.
Intrafamilial clinical variability is common, particularly when some affected family members have a heterozygous ALPL pathogenic variant and other affected family members have biallelic pathogenic variants. Individuals with severe perinatal, childhood, and adult forms of hypophosphatasia may be seen in families segregating two ALPL pathogenic variants.
Reliable assessment of recurrence risk requires identification of the causative pathogenic variant(s) in the proband and molecular genetic testing of the proband's parents to confirm their genetic status.
Autosomal Recessive Inheritance (Proband with Biallelic Pathogenic Variants) – Risk to Family Members
Parents of a proband
If a
pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the
proband occurred as a
de novo event in the proband [
Taillandier et al 2005,
Zhang et al 2012] or as a
postzygotic de novo event in a mosaic parent. If the proband appears to have
homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
Sibs of a proband
If both parents are known to be
heterozygous for an
ALPL pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting
biallelic pathogenic variants, a 50% chance of being heterozygous, and a 25% chance of inheriting neither of the
familial pathogenic variants.
Sibs who inherit
biallelic pathogenic variants tend to have similar disease severity; however, growth differences, nutrition, activity level, and earlier age of diagnosis all may influence
phenotype. Sibs with
compound heterozygous variants tend to display less intrafamilial clinical variability at the severe end of the spectrum and more variability at the milder end of the spectrum.
Offspring of a proband. Unless an individual with autosomal recessive hypophosphatasia has children with an affected individual or a heterozygote, offspring will be obligate heterozygotes for a pathogenic variant in ALPL. Note: In the Canadian Mennonite population, the prevalence of the perinatal (severe) form is 1:2,500, with a carrier frequency of 1:25, due to a founder variant (see Prevalence).
Other family members. Each sib of the proband's parents is at a 50% risk of being heterozygous for a pathogenic variant in ALPL.
Heterozygote Detection
Heterozygote testing for at-risk relatives requires prior identification of the ALPL pathogenic variants in the family.
Autosomal Dominant Inheritance (Proband with a Heterozygous ALPL Variant with a Dominant-Negative Effect) – Risk to Family Members
Parents of a proband
All individuals reported to date with hypophosphatasia caused by a
heterozygous ALPL variant with a
dominant-negative effect inherited the
ALPL pathogenic variant from a parent (who may or may not have clinical manifestations of hypophosphatasia).
Recommendations for the evaluation of parents of a
proband include review of clinical history and laboratory evaluations for signs of hypophosphatasia. Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable
recurrence risk counseling.
If the
pathogenic variant identified in the
proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
Evaluation of parents may determine that a parent is affected but has escaped previous diagnosis because of failure by health care professionals to recognize the disorder, reduced
penetrance, and/or a milder phenotypic presentation. Therefore, an apparently negative family history cannot be confirmed unless
molecular genetic testing has demonstrated that neither parent is
heterozygous for the
ALPL pathogenic variant identified in the
proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the
proband is known to have the
pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
Offspring of a proband. Each child of an individual with a heterozygous ALPL pathogenic variant has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has an ALPL pathogenic variant, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Pregnancy with high a priori risk (pregnancy known to be at increased risk based on family history)
Fetal ultrasonography. Recurrence of perinatal hypophosphatasia may reliably be identified by prenatal ultrasound examination. Undermineralization, small thoracic cavity, shortened long bones, and bowing are typical features of
autosomal recessive and severe hypophosphatasia. Long bone bowing has been reported prenatally in affected sibs and in children of individuals with childhood (juvenile) or adult hypophosphatasia, but the finding is not diagnostic of perinatal severe hypophosphatasia, since it may also be seen in perinatal benign hypophosphatasia, a clinical form that can improve during later stages of pregnancy and result in nonlethal hypophosphatasia [
Wenkert et al 2011]. Established information on the functional effect of some
ALPL pathogenic variants can assist in distinguishing lethal and nonlethal hypophosphatasia prenatally [
Sperelakis-Beedham et al 2021].
Pregnancy with low a priori risk (pregnancy not known to be at risk)
Fetal ultrasonography. Although perinatal hypophosphatasia may be distinguished from other skeletal dysplasias by prenatal ultrasonography, care must be taken in the interpretation of bowed long bones. Undermineralization, small thoracic cavity, shortened long bones, and bowing are typical features of
autosomal recessive and severe hypophosphatasia. However, prognosis is difficult to predict based on ultrasound findings alone: bowed and shortened long bones have been observed on prenatal ultrasound in individuals who ultimately were shown to have – variably – perinatal (benign), childhood (juvenile), or adult hypophosphatasia. The bowing resolves postnatally. In 50% of individuals, when
ALPL molecular testing has been performed, a single
pathogenic variant in
ALPL has been identified, confirming the benign nature of the
phenotype and excluding perinatal (severe) hypophosphatasia.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hypophosphatasia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
ALPL encodes alkaline phosphatase, tissue-nonspecific isozyme (TNSALP), the isozyme present in liver, kidney, and bone. It is functional as a homodimer. The enzyme acts as a (lipid) membrane-bound ectophosphatase with inorganic pyrophosphate (PPi), pyridoxal 5'-phosphate (PLP), and phosphoethanolamine (PEA) as natural substrates.
ALPL pathogenic variants are distributed throughout the 12 exons of the gene. Pathogenic missense variants account for 74.6% of variants; the remainder comprise microdeletions/insertions (13.3%), pathogenic splice site variants (6.0%), pathogenic nonsense variants (3.7%), gross deletions (1.3%), and a nucleotide substitution affecting the major transcription initiation site. This variety of pathogenic variants results in highly variable clinical expression and in a great number of compound heterozygous genotypes.
Genotype-phenotype correlations have been studied using site-directed mutagenesis and 3D enzyme modeling. These studies have allowed the characterization of severe and moderate alleles (alleles producing significant residual enzymatic activity) and alleles with a dominant-negative effect responsible for dominant inheritance [Fukushi et al 1998, Shibata et al 1998, Zurutuza et al 1999, Mornet et al 2001, Watanabe et al 2002, Nasu et al 2006, Brun-Heath et al 2007, Fauvert et al 2009, Mornet et al 2021]. However, such tools do not always predict the severity of pathogenic variants.
Mechanism of disease causation. Pathogenic variants may result in various consequences, sometimes cumulative: decrease or abolition of the catalytic activity, inability to form homodimers, and sequestration of mutated proteins in cell compartments resulting in an inability to reach the cell membrane [Cai et al 1998, Fukushi et al 1998, Shibata et al 1998, Watanabe et al 2002, Brun-Heath et al 2007, Sultana et al 2013, Numa-Kinjoh et al 2015].
Table 12.
Notable ALPL Pathogenic Variants
View in own window
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.