Clinical Description
The classic clinical tetrad of nail-patella syndrome (NPS) involves changes in the nails, knees, and elbows and the presence of iliac horns (see Diagnosis). Many other features may be seen in NPS, including kidney disease and glaucoma [Sweeney et al 2003]. The clinical manifestations are extremely variable in both frequency and severity, with inter- and intrafamilial variability. Individuals may be severely affected by one aspect of NPS but have much milder or no manifestations elsewhere. Though the skeleton is affected in NPS, affected individuals are of average stature.
To date, more than 170 pathogenic variants in LMX1B have been reported in individuals identified to have NPS [Lichter et al 1997, Bongers et al 2002, Sweeney et al 2003, Dunston et al 2005, Lemley 2009, López-Arvizu et al 2011, Boyer et al 2013, Ghoumid et al 2016, Harita et al 2020]. However, approximately 5%-10% of individuals with clinical and radiographic findings of NPS do not have a detectable pathogenic variant in LMX1B. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Nail-Patella Syndrome
View in own window
NPS = nail-patella syndrome
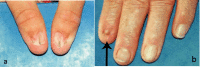
Nail changes are the most constant feature of NPS.
Nail changes may be observed at birth and are most often bilateral and symmetric.
The thumbnails are the most severely affected; the severity of the nail changes tends to decrease from the index finger toward the little finger.
Each individual nail is usually more severely affected on its ulnar side.
Dysplasia of the toenails is usually less marked and less frequent than that of the fingernails; if the toenails are involved, it is often the fifth toenail that is affected.
Digital changes. A reduction in flexion of the distal interphalangeal (DIP) joints is associated with loss of the creases in the skin overlying the dorsal surface of the DIP joints of the fingers.
The gradient of severity is the same as seen in the nails; therefore, the index fingers are the most affected.
Hyperextension of the proximal interphalangeal (PIP) joints with flexion of the DIP joints (resulting in "swan-necking") and fifth-finger clinodactyly may also be seen.
Knee abnormalities. Symptoms include pain, instability, locking, clicking, patella dislocation, and inability to straighten the knee joint. Knee involvement may also be associated with poor development of the vastus medialis muscle.
Patellae:
Findings may be asymmetric.
The patellae may be small, irregularly shaped, or absent.
The displacement of the patella is lateral and superior; the hypoplastic patella is often located laterally and superiorly even when not actually dislocated.
There may be prominent medial femoral condyles, hypoplastic lateral femoral condyles, and prominent tibial tuberosities.
These changes together with a hypoplastic or absent patella give the knee joint a flattened profile.
Flexion contractures of the knees may occur as a result of tight hamstring muscles.
Osteochondritis dissecans, synovial plicae, and absence of the anterior cruciate ligament may also occur.
Early degenerative arthritis is common.
Elbow involvement can include:
Affected individuals may experience dislocation of the radial head, usually posteriorly. Elbow involvement may be asymmetric.
Illiac horns are considered pathognomonic of NPS [Sweeney et al 2003].
Pelvic radiograph is usually necessary for their detection ().
Although large horns may be palpable, they are typically asymptomatic.
In children, iliac horns may have an epiphysis at the apex.
Involvement of the ankles and feet
Talipes equinovarus, calcaneovarus, calcaneovalgus, equinovalgus, and hyperdorsiflexion of the foot may occur.
Tight Achilles tendons are common, contributing to talipes equinovarus and to toe-walking.
Pes planus is common.
Arthrogryposis. Though contractures of the elbows, knees, and calcaneovarus/calcaneovalgus are recognized in individuals with NPS, the term "arthrogryposis" is not often used in the clinical description of this condition.
Spinal and chest wall problems. Back pain occurs in half of individuals with NPS. There may be an increased lumbar lordosis, scoliosis (usually mild), spondylolisthesis, spondylolysis, or pectus excavatum.
Osteoporosis. Bone mineral density is reduced by 8%-20% in the hips of individuals with NPS. An increased rate of fractures has also been reported.
General appearance. A lean body habitus may be associated with NPS and affected individuals often have difficulty putting on weight (particularly muscle) despite adequate dietary intake and exercise.
In particular, muscle mass in the upper arms and upper legs tends to be decreased.
The tendency to be very lean is most evident in adolescents and young adults and becomes less apparent after middle age.
Increased lumbar lordosis may make the buttocks appear prominent.
The high forehead and hairline, particularly at the temples, resembles a receding male pattern hairline when seen in women.
Renal involvement
The first sign of renal involvement is usually proteinuria, with or without hematuria.
Proteinuria may present at any age from birth onwards and may be intermittent.
Renal problems may present during (or be exacerbated by) pregnancy.
Once proteinuria is present, it may remit spontaneously, remain asymptomatic, or progress to nephrotic syndrome and occasionally to ESKD.
Progression to kidney failure may appear to occur rapidly or after many years of asymptomatic proteinuria. The factors responsible for this progression are yet to be identified but the presence and severity of proteinuria appears to be predictive of progression [
Harita et al 2020]. In individuals with ESKD, kidney transplantation may be considered and typically has a favorable outcome (see Management,
Treatment of Manifestations).
Nephritis may also occur in NPS.
Ultrastructural (electron microscopic) renal abnormalities are the most specific histologic changes seen in individuals with NPS and include irregular thickening of the glomerular basement membrane with electron-lucent areas giving a mottled "moth-eaten" appearance, and the presence of collagen-like fibers within the basement membrane and the mesangial matrix.
Ophthalmologic findings
Gastrointestinal involvement. One third of individuals with NPS have problems with constipation (often from birth) or irritable bowel syndrome [Sweeney et al 2003].
Neurologic problems. Many individuals with NPS exhibit reduced sensation to pain and temperature in the hands and feet, most likely because of the inability of Aδ and C fibers to connect with interneurons in the dorsal spinal cord [Dunston et al 2005]. Some affected individuals report intermittent numbness, tingling, and burning sensations in the hands and feet, with no obvious precipitant.
Rarely, these symptoms may be secondary to local orthopedic problems or neurologic compromise from the spine or cervical ribs.
In most cases, the paresthesia follows a glove and stocking pattern rather than the distribution of a particular dermatome or peripheral nerve.
Epilepsy was reported in 6% of affected individuals in one large study [Sweeney et al 2003].
Dental problems. Dental problems may include weak, crumbling teeth and thin dental enamel [Sweeney et al 2003].
Vasomotor problems. Some individuals have symptoms of a poor peripheral circulation, such as very cold hands and feet, even in warm weather. Some may be diagnosed with Raynaud's phenomenon [Sweeney et al 2003].
Vascular anomalies. There are limited reports of vascular anomalies in individuals with NPS, including internal carotid artery aplasia [Kraus et al 2020] and spontaneous coronary artery dissection [Nizamuddin et al 2015, Kaadan et al 2018]. The prevalence of vascular issues in a large population of individuals with NPS and the function of LMX1B in vessel formation must be studied further to determine if these findings are rare coincidental occurrences or part of the NPS phenotype. At this point, it is too early to know if this is part of the NPS phenotype.
Other. Congenital hip dislocation has been rarely reported [Jacofsky et al 2003, West & Louis 2015].