CLCN7-Related Osteopetrosis
Cristina Sobacchi, MS, Anna Villa, MD, PhD, Ansgar Schulz, MD, and Uwe Kornak, MD, PhD.
Author Information and AffiliationsInitial Posting: February 12, 2007; Last Update: January 20, 2022.
Estimated reading time: 31 minutes
Summary
Clinical characteristics.
The spectrum of CLCN7-related osteopetrosis includes infantile malignant CLCN7-related autosomal recessive osteopetrosis (ARO), intermediate autosomal osteopetrosis (IAO), and autosomal dominant osteopetrosis type II (ADOII; Albers-Schönberg disease).
ARO. Onset is at birth. Findings may include: fractures; reduced growth; sclerosis of the skull base (with or without choanal stenosis or hydrocephalus) resulting in optic nerve compression, facial palsy, and hearing loss; absence of the bone marrow cavity resulting in severe anemia and thrombocytopenia; dental abnormalities, odontomas, and risk for mandibular osteomyelitis; and hypocalcemia with tetanic seizures and secondary hyperparathyroidism. Without treatment maximal life span in ARO is ten years.
IAO. Onset is in childhood. Findings may include: fractures after minor trauma, characteristic skeletal radiographic changes found incidentally, mild anemia, and occasional visual impairment secondary to optic nerve compression. Life expectancy in IAO is usually normal.
ADOII. Onset is usually late childhood or adolescence. Findings may include: fractures (in any long bone and/or the posterior arch of a vertebra), scoliosis, hip osteoarthritis, and osteomyelitis of the mandible or septic osteitis or osteoarthritis elsewhere. Cranial nerve compression is rare.
Diagnosis/testing.
The diagnosis of a CLCN7-related osteopetrosis is established in a proband with suggestive findings and biallelic pathogenic variants or a heterozygous pathogenic variant in CLCN7 identified by molecular genetic testing.
Management.
Treatment of manifestations:
ARO. Calcium supplementation for hypocalcemic convulsions; management of calcium homeostasis per individual needs; erythrocyte or platelet transfusions as needed; antibiotics for leukocytopenia; immunoglobulins for hypogammaglobulinemia. Newly diagnosed individuals should be transferred as soon as possible to a pediatric center experienced in allogeneic stem cell transplantation in this disease. The collaboration of pediatricians, pediatric neurologists, ophthalmologists, and psychologists is required to determine best treatment of neurologic and ophthalmic issues, which may include surgical decompression of the optic nerve and hearing aids. Treatment of fractures by an experienced orthopedist and dental care with attention to tooth eruption, ankylosis, abscesses, cysts, and fistulas.
ADOII. Orthopedic treatment for fractures and arthritis with attention to potential post-surgical complications (delayed union or non-union of fractures, infection); fractures near joints may require total joint arthroplasty. Medical treatment for arthritis with anti-inflammatory agents; transfusions for anemia and thrombocytopenia; surgical optic nerve decompression, hearing aids, and regular dental care and good oral hygiene.
Prevention of primary manifestations: In ARO, hematopoietic stem cell transplantation (HSCT) can be curative; however, cranial nerve dysfunction is usually irreversible, and progressive neurologic sequelae occur in children with the neuronopathic form even after successful HSCT.
Surveillance: Complete blood count, ophthalmologic examination, and audiologic evaluation at least once a year; dental evaluation every 6-12 months or as directed. For ARO, follow up as directed by the transplantation center following HSCT.
Agents/circumstances to avoid: In ADOII, activities with high fracture risk. Orthopedic surgery should only be performed when absolutely necessary.
Genetic counseling.
CLCN7-related osteopetrosis is inherited in an autosomal recessive or autosomal dominant manner: ARO is inherited in an autosomal recessive manner; about 40% of IAO is inherited in an autosomal recessive manner and about 60% in an autosomal dominant manner; and ADOII is inherited in an autosomal dominant manner.
Autosomal recessive inheritance. If both parents are known to be heterozygous for a CLCN7 pathogenic variant associated with autosomal recessive osteopetrosis, each sib of an affected individual has at conception a 25% chance of inheriting biallelic pathogenic variants and being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial CLCN7 pathogenic variants. Carrier testing for at-risk relatives requires prior identification of the CLCN7 pathogenic variants in the family.
Autosomal dominant inheritance. Most individuals diagnosed with autosomal dominant CLCN7-related osteopetrosis have an affected parent. Each child of an individual with autosomal dominant osteopetrosis has a 50% chance of inheriting the pathogenic variant.
Once the CLCN7 pathogenic variant(s) have been identified in an affected family member, prenatal testing and preimplantation genetic testing for CLCN7-related osteopetrosis are possible.
GeneReview Scope
CLCN7-Related Osteopetrosis: Included Phenotypes
View in own window
| Phenotype 1 | Synonym |
|---|
| Autosomal recessive osteopetrosis (ARO) | Infantile malignant CLCN7-related autosomal recessive osteopetrosis |
| Intermediate autosomal osteopetrosis (IAO) | |
| Autosomal dominant osteopetrosis type II (ADOII) | Albers-Schönberg disease |
Diagnosis
The spectrum of CLCN7-related osteopetrosis includes the following:
Autosomal recessive osteopetrosis (ARO; infantile malignant CLCN7-related autosomal recessive osteopetrosis)
Intermediate autosomal osteopetrosis (IAO)
Autosomal dominant osteopetrosis type II (ADOII; Albers-Schönberg disease)
Suggestive Findings
A CLCN7-related osteopetrosis should be suspected in individuals with osteosclerosis noted on radiographs, which may be accompanied by hypocalcemia and resulting convulsions, anemia, thrombocytopenia, visual impairment, and/or central nervous system involvement (Table 1).
Table 1.
Diagnostic Features of the Subtypes of CLCN7-Related Osteopetrosis
View in own window
| Finding | Subtype of CLCN7-Related Osteopetrosis |
|---|
| ARO | IAO | ADOII |
|---|
| Radiographic changes | Pathognomonic 1 | Characteristic 2 | Characteristic 3 |
| Hypocalcemia | Severe to absent | Absent | Absent |
| Anemia | Severe to moderate | Mild to absent | Absent |
| Thrombocytopenia | Severe to absent | Absent | Absent |
| Visual impairment | Frequent | Rare | Very rare |
| CNS involvement | Severe to absent 4 | Absent | Absent |
| Age of onset of symptoms | Birth | First 2 years | First 10 yrs |
| Inheritance | AR | AR or AD | AD |
ADOII = autosomal dominant osteopetrosis type II; ARO = infantile malignant CLCN7-related autosomal recessive osteopetrosis; CNS = central nervous system; IAO = intermediate autosomal osteopetrosis
- 1.
Generalized osteosclerosis, club-shaped long bones, sclerosis of the skull base, bone-within-bone appearance; these signs are observed in all types of ARO.
- 2.
Findings similar to ARO, already present in early childhood, but less severe
- 3.
Findings:
• Osteosclerosis of the spine ("sandwich vertebrae")
• Bone-within-bone appearance, mainly in iliac wings
• Erlenmeyer-shaped femoral metaphysis
• Mild osteosclerosis of the skull base
• Transverse bands of osteosclerosis in long bones
- 4.
Typical signs of CNS involvement:
• Delayed psychomotor development
• Loss of abilities
• Seizures (can be also due to hypocalcemia)
• On MRI: global brain atrophy, cortical abnormalities, and in rare individuals, heterotopias
Establishing the Diagnosis
The diagnosis of a CLCN7-related osteopetrosis is established in a proband with suggestive findings and biallelic pathogenic variants or a heterozygous pathogenic variant in CLCN7 identified by molecular genetic testing (see Table 2).
Note: Identification of biallelic CLCN7 variants of uncertain significance, of one known CLCN7 pathogenic variant and one CLCN7 variant of uncertain significance, or of a heterozygous CLCN7 variant of uncertain significance does not establish or rule out a diagnosis of this disorder.
Because the phenotype of a CLCN7-related osteopetrosis may be indistinguishable from other inherited sclerosing bone dysplasias resulting from impaired osteoclast function, recommended molecular genetic testing approaches include use of a multigene panel or comprehensive genomic testing.
Note: Single-gene testing (sequence analysis of CLCN7, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
An osteopetrosis
multigene panel that includes CLCN7 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing does not require the clinician to determine which genes is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in CLCN7-Related Osteopetrosis
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
CLCN7
| Sequence analysis 3 | ~99% 4, 5 |
| Gene-targeted deletion/duplication analysis 6 | Rare 7 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
Data derived from the subscription-based professional view of Human Gene Mutation Database [Stenson et al 2020]
- 5.
A noncoding deep intronic variant in CLCN7 causing autosomal recessive osteopetrosis was reported to have been missed on genome sequencing [Chorin et al 2020].
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
A homozygous 101-kb contiguous gene deletion including exons 7-25 of CLCN7 has been reported [Pangrazio et al 2012].
Clinical Characteristics
Clinical Description
To date, approximately 300 individuals have been identified with a pathogenic variant in CLCN7 [Stenson et al 2020]. The three different associated phenotypes are described in this section.
Autosomal Dominant Osteopetrosis Type II (ADOII)
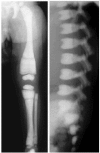
Although ADOII is sometimes called "benign osteopetrosis," as many as 60%-80% of individuals with radiologic signs of ADOII experience clinical problems ().
Autosomal dominant osteopetrosis type II radiographs Reprinted from Bénichou et al [2000] with permission from Elsevier
Onset of radiologic and clinical manifestations of ADOII is usually in late childhood or adolescence, although earlier occurrence has been reported. Osteosclerosis of the spine predominates, with a "sandwich vertebra" appearance, a diagnostic criterion for ADOII. Most affected individuals have a "bone-within-bone" appearance primarily in the iliac wings, but also in other bones. Transverse bands of sclerosis, perpendicular to the main axis, are often observed in long bones. Increase in the skull base density can be seen [Bénichou et al 2000, Cleiren et al 2001].
Clinical findings vary even within the same family [Waguespack et al 2007]. In three families in which most affected individuals had mild ADOII, early-onset disease, anemia, and blindness caused by optic nerve compression were observed in some affected family members; this phenotype has been called "intermediate osteopetrosis" because of its overlap with mild ARO.
The main complications affect the skeleton:
Cranial nerve compression caused by osteosclerosis of the skull base is rare. Hearing loss and visual loss occurs in up to 19% of affected individuals.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Prevalence
The prevalence of ADOII has been estimated at up to 1:20,000 [Bénichou et al 2000]. The disease is probably underdiagnosed in those with milder phenotypes.
ARO is less common, with an estimated prevalence of 1:250,000.
Differential Diagnosis
Pathogenic variants in CLCN7 account for 13% of the infantile autosomal recessive osteopetrosis (ARO), 40% of the recessive intermediate autosomal osteopetrosis (IAO), 60% of the dominant IAO, and 99% of the autosomal dominant osteopetrosis type II (ADOII) [Authors, personal observation].
ARO
Table 3.
Other Types of Autosomal Recessive Osteopetrosis
View in own window
| Gene | Disorder | Clinical Characteristics | Features Distinguishing this Disorder from CLCN7-Related ARO |
|---|
|
CA2
| ARO w/renal tubular acidosis (RTA) (OMIM 259730) | Generalized osteosclerosis. Cerebral calcifications are typical & may be assoc w/ID. 1 | Onset of ARO w/RTA is usually later than in infantile malignant form of ARO & disease course is milder. |
|
OSTM1
| OSTM1-related ARO (OMIM 259720) | ~4% of ARO is caused by pathogenic variants in OSTM1. Extremely severe form of ARO w/CNS involvement 2 that is indistinguishable from most severe forms of CLCN7-related ARO. | OSTM1-related ARO is frequently assoc w/structural brain anomalies. |
|
PLEKHM1
| PLEKHM1-related ARO (OMIM 611497) | Very rare, can look like ADOII | PLEKHM1-related ARO appears to be very mild & can regress w/↑ age. 3 One person w/PLEKHM1-related ARO caused by a heterozygous pathogenic variant has been described. 4 |
|
SNX10
| SNX10-related ARO (OMIM 615085) | ~4% of ARO is caused by pathogenic variants in SNX10; in particular, "Västerbottenian osteopetrosis" is caused by SNX10 pathogenic variants. Loss of vision, anemia, & bone fragility are frequently observed, warranting use of HSCT. 5 | SNX10-related ARO appears to be slightly less severe than CLCN7-related ARO. |
|
TCIRG1
| TCIRG1-related ARO (OMIM 259700) | >50% of ARO is caused by pathogenic variants in TCIRG1. | Higher frequency of neurodevelopmental delay & seizures in CLCN7-related ARO than in TCIRG1-related ARO. 6 Noncoding TCIRG1 variants can cause milder phenotype that resembles ADOII. |
|
TNFRSF11A
| Osteoclast-poor ARO (OMIM 612301) | Characterized by onset w/in 1st yr of life & typical ARO manifestations. Investigation of bone biopsy is prerequisite for reliable diagnosis. | TNFSF11 pathogenic variants cause a slight T-cell defect & TNFRSF11A pathogenic variants can lead to hypogammaglobulinemia similar to a common variable immune deficiency. 7 It is crucial to rule out TNFSF11- & TNFRSF11A-related ARO, as HSCT is not successful in these persons. |
|
TNFSF11
| Osteoclast-poor ARO (OMIM 259710) |
ADOII = autosomal dominant osteopetrosis type II; ARO = autosomal recessive osteopetrosis; CNS = central nervous system; ID = intellectual disability; HSCT = hematopoietic stem cell transplantation
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
A mild form of ARO can be caused by deep intronic or splice site variants in TCIRG1 (OMIM 259700) [Sobacchi et al 2014, Palagano et al 2015]. This form can be very similar to CLCN7-related IAO or ADOII.
Autosomal Dominant Osteopetrosis (ADO)
Heterozygous pathogenic variants in LRP5 are associated with ADO type I (ADOI) (OMIM 607634). In ADOI, osteosclerosis is most pronounced in the skull vault and does not lead to sandwich vertebrae. ADOI is not associated with an increased fracture rate. (Note: It is debated whether this disease entity should be called "endosteal hyperostosis" or "high bone mass disorder," as "osteopetrosis" should be reserved for osteoclast-related disorders.)
Other
Table 4.
Other Disorders to Consider in the Differential Diagnosis of CLCN7-Related Osteopetrosis
View in own window
| Gene | Disorder/Phenotype | MOI | Comment |
|---|
|
ANKH
|
AD craniometaphyseal dysplasia
| AD | Clinical hallmark is skull hyperostosis → deep-set eyes & paranasal bossing. Facial nerve palsy is common & more frequent than optic nerve compression. The femur shows a modeling defect, but no osteosclerosis. Susceptibility to fractures is not ↑.
|
|
CSF1R
| Brain anomalies, neurodegeneration, & dysosteosclerosis (BANDDOS) (OMIM 618476) | AR | Characterized by structural brain anomalies & a primary neurodegenerative phenotype. Like other forms of dysosteosclerosis, affected persons have platyspondyly. |
|
CTSK
|
Pycnodysostosis
| AR | Short-limbed short stature, typical facial appearance (convex nasal ridge & small jaw w/obtuse mandibular angle), osteosclerosis w/↑ bone fragility, acroosteolysis of distal phalanges, delayed closure of cranial sutures, & dysplasia of clavicle In some affected persons clinical presentation can resemble IAO. 1
|
|
FERMT3
| Leukocyte adhesion deficiency type III (OMIM 612840) | AR | Affected persons present w/recurrent infections & bleeding diathesis regardless of platelet or leukocyte count. In some, high bone density can be found, since fermitin family homolog 3 signaling is required for osteoclast-mediated bone resorption. 2 |
|
GJA1
| AR craniometaphyseal dysplasia (OMIM 218400) | AR | Typical features of craniometaphyseal dysplasia (macrocephaly, hearing loss, skull hyperostosis w/paranasal bossing, metaphyseal widening) but less pronounced calvarial thickening 3 Due to diaphyseal osteosclerosis; can occasionally resemble mild forms of osteopetrosis
|
|
LRP6
| High bone mass phenotype 4 | AD | Endosteal hyperostosis similar to ADOI but w/hypodontia |
|
LRRK1
| Osteosclerotic metaphyseal dysplasia (OSMD) 5 | AR | While the disorder name differs, LRRK1-related OSMD is a typical recessive osteopetrosis w/dysfunctional osteoclasts. No neurologic complications. LRRK1-related OSMD is milder than CLCN7-related ARO & can be best compared to IAO.
|
|
SLC29A3
| Dysosteosclerosis 6 | AR | Assoc w/platyspondyly (a distinguishing feature). Disorder appears to be osteoclast-poor. No neurologic complications. SLC39A3-related dysosteosclerosis is milder than CLCN7-related ARO & is best compared to IAO.
|
|
SOST
| SOST-related sclerosing bone dysplasias, incl van Buchem disease & sclerosteosis | AR | Moderate-to-gross skull hyperostosis → cranial nerve dysfunction, mandibular enlargement, & generalized osteosclerosis. Sclerosteosis also comprises syndactyly & tall stature & can be lethal as a result of ↑ intracranial pressure. |
AD = autosomal dominant; ADO = autosomal dominant osteopetrosis; AR = autosomal recessive; ARO = autosomal recessive osteopetrosis; IAO = intermediate autosomal osteopetrosis; MOI = mode of inheritance
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
Management
General guidelines for diagnosis, therapy, and follow up are available for all forms of osteopetrosis; see Author Information.
Due to the difference in severity, the evaluation, treatment, and surveillance recommendations for infantile malignant CLCN7-related autosomal recessive osteopetrosis (ARO) and CLCN7-related autosomal dominant osteopetrosis type II (ADOII) differ. The CLCN7-related intermediate autosomal osteopetrosis (IAO) phenotype lies between these two forms and has variable manifestations and prognosis. Therefore, management options for CLCN7-related IAO must be evaluated on an individual basis.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with ARO, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Infantile Malignant CLCN7-Related Autosomal Recessive Osteopetrosis (ARO)
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Osteosclerosis
| Baseline radiographic skeletal survey | |
|
Choanal stenosis
| Otorhinolaryngologic exam to evaluate for choanal stenosis assoc w/sclerosis of skull base | |
Hypocalcemia &
secondary hyper-
parathyroidism
| Calcium concentrations in blood & urine | |
Anemia &
thrombocytopenia
| CBC w/reticulocyte count | |
| Abdominal ultrasound | Evaluate for hepatosplenomegaly assoc w/extrameduallry hematopoeisis. |
Neurologic
complications
| MRI &/or CT of neurocranium to evaluate for narrowed neuroforamina, hydrocephalus, & brain abnormalities in neuronopathic form of osteopetrosis | |
| EEG | Evaluate for evidence of neurodegeneration. |
| Baseline neurodevelopmental assessment | |
|
Visual impairment
| Ophthalmologic exam incl VEPs to evaluate for optic nerve atrophy assoc w/optic nerve compression w/in skull base | |
|
Hearing loss
| Baseline audiologic assessment | |
Genetic
counseling
| By genetics professionals 1 | To inform affected persons & their families re nature, MOI, & implications of CLCN7-related ARO to facilitate medical & personal decision making |
Family support
& resources
| Assess need for:
| |
ARO = autosomal recessive osteopetrosis; MOI = mode of inheritance; VEP = visual evoked potential
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
To establish the extent of disease and needs in an individual diagnosed with ADOII, the evaluations summarized in Table 6 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 6.
Recommended Evaluations Following Initial Diagnosis in Individuals with CLCN7-Related Autosomal Dominant Osteopetrosis Type II
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Osteosclerosis
| Baseline radiographic skeletal survey | |
|
Anemia & thrombocytopenia
| CBC w/reticulocyte count | |
|
Neurologic complications
| MRI &/or CT of neurocranium to evaluate for narrowed neuroforamina | |
|
Visual impairment
| Ophthalmologic exam incl VEPs to evaluate for optic nerve atrophy assoc w/optic nerve compression w/in skull base | |
|
Hearing loss
| Baseline audiologic assessment | |
|
Genetic counseling
| By genetics professionals 1 | To inform affected persons & their families re nature, MOI, & implications of CLCN7-related ADOII to facilitate medical & personal decision making |
ADOII = autosomal dominant osteopetrosis type II; MOI = mode of inheritance; VEP = visual evoked potential
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Due to the difference in severity, treatment strategies for ARO and ADOII differ. IAO lies between these two forms and has a variable prognosis. Therefore, treatment options must be evaluated on an individual basis.
Table 7.
Treatment of Manifestations in Individuals with Infantile Malignant CLCN7-Related Autosomal Recessive Osteopetrosis (ARO)
View in own window
Manifestation/
Concern | Treatment | Considerations/Other |
|---|
Hypocalcemic
convulsions
| Calcium supplementation 1 | |
Bone marrow
failure
| Erythrocyte or platelet transfusions (irradiated products) as needed | In case of leukocytopenia &/or hypogammaglobulinemia, which may develop in a subset of persons, antibiotics & immunoglobulins may be given in a prophylactic or therapeutic manner. |
| Newly diagnosed persons should be transferred as soon as possible to pediatric center experienced in allogeneic HSCT in this disease. | See Prevention of Primary Manifestations. |
Neurologic
issues
| The collaboration of pediatricians, pediatric neurologists, ophthalmologists, & psychologists is required to determine best treatment. | |
Visual
impairment
| Surgical decompression of optic nerve, a difficult procedure, has been performed w/some success to prevent vision loss in those w/ARO in general [Hwang et al 2000]. | |
|
Hearing loss
| Hearing aids | |
|
Fractures
| Treatment by orthopedist w/experience w/ARO in collaboration w/treating pediatrician | |
|
Dental issues
| Treatment as needed by dentist | Oral surgery may be needed for defective tooth eruption, ankylosis, abscesses, & formation of cysts & fistulas. |
ARO = autosomal recessive osteopetrosis; HSCT = hematopoietic stem cell transplantation
- 1.
The management of calcium homeostasis may be difficult and recommendations are conflicting: whereas physiologic doses of calcium and vitamin D have been used to treat children with osteopetrosis who have rickets, restriction of calcium and vitamin D has been used to prevent progression of disease and hypercalcemic crisis following HSCT. Treatment needs to take into account the particular situation of the affected individual.
Table 8.
Treatment of Manifestations in Individuals with CLCN7-Related Autosomal Dominant Osteopetrosis Type II (ADOII)
View in own window
| Manifestation/Concern | Treatment | Considerations/Other |
|---|
|
Fractures
| Treatment by orthopedist w/experience w/ADOII | Fractures near joints may require total joint arthroplasty [Strickland & Berry 2005]. |
|
Arthritis
| Medical treatment w/anti-inflammatory agents | |
|
Anemia & thrombocytopenia
| Transfusions | |
|
Visual impairment
| Surgical optic nerve decompression | |
|
Hearing loss
| Hearing aids | |
|
Dental issues
| Regular dental care & good oral hygiene | |
ADOII = autosomal dominant osteopetrosis type II
Prevention of Primary Manifestations
Surveillance
Table 9.
Recommended Surveillance for Individuals with Infantile Malignant CLCN7-Related Autosomal Recessive Osteopetrosis (ARO)
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Bone marrow failure
| CBC | Annually at a minimum |
|
Visual loss
| Ophthalmic exam |
|
Hearing loss
| Audiologic eval |
|
Dental issues
| Dental eval | Every 6-12 mos or as directed |
|
Potential for graft failure after HSCT
| Transplant center surveillance | As directed by specialists |
CBC = complete blood count; HSCT = hematopoietic stem cell transplantation
Table 10.
Recommended Surveillance for Individuals with CLCN7-Related Autosomal Dominant Osteopetrosis Type II (ADOII)
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Anemia & thrombocytopenia
| CBC | At least annually |
|
Visual loss
| Ophthalmic exam |
|
Hearing loss
| Audiologic eval |
|
Dental issues
| Dental eval | Every 6-12 mos or as directed |
Agents/Circumstances to Avoid
ADOII
Activities with high fracture risk should be avoided.
Orthopedic surgery should only be performed when absolutely necessary and the surgeon should be aware of potential complications and difficulties in handling osteopetrotic bone.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger sibs of an affected individual by molecular genetic testing of the CLCN7 pathogenic variant(s) in the family in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures.
For hematopoietic stem cell donation (relatives of a proband with CLCN7-related ARO). Any relative considering stem cell donation should undergo molecular genetic testing to clarify their genetic status. Whenever possible, related donors who do not have a familial CLCN7 pathogenic variant are preferred.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
CLCN7-related osteopetrosis is inherited in an autosomal recessive or autosomal dominant manner:
Infantile malignant CLCN7-related autosomal recessive osteopetrosis (ARO) is inherited in an autosomal recessive manner.
About 40% of CLCN7-related intermediate autosomal osteopetrosis (IAO) is inherited in an autosomal recessive manner and about 60% in an autosomal dominant manner.
CLCN7-related autosomal dominant osteopetrosis type II (ADOII) is inherited in an autosomal dominant manner.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are presumed to be heterozygous for a CLCN7 pathogenic variant.
Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a CLCN7 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
Individuals who are heterozygous for a CLCN7 pathogenic variant associated with ARO are asymptomatic; however, no systematic studies have been performed to evaluate for subtle changes in bone mass.
Sibs of a proband
If both parents are known to be heterozygous for a CLCN7 pathogenic variant associated with autosomal recessive osteopetrosis, each sib of an affected individual has at conception a 25% chance of inheriting biallelic CLCN7 pathogenic variants and being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial CLCN7 pathogenic variants.
The limited data available suggest that in the presence of ARO, a similar presentation of the disorder is expected in individuals with biallelic CLCN7 pathogenic variants in the same family; in particular, if the neuronopathic form of the disorder is present in the proband, a primary central nervous system involvement is likely to be present in other affected sibs [Sobacchi, unpublished observation].
Individuals who are heterozygous for a CLCN7 pathogenic variant associated with autosomal recessive osteopetrosis are asymptomatic; however, no systematic studies have been performed to evaluate for subtle changes in bone mass.
Offspring of a proband
The offspring of an individual with autosomal recessive CLCN7-related osteopetrosis are obligate heterozygotes (carriers) for a pathogenic variant in CLCN7.
In general, individuals with ARO reproduce only if successfully treated by hematopoietic stem cell transplantation.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a CLCN7 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the CLCN7 pathogenic variants in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Most individuals diagnosed with autosomal dominant CLCN7-related osteopetrosis have an affected parent.
A proband with autosomal dominant CLCN7-related osteopetrosis may have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with CLCN7-related osteopetrosis caused by a de novo pathogenic variant is unknown.
If the proband appears to be the only affected family member (i.e., a simplex case), recommendations for the evaluation of parents include x-ray investigation of the skeleton and molecular genetic testing for the pathogenic variant identified in the proband. Evaluation of the parents is used to confirm their genetic status and to allow reliable recurrence risk counseling.
If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
The proband has a de novo pathogenic variant.
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Evaluation of parents may determine that one is affected but has escaped previous diagnosis because of reduced penetrance, failure by health care professionals to recognize the syndrome, and/or a milder phenotypic presentation. Therefore, an apparently negative family history cannot be confirmed without appropriate clinical evaluation of the parents and/or molecular genetic testing (to establish that neither parent is heterozygous for the pathogenic variant identified in the proband).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the proband is affected and/or is known to have the
CLCN7 pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Because ADOII is associated with both intrafamilial clinical variability and reduced penetrance (see
Penetrance), a sib who inherits a
CLCN7 pathogenic variant associated with autosomal dominant
CLCN7-related osteopetrosis may be more or less severely affected than the proband.
If the proband has a known
CLCN7 pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the CLCN7 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for CLCN7-related osteopetrosis because of the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant CLCN7-related osteopetrosis has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents. If a parent has the CLCN7 pathogenic variant, his or her family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the CLCN7 pathogenic variant(s) have been identified in an affected family member, prenatal testing and preimplantation genetic testing for CLCN7-related osteopetrosis are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
The OsteoPETrosis Society (OPETS)
Email: osteopetrosispatient@gmail.com; janecastello.opets@gmail.com
MedlinePlus
National Institute of Arthritis and Musculoskeletal and Skin Diseases
Phone: 877-226-4267
Email: niamsinfo@mail.nih.gov
Network for Rare Osteopathies (NetsOs)
CeSER
Alexandrinenstraße 5
Germany
Phone: 49 (0)234 509-2601
Fax: 49 (0)234 509-2688
Email: ceser@klinikum-bochum.de
European Society for Immunodeficiencies (ESID) Registry
Email: esid-registry@uniklinik-freiburg.de
UCLA International Skeletal Dysplasia Registry (ISDR)
Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
CLCN7-Related Osteopetrosis: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
CLCN7-related osteopetrosis is caused by osteoclast dysfunction. The osteoclast is a highly specialized cell with the unique ability to resorb large amounts of mineralized bone tissue. After attaching to the bone surface, a sealing zone that isolates the resorption lacuna from the extracellular environment is formed. Large quantities of acidic vesicles then fuse with the plasma membrane juxtaposed to the bone surface to create the ruffled membrane. This structure is exclusively found in osteoclasts and secretes large amounts of acid into the resorption lacuna, which therefore is also referred to as an "extracellular lysosome" [Sobacchi et al 2013]. The low pH is required to dissolve the bone mineral and for the optimal activity of acid hydrolases that degrade the bone matrix.
CLCN7 encodes the ClC-7 chloride channel, which resides in lysosomal vesicles and in the ruffled membrane and acts as a 2Cl-/1H+ exchanger transporting negative charges into the resorption lacuna in parallel to the protons pumped in this extracellular space by the ruffled membrane v-type H+-ATPase [Kornak et al 2001, Leisle et al 2011].
Mechanism of disease causation. Pathogenic variants in CLCN7 lead to a loss of chloride channel function of varying degree. In the most severe autosomal recessive osteopetrosis phenotype, the ClC-7 chloride channel is absent, while the autosomal dominant osteopetrosis type II phenotype-causing pathogenic variants do not necessarily impair chloride currents, but do result in abnormal gating behavior of the channel [Leisle et al 2011].
Table 11.
Notable CLCN7 Pathogenic Variants
View in own window
| Reference Sequences | DNA Nucleotide Change | Predicted Protein Change | Comment [Reference] |
|---|
NM_001287.6
NP_001278.1
| c.643G>A | p.Gly215Arg | Recurrent pathogenic variants assoc w/ADOII [Cleiren et al 2001] |
| c.856C>T | p.Arg286Trp |
ADOII = autosomal dominant osteopetrosis type II
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Chapter Notes
Author History
Marie-Christine de Vernejoul, MD, PhD; Hôpital Lariboisière (2007-2016)
Uwe Kornak, MD, PhD (2007-present)
Ansgar Schulz, MD (2007-present)
Cristina Sobacchi, MS (2016-present)
Anna Villa, MD, PhD (2016-present)
Revision History
20 January 2022 (ha) Comprehensive update posted live
9 June 2016 (ha) Comprehensive update posted live
20 June 2013 (me) Comprehensive update posted live
14 October 2010 (me) Comprehensive update posted live
12 February 2007 (me) Review posted live
8 September 2006 (uk) Original submission
References
Literature Cited
Aker M, Rouvinski A, Hashavia S, Ta-Shma A, Shaag A, Zenvirt S, Israel S, Weintraub M, Taraboulos A, Bar-Shavit Z, Elpeleg O. An SNX10 mutation causes malignant osteopetrosis of infancy.
J Med Genet. 2012;49:221–6. [
PubMed: 22499339]
Bénichou OD, Laredo JD, de Vernejoul MC. Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestations in 42 patients.
Bone. 2000;26:87–93. [
PubMed: 10617161]
Bo T, Yan F, Guo J, Lin X, Zhang H, Guan Q, Wang H, Fang L, Gao L, Zhao J, Xu C. Characterization of a relatively malignant form of osteopetrosis caused by a novel mutation in the PLEKHM1 gene.
J Bone Miner Res. 2016;31:1979–87. [
PubMed: 27291868]
Brance ML, Brun LR, Cóccaro NM, Aravena A, Duan S, Mumm S, Whyte MP. High bone mass from mutation of low-density lipoprotein receptor-related protein 6 (LRP6).
Bone. 2020;141:115550. [
PubMed: 32730923]
Campos-Xavier AB, Saraiva JM, Ribeiro LM, Munnich A, Cormier-Daire V. Chloride channel 7 (CLCN7) gene mutations in intermediate autosomal recessive osteopetrosis.
Hum Genet. 2003;112:186–9. [
PubMed: 12522560]
Chorin O, Yachelevich N, Mohamed K, Moscatelli I, Pappas J, Henriksen K, Evrony GD. Transcriptome sequencing identifies a noncoding, deep intronic variant in CLCN7 causing autosomal recessive osteopetrosis.
Mol Genet Genomic Med. 2020;8:e1405. [
PMC free article: PMC7549584] [
PubMed: 32691986]
Cleiren E, Bénichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, deVernejoul MC, Van Hul W. Albers-Schonberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene.
Hum Mol Genet. 2001;10:2861–7. [
PubMed: 11741829]
Corbacioglu S, Hönig M, Lahr G, Stöhr S, Berry G, Friedrich W, Schulz AS. Stem cell transplantation in children with infantile osteopetrosis is associated with a high incidence of VOD, which could be prevented with defibrotide.
Bone Marrow Transplant. 2006;38:547–53. [
PubMed: 16953210]
Crazzolara R, Maurer K, Schulze H, Zieger B, Zustin J, Schulz AS. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III.
Pediatr Blood Cancer. 2015;62:1677–9. [
PubMed: 25854317]
Dozier TS, Duncan IM, Klein AJ, Lambert PR, Key LL Jr. Otologic manifestations of malignant osteopetrosis.
Otol Neurotol. 2005;26:762–6. [
PubMed: 16015181]
Even-Or E, Schiesel G, Simanovsky N. NaserEddin A, Zaidman I, Elpeleg O, Mor-Shaked H, Stepensky P. Clinical presentation and analysis of genotype-phenotype correlations in patients with malignant infantile osteopetrosis.
Bone. 2022;154:116229. [
PubMed: 34624559]
Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson JP, Keeling DJ, Andersson AK, Wallbrandt P, Zecca L, Notarangelo LD, Vezzoni P, Villa A. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis.
Nat Genet. 2000;25:343–6. [
PubMed: 10888887]
Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, Andolina M, Flanagan A, Horwitz EM, Mihci E, Notarangelo LD, Ramenghi U, Teti A, Van Hove J, Vujic D, Young T, Albertini A, Orchard PJ, Vezzoni P, Villa A. Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis.
J Bone Miner Res. 2003;18:1740–7. [
PubMed: 14584882]
Gaytán-Morales JF, Castorena-Villa I, Mendoza-Camargo FO, Cortés-Flores DC, Gómez-Domíguez YA, Montenegro-Chahar PD, García-Maldonado P, Parra-Ortega I. Hematopoietic stem cell transplantation in a patient with osteopetrosis and mutation in CLCN7: long-term follow-up.
Bol Med Hosp Infant Mex. 2021;78:225–33. [
PubMed: 33651788]
Guerrini MM, Sobacchi C, Cassani B, Abinun M, Kilic SS, Pangrazio A, Moratto D, Mazzolari E, Clayton-Smith J, Orchard P, Coxon FP, Helfrich MH, Crockett JC, Mellis D, Vellodi A, Tezcan I, Notarangelo J, Rogers MJ, Vezzoni P, Villa A, Frattini A. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations.
Am J Hum Genet. 2008;83:64–76. [
PMC free article: PMC2443850] [
PubMed: 18606301]
Howaldt A, Nampoothiri S, Quell LM, Ozden A, Fischer-Zirnsak B, Collet C, de Vernejoul MC, Doneray H, Kayserili H, Kornak U. Sclerosing bone dysplasias with hallmarks of dysosteosclerosis in four patients carrying mutations in SLC29A3 and TCIRG1.
Bone. 2019;120:495–503. [
PubMed: 30537558]
Howaldt A, Hennig AF, Rolvien T, Rössler U, Stelzer N, Knaus A, Böttger S, Zustin J, Geißler S, Oheim R, Amling M, Howaldt HP, Kornak U. Adult osteosclerotic metaphyseal dysplasia with progressive osteonecrosis of the jaws and abnormal bone resorption pattern due to a LRRK1 splice site mutation.
J Bone Miner Res. 2020;35:1322–32. [
PubMed: 32119750]
Hu Y, Chen IP, de Almeida S, Tiziani V, Do Amaral CM, Gowrishankar K, Passos-Bueno MR, Reichenberger EJ. A novel autosomal recessive GJA1 missense mutation linked to Craniometaphyseal dysplasia.
PLoS One. 2013;8:e73576. [
PMC free article: PMC3741164] [
PubMed: 23951358]
Hwang JM, Kim IO, Wang KC. Complete visual recovery in osteopetrosis by early optic nerve decompression.
Pediatr Neurosurg. 2000;33:328–32. [
PubMed: 11182645]
Jacquemin C, Mullaney P, Svedberg E. Marble brain syndrome: osteopetrosis, renal acidosis and calcification of the brain.
Neuroradiology. 1998;40:662–3. [
PubMed: 9833897]
Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature. 2017;549:519–22. [
PubMed: 28959963]
Kantaputra PN, Thawanaphong S, Issarangporn W, Klangsinsirikul P, Ohazama A, Sharpe P, Supanchart C. Long-term survival in infantile malignant autosomal recessive osteopetrosis secondary to homozygous p.Arg526Gln mutation in CLCN7.
Am J Med Genet A. 2012;158A:909–16. [
PubMed: 22419446]
Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man.
Cell. 2001;104:205–15. [
PubMed: 11207362]
Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch TJ, Kubisch C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis.
Hum Mol Genet. 2000;9:2059–63. [
PubMed: 10942435]
Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, Morris EC, Moshous D, Neven B, Schulz A, Slatter M, Veys P, et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity.
Bone Marrow Transplant. 2021;56:2052–62. [
PMC free article: PMC8410590] [
PubMed: 34226669]
Leisle L, Ludwig CF, Wagner FA, Jentsch TJ, Stauber T. ClC-7 is a slowly voltage-gated 2Cl(-)/1H(+)-exchanger and requires Ostm1 for transport activity.
EMBO J. 2011;30:2140–52. [
PMC free article: PMC3117652] [
PubMed: 21527911]
Natsheh J, Drozdinsky G, Simanovsky N, Lamdan R, Erlich O, Gorelik N, Or R, Weintraub M, Stepensky P. Improved outcomes of hematopoietic stem cell transplantation in patients with infantile malignant osteopetrosis using fludarabine-based conditioning.
Pediatr Blood Cancer. 2016;63:535–40. [
PubMed: 26485304]
Nicoli ER, Weston MR, Hackbarth M, Becerril A, Larson A, Zein WM, Baker PR 2nd, Burke JD, Dorward H, Davids M, Huang Y, Adams DR, Zerfas PM, Chen D, Markello TC, Toro C, Wood T, Elliott G, Vu M. Undiagnosed Diseases Network, Zheng W, Garrett LJ, Tifft CJ, Gahl WA, Day-Salvatore DL, Mindell JA, Malicdan MCV. Lysosomal storage and albinism due to effects of a de novo CLCN7 variant on lysosomal acidification.
Am J Hum Genet. 2019;104:1127–38. [
PMC free article: PMC6562152] [
PubMed: 31155284]
Orchard PJ, Fasth AL, Le Rademacher J, He W, Boelens JJ, Horwitz EM, Al-Seraihy A, Ayas M, Bonfim CM, Boulad F, Lund T, Buchbinder DK, Kapoor N, O'Brien TA, Perez MA, Veys PA, Eapen M. Hematopoietic stem cell transplantation for infantile osteopetrosis.
Blood. 2015;126:270–6. [
PMC free article: PMC4497967] [
PubMed: 26012570]
Palagano E, Blair HC, Pangrazio A, Tourkova I, Strina D, Angius A, Cuccuru G, Oppo M, Uva P, Van Hul W, Boudin E, Superti-Furga A, Faletra F, Nocerino A, Ferrari MC, Grappiolo G, Monari M, Montanelli A, Vezzoni P, Villa A, Sobacchi C. Buried in the middle but guilty: intronic mutations in the TCIRG1 gene cause human autosomal recessive osteopetrosis.
J Bone Miner Res. 2015;30:1814–21. [
PubMed: 25829125]
Pangrazio A, Fasth A, Sbardellati A, Orchard PJ, Kasow KA, Raza J, Albayrak C, Albayrak D, Vanakker OM, De Moerloose B, Vellodi A, Notarangelo LD, Schlack C, Strauss G, Kühl JS, Caldana E, Iacono NL, Susani L, Kornak U, Schulz A, Vezzoni P, Villa A, Sobacchi C. SNX10 mutations define a subgroup of human autosomal recessive osteopetrosis with variable clinical severity.
J Bone Miner Res. 2013;28:1041–9. [
PubMed: 23280965]
Rössler U, Hennig AF, Stelzer N, Bose S, Kopp J, Søe K, Cyganek L, Zifarelli G, Ali S, von der Hagen M, Strässler ET, Hahn G, Pusch M, Stauber T, Izsvák Z, Gossen M, Stachelscheid H, Kornak U. Efficient generation of osteoclasts from human induced pluripotent stem cells and functional investigations of lethal CLCN7-related osteopetrosis.
J Bone Miner Res. 2021;36:1621–35. [
PubMed: 33905594]
Pangrazio A, Frattini A, Valli R, Maserati E, Susani L, Vezzoni P, Villa A, Al-Herz W, Sobacchi C. A homozygous contiguous gene deletion in chromosome 16p13.3 leads to autosomal recessive osteopetrosis in a Jordanian patient.
Calcif Tissue Int. 2012;91:250–4. [
PubMed: 22847576]
Pangrazio A, Poliani PL, Megarbane A, Lefranc G, Lanino E, Di Rocco M, Rucci F, Lucchini F, Ravanini M, Facchetti F, Abinun M, Vezzoni P, Villa A, Frattini A. Mutations in OSTM1 (grey lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement.
J Bone Miner Res. 2006;21:1098–105. [
PubMed: 16813530]
Pangrazio A, Pusch M, Caldana E, Frattini A, Lanino E, Tamhankar PM, Phadke S, Lopez AG, Orchard P, Mihci E, Abinun M, Wright M, Vettenranta K, Bariae I, Melis D, Tezcan I, Baumann C, Locatelli F, Zecca M, Horwitz E, Mansour LS, Van Roij M, Vezzoni P, Villa A, Sobacchi C. Molecular and clinical heterogeneity in CLCN7-dependent osteopetrosis: report of 20 novel mutations.
Hum Mutat. 2010;31:E1071–80. [
PubMed: 19953639]
Pangrazio A, Puddu A, Oppo M, Valentini M, Zammataro L, Vellodi A, Gener B, Llano-Rivas I, Raza J, Atta I, Vezzoni P, Superti-Furga A, Villa A, Sobacchi C. Exome sequencing identifies CTSK mutations in patients originally diagnosed as intermediate osteopetrosis.
Bone. 2014;59:122–6. [
PMC free article: PMC3885796] [
PubMed: 24269275]
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation.
Nat Genet. 2016;48:126–33. [
PMC free article: PMC4731925] [
PubMed: 26656846]
Shroff R, Beringer O, Rao K, Hofbauer L, Schulz A. Denosumab for post-transplantation hypercalcemia in osteopetrosis.
N Engl J Med. 2012;367:1766–7. [
PubMed: 23113501]
Sobacchi C, Frattini A, Guerrini MM, Abinun M, Pangrazio A, Susani L, Bredius R, Mancini G, Cant A, Bishop N, Grabowski P, Del Fattore A, Messina C, Errigo G, Coxon FP, Scott DI, Teti A, Rogers MJ, Vezzoni P, Villa A Helfrich MH. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL.
Nat Genet. 2007;39:960–2. [
PubMed: 17632511]
Sobacchi C, Pangrazio A, Lopez AG, Gomez DP, Caldana ME, Susani L, Vezzoni P, Villa A. As little as needed: the extraordinary case of a mild recessive osteopetrosis owing to a novel splicing hypomorphic mutation in the TCIRG1 gene.
J Bone Miner Res. 2014;29:1646–50. [
PMC free article: PMC4258090] [
PubMed: 24535816]
Sobacchi C, Schulz A, Coxon FP, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function.
Nat Rev Endocrinol. 2013;9:522–36. [
PubMed: 23877423]
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting.
Hum Genet. 2020;139:1197–207. [
PMC free article: PMC7497289] [
PubMed: 32596782]
Steward CG. Neurological aspects of osteopetrosis.
Neuropathol Appl Neurobiol. 2003;29:87–97. [
PubMed: 12662317]
Steward CG, Pellier I, Mahajan A, Ashworth MT, Stuart AG, Fasth A, Lang D, Fischer A, Friedrich W, Schulz AS. Severe pulmonary hypertension: a frequent complication of stem cell transplantation for malignant infantile osteopetrosis.
Br J Haematol. 2004;124:63–71. [
PubMed: 14675409]
Strickland JP, Berry DJ. Total joint arthroplasty in patients with osteopetrosis: a report of 5 cases and review of the literature.
J Arthroplasty. 2005;20:815–20. [
PubMed: 16139724]
Van Wesenbeeck L, Odgren PR, Coxon FP, Frattini A, Moens P, Perdu B, MacKay CA, Van Hul E, Timmermans JP, Vanhoenacker F, Jacobs R, Peruzzi B, Teti A, Helfrich MH, Rogers MJ, Villa A, Van Hul W. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans.
J Clin Invest. 2007;117:919–30. [
PMC free article: PMC1838941] [
PubMed: 17404618]
Waguespack SG, Hui SL, Dimeglio LA, Econs MJ. Autosomal dominant osteopetrosis: clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation.
J Clin Endocrinol Metab. 2007;92:771–8. [
PubMed: 17164308]
Wang H, Pan M, Ni J, Zhang Y, Zhang Y, Gao S, Liu J, Wang Z, Zhang R, He H, Wu B, Duan X. ClC-7 deficiency impairs tooth development and eruption.
Sci Rep. 2016;6:19971. [
PMC free article: PMC4734291] [
PubMed: 26829236]