By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2000, Sinauer
Associates.
Bookshelf ID: NBK9967

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Gilbert SF. Developmental Biology. 6th edition. Sunderland (MA): Sinauer Associates; 2000.

Primary sex determination is the determination of the gonads. In mammals, primary sex determination is strictly chromosomal and is not usually influenced by the environment. In most cases, the female is XX and the male is XY. Every individual must have at least one X chromosome. Since the female is XX, each of her eggs has a single X chromosome. The male, being XY, can generate two types of sperm: half bear the X chromosome, half the Y. If the egg receives another X chromosome from the sperm, the resulting individual is XX, forms ovaries, and is female; if the egg receives a Y chromosome from the sperm, the individual is XY, forms testes, and is male. The Y chromosome carries a gene that encodes a testis-determining factor. This factor organizes the gonad into a testis rather than an ovary. Unlike the situation in Drosophila (discussed below), the mammalian Y chromosome is a crucial factor for determining sex in mammals. A person with five X chromosomes and one Y chromosome (XXXXXY) would be male. Furthermore, an individual with only a single X chromosome and no second X or Y (i.e., XO) develops as a female and begins making ovaries, although the ovarian follicles cannot be maintained. For a complete ovary, a second X chromosome is needed.
In mammalian primary sex determination, there is no “default state.” The formation of ovaries and testes are both active, gene-directed processes. Moreover, as we shall see, both diverge from a common precursor, the bipotential gonad.
Secondary sex determination affects the bodily phenotype outside the gonads. A male mammal has a penis, seminal vesicles, and prostate gland. A female mammal has a vagina, cervix, uterus, oviducts, and mammary glands. In many species, each sex has a sex-specific size, vocal cartilage, and musculature. These secondary sex characteristics are usually determined by hormones secreted from the gonads. However, in the absence of gonads, the female phenotype is generated. When Jost (1953) removed fetal rabbit gonads before they had differentiated, the resulting rabbits had a female phenotype, regardless of whether they were XX or XY. They each had oviducts, a uterus, and a vagina, and each lacked a penis and male accessory structures.
The general scheme of mammalian sex determination is shown in Figure 17.2. If the Y chromosome is absent, the gonadal primordia develop into ovaries. The ovaries produce estrogen, a hormone that enables the development of the Müllerian duct into the uterus, oviducts, and upper end of the vagina. If the Y chromosome is present, testes form and secrete two major hormones. The first hormone—anti-Müllerian duct hormone (AMH; also referred to as Müllerian-inhibiting substance, MIS)—destroys the Müllerian duct. The second hormone—testosterone—masculinizes the fetus, stimulating the formation of the penis, scrotum, and other portions of the male anatomy, as well as inhibiting the development of the breast primordia. Thus, the body has the female phenotype unless it is changed by the two hormones secreted by the fetal testes. We will now take a more detailed look at these events.

Postulated cascades leading to the formation of the sexual phenotypes in mammals. The conversion of the genital ridge into the bipotential gonad requires the LHX9, SF1 and WT1 genes, since mice lacking either of these genes lack gonads. The bipotential (more...)
The gonads embody a unique embryological situation. All other organ rudiments can normally differentiate into only one type of organ. A lung rudiment can become only a lung, and a liver rudiment can develop only into a liver. The gonadal rudiment, however, has two normal options. When it differentiates, it can develop into either an ovary or a testis. The path of differentiation taken by this rudiment determines the future sexual development of the organism. But, before this decision is made, the mammalian gonad first develops through a bipotential (indifferent) stage, during which time it has neither female nor male characteristics.
In humans, the gonadal rudiments appear in the intermediate mesoderm during week 4 and remains sexually indifferent until week 7. The gonadal rudiments are paired regions of the intermediate mesoderm; they form adjacent to the developing kidneys. The ventral portions of the gonadal rudiments are composed of the genital ridge epithelium. During the indifferent stage, the genital ridge epithelium proliferates into the loose connective mesenchymal tissue above it (Figure 17.3A,B). These epithelial layers form the sex cords. The germ cells migrate into the gonad during week 6, and are surrounded by the sex cords. In both XY and XX gonads, the sex cords remain connected to the surface epithelium.

Differentiation of human gonads shown in transverse section. (A) Genital ridge of a 4-week embryo. (B) Genital ridge of a 6-week indifferent gonad showing primitive sex cords. (C) Testis development in the eighth week. The sex cords lose contact with (more...)
If the fetus is XY, the sex cords continue to proliferate through the eighth week, extending deeply into the connective tissue. These cords fuse, forming a network of internal (medullary) sex cords and, at its most distal end, the thinner rete testis (Figure 17.3C,D). Eventually, the sex cords—now called testis cords—lose contact with the surface epithelium and become separated from it by a thick extracellular matrix, the tunica albuginea. Thus, the germ cells are found in the cords within the testes. During fetal life and childhood, the testis cords remain solid. At puberty, however, the cords will hollow out to form the seminiferous tubules, and the germ cells will begin to differentiate into sperm.
The cells of the seminiferous tubule are called Sertoli cells. The Sertoli cells of the testis cords nurture the sperm and secrete anti-Müllerian duct hormone. The sperm are transported from the inside of the testis through the rete testis, which joins the efferent ducts. These efferent tubules are the remnants of the mesonephric kidney, and they link the testis to the Wolffian duct, which used to be the collecting tube of the mesonephric kidney (see Chapter 15). In males, the Wolffian duct differentiates to become the epididymis (adjacent to the testis) and the vas deferens, the tube through which the sperm pass into the urethra and out of the body. Meanwhile, during fetal development, the interstitial mesenchyme cells of the testes differentiate into Leydig cells, which make testosterone.
Mammalian gonads. The histology of the mammalian ovary and testis can be seen in labeled photographs that show progressively smaller regions at higher magnifications. [Click on Gametogenesis]
In females, the germ cells will reside near the outer surface of the gonad. Unlike the sex cords in males, which continue their proliferation, the initial sex cords of XX gonads degenerate. However, the epithelium soon produces a new set of sex cords, which do not penetrate deeply into the mesenchyme, but stay near the outer surface (cortex) of the organ. Thus, they are called cortical sex cords. These cords are split into clusters, with each cluster surrounding a germ cell (Figure 17.3E,F). The germ cells will become the ova, and the surrounding cortical sex cords will differentiate into the granulosa cells. The mesenchyme cells of the ovary differentiate into the thecal cells. Together, the thecal and granulosa cells will form the follicles that envelop the germ cells and secrete steroid hormones. Each follicle will contain a single germ cell. In females, the Müllerian duct remains intact, and it differentiates into the oviducts, uterus, cervix, and upper vagina. The Wolffian duct, deprived of testosterone, degenerates. A summary of the development of mammalian reproductive systems is shown in Figure 17.4.
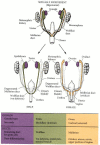
Summary of the development of the gonads and their ducts in mammals. Note that both the Wolffian and Müllerian ducts are present at the indifferent gonad stage.
Several genes have been found whose function is necessary for normal sexual differentiation. Unlike those that act in other developing organs, the genes involved in sex determination differ extensively between phyla, so one cannot look at Drosophila sex-determining genes and expect to see their homologues directing mammalian sex determination. However, since the phenotype of mutations in sex-determining genes is often sterility, clinical studies have been used to identify those genes that are active in determining whether humans become male or female. Experimental manipulations to confirm the functions of these genes can be done in mice.
In humans, the major gene for the testis-determining factor resides on the short arm of the Y chromosome. Individuals who are born with the short arm but not the long arm of the Y chromosome are male, while individuals born with the long arm of the Y chromosome but not the short arm are female. By analyzing the DNA of rare XX men and XY women, the position of the testis-determining gene has been narrowed down to a 35,000-base-pair region of the Y chromosome located near the tip of the short arm. In this region, Sinclair and colleagues (1990) found a male-specific DNA sequence that could encode a peptide of 223 amino acids. This peptide is probably a transcription factor, since it contains a DNA-binding domain called the HMG (high-mobility group) box. This domain is found in several transcription factors and nonhistone chromatin proteins, and it induces bending in the region of DNA to which it binds (Figure 17.5; Giese et al. 1992). This gene is called SRY (sex-determining region of the Y chromosome), and there is extensive evidence that it is indeed the gene that encodes the human testis-determining factor. SRY is found in normal XY males and in the rare XX males, and it is absent from normal XX females and from many XY females. Another group of XY females was found to have point or frameshift mutations in the SRY gene; these mutations prevent the SRY protein from binding to or bending DNA (Pontiggia et al. 1994; Werner et al. 1995). It is thought that several testis-specific genes contain SRY-binding sites in their promoters or enhancers, and that the binding of SRY to these sites begins the developmental pathway to testis formation (Cohen et al. 1994).

Association of DNA with the SRY protein can cause the DNA to bend 70–80 degrees. The black structure represents the HMG box of the SRY protein. The red coil is the double helix of DNA specifically bound by SRY. (After Haqq et al. 1994 and Werner (more...)
If SRY actually does encode the major testis-determining factor, one would expect that it would act in the genital ridge immediately before or during testis differentiation. This prediction has been met in studies of the homologous gene found in mice. The mouse gene (Sry) also correlates with the presence of testes; it is present in XX males and absent in XY females (Gubbay et al. 1990; Koopman et al. 1990). The Sry gene is expressed in the somatic cells of the bipotential mouse gonad immediately before or during its differentiating into a testis; its expression then disappears (Hacker et al. 1995).
The most impressive evidence for Sry being the gene for testis-determining factor comes from transgenic mice. If Sry induces testis formation, then inserting Sry DNA into the genome of a normal XX mouse zygote should cause that XX mouse to form testes. Koopman and colleagues (1991) took the 14-kilobase region of DNA that includes the Sry gene (and presumably its regulatory elements) and microinjected this sequence into the pronuclei of newly fertilized mouse zygotes. In several instances, the XX embryos injected with this sequence developed testes, male accessory organs, and penises (Figure 17.6). (Functional sperm were not formed, but they were not expected, either, because the presence of two X chromosomes prevents sperm formation in XXY mice and men, and the transgenic mice lacked the rest of the Y chromosome, which contains genes needed for spermatogenesis.) Therefore, there are good reasons to think that Sry/SRY is the major gene on the Y chromosome for testis determination in mammals.
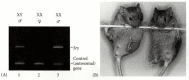
An XX mouse transgenic for Sry is male. (A) Polymerase chain reaction followed by electrophoresis shows the presence of the Sry gene in normal XY males and in a transgenic XX Sry mouse. The gene is absent in a female XX littermate. (B) The external genitalia (more...)
Sry/SRY is necessary, but not sufficient, for the development of the mammalian testis. Studies on mice (Eicher and Washburn 1983; Washburn and Eicher 1989; Eicher et al. 1996) have shown that the Sry gene of some strains of mice failed to produce testes when placed into a different strain of mouse. When the Sry protein binds to its sites on DNA, it probably creates large conformational changes. It unwinds the double helix in its vicinity and bends the DNA as much as 80 degrees (Pontiggia et al. 1994; Werner et al. 1995). This bending may bring distantly bound proteins of the transcription apparatus into close contact, enabling them to interact and influence transcription. The identities of these proteins are not yet known, but they, too, are needed for testis determination.
SRY may have more than one mode of action in converting the bipotential gonads into testes. It had been assumed for the past decade that SRY worked directly in the genital ridge to convert the epithelium into male-specific Sertoli cells. Recent studies (Capel et al. 1999), however, have suggested that SRY works via an indirect mechanism: SRY in the genital ridge cells induces the cells to secrete a chemotactic factor that permits the migration of mesonephric cells into the XY gonad. These mesonephric cells induce the gonadal epithelium to become Sertoli cells with male-specific gene expression patterns. The researchers found that when they cultured XX gonads with either XX or XY mesonephrons, the mesonephric cells did not enter the gonads. However, when they cultured XX or XY mesonephrons with XY gonads, or with gonads from XX mice containing the Sry transgene, the mesonephric cells did enter the gonads (Figure 17.7). There was a strict correlation between the presence of Sry in the gonadal cells, mesonephric cell migration, and the formation of testis cords. Tilmann and Capel (1999) showed that mesonephric cells are critical for testis cord formation and that the migrating mesonephric cells can induce XX gonadal cells to form testis cords. It appears, then, that Sry may function indirectly to create testes by inducing mesonephric cell migration into the gonad.

Migration of the mesonephric cells into Sry+ gonadal rudiments. In the experiment diagrammed, urogenital ridges (containing both the mesonephric kidneys and gonadal rudiments) were collected from 12-day embryonic mice. One of the mice was marked with (more...)
17.2 Finding the male-determining genes. The mapping of the testis-determining factor to the SRY region took scientists more than 50 years to accomplish. Moreover, other testis-forming genes have been found on the autosomes. http://www.devbio.com/chap17/link1702.shtml
One of the autosomal genes involved in sex determination is SOX9, which encodes a putative transcription factor that also contains an HMG box. XX humans who have an extra copy of SOX9 develop as males, even though they have no SRY gene (Huang et al. 1999). Individuals having only one functional copy of this gene have a syndrome called campomelic dysplasia, a disease involving numerous skeletal and organ systems. About 75% of XY patients with this syndrome develop as phenotypic females or hermaphrodites (Foster et al. 1994; Wagner et al. 1994; Mansour et al. 1995). It appears that SOX9 is essential for testis formation. The mouse homologue of this gene, Sox9, is expressed only in male (XY) but not in female (XX) genital ridges. Moreover, Sox9 expression is seen in the same genital ridge cells as Sry, and it is expressed just slightly after Sry expression (Wright et al. 1995; Kent et al. 1996). The Sox9 protein binds to a promoter site on the Amh gene, providing a critical link in the pathway toward a male phenotype (Figure 17.8; Arango et al. 1999).
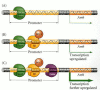
Synergism of Sox9 and Sf1 to activate the expression of the Amh gene. (A) The binding of Sox9 to the Amh promoter initiates transcription of the Amh gene in the Sertoli cells. (B) After Sox9 binds, the expression of AMH is upregulated by the binding of (more...)
While Sry is found specifically in mammals, Sox9 is found throughout the vertebrates. Sox9 may be the older and more central sex determination gene, although in mammals it became activated by its relative, Sry.
Another protein that may be directly or indirectly activated by SRY is the transcription factor SF1 (steroidogenic factor 1). Sf1 is necessary to make the bipotential gonad; but while Sf1 levels decline in the genital ridge of XX mouse embryos, the Sf1 gene stays on in the developing testis. Sf1 appears to be active in masculinizing both the Leydig and the Sertoli cells. In the Sertoli cells, Sf1, working in collaboration with Sox9, is needed to elevate the levels of AMH transcription (see Figure 17.8; Shen et al. 1994; Arango et al. 1999). In the Leydig cells, Sf1 activates the genes encoding the enzymes that make testosterone. The importance of SF1 for testis development and AMH regulation in humans is demonstrated by an XY patient who is heterozygous for SF1. Although the genes for SRY and SOX9 are normal, this individual has malformed fibrous gonads and retains fully developed Müllerian duct structures (Achermann et al. 1999). It is thought that SRY (directly or indirectly) activates the SF1 gene, and the SF1 protein then activates both components of the male sexual differentiation pathway (Sertoli AMH and Leydig testosterone).
In 1980, Bernstein and her colleagues reported two sisters who were genetically XY. Their Y chromosomes were normal, but they had a duplication of a small portion of the short arm of the X chromosome. Subsequent cases were found, and it was concluded that if there were two copies of this region on the active X chromosome, the SRY signal would be reversed (Figure 17.9). Bardoni and her colleagues (1994) proposed that this region contains a gene for a protein that competes with the SRY factor and that is important in directing the development of the ovary. In testicular development, this gene would be suppressed, but having two active copies of the gene would override this suppression. This gene, DAX1, has been cloned and shown to encode a member of the nuclear hormone receptor family (Muscatelli et al. 1994; Zanaria 1994). Dax1 is expressed in the genital ridges of the mouse embryo, shortly after Sry expression. Indeed, in XY mice, Sry and Dax1 are expressed in the same cells. DAX1 appears to antagonize the function of SRY, and it down-regulates SF1 expression (Nachtigal et al. 1998; Swain et al. 1998). Thus, DAX1 is probably a gene that is involved in ovary determination.
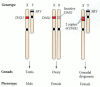
Phenotypic sex reversal in humans having two copies of the DAX1 locus. DAX1 (on the X chromosome) plus SRY (on the Y chromosome) produces testes. DAX1 without SRY (since the other DAX1 locus is on the inactive X chromosome) produces ovaries. Two active (more...)
The WNT4 gene is another gene that may be critical in ovary determination. This gene is expressed in the mouse genital ridge while it is still in its bipotential stage. Wnt4 expression then becomes undetectable in XY gonads (which become testes), whereas it is maintained in XX gonads as they begin to form ovaries. In transgenic XX mice that lack the Wnt4 genes, the ovary fails to form properly, and its cells express testis-specific markers, including AMH- and testosterone-producing enzymes (Vainio et al. 1999). Sry may form testes by repressing Wnt4 expression in the genital ridge, as well as by promoting Sf1. One possible model is shown in Figure 17.10.

Possible mechanism for primary sex determination in mammals. While we do not know the specific interactions involved, this model attempts to organize the data into a coherent sequence. Other models are possible. In this model, SRY competes with the DAX1 (more...)
It should be realized that both testis and ovary development are active processes. In mammalian primary sex determination, neither is a “default state” (Eicher and Washburn 1986). Although remarkable progress has been made in recent years, we still do not know what the testis- or ovary-determining genes are doing, and the problem of primary sex determination remains (as it has since prehistory) one of the great unsolved problems of biology.
Primary sex determination involves the formation of either an ovary or a testis from the bipotential gonad. This, however, does not give the complete sexual phenotype. Secondary sex determination in mammals involves the development of the female and male phenotypes in response to hormones secreted by the ovaries and testes. Both female and male secondary sex determination have two major temporal phases. The first occurs within the embryo during organogenesis; the second occurs during adolescence.
As mentioned earlier, if the bipotential gonads are removed from an embryonic mammal, the female phenotype is realized: the Müllerian ducts develop while the Wolffian duct degenerates. This pattern also is seen in certain humans who are born without functional gonads. Individuals whose cells have only one X chromosome (and no Y chromosome) originally develop ovaries, but these ovaries atrophy before birth, and the germ cells die before puberty. However, under the influence of estrogen, derived first from the ovary but then from the mother and placenta, these infants are born with a female genital tract (Langman and Wilson 1982).
The formation of the male phenotype involves the secretion of two testicular hormones. The first of these hormones is AMH, the hormone made by the Sertoli cells that causes the degeneration of the Müllerian duct. The second is the steroid testosterone, which is secreted from the fetal Leydig cells. This hormone causes the Wolffian duct to differentiate into the epididymis, vas deferens, and seminal vesicles, and it causes the urogenital swellings to develop into the scrotum and penis.
The existence of these two independent systems of masculinization is demonstrated by people having androgen insensitivity syndrome. These XY individuals have the SRY gene, and thus have testes that make testosterone and AMH. However, they lack the testosterone receptor protein, and therefore cannot respond to the testosterone made by their testes (Meyer et al. 1975). Because they are able to respond to estrogen made in their adrenal glands, they develop the female phenotype (Figure 17.11). However, despite their distinctly female appearance, these individuals do have testes, and even though they cannot respond to testosterone, they produce and respond to AMH. Thus, their Müllerian ducts degenerate. These people develop as normal but sterile women,* lacking a uterus and oviducts and having testes in the abdomen.

An XY individual with androgen insensitivity syndrome. Despite the XY karyotype and the presence of testes, such individuals develop female secondary sex characteristics. Internally, however, these women lack the Müllerian duct derivatives and (more...)
Although testosterone is one of the two primary masculinizing hormones, there is evidence that it might not be the active masculinizing hormone in certain tissues. Testosterone appears to be responsible for promoting the formation of the male reproductive structures (the epididymis, seminal vesicles, and vas deferens) that develop from the Wolffian duct primordium. However, it does not directly masculinize the male urethra, prostate, penis, or scrotum. These latter functions are controlled by 5a-dihydrotestosterone (Figure 17.12). Siiteri and Wilson (1974) showed that testosterone is converted to 5α-dihydrotestosterone in the urogenital sinus and swellings, but not in the Wolffian duct. 5α-dihydrotestosterone appears to be a more potent hormone than testosterone.

Testosterone- and dihydrotestosterone-dependent regions of the human male genital system. (After Imperato-McGinley et al. 1974.)
The importance of 5α-dihydrotestosterone was demonstrated by Imperato-McGinley and her colleagues (1974). They found a small community in the Dominican Republic in which several inhabitants had a genetic deficiency of the enzyme 5α-ketosteroid reductase 2, the enzyme that converts testosterone to dihydrotestosterone. These individuals lack a functional gene for this enzyme (Andersson et al. 1991; Thigpen et al. 1992). Although XY children with this syndrome have functioning testes, they have a blind vaginal pouch and an enlarged clitoris. They appear to be girls and are raised as such. Their internal anatomy, however, is male: they have testes, Wolffian duct development, and Müllerian duct degeneration. Thus, it appears that the formation of the external genitalia is under the control of dihydrotestosterone, whereas Wolffian duct differentiation is controlled by testosterone itself. Interestingly, when the testes of these children produce more testosterone at puberty, the external genitalia are able to respond to the higher levels of the hormone, and they differentiate. The penis enlarges, the scrotum descends, and the person originally thought to be a girl is shown to be a young man.
17.3 Dihydrotestosterone in adult men. The drug finasteride, which inhibits the conversion of testosterone to dihydrotestosterone, is being used to treat prostate growth and male pattern baldness. http://www.devbio.com/chap17/link1703.shtml
17.4 Insulin-like hormone 3. In addition to testosterone, the Leydig cells secrete another hormone, insulin-like hormone 3 (Insl3). This hormone is required for the descent of the gonads into the scrotum. Males lacking this hormone are infertile because the testes do not descend. In females, lack of this hormone deregulates the menstrual cycle. http://www.devbio.com/chap17/link1704.shtml
Anti-Müllerian duct hormone (AMH), the hormone that causes the degeneration of the Müllerian duct, is a 560-amino acid glycoprotein secreted from the Sertoli cells (Tran et al. 1977; Cate et al. 1986). When fragments of fetal testes or isolated Sertoli cells are placed adjacent to cultured tissue segments containing portions of the Wolffian and Müllerian ducts, the Müllerian duct atrophies even though no change occurs in the Wolffian duct (Figure 17.13). AMH is thought to bind to the mesenchyme cells surrounding the Müllerian duct and to cause these cells to secrete a paracrine factor that induces apoptosis in the Müllerian duct epithelium (Trelstad et al. 1982; Roberts et al. 1999).

Assay for AMH activity in the anterior segment of a 14.5-day fetal rat reproductive tract. (A) At the start of the experiment, both the Müllerian duct (arrow at left) and Wolffian duct (arrow at right) are open. (B) After 3 days in culture with (more...)
17.5 Roles of AMH. AMH may have other roles in sex determination besides causing the breakdown of the Müllerian ducts. It may cause sex reversal in some mammals, and may become useful as an anti-tumor drug. http://www.devbio.com/chap17/link1705.shtml
Estrogen is needed for the complete development of both the Müllerian and the Wolffian ducts. In females, estrogen secreted from the fetal ovaries appears sufficient to induce the differentiation of the Müllerian duct into its various components: the uterus, oviducts, and cervix. The extreme sensitivity of the Müllerian duct to estrogenic compounds is demonstrated by the teratogenic effects of diethylstilbesterol (DES), a powerful synthetic estrogen that can cause infertility by changing the patterning of the Müllerian duct (see Mittendorf 1995). In mice, DES can cause the oviduct epithelium to take on the appearance of the uterus, and the uterine epithelium to resemble that of the cervix (Ma et al. 1998).
In males, estrogen is actually needed for fertility. One of the functions of the efferent duct (vas efferens) cells is to absorb about 90% of the water from the lumen of the rete testis. This concentrates the sperm, giving them a longer lifespan and providing more sperm per ejaculate. This absorption of water is regulated by estrogen. If estrogen or its receptor is absent in mice, this water is not absorbed, and the mouse is sterile (Hess et al. 1997). While blood concentrations of estrogen are higher in females than in males, the concentration of estrogen in the rete testis is even higher than that in female blood.
17.6 The mechanisms of breast development. Breast tissue has a sexually dimorphic mode of development. Testosterone inhibits breast development, while estrogen promotes it. Most breast development is accomplished after birth, and different hormones act during puberty and pregnancy to cause breast enlargement and differentiation. http://www.devbio.com/chap17/link1706.shtml
17.7 The action of DES. Diethylstilbesterol was a drug given to women to ease their pregnancies. Unfortunately, it was later found to alter the reproductive tract of female fetuses. The mechanism of DES action is thought to involve the repression of the Hox genes that instruct the regional specificity of the Müllerian duct. http://www.devbio.com/chap17/link1707.shtml
Sex Determination and Behaviors.
Androgen insensitivity syndrome is one of several conditions called pseudohermaphroditism. In a pseudohermaphrodite, there is only one type of gonad, but the secondary sex characteristics differ from what would be expected from the gonadal sex. In humans, male pseudohermaphroditism can be caused by mutations in the androgen receptor or by mutations affecting testosterone synthesis (Geissler et al. 1994). Female pseudohermaphroditism can be caused by an overproduction of testosterone.
True hermaphrodites (rare in humans, but the norm in some invertebrates such as nematodes and earthworms) contain both male and female gonadal tissue. Mammalian true hermaphrodites result from abnormalities of primary sex determination. Such abnormalities can occur when the Y chromosome is translocated to the X chromosome. In those tissues where the translocated X chromosome is inactivated during dosage compensation, the SRY gene will be turned off. However, in those tissues where the translocated X chromosome is not inactivated, the SRY gene will be on (Berkovitz et al. 1992; Margarit et al. 2000).
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...