By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2000, Geoffrey M Cooper.
Bookshelf ID: NBK9894

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000.

The activation of cellular oncogenes represents only one of two distinct types of genetic alterations involved in tumor development; the other is inactivation of tumor suppressor genes. Oncogenes drive abnormal cell proliferation as a consequence of genetic alterations that either increase gene expression or lead to uncontrolled activity of the oncogene-encoded proteins. Tumor suppressor genes represent the opposite side of cell growth control, normally acting to inhibit cell proliferation and tumor development. In many tumors, these genes are lost or inactivated, thereby removing negative regulators of cell proliferation and contributing to the abnormal proliferation of tumor cells.
The first insight into the activity of tumor suppressor genes came from somatic cell hybridization experiments, initiated by Henry Harris and his colleagues in 1969. The fusion of normal cells with tumor cells yielded hybrid cells containing chromosomes from both parents (Figure 15.32). In most cases, such hybrid cells were not capable of forming tumors in animals. Therefore, it appeared that genes derived from the normal cell parent acted to inhibit (or suppress) tumor development. Definition of these genes at the molecular level came, however, from a different approach—the analysis of rare inherited forms of human cancer.

Suppression of tumorigenicity by cell fusion. Fusion of tumor cells with normal cells yields hybrids that contain chromosomes from both parents. Such hybrids are usually nontumorigenic.
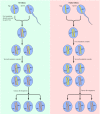
The first tumor suppressor gene was identified by studies of retinoblastoma, a rare childhood eye tumor. Provided that the disease is detected early, retinoblastoma can be successfully treated, and many patients survive to have families. Consequently, it was recognized that some cases of retinoblastoma are inherited. In these cases, approximately 50% of the children of an affected parent develop retinoblastoma, consistent with Mendelian transmission of a single dominant gene that confers susceptibility to tumor development (Figure 15.33).

Inheritance of retinoblastoma. Susceptibility to retinoblastoma is transmitted to approximately 50% of offspring. Affected and normal indivi- duals are indicated by purple and green symbols, respectively.
Although susceptibility to retinoblastoma is transmitted as a dominant trait, inheritance of the susceptibility gene is not sufficient to transform a normal retinal cell into a tumor cell. All retinal cells in a patient inherit the susceptibility gene, but only a small fraction of these cells give rise to tumors. Thus, tumor development requires additional events beyond inheritance of tumor susceptibility. In 1971, Alfred Knudson proposed that the development of retinoblastoma requires two mutations, which are now known to correspond to the loss of both of the functional copies of the tumor susceptibility gene (the Rb tumor suppressor gene) that would be present on homologous chromosomes of a normal diploid cell (Figure 15.34). In inherited retinoblastoma, one defective copy of Rb is genetically transmitted. The loss of this single Rb copy is not by itself sufficient to trigger tumor development, but retinoblastoma almost always develops in these individuals as a result of a second somatic mutation leading to the loss of the remaining normal Rb allele. Noninherited retinoblastoma, in contrast, is rare, since its development requires two independent somatic mutations to inactivate both normal copies of Rb in the same cell.
Mutations of Rb during retinoblastoma development. In hereditary retinoblastoma, a defective copy of the Rb gene (Rb-) is inherited from the affected parent. A second somatic mutation, which inactivates the single normal Rb+ copy in a retinal cell, then (more...)
The functional nature of the Rb gene as a negative regulator of tumorigenesis was initially indicated by observations of chromosome morphology. Visible deletions of chromosome 13q14 were found in some retinoblastomas, suggesting that loss (rather than activation) of the Rb gene led to tumor development (Figure 15.35). Gene-mapping studies further indicated that tumor development resulted from loss of normal Rb alleles in the tumor cells, consistent with the function of Rb as a tumor suppressor gene. Isolation of the Rb gene as a molecular clone in 1986 then firmly established that Rb is consistently lost or mutated in retinoblastomas. Gene transfer experiments also demonstrated that introduction of a normal Rb gene into retinoblastoma cells reverses their tumorigenicity, providing direct evidence for the activity of Rb as a tumor suppressor.

Rb deletions in retinoblastoma. Many retinoblastomas have deletions of the chromosomal locus (13q14) that contains the Rb gene.
Although Rb was identified in a rare childhood cancer, it is also involved in some of the more common tumors of adults. In particular, studies of the cloned gene have established that Rb is lost or inactivated in many bladder, breast, and lung carcinomas. The significance of the Rb tumor suppressor gene thus extends beyond retinoblastoma, apparently contributing to development of a substantial fraction of more common human cancers. In addition, as noted earlier in this chapter, the Rb protein is a key target for the oncogene proteins of several DNA tumor viruses, including SV40, adenoviruses, and human papillomaviruses, which bind to Rb and inhibit its activity (Figure 15.36). Transformation by these viruses thus results, at least in part, from inactivation of Rb at the protein level rather than from mutational inactivation of the Rb gene.

Interaction of Rb with oncogene proteins of DNA tumor viruses. The oncogene proteins of several DNA tumor viruses (e.g., SV40 T antigen) induce transformation by binding to and inactivating Rb protein.
Characterization of Rb as a tumor suppressor gene served as the prototype for the identification of additional tumor suppressor genes that contribute to the development of many different human cancers (Table 15.5). Some of these genes were identified as the causes of rare inherited cancers, playing a role similar to that of Rb in hereditary retinoblastoma. Other tumor suppressor genes have been identified as genes that are frequently deleted or mutated in common noninherited cancers of adults, such as colon carcinoma. In either case, it appears that most tumor suppressor genes are involved in the development of both inherited and noninherited forms of cancer. Indeed, mutations of some tumor suppressor genes appear to be the most common molecular alterations leading to human tumor development.
Tumor Suppressor Genes.
The second tumor suppressor gene to have been identified is p53, which is frequently inactivated in a wide variety of human cancers, including leukemias, lymphomas, sarcomas, brain tumors, and carcinomas of many tissues, including breast, colon, and lung. In total, mutations of p53 may play a role in up to 50% of all cancers, making it the most common target of genetic alterations in human malignancies. It is also of interest that inherited mutations of p53 are responsible for genetic transmission of a rare hereditary cancer syndrome, in which affected individuals develop any of several different types of cancer. In addition, the p53 protein (like Rb) is a target for the oncogene proteins of SV40, adenoviruses, and human papillomaviruses.
Like p53, the INK4 and PTEN tumor suppressor genes are very frequently mutated in several common cancers, including lung cancer, prostate cancer, and melanoma. Two other tumor suppressor genes (APC and MADR2) are frequently deleted or mutated in colon cancers. In addition to being involved in noninherited cases of this common adult cancer, inherited mutations of the APC gene are responsible for a rare hereditary form of colon cancer, called familial adenomatous polyposis. Individuals with this condition develop hundreds of benign colon adenomas (polyps), some of which almost inevitably progress to malignancy. Inherited mutations of two other tumor suppressor genes, BRCA1 and BRCA2, are responsible for hereditary cases of breast cancer, which account for 5 to 10% of the total breast cancer incidence.
Additional tumor suppressor genes have been implicated in the development of brain tumors, pancreatic cancers, and basal cell skin carcinomas, as well as in several rare inherited cancer syndromes, such as Wilms' tumor. The number of identified tumor suppressor genes is rapidly expanding, and the characterization of these genes remains an active area of cancer research.
In contrast to proto-oncogene and oncogene proteins, the proteins encoded by most tumor suppressor genes inhibit cell proliferation or survival. Inactivation of tumor suppressor genes therefore leads to tumor development by eliminating negative regulatory proteins. In several cases, tumor suppressor proteins inhibit the same cell regulatory pathways that are stimulated by the products of oncogenes.
The protein encoded by the PTEN tumor suppressor gene is an interesting example of antagonism between oncogene and tumor suppressor gene products (Figure 15.37). The PTEN protein is a lipid phosphatase that dephosphorylates the 3 position of phosphatidylinositides, such as phosphatidylinositol 3,4,5-bisphosphate (PIP3). By dephosphorylating PIP3, PTEN antagonizes the activities of PI 3-kinase and Akt, which can act as oncogenes by promoting cell survival. Conversely, inactivation or loss of the PTEN tumor suppressor protein can contribute to tumor development as a result of increased levels of PIP3, activation of Akt, and inhibition of programmed cell death.
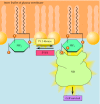
Suppression of cell survival by PTEN. The tumor suppressor protein PTEN is a lipid phosphatase that dephosphorylates PIP3 at the 3 position of inositol, yielding PIP2. PTEN thus counters the action of the oncogenes PI 3-kinase and Akt, which promote cell (more...)
Proteins encoded by both oncogenes and tumor suppressor genes also function in the Hedgehog signaling pathway (see Figure 13.44). The receptor Smoothened is an oncogene in basal cell carcinomas, whereas Patched (the negative regulator of Smoothened) is a tumor suppressor gene. In addition, the Gli proteins (the mammalian homologs of the Drosophila Ci transcription factor activated by Smoothened) were first identified as the products of an amplified oncogene.
Several tumor suppressor genes encode transcriptional regulatory proteins. A good example is provided by the product of WT1, which is frequently inactivated in Wilms' tumors (a childhood kidney tumor). The WT1 protein is a repressor that appears to suppress transcription of a number of growth factor-inducible genes. One of the targets of WT1 is thought to be the gene that encodes insulin-like growth factor II, which is overexpressed in Wilms' tumors and may contribute to tumor development by acting as an autocrine growth factor. Inactivation of WT1 may thus lead to abnormal growth factor expression, which in turn drives tumor cell proliferation. Two other tumor suppressor genes, DPC4 and MADR2, encode SMAD family transcription factors that are activated by TGF-β signaling and lead to inhibition of cell proliferation.
The products of the Rb and INK4 tumor suppressor genes regulate cell cycle progression at the same point as that affected by cyclin D1 (Figure 15.38). Rb inhibits passage through the restriction point in G1 by repressing transcription of a number of genes involved in cell cycle progression and DNA synthesis (see Figure 14.20). In normal cells, passage through the restriction point is regulated by Cdk4/cyclin D complexes, which phosphorylate and inactivate Rb. Mutational inactivation of Rb in tumors thus removes a key negative regulator of cell cycle progression. The INK4 tumor suppressor gene, which encodes the Cdk inhibitor p16, also regulates passage through the restriction point. As discussed in Chapter 14, p16 inhibits Cdk4/cyclin D activity. Inactivation of INK4 therefore leads to elevated activity of Cdk4/cyclin D complexes, resulting in uncontrolled phosphorylation of Rb.
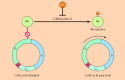
Inhibition of cell cycle progression by Rb and p16. Rb inhibits progression past the restriction point in G1. Cdk4/cyclin D complexes promote passage through the restriction point by phosphorylating and inactivating Rb. The activity of Cdk4/cyclin D is (more...)
The p53 gene product regulates both cell cycle progression and apoptosis (Figure 15.39). DNA damage leads to rapid induction of p53, which activates transcription of the Cdk inhibitor p21 (see Figure 14.21). The inhibitor p21 blocks cell cycle progression, both by acting as a general inhibitor of Cdk/cyclin complexes and by inhibiting DNA replication by binding to PCNA (proliferating cell nuclear antigen). The resulting cell cycle arrest presumably allows time for damaged DNA to be repaired before it is replicated. Loss of p53 prevents this damage-induced cell cycle arrest, leading to increased mutation frequencies and a general instability of the cell genome. Such genetic instability is a common property of cancer cells, and it may contribute to further alterations in oncogenes and tumor suppressor genes during tumor progression.
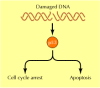
Action of p53. Wild-type p53 is required for both cell cycle arrest and apoptosis induced by DNA damage.
In addition to mediating cell cycle arrest, p53 is required for apoptosis induced by DNA damage. Unrepaired DNA damage normally induces apoptosis of mammalian cells, a response that is presumably advantageous to the organism because it eliminates cells carrying potentially deleterious mutations (e.g., cells that might develop into cancer cells). Cells lacking p53 fail to undergo apoptosis in response to agents that damage DNA, including radiation and many of the drugs used in cancer chemotherapy. This failure to undergo apoptosis in response to DNA damage contributes to the resistance of many tumors to chemotherapy. In addition, loss of p53 appears to interfere with apoptosis induced by other stimuli, such as growth factor deprivation and oxygen deprivation. These effects of p53 inactivation on cell survival are thought to account for the high frequency of p53 mutations in human tumors.
As discussed earlier, the development of cancer is a multistep process in which normal cells gradually progress to malignancy. The complete sequence of events required for the development of any human cancer is not yet known, but it is clear that both the activation of oncogenes and the inactivation of tumor suppressor genes are critical steps in tumor initiation and progression. Accumulated damage to multiple genes eventually results in the increased proliferation, invasiveness, and metastatic potential that are characteristic of cancer cells.
The role of multiple genetic defects is best understood in the case of colon carcinomas, which have been studied extensively by Bert Vogelstein and his colleagues. These tumors frequently involve mutation of rasK oncogenes and inactivation or deletion of three distinct tumor suppressor genes—APC, MADR2, and p53. Lesions representing multiple stages of colon cancer development are regularly obtained as surgical specimens, so it has been possible to correlate these genetic alterations with discrete stages of tumor progression (Figure 15.40).

Genetic alterations in colon carcinomas. Inactivation of APC is an early event in tumor development, giving rise to a proliferative cell population. Mutations of rasK then frequently occur and are found in early-stage adenomas. Subsequent mutations of (more...)
These studies indicate that inactivation of APC is an early event in tumor development. Genetic transmission of mutant APC genes in patients with familial adenomatous polyposis results in abnormal colon cell proliferation, leading to the outgrowth of multiple adenomas in the colons of affected patients. Mutations of APC also occur frequently in patients with noninherited colon carcinomas and are generally detected at early stages of the disease process. Mutations of rasK genes then appear to occur, and rasK oncogenes are also frequently present in small and intermediate-size adenomas. In contrast, the MADR2 and p53 tumor suppressor genes are inactivated usually at later stages of tumor progression. Mutations in these genes are only rarely found in early-stage adenomas, but they are frequently present in advanced adenomas and malignant carcinomas. The loss of these tumor suppressor genes in colon cancers thus appears to be involved in later stages of progression to malignancy, rather than in the initial stages of tumor formation.
Although these genetic alterations most often occur in the order described here, this is not an obligatory sequence of events. For example, mutations in p53 are sometimes detected in early adenomas. More important than the order in which mutations occur is the fact that colon cancer ultimately results from accumulated damage to multiple genes. Accumulated damage to both oncogenes and tumor suppressor genes similarly appears to be responsible for the development of other types of cancer, including breast and lung carcinomas. The progressive loss of growth control that is characteristic of cancer cells is thus thought to be the end result of abnormalities in the products of multiple genes that normally regulate cell proliferation, differentiation, and survival.
Molecular Medicine: Herception: Cancer Treatment Targeted against the erbB-2 Oncogene.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...