

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Gruber A, Durham AM, Huynh C, et al., editors. Bioinformatics in Tropical Disease Research: A Practical and Case-Study Approach [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2008.

Bioinformatics in Tropical Disease Research: A Practical and Case-Study Approach [Internet].
Show details1. Why Is It Important to Study Proteins?
In the drama of life on a molecular scale, proteins are where the action is (1).
Proteins are molecular devices, in the nanometer scale, where biological function is exerted (1). They are the building blocks of all cells in our bodies and in all living creatures of all kingdoms. Although the information necessary for life to go on is encoded by the DNA molecule, the dynamic process of life maintenance, replication, defense and reproduction are carried out by proteins.
There are twenty natural amino acids, whose frequency is higher than other special ones with particular functions. These twenty amino acids can be grouped together forming polypeptide chains, or proteins, in different ways determined by the genetic code and limited by stereochemical properties. These proteins may have a constitutive or transient cell expression in regard to its functions. It is worth mentioning that efforts are underway to make proteins of unnatural amino acids as well (2, 3).
The Gene Ontology Project (4) uses three categories to describe a gene product or protein. Molecular Function is the category that describes the tasks performed by individual proteins and can be broadly divided into twelve subcategories; namely cellular processes, metabolism, DNA replication/modification, transcription/translation, intracellular signaling, cell-cell communication, protein folding/degradation, transport, multifunctional proteins, cytoskeletal/structural, defense and immunity, and miscellaneous functions. Structural proteins, for example, are responsible for the cell integrity and cell overall shape (5), their functions varies from composing the cytoskeleton (6) to assembling transmembrane ion channels (7,8), a structure essential for cell osmolarity and even for synaptic information flux. Not only the DNA molecule cannot replicate itself without protein machinery such as the transcription complex, or transcription bubble (9); but also the mitosis and meiosis events of cell duplication and gamete production cannot go on without proteins performing crossing-over events and chromosome segregation (10). The immune system, responsible for our body's defense, is based on structure recognition, on differentiation of self from non-self; this can only be possible through specialized cells that can bind and identify what is foreign to the body. Such recognition processes occurs via protein-protein interactions on the surface of the immune system's cells, where binding affinity can determine if an immune response will or will not be initiated, and also where non-appropriated protein interactions or recognition can cause auto-immune diseases (11, 12). Biochemical reactions of cell breathing, oxygen and carbonic gas transport, food absorption, energy usage, energy storage, heat or cold physiological reactions, or any life process one can figure out, is basically carried out by a protein, or a protein complex. All processes taking place in a living organism have proteins acting somewhere, in a precise way, developed under the pressures of natural selection.
These are only some examples of protein function. A remarkable fact is that all tasks they can perform are based on a common principle, the twenty amino acids that can form a protein. That is the reason why studying proteins, their composition, structure, dynamics and function, is so important. We must understand how these molecules fold, how they assemble into complexes, how they function if we wish to answer questions such as why we have cancer, why we grow old, why we get sick, how can we find cures for many diseases, why life as we know it has evolved in this way and on this planet and not anywhere else, at least for the moment.
All proteins functions are dependent on their structure, which, in turn, depends on physical and chemical parameters. This is other important fact on studying these molecules; classical biological, physical, chemical, mathematical and informatics sciences have been working together in a new area known as bioinformatics to allow a new level of knowledge about life organization (13).
2. Explosion of Biological Sequence and Structure Data
Proteins have traditionally being studied individually. A protein of interest had its coding sequence identified and cloned in a proper expression vector. Hence, provided that cloning, expression and purification were successful, enough quantities of pure proteins could be employed in biochemical experiments or used to prepare solutions for NMR spectroscopy or to grow crystals for structure determination by X-ray crystallography. With the sequencing technology advances in the early 80's, the paradigm shifted from studying single proteins to whole set of proteins of an organism (1, 14).
From the second half of the 90's we can observe an exponential growth of the biological sequence and structure data, mainly due to the Genome Projects underway in different countries around the world, both on public and private industries (Fig. 1). As of April 2007, there were 1952 genome projects, of which 343 are of published complete genomes, 993 and 588 are of ongoing prokaryotic and eukaryotic genomes, respectively, and 28 of metagenomes (www.genomesonline.org(15)).
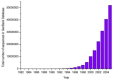
Figure 1
Number of sequences available in the GenBank as of December 15, 2005. The biological data explosion in mid 90's can be easily seen with the exponential growth from 1995. For a detailed description of the complete data set, please see http://www.ncbi.nlm.nih.gov/Genbank/genbankstats.html. (more...)
These data area stored in databases or data banks whose number increases every year. According to Galperin (16) there are 858 databases, 139 more than the previous year, available to the public. The amount of data produced urged the necessity for fast and reliable ways of accessing, retrieving, researching and understanding these data (13, 17). Bioinformatics tools are used to perform such tasks in all biological databases, of which GenBank (18) at NCBI and the RCSB/PDB (19) are the most representative for sequences and structures, respectively.
2.1 The GenBank® Database
GenBank, possibly the most widely known biological primary database, contains publicly available DNA sequences for more than 205,000 different species, obtained largely through submissions from individual laboratories and batch submissions from large-scale sequencing projects. Together with the EMBL Data Library in Europe and the DNA Data Bank of Japan, they make the three most important sequence databases with daily exchange of data, designed to provide and encourage access within the scientific community to the most up to date and comprehensive DNA sequence information, ensuring worldwide coverage (18).
As of December 15, 2005 there are 52,016,762 sequences in GenBank®. A BLAST (20) search on February 8, 2006 revealed that out of the number above 3,284,262 sequences are non-redundant (nr) coding sequences (CDS), excluding environmental samples. The nr sequences are updated regularly; hence its value can change from one week to another.
2.2 The RCSB/PDB Database
The Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) provides an information web resource to biological macromolecular structures (19). It includes tools and resources for understanding the relationship between sequence, structure and function of biological macromolecules. As of February 7, 2006 there were 35,026 structures in the RCSB/PDB, of which 33,429 are proteins (Fig. 2), including those making up complexes with nucleic acids [Summary Table of Released Entries].

Figure 2
Growth of the total number of structures in the RCSB/PDB data base (Kouranov et al., 2006). The exponential growth follows the same pattern of Fig 1.
As in the GenBank nr sequences, if we use a 90% sequence identity cutoff to eliminate redundancy in the protein structure data bank we end up with 12,611 clusters or distinct sets of structures [Current Statistics on Redundancy in the RCSB Protein Data Bank]. If we now assume this number represents the different types of known protein folds and compare it to the different types of known protein sequences, from the nr database of GenBank®, we conclude that only 0.38% of the available protein sequences have known folds. Using 1028, the current number of folds determined by SCOP [As Folds Defined By SCOP, (21, 22)], the number of protein sequences with known folds, 0,03%, is considerably smaller. This is in part due to the faster rate with which protein sequences can be determined (from CDS) as compared to the determination of novel protein 3D structures. [Comment: Also, many protein sequences have similar 3D folds, and many of these sequences have little sequence similarity to other sequences with the same 3D fold. This is one main reason why the number of SCOP folds is much less than that predicted by simple sequence similarity.] Clearly, computational methods that are complementary to experimental methods for the determination of protein structures are necessary, and these can be provided by structural bioinformatics efforts [reviewed by 23–25].
According to the Oxford English Dictionary, Structural Bioinformatics is conceptualizing biology in terms of molecules, in the sense of physical chemistry and applying informatics techniques, derived from disciplines such as mathematics, computer science and statistics, to understand, organize and explore the structural information associated to these molecules on a large scale (13). There are several computational methods for protein structure determination, including homology modeling (26), fold recognition via threading (27), and ab initio methods (28).
The huge increase in the amount of sequence and structure data of proteins together with advances in experimental and computational, bioinformatics methods, are improving our knowledge about the relationship between protein sequence, structure, dynamics and function. This knowledge, in turn, is being used to understand how proteins interact with other molecules such as small molecules or ligands that can become a drug candidate. In the following sections the hierarchy of protein structure and its application to drug design in tropical disease will be described. The last section is an exercise that will enable the reader or student to apply several bioinformatics tools starting from a partial DNA sequence or protein accession number and yielding a model of its structure and function.
3. Basic Principles of Protein Structure
When an all atom model of a protein structure is seen for the first time (be it a figure, a three-dimensional model or a computer graphics representation) it may appear a daunting task to decipher any underlying pattern within it. After all, such structures typically contain thousands if not tens- or hundreds-of-thousands of atoms. For this reason, it is often considered convenient to simplify the problem by using a hierarchical description of protein structure in which successive layers of the hierarchy describe increasingly more complex levels of organization. Typically four levels are used and referred to as the primary, secondary, tertiary and quaternary structure of a protein. It is often useful to include two additional intervening levels between the secondary and tertiary structures, which are referred to as super secondary structures and domains. Many excellent textbooks exist on the subject which should be consulted for more detailed information (1, 29–31). Here, we will attempt only to give an outline in order to emphasize the importance of the study of the three-dimensional structure of proteins for the drug design process which will be described towards the end of this chapter.
3.1 Primary structure
The primary structure of a protein originally referred to its complete covalent structure but is more frequently interpreted as being the sequence of amino acids of each polypeptide chain of which the protein is composed. These are often one and the same thing but disulphide bonds and other rarer types of covalent bond formed between amino acid side chains are not directly encoded by the sequence itself.
A polypeptide chain is a unidimensional heteropolymer composed of amino acid residues. There are basically only twenty naturally occurring amino acids which are directly encoded by the corresponding gene, although in exceptional cases stop codons can be used for the incorporation of two additional amino acids (selenocysteine and pyrrolysine (32)). All of these amino acids are α-amino acids which possess the generic structure given in Figure 3a. Common to all such amino acids is the amino group, carboxylic acid group and hydrogen bound to the central carbon atom (the α carbon). Only the R group (also known as the side chain) differs from one amino acid to another and it varies in terms of size, polarity, hydrophobicity, charge, shape, volume etc. With the twenty different amino acids available, nature is able to produce the wide diversity of functions which proteins perform in living organisms.
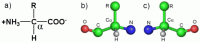
It should be noted that the α-carbon is tetrahedral and in general is bound to four different chemical moieties. As such it is asymmetric (a chiral center) and two different enantiomers (D and L) for each amino acid exist (Fig 3b). Amongst the naturally occurring amino acids, the only exception is the amino acid glycine, whose R-group is a hydrogen atom, making the α-carbon symmetric. This confers a series of important conformational properties on this amino acid which are often essential for the maintenance of a given structure. For this reason, critical glycines are often conserved amongst members of a given protein family. The remaining amino acids are (with very few exceptions) always found to be L-amino acids. This has important consequences for the chiral structures observed in proteins at all levels of the hierarchy. Only two other chiral centers exist within the twenty amino acids, these are the β-carbons within the side-chains of the amino acids threonine and isoleucine, which also exist as only one of the two possible enantiomers.
A polypeptide chain is generated by a series of condensation reactions between the carboxyl group of one amino acid and the amino group of the next, yielding a covalent bond (Fig. 4). This is an amide bond which is given the trivial name of a peptide bond in the case of polypeptide chains. The union of two amino acids results in a dipeptide, which possesses a free amino group (N-terminus) and carboxyl group (C-terminus) allowing the condensation reaction to continue ad infinitum in principle, in both directions (Fig. 4). After undergoing condensation, the amino acid is termed a residue (to denote `that which is left' after condensation). Given the existence of the 20 naturally occurring amino acids, for a polypeptide of length n there are 20n possible amino acid sequences. Since n is typically of the order of hundreds and may even be tens-of-thousands, the number of theoretically possible polypeptide sequences is literally astronomical. The vast majority do not currently exist (indeed, there is insufficient matter in the universe) and never have existed before (as there has been insufficient time to generate them). The polypeptides we observe in the extant species are the products selected from a tiny fraction of all possible combinations that were expressed along the evolutive process.
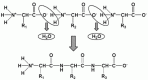
3.1.1 The conformational properties of a polypeptide chain
Due to the chemical nature of the amide bond, the peptide linkage between two amino acids is subject to the phenomenon of resonance, meaning that it acquires the characteristics of a partial double bond. The C-N distance (1.32Å) is shorter than a normal single bond and longer than a normal double bond. Furthermore, this introduces rigidity into the structure as the bond is no longer freely rotatable. The consequence is that the α-carbon atoms of two adjacent amino acids, together with the carbonyl (C=O) and NH groups of the peptide group itself, all lie within the same plane (Fig. 5). The torsion (dihedral) angle associated with this bond is termed ω, which in order to be planar must be either 180o (trans) or 0o (cis). Cis peptide bonds are only common when the amino acid proline lies on the C-terminal side of the bond. This is due to the unusual nature of the proline side chain which forms a covalent bond with its own mainchain nitrogen generating a closed ring. As such proline is strictly speaking a secondary amino acid and neither trans nor cis are particularly energetically favorable. Approximately one third adopt the cis conformation and these are frequently conserved amongst protein family members (33).
Figure 5
A short section of polypeptide chain showing the planar peptide groups and identifying the torsion angles ϕ and ψ.
Polypeptides necessarily obey standard stereochemistry and therefore all bond lengths and angles are effectively fixed, varying only minimally around their standard values. Therefore, the only source of conformational freedom that the polypeptide possesses comes from the torsional rotation around its single bonds. We have already seen that the peptide bond is not freely rotable and therefore ω is effectively fixed, normally in the trans configuration. There are only two remaining single bonds per residue along the main chain, and from these bonds one may associate the torsion angles ϕ and ψ (Fig. 5), which are defined to be 0o when in the conformation trans. The conformation of the mainchain of a given residue may therefore be described in terms of these two parameters, which may be conveniently represented in terms of a two-dimensional coordinate system (the Ramachandran plot), in which both ϕ and ψ vary between -180 and +180o.
3.2 Secondary structure
Not all combinations of ϕ and ψ are stereochemically possible, since many (such as 0o/0o for example) lead to steric hindrance. In fact, as shown originally by Ramachandran, only about one third of ϕ/ψ space is stereochemically accessible to amino acid residues in a real polypeptide (34).
If the ϕ and ψ angles are repeated systematically for all residues within a stretch of polypeptide, the result will inevitably be a helix. An infinite number of types of helix are theoretically possible, depending on the combination of ϕ and ψ, and they may be conveniently characterized by two parameters, n and d, corresponding respectively to the number of residues per helical turn and the shift parallel to the helical axis per residue. There are convenient mathematical relationships which relate ϕ and ψ to n and d.
However, of all the theoretically possible helices, only a limited number occur with any frequency in protein structures. Those which are most important in the case of globular proteins correspond to the α-helix (ϕ ≈ -63o, ψ ≈ -42o, n = +3.6, d = 1.5Å) and the β-strand (ϕ ≈ -120o and ψ ≈ +135o, n = -2.3, d = 3.3Å). These are the most important types of secondary structure and correspond to small stretches of consecutive residues. The negative n value in the case of the β-strand indicates that it is a left-handed helix, whereas the α-helix is right-handed. The fact that the n parameter is close to 2 for β-strands (the minimum value theoretically possible) means that strands are in fact very stretched out helices with a narrow main chain diameter. Consequently the side chains emerge alternately on opposite sides of the strand, giving rise to a zig-zag or pleated appearance.
The α-helix (Fig. 6) is particularly abundant in both globular and some fibrous proteins due to its inherent stability. This arises as a consequence of several factors including the presence of consecutive hydrogen bonds between the carbonyl of residue i and the NH of residue i+4, the radius of the helix which is compatible with Van de Waals contacts across the helical axis and the possibility of forming ion pairs (salt bridges) between oppositely charged residues separated by 3 or 4 residues along the sequence. The hydrogen bonds are particularly strong due to the ideal separation between donor (nitrogen) and acceptor (oxygen) atoms as well as due to their linearity and dipolar alignment. Alternative but similar helices, such as the 310 and π helices are much less common due to the weaker nature of the stabilizing forces described above. Autonomous 310 helices do exist, but they are much more commonly observed as the first or final turn of an α-helix (35). π-helices may be a little more common than originally thought (36).
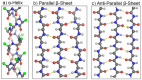
Figure 6
The most common types of secondary structure.
α-helices in general present a series of common physicochemical and structural properties. They tend to be amphipathic, with a hydrophobic face pointing towards the protein core and a hydrophilic face directed towards aqueous solvent (this may be reversed in the case of membrane proteins). Competition between water and the amide groups of the protein backbone tends to lengthen the hydrogen bonds on the hydrophilic face, leading to helix curvature (37). The alignment of the individual dipoles of the peptide units leads to a macroscopic dipole associated with the whole helix which is typically of the order of 0.5 units of positive charge at the N-terminus and 0.5 units of negative charge at the C-terminus (38). Together with the free NH groups within the first turn, this makes the N-termini of α-helices ideal binding sites for anions such as phosphate. In the absence of such an anion, frequently the N-terminus is capped by the side chain of the first helical residue (the N-cap) which is most often an asparagine or aspartic acid (39). The most common amino acid encountered at the second position is a proline, meaning that the sequence Asn-Pro would, in principle, be a strong helix initiation signal.
β-strands are unable to form internal hydrogen bonds. They assemble into sheets by the formation of hydrogen bonds between strands (Fig. 6). Sheets may be parallel, antiparallel or mixed in nature depending on the relative orientation of the strands of which they are composed. The ϕ and ψ angles vary from one type to another and β-sheets are capable of assuming a wide array of architectures (40). One characteristic common to almost all β-sheets is that they are twisted. When viewed perpendicular to the strands, the sheet has a left-handed twist and when viewed parallel to the strands it is right-handed. The latter may appear a contradiction given the left-handed twist of the individual strands (see above), but results from the fact that only every second residue (1, 3, 5, 7 etc or 2, 4, 6 etc.) contribute hydrogen bonds to the same side of the strand. Planar sheets (with n = 2) do exist but are rare compared with twisted ones. Figure 7 shows examples of both flat and twisted β-sheets.

Figure 7
Flat (n=2) and twisted β-sheets (left and right respectively)
Amongst the different architectures that β-sheets form are parallel (rare) and anti-parallel (common) coiled coils, parallel and anti-parallel saddles (hyperbolic paraboloids), 8-stranded parallel barrels and anti-parallel barrels with a highly variable number of coiled strands, helicoids of three strands etc. (40)
In globular proteins the α-helices and β-strands will often traverse the structure. Since both have approximately linear axes, clearly other types of structure are needed in order to cause chain reversal. These are termed turns and loops. Turns are composed of a well defined number of residues. δ-turns are composed of two residues, γ-turns three, β-turns four, α-turns five and π-turns six (41). The most well-known and widely described are the β-turns, which come in several different forms distinguishable by the ϕ and ψ angles of the second and third residues of the turn. In most (but not all) definitions, a hydrogen bond exists between the carbonyl of the first residue and the NH of the fourth. Several classifications have been proposed for β-turns and types I and II are the most common (see 41). However types I' and II' are observed in β-hairpins (a super-secondary structure composed of two anti-parallel β-strands) due to the compatibility of the sheet twist with that of the turn itself (42). In general, turns can not be represented by a single point in ϕ/ψ space (unlike the helices described above). Indeed many of the β-turns require one of the internal residues (2 or 3) to adopt `prohibited' ϕ/ψ angles, imposing a requirement for a glycine at specific positions (41). The type of turn may sometimes therefore, be easily predicted from the sequence of its component residues.
Loops are more difficult to classify as they are larger than turns and can therefore adopt a wide variety of conformations. Different to turns, they generally present a hydrophobic core and, at least in the case of Ω-loops, are as compact as other parts of the structure.
3.3 Tertiary structure
In order to form globular structures, the individual elements of secondary structure of a polypeptide must pack against one another in order to form a stable, compact and biologically active tertiary structure. The ways in which secondary structures most commonly pack has been widely studied (43–45). The most simple models which aim to explain experimental observations are based on the interdigitation of side chains. For example, the knobs-into-holes approach or ridges-into-grooves model (43) explain equally well the most commonly observed packing angles between two α-helices (Ω = -50o and 20o). A similar approach can be used for the packing of β-sheets against one another or of helices against sheets (44, 45).
The final result of the folding process is the full three-dimensional structure of the polypeptide. Clearly, this depends on the amino acid sequence and therefore on the atomic details of the structure. However, for soluble proteins this will almost inevitably result in the generation of a hydrophobic core in which apolar residues tend to cluster their side chains within the protein's interior, leaving hydrophilic residues exposed to solvent. For descriptive purposes it is useful to simplify the structure by removing the atomic detail and considering only the trace of the mainchain in space (see Fig. 8). This is often referred to as the fold of the protein. Many classifications of protein folds exist and two of the most widely used are SCOP (22) and CATH (46), both of which use a hierarchical approach. The highest level of the hierarchy classifies folds into classes, based on their overall secondary structure content (mainly α, mainly β, α/β, irregular). In the case of the CATH classification (Fig. 9), the second layer of the hierarchy deals with different architectures within a given class, of which there are currently approximately 30 major types. For example, within the α-class folds may be classified at the architectural level as α-bundles, α-non-bundles, α-horseshoes, α-solenoids and α/α barrels. Within the β-class, there are β-rolls, β-barrels, β-clams, β-sandwiches β-trefoils, β-prisms (parallel and orthogonal) β-propellors (with different numbers of blades), different types of β-solenoids (or β-helices) etc. Within the α/β-class we find α/β barrels (TIM barrels), α/β sandwiches (with varying numbers of layers and types), the α/β-box, α/β-horseshoe, α/β-propellor etc. The architecture describes the overall arrangement of secondary structural elements but without concern for their connectivity.

Figure 8
Different representations of the same protein structure (Alzheimer's amyloid precursor protein). All atoms except hydrogens (left), Cα- trace (center) and ribbon (right).

Figure 9
Part of the CATH classification. Examples from the upper most levels in the hierarchy (Class, Architecture and Topology) are shown.
At the level of topology, a finer description is given within a given architecture in which the connectivity of the secondary structures is taken into account. For example, even considering a relatively simple architecture, that of an α-bundle, for example a four-helix bundle, there is a surprising number of different topologies (ways of orientating the helices with respect to each other and then connecting them). In fact there are 48 different topologies theoretically possible for a four-helix bundle (47). However, probably for reasons of simplicity of folding, nature has favored the all anti-parallel bundle in which two diagonally related helices are orientated parallel and the remainder anti-parallel. These topologies are more common than one would expect by chance.
There is a general belief that the total number of folds is relatively limited (perhaps a few thousand) and it is expected that as the result of structural genomics initiatives, within the next few years our knowledge of the universe of possible folds used by soluble proteins at least will be reasonably complete (48). Already it is clear that some folds are more common than others and these have been termed superfolds by some authors (49). They include the TIM barrel, the jelly-roll and the up-and-down 4-helix bundle.
With the accumulation of new protein structures at an ever increasing rate, determined by X-ray diffraction and multi-dimensional NMR, it has been possible to derive a series of empirical rules which govern the possible ways in which a polypeptide may fold. Although many such rules remain founded on a purely empirical basis, some of them can probably be explained by appealing to the need for efficient folding kinetics. The most well established rule is that which determines that connections between elements of secondary structure of the type βxβ (where β refers to a β-strand and x may be anything, an α-helix, a loop or another β-strand) should be right-handed (50). There are very few exceptions to this rule, but one spectacular example is the left-handed β-helix as seen for example in UDP-N-acetylglucosamine acyltransferase (51). Other connectivity rules prohibit the crossing-over of loop connections and the formation of knots for example. However, as with most empirical rules, exceptions can always be found and recently some dramatically knotted structures have been described (52).
It should be noted that proteins are inherently chiral molecules and this has important consequences for the interactions they make. Many enzymes for example are stereoselective with respect to their substrates due to inherent asymmetry in their active sites. For example, when an artificial HIV protease was synthesized from D instead of L amino acids, the result was an active protease which was specific for a substrate of the opposite enantiomer (53). The consequences of the asymmetric α-carbon atom can be seen at most if not all levels of protein structure. It is not by chance that the right-handed α-helix is energetically favored over its left-handed partner, nor that β-strands are left-handed (right handed when only every second residue is viewed) or their sheets left-handed (when viewed perpendicular to the strands). Furthermore, the packing of right-handed α-helices using side chain interdigitation leads to the common packing angle of 20o, meaning that when these helices coil around one another to form coiled coils (as seen in keratin, myosin and leucine zippers for example) inevitably such structures will be left-handed. At the next level up, the right handed connections found in βxβ units together with the twist on β-strands themselves means that all (βα)8 barrels (TIM barrels) have the same chirality. Indeed, the barrel will always `roll up' the same way because of the combination of strand twist and sheet shear (the shifting of one strand with respect to its neighbors which results in the inclination of the strands with respect to the barrel axis) (1, 40).
3.4 Quaternary structure
Some proteins (a minority) are composed only of a single polypeptide chain. The remainder are homo- or hetero-oligomers or macromolecular assemblies or polymers. In a recent evaluation of the proteins encoded by the E. coli genome, only about 20% were expected to be monomeric (54). Dimers were the most common, representing nearly 40% of all proteins, followed by tetramers (21%). When two subunits (polypeptide chains) interact to form an interface, two possibilities exist; the interface may be either isologous or heterologous. In the first case, the association leads to the formation of a symmetric dimer possessing a two-fold (diad) axis, in which if a region A on the first subunit interacts with a region B on the second, then region A of the second will necessarily interact with region B on the first (1). In the case of heterologous interactions this is not the case. They may be either symmetric, leading to ring-like structures possessing symmetry axes of higher order (>2) or asymmetric.
Almost all oligomeric structures possess internal symmetry. Given that the individual subunits themselves are asymmetric and chiral (because they are made of L-amino acids) this means that the symmetry can only arise by the association of subunits around pure rotation axes. In principle, these can be of any order and many examples including 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 9-fold and 11-fold axes (and maybe others) have been observed in nature. Three fundamentally different arrangements are possible. Cyclic structures (built up of a ring of subunits employing one type of heterologous interface generally lead to pore-like structures, particularly when the number of subunits becomes large. Examples include neuraminidase (4-fold) and the light-harvesting complex (11-fold). Dihedral structures, besides possessing one principle axis, also have a series of perpendicular 2-fold axes. For example, most tetramers are built using 222 symmetry (three mutually perpendicular 2-fold axes) rather than a single 4-fold axis, as found in neuraminidase. Dihedral symmetry gives more options for creating different interfaces between subunits, and this richness is often taken advantage of in allosterically regulated enzymes. Together with a higher order principal axis, dihedral symmetry can be used for creating proteins with internal cavities accessible via channels. Examples include the chaperone GroEL (55) and the proteasome (56), both of which have 7-fold axes.
The third option is the use of cubic or icosahedral symmetry in which a series of three fold axes are accompanied by non-perpendicular two-fold, four-fold or five-fold axes (or combinations of these). The resulting structures are generally approximately spherical and are used as storage molecules (such as ferritin for example) or as viral capsids. Furthermore, translational symmetry can be added to the rotation axes to produce helical symmetry as seen in microtubles, tobacco mosaic virus and other filamentous structure.
What are the advantages of building oligomeric proteins and why are they so common? Several answers exist. Sometimes it is just a question of being big, as for example in the case of plasma proteins which, in order to avoid ultra-filtration by the kidneys, must be greater than 60KDa. Being large can often also increase stability and reduce the surface area/volume ratio, thus making the protein less susceptible to proteolysis. Being oligomeric also gives rise to the potential for cooperativity in enzymes and transport molecules, as well as being an efficient way of building large structures with limited genetic material, such as in spherical viruses for example. The potential of having multiple identical binding sites within a single molecule (as in immunoglobulins for example) introduces the possibility for a cross-linking activity and as mentioned previously many oligomeric structures serve to generate cavities with a specific biological function.
3.5 A final comment on protein structure
The reason for interest in the full three-dimensional structure of a protein is generally to understand its biological activity or to interfere with it, as for example in the case of drug design (see below). In both cases a merely topological description is clearly insufficient because biological activity depends on atomic detail and often apparently subtle modifications to a structure can dramatically affect its activity. Here we do not have space for full descriptions of even a limited number of protein families. However, in the following section it should become evident that the knowledge of a high resolution atomic structure of a target protein is an essential pre-requisite for structure based drug design.
4. The Use of Protein Structures in Inhibitor Discovery and Design
Before the development of techniques for obtaining the 3D structure of proteins, drugs were discovered by systematic modification of compounds with known biological activities that were discovered by chance or random screening, based on trial and error. It was a time and money consuming approach, and even when employing all medicinal chemistry's methods to reduce the amount of compounds to be synthesized and tested, these approaches required exhaustive procedures of syntheses and evaluation. Traditional medicinal chemists focused their attention in understanding the relationships between structures and activities of known active compounds and proposing modifications that would improve some biological property of the molecule (57, 58).
Medicinal Chemists took immediate advantage of the rapid development in computing and computer graphics in particular, and developed techniques for visualizing, manipulating and superposing 3D structures of active compounds in order to obtain faster information about the structure-activities relationship of known active compounds. Different computer based Quantitative Structure-Activities Relationship (QSAR) approaches like HQSAR, CoMFA and related 3D QSAR approaches became popular amongst medicinal chemists.
Simultaneously, significant advances in molecular and structural biology were made and currently tens of thousands of 3D protein structures are available. This represents an enormous amount of structural information about medicinally relevant receptors, furthermore structures of protein complexes with small ligands are also an invaluable source of information about the favorable interactions that stabilize the ligand's association with the binding pocket of the protein (59).
Recently, powerful desktop computers have become affordable, together with an ever increasing diversity of free software for academic purposes as well as a marked improvement in data transfer through internet access. Thus, it is now feasible to work at home on powerful workstations, in close contact with academic research groups throughout the world, and taking full advantage of up-to-date software for visualizing molecules, performing molecular modeling and even to process crystallographic data and solve structures.
In spite of such unquestionable progress, structure-based ligand design still faces several major challenges and limitations such as a lack of availability of 3D structures for many important receptors, particularly membrane-bound receptors, adequate computational approaches to deal with induced-fit in ligand-receptor interactions or water molecules in the binding site and selectivity between different isoforms of the same receptor or enzyme. Furthermore, collaboration with synthetic chemists and experimental biologists will probably continue to be a critical determinant of success (59).
Drugs are ligands that not only fit into the binding pocket of the target protein, but also are absorbed, transported, distributed to the right body compartment, as well as ideally being stable to metabolization, non-toxic, free of side effects and chemically stable in the formulation. So it is clear that the ligand design stage of the drug discovery process is just one of the initial steps in a multidisciplinary effort that usually requires several rounds of refinement between the identification of an active principle and the selection of a viable drug candidate. Structural based design can assist this continuous process by pinpointing opportunities for structural modifications that do not interfere with binding, but may improve affinity and selectivity, and which may favorably alter the properties of the molecule, such as solubility, hydrophobicity and ionization state (59).
Finding a lead compound, optimizing its properties and turning it into an approved drug takes an enormous amount of time and money. It is a quest that demands anintense and cooperative effort between multidisciplinary discovery teams (60).
In some cases the careful analysis of a protein-ligand structure can be enough to suggest modifications in the ligand to improve affinity. More structures of the same protein with different ligands can make it easier. Molecular modeling tools can model a protein's structure based on a previously known homologous structure, so it is possible to use the structural information to design ligands for proteins for which no structure is known. Virtual screening can provide ligand structures completely different from the already known ligands. QSAR models can aid the design of modifications to improve a biological property of a ligand and can predict activities with a reasonable error. Docking procedures can be used to model binding modes and estimate binding energies.
4.1 Applications of Protein Structures
Predicting the binding modes and affinities of different compounds as they interact with protein binding sites is the main goal of structure-based drug design. Computational approaches to this problem are usually called docking. Figure 10 shows schematically one approach to this problem as implemented in the program DOCK (61).

The key characteristic of a good docking program is its ability to reproduce the experimental binding modes of ligands (62). A large number of programs have been written to address this area. In general, the programs operate as follows: A large number of conformations are generated for the small molecule, either prior to docking or during the docking routine. Each conformation is positioned in the active site in a variety of orientations, the combination of conformation and orientation being known as a "pose". This sampling procedure is usually based genetic algorithms, Monte Carlo simulation, simulated annealing or distance geometry (61, 62). Many poses are selected and ranked by a scoring function, in order to determine the best overall pose.
In general, the potential flexibility within both the ligand and residues of the binding site represents a significant computational challenge. To make the docking problem tractable, most programs address adequately the question of ligand flexibility, but assume that the binding site is rigid. Also, some programs partially address protein flexibility. The scoring functions developed to date usually use either a classical molecular mechanics force field, an empirical function describing terms such as hydrogen-bonding and lipophilicity, or a "knowledge-based" potential derived from analysis of protein-ligand complexes (62, 63).
Another important characteristic of a good docking program is the ability of its scoring functions to score and rank multiple ligands against a single target according to their experimental binding affinities (62). This task is more problematic (64), the scoring functions used in docking programs must contain many approximations, since they have to be fast enough to evaluate extremely large numbers of possible poses. For instance, they do not usually take explicit account of entropic changes due to displacement of waters from the active site or of reduction in degrees of freedom in the ligand. Usually a scoring function that can select the correct pose for one ligand will not necessarily rank a series of potential ligands in the correct order (63).
An important use of protein–ligand docking programs is virtual screening, in which large libraries of compounds are docked into a target binding site and scored. For this purpose, the dockings need to be even quicker. Speeding up a docking protocol is often done at the cost of sampling fewer binding modes, and, as a result, reduces the success rates. It is therefore important that search parameters are chosen that give docking speeds useful for virtual screening applications, with an acceptable loss in docking accuracy (63).
To overcome the "ranking problem" researchers have employed different methods ranging from brute force, which means using highly expensive computer clusters to do all the heavy computational work, to using more elegant approaches like consensus scoring. This method consists of using a docking program to do the conformational search and rank the compounds, and employ other docking tools' scoring functions to reevaluate the top results. Only the top scored compounds common to each scoring function will be identified as candidates for bioassay. Compared to single scoring procedures, it was shown that false positives in virtual library screening were largely reduced and the hit-rates were improved. There are many different ways of performing consensus scoring (64).
Some docking tools also have another useful characteristic, the estimation of binding energy. However, this technique still demands a very high computational power and most projects can not afford it. Usually only the best ligands found on virtual screening are submitted to this procedure. Note that even though these calculations are not very accurate, if it can provide the correct rankings of candidate molecules, it will provide valuable information.
4.1.1 Getting started with Virtual Screening
Virtual screening results are highly dependent on database and target preparation. Databases need to be tuned, and that means a considerable effort should be put into filtering out molecules with undesirable physical properties and chemical functionalities such as high molecular weight, reactive groups, too many rotating bonds and non suitable lipophilicity (65).Tautomeric states, ionization and charges should be also observed, as well as the possible enantiomers arising from the chiral centers. Finally, one must decide if one wants a database of drug-like or lead-like compounds.
Before preparing the receptor, it is necessary to check if it is adequate for a virtual screening procedure. It is common to have available more than one structure of the same protein in complex with different ligands or without any ligand bound. It is up to the medicinal chemist to choose between these different options. As a guideline, the resolution must be as high as possible. Another important step is to read the pdb file of the receptor. Useful information can be found about the crystallographic parameters employed, notes from the author about residues with low electron density and even mutations in the protein. Also programs such as Procheck and Whatcheck can be used to analyze the stereochemistry and chemical environment of each residue of the protein.
After choosing the most appropriate receptor structure, usually the addition of hydrogen atoms is required, followed by minimization of the potential energy to avoid steric clashes. Some changes must be carefully done on the binding site, like assigning the right protonation state to ionizable residues, checking the tautomeric states of the histidines, the possible flips of the glutamines and asparagines and the orientations of the hydrogen atoms of the hydroxyl groups on the serine and threonine residues (65).
Remember that virtual screening procedures usually take days to finish and if a mistake is made in the preparation stages, the entire work may be compromised. Also one of the most important procedures is the visual inspection of the best ranked compounds in the context of the binding site. Serendipity, the principle that "chance only favors the prepared mind" in the words of Louis Pasteur, plays an important role in the whole virtual screening process especially during the visual inspection stage.
4.1.2 Protein structure and 3D-QSAR
Protein structures can be used in many ways to improve the results of Quantitative Structure-Activity Relationship (QSAR) techniques. Two of the most common techniques are the Comparative Molecular Field Analysis (CoMFA) and the Comparative Molecular Similarity Indices Analysis (CoMSIA). Both methods assume that the ligands have a common binding mode, their 3D structures are modeled and superposed and Partial Least Square (PLS) analysis is performed to try to find a correlation between the compounds' biological activities and their 3D structures (66).
These methods strongly rely on a correct superposition of the ligand structures and, if the structure of the target protein is known, a valid approach is to dock the ligands into the protein biding site and use the result as a "structural alignment". The protein structural information can also be used to aid the interpretation of the results.
Another QSAR method, Comparative Binding Energy (COMBINA) requires at least one structure of a ligand-protein complex and experimental binding data for the training set of ligand-protein complexes. The training set is used for model development only and not for evaluation of the model, which must be performed using an independent set of data. The structure of the known ligand-protein complex is used to model all remaining complexes of the training set (in case of no structural data bPLSeing available), these complexes are superimposed and energy is minimized. The interaction energy between ligand and receptor in each complex is computed and decomposed on the basis of physical properties and location in the molecule. PLS analysis is then used to build a model correlating the biological binding data with the sum of weighted, selected components of the interaction energy (66).
4.2 Structure-based Drug design targeting Tropical Diseases
Tropical diseases are responsible for some of the most severe worldwide health problems. Hundreds of millions individuals are affected by these diseases (67–71) killing millions of people annually. Structural differences between host and parasite can be exploited aiming to develop a selective drug against the parasites, without causing damage to the host.
An example of this approach is the dihydrofolate reductase-thymidylate synthase (DHFR-TS) enzyme (Fig. 11). Although the human host presents this enzyme in its biochemical pathway, the human enzyme differs structurally from the parasite enzyme, and these differences can be exploited in order to design selective inhibitors against the parasites′ homologous enzyme. The bifunctional enzyme DHFR-TS from parasites such as Toxoplasma gondii, Cryptosporidium parvum and four different species of Plasmodium plays a crucial role in pyrimidine biosynthesis and has been used as a target for structure-based drug design.

Figure 11
Docking of the synthetic ligand Wr99210 into the binding site of wild-type Plasmodium falciparum DHFR (pdb ID: 1j3i). The program GOLD (red) was able to reproduce the crystallographic position of the ligand (yellow) (62).
The structure of DHFR-TS from Leishmania major, and the recent determination of the structures from Cryptosporidium hominis and P. falciparum (67) can now shed some light on the binding mode of wild type DHFR-TS and mutants resistant to current inhibitors, such as methotrexate and dapsone (Lapdap ™). These structural data offer the possibility to develop novel inhibitors with higher potency and selectivity, and these data would be useful for molecular modeling DHFRs from other species of parasites for which structures are not available yet (72). Furthermore, the widespread resistance to currently available antimalarial drugs has prompted an increase in the search for new targets, some of them already in the validation stages (72).
DHFR inhibitors are largely ineffective for the control of trypanosomatid infections, partly due to the presence of pteridine reductase (PTR1), which is involved in folate/pterin metabolism. Crystal structures of the PTR1 from Leishmania tarentolae and Trypanosoma cruzi, and a series of crystallographic analyses of the Leishmania major's enzyme in binary and ternary complexes identified important interactions that can be exploited in a structure-based development of novel enzyme inhibitors with potential therapeutic value (73).
Moreover, other approaches are being considered in order to tackle the development of novel drugs against tripanosomatides. The crystal structure and mechanism of trans-sialidase, an enzyme that appears to be critical for T. cruzi survival and cell invasion capability is now available. The structure provides the first step to the structure-based design of novel inhibitors with potential therapeutic applications (74).
Also, the 2.0Å resolution structure of UDP-galactose 4′-epimerase from the protozoan parasite Trypanosoma bruce, the causing agent of the African sleeping sickness in humans, and the wasting disease nagana in cattle, was recently determined. A structural comparison with the human enzyme identified a potentially important difference in the substrate-binding cleft that might assist a structure-based approach to the development of TbGalE specific inhibitors (68).
Other enzymes that have been considered as good targets for antiparasitic drug development are the Glutathione S-transferases, a family of detoxification enzymes that catalyze the conjugation of glutathione with various endogenous and xenobiotic electrophiles. Studies have identified antischistosomal (75), antimalarial and antifilarial (69) activity of compounds known for their GST inhibiting activity (70).
Crystal structures of the Glutathione S-transferase of the filarial worm Onchocerca volvulus, responsible for the debilitating disease onchocerciasis, were recently obtained in high resolution (1.5Å and 1.8Å). The structural analysis of the enzyme shows that it is a suitable drug target for intervention in filarial infections, with the potential advantage of not compromising the human host (69).
Other structure-based efforts to change the tropical diseases panorama are being made. An example is the recent study that used available structures and gene sequences of the enzyme glyoxalase I to create a model of the Leishmania donovani enzyme (76). This work suggested that the differences between the human and the L. donovani structures could be exploited for structure-based drug designing of selective inhibitors against the parasite's enzyme.
Also, structural analysis of the Mycobacterium tuberculosis type II dehydroquinase suggested modifications on known inhibitors that led to the synthesis of a new inhibitor over than 180 times more potent than the prototype inhibitor. Docking studies indicated key electrostatic binding interactions between the inhibitor and the enzyme. Structural studies of enzyme-inhibitor complexes are underway and should be considered for the design of the next generation of inhibitors (71).
5. Practical Section: Computational Characterization of a Gene Product from a Partial Gene Sequence Only
The objective of this exercise is to bring the reader or student in contact with freely available online bioinformatics tools used to computationally characterize a complete protein starting with only a partial DNA sequence of its corresponding gene. Through a set of guiding questions, with suggestions, this exercise will involve searching the GenBank for the whole gene sequence, translating it into the corresponding protein sequence, and searching for the protein structure (here it can either be assumed that the student is already familiar with the outputs from the distinct BLAST programs or that learning will be carried out through the exercise). If no structure is available, then the student should search for similar sequences with 30% or greater identity that have known 3D structures. Depending on the course length, the student can proceed by determining the protein structure by homology modelling or simply by using the known 3D structure to infer structural similarity and function for his/her protein. The student is encouraged to download any known protein 3D structure file to his/her computer and visualize it using a locally installed biological macromolecule viewer such as SwissPDBViewer (77) or VMD (78). This will be a unique opportunity for the student to get familiar with protein architecture. The same experiment can be easily changed to investigate a whole genome as well. The set of questions can be used to guide the student through the writring up of an article describing the gene and its product. This exercise should be conducted by a lecturer or tutor in bioinformatics.
5.1 Molecular Biology or Genomics Laboratory Experiment
In a molecular biology or genomics laboratory a DNA fragment, supposedly corresponding to a gene, was identified. Its first 30 base pairs have been sequenced and determined to be 5'- TGACACAACACAAGGACGCACATGACAGGA -3'. Answer the questions below through Bioinformatics experiments.
5.1.1 Gene Characterization
1 - Can the putative gene be completely and unambiguously characterized from the partial DNA sequence given above? How?
Yes. The gene can be completely and unambiguously characterized, but the solution will require a more careful analysis than it did a couple of years ago. The reason is simple; there were much less sequences than there are now.
Solution:
Since we have a nucleotide sequence as our initial data set, we must go to the NCBI BLAST page, select the nucleotide blast program and search the nucleotide database using a nucleotide query. The page will look like this:

This BLASTN interface is easy to understand. It is divided into four major sections.
- - The first allows the user to paste the query sequence for analysis. Our query sequence containing 30 nucleotides, highlighted in the red rectangle, was pasted into the search box.
- - The second section permits the choice of a database to be searched and optional sequence range coordinates to the search. We will search the non-redundant (nr) nucleotide (nt) database in this exercise. Note that this is no longer the default option. That is why it is highlighted in yellow.
- - The third section offers optimization alternatives to the search. We will use standard parameters and settings for our search. We will search for highly similar sequences using megablast.
- - This section shows a summary of our search features that were defined in the previous sections. We will now mark the option to show the results in a new window. This option is very useful, since it allows us to re-run BLAST searches with different sequences and/or parameters while maintaining the former results.
Now we are ready to perform our search, just click on BLAST icon. After a few seconds the results page will appear in a new window. Assuming that you are familiar with a BLAST output, only the part of the output list of hits and alignments will be illustrated here.
Output list of hits:
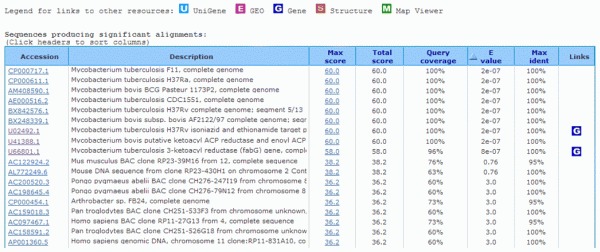
You may notice that only the first 11 hits present very low E-values (below 10–5), which are most possibly significant. If we wish to be more stringent, demanding a 100% coverage in the sequence alignment, only the first eight hits are relevant for our search and the characterization of our sequence.
The first six hits are related to complete genome sequences which makes it more difficult for us to identify our target sequence. We hope to find the putative gene directly or DNA sequences containing the putative gene. Hence, the only direct and easy options left are hits seven and eight.
This straightforward analysis have shown that the partial DNA sequence we are seeking to identify most likely is part of a gene belonging to either Mycobacterium tuberculosis or to Mycobacterium bovis, or both.
How to solve this possible ambiguity? We must now look carefully at the sequence alignments that follow the list of hits for these two hits we have selected. The results are:
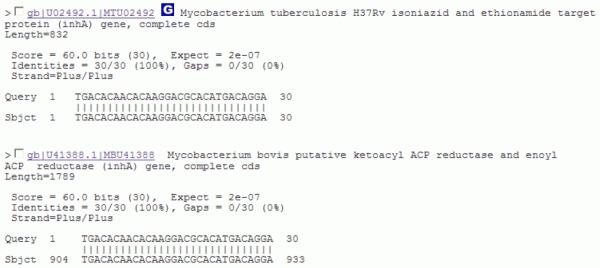
The alignment statistics is identical for both hits.
For the hit whose GenBank accession number is U02492.1, our query sequence begins exactly at the first position of the hit sequence.
For the hit whose GenBank accession number is U41388.1 the 5' end of our query sequence matches residue 904 on the hit sequence.
If you click on the accession number links, you will be redirected to the corresponding annotation of these sequences. As suggested by the annotation, the second one is a much longer nucleotide sequence (1789 bp) containing not only one, but two putative genes, one of them starting at base 904 which is similar to that found in the first hit. Also, the second hit corresponds to genes that are putative, that is, with no experimental confirmation.
From these analyses we can now proceed to the next question.
2 – Does the partial DNA sequence above correspond to a gene? If so, what is its GenBank accession number?
Yes. According to the analyses in 1, the 30-nucleotide partial DNA sequence most likely corresponds to a confirmed gene. Its accession number is best represented by U02492.1.
3 – What is the gene name?
The gene name can be obtained from the annotation in the hit list or by searching the NCBI nucleotide database with the GenBank accession number U02492.1. Another way is to click on the link associated with the entry U02492.1 with the mouse right hand button and open it in a new window. The top of the resulting page will be as follow:

Looking carefully at this GenBank flat file we can obtain all the information about the nucleotide entry U02492.1. We can clearly see all over, but more specifically in the FEATURES section that the gene name is inhA.
4 – What is the gene size in base pairs?
In the FEATURE section, in the ninth line, the link gene shows that the inhA gene starts at base 22 and ends at base 831, thus totaling 810 base pairs.
5 – Which organism(s) does the gene belong to?
The analyses showed that the gene can be found in both bacteria M. tuberculosis and M. bovis.
5.1.2 Gene Product or Protein Characterization
6 – How many amino acids does the gene product contain?
Again, looking carefully at the U02492.1 entry, we can easily find the answer to this question or by a simple mathematical calculation. For example, if the gene contains 810 bp, the last three bases compose the stop codon, so the protein coding region is 807 bp long. After dividing this number by three (the number of bases in a codon) we obtain 269 amino acids. However, very often the annotation information is not available, and the only information we have is the nucleotide sequence. Thus, how can we find out the protein sequence and, consequently, its corresponding number of amino acids?
We need to translate our inhA gene sequence into its corresponding protein. There are several different ways to perform this task. We will use one of them, a tool called Translate (http://us.expasy.org/tools/dna.html), which can be accessed through the ExPASy web site. This tool translates a DNA (RNA) sequence into its six ( three in each direction) different possible open reading frames (ORFs). The Translate interface looks like this:

Our nucleotide sequence, corresponding to the GenBank entry U02492.1, was copied and pasted for analysis (red rectangle). After clicking on TRANSLATE SEQUENCE we obtain:

Our task now is to find out which of the six frames corresponds to the inhA gene product. In other words, we are asking what is the correct ORF. Often the correct ORF is the longest amongst the six possible ones. If we follow this assumption (which not always is correct!), the longest ORF amongst the six found by the Translate tool ORF 1 (5'-3'Frame 1) is the right answer. Clicking over the link 5'-3'Frame 1 will show us the conceptual translation in one-letter amino acid code, as follows:
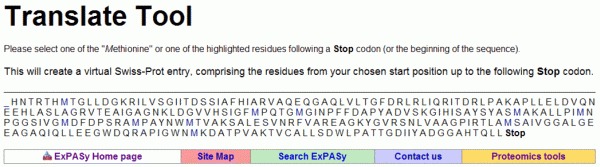
Once identified, the ORF can be translated into its corresponding protein or amino acid sequence. In most prokaryote species, an ORF starts with an ATG coding for methionine (Met or M) and ends with a stop codon (TAA, TAG or TGA). Now, clicking on the first Met residue will give us our protein sequence in a format similar to the GenBank flatfile, including the number of amino acids. In this case, 269 (see figure below).
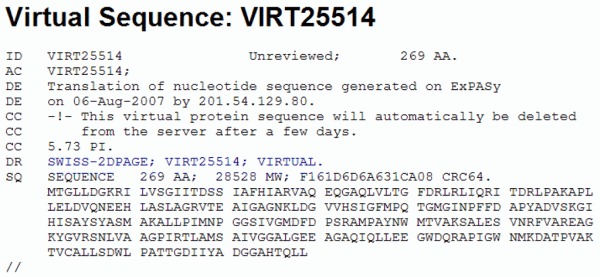
If we continue and click on the FASTA format link at the bottom of the page, we obtain our protein sequence in the the FASTA format, ready to be used by other programs.

7 – What is the gene product name and possible function?
So far we have worked with a DNA sequence. Now we will query the NCBI protein databases with the protein sequence obtained above and discover its possible function. For this puropose, we will go back to the basic BLAST page at NCBI (http://www.ncbi.nlm.nih.gov/BLAST/) but, at this time, we will select the link "protein blast", which will lead us to the BLASTp page. The BLASTp page at NCBI will look like this:

Our query sequence is already inserted in the search box. BLASTp standard parameters for protein-protein comparison are being used such as the substitution matrix BLOSUM62 and the nr database. For further details click in Algorithm parameters link at the bottom of the page.
The output list of results is shown for the top-21 hits:

The bit score and the E values indicate that we found our protein. Moreover, the red squares on the right with the letter "S" inside indicate that our proteins most likely have a 3D structure (S) which will help to answer the next questions.
To increase our confidence that the hits correspond to homologs of our query protein, we must check if the alignment covers the whole query protein sequence. The figure below shows the first alignment in the list of results.

Inspecting the annotation, we can see the our query protein is called Enoyl (Acyl Carrier Protein or ACP) Reductase of Mycobacterium tuberculosis strain H37Rv or InhA. To obtain a more complete answer we can access the protein annotation through its GenBank accession number NP_21600, similarly as we did with the gene accession number. The FEATURES section of the resulting archive is:
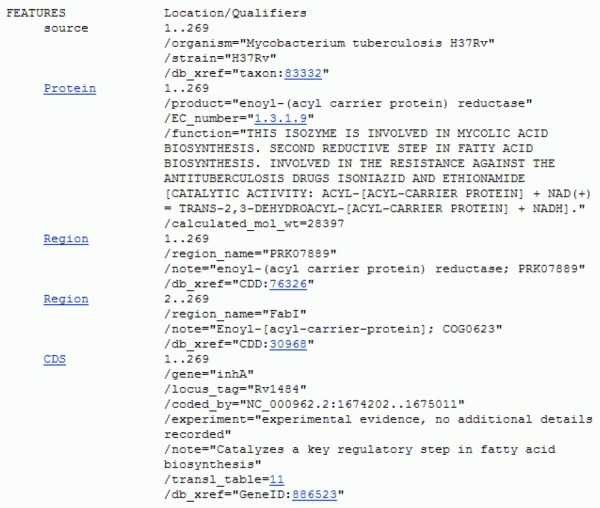
Since M. tuberculosis is a bacterium, this enzyme most likely functions in the cytoplasm and is involved in the biosynthesis of mycolic acid, an essential component of its cell wall. The blue links of the qualifiers can provide the curious reader with even more information about this enzyme.
8 – Is this gene present in humans? What consequences could this fact have for a possible application of gene product for a structure-based drug discovery?
To answer this question, you can run a BLASTp similarity search of this protein sequence against a database of human RefSeq proteins. Also, it is necessary to read about the fatty acid biosynthesis in humans and bacteria. A quick answer is: no, it is not present in humans. Hence, it is theoretically an ideal drug target against tuberculosis. In fact, the reader will find out that this enzyme has been proven to be a bonafide target of the drug isoniazid (INH).
9 – Does the gene product have a 3D structure? If so, what is its RCSB/PDB identification number? Describe the gene product architecture.
As we can see from the BLASTp output above, there are now several 3D structures for the InhA enzyme, enzyme-NADH complex, tertiary complexes involving drug candidates, and with the NADH-INH adduct which inhibits the enzyme. Historically, the first InhA structure to be determined was that with PDB ID 1ENY (the 8th entry in BLASTp's hit list), so we will investigate this particular one. To see the 3D structure, you should now go to PDBs search page (http://www.rcsb.org/pdb/home/home.do). To do this, enter the PDB code of the protein (1ENY) in the search bar at the top of the page and click the button "Site Search". The initial page of the result will look as follows:


We can obtain all the available information about the 3D structure of this enzyme by browsing through the links or download the PDB file to a local directory in our computer and work with our preferred molecular modeling and visualization package. For instance, we can see in the classification section that this enzyme has a 3-layer (αβα) sandwich architecture according to the CATH classification. We will visualize this next.
Some of the visualization software can be accessed directly from the page illustrated above. They can be located in the bottom of the section "Images and Visualization". Another alternative is the site First Glance in Jmol (http://molvis.sdsc.edu/fgij/index.htm). Go to the site, enter 1ENY in the blank box and click the Submit button:

As the name suggests, we can do many simple visualization manipulations that will help us to understand the molecule structure, interactions and function. Note: the use of molecular visualization software, no matter how simple it is, demands at least a general knowledge of the object to be visualized. Hence, to be able to run this software, the user must understand the basic principles of protein structure and function.
After submitting the 1ENY PDB code, and waiting a little bit to load the JAVA machine, we obtain:
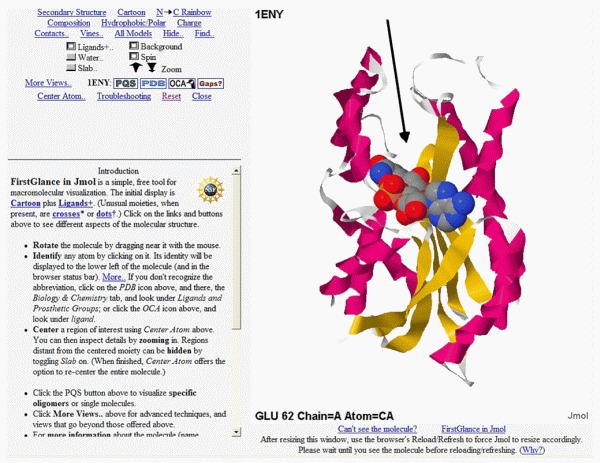
Here the InhA structure appears static, but the site will initially display the image rolling in order to present an even better idea of its three-dimensionality. Pressing the left mouse button and moving it left and right and up and down allows the user to keep full control of the rotation of the molecule. Also, pressing the left mouse button at the same time as the control or alt keys, and moving the mouse, will change the zoom.
It's possible to visualize the molecule using different representations. Above, the enzyme backbone is represented as ribbons where α helices and strands of the β sheet are colored red and yellow, respectively. Turns and loops are in grey. In this view angle, we can see α helices at left, a central β sheet, and more α helices at right. That is why this enzyme has an architecture of the 3-layer-αβα sandwich type. The two α helices layers are the "bread slices" and the β sheet, the sandwich's filling!
We can also observe the space-filling model of the NADH coenzyme colored according to the CPK rules (nitrogen in blue, carbon in grey, oxygen in red, phosphorus in orange). This coenzyme is the key to the InhA function.
In the figure, the added black arrow points to the position where the substrate will fit inside the enzyme. Drugs that inhibit this enzyme will most likely bind to this region.
10 – If the gene product does not have a 3D structure, would it be possible to infer its structure from the closest homologues with a known 3D structure? Explain the answer in details.
Remember, all the information we gathered so far started from a very small nucleotide sequence. Some basic knowledge of molecular biology and the usage of only a few of over 1200 bioinformatics websites are available in a Web Server Edition of the Nucleic Acids Research journal (http://nar.oxfordjournals.org/content/vol35/suppl_2/index.shtml?etoc).
Fortunately, we found a 3D structure for our 30 nucleotide sequence. If our protein did not have a structure, we could use the principle of comparative or homology modeling to build a 3D structure for it. This principle states that if a protein sequence of unknown structure (target protein) has a sequence identity greater than or equal to 30% with another protein with known structure (template), over its whole length, then the target protein 3D structure can be modeled based on one or on a combination of several template molecules (26). This is possible because homolog proteins descend from a common ancestor and are likely to present the same structure and function. Caution note: This is correct in general, but there are exceptions though.
References
- (1)
- Lesk, A. M. Introduction to Protein Architecture. OUP, Oxford, 2001.
- (2)
- Xie J. , Schultz P. G. An expanding genetic code. Methods. 2005;36:226–238. [PubMed: 16076448]
- (3)
- Anthony-Cahill S. J. , Griffith M. C. , Noren C. J. , Suich D. J. , Schultz P. G. Site-specific mutagenesis with unnatural amino acids. Trends Biochem. Sci. 1989;14:400–403. [PubMed: 2683258]
- (4)
- Ashburner M. , Ball C.A. , Blake J. A. , Botstein D. , Butler H. , Cherry J. M. , Davis A. P. , Dolinski K. , Dwight S. S. , Eppig J. T. , Harris M. A. , Hill D. P. , Issel-Tarver L. , Kasarskis A. , Lewis S. , Matese J. C. , Richardson J. E. , Ringwald M. , Rubin G. M. , Sherlock G. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genet. 2000;25:25–29. [PMC free article: PMC3037419] [PubMed: 10802651]
- (5)
- Cabeen M.T. , Jacobs-Wagner C. Bacterial cell shape. Nat. Rev. Microbiol. 2005;3:601–610. [PubMed: 16012516]
- (6)
- Kostin S. , Hein S. , Arnon E. , Scholz D. , Schaper J. The Cytoskeleton and Related Proteins in the Human Failing Heart. Heart Failure Reviews. 2000;5:271–280. [PubMed: 16228910]
- (7)
- Kunzelmann K. Ion Channels and Cancer. J. Membr. Biol. 2005;205:159–73. [PubMed: 16362504]
- (8)
- Hille B. , Armstrong C. M. , MacKinnon R. Ion channels: from idea to reality. Nat. Med. 1999;5:1105–1109. [PubMed: 10502800]
- (9)
- Green M. R. Eukaryotic transcription activation: right on target. Mol. Cell. 2005;18:399–402. [PubMed: 15893723]
- (10)
- Bloom K. Chromosome segregation: seeing is believing. Curr. Biol. 15:R500–503. [PubMed: 16005281]
- (11)
- Delves P. J. , Roitt I. M. The immune system. Second of two parts. N. Engl. J. Med. 2000a;343:108–117. [PubMed: 10891520]
- (12)
- Delves P. J. , Roitt I. M. The immune system. First of two parts. N. Engl. J. Med. 2000b;343:37–49. [PubMed: 10882768]
- (13)
- Luscombe N.M. , Greenbaum D. , Gerstein M. What is Bioinformatics? A proposed definition and overview of the field. Method Inform. Med. 2001;40:346–358. [PubMed: 11552348]
- (14)
- Burley S. K. , Almo S. C. , Bonanno J. B. , Capel M. , Chance M. R. , Gaasterland T. , Lin D. , Sali A. , Studier F. W. , Swaminathan S. Structural genomics: beyond the Human Genome Project. Nat. Genet. 1999;23:151–157. [PubMed: 10508510]
- (15)
- Liolios K. , Tavernarakis T. , Hugenholtz P. , Kyrpides N. C. The Genomes On Line Database (GOLD) v.2: a monitor of genome projects worldwide. Nucl. Acids Res. 2006;34:D332–D334. [PMC free article: PMC1347507] [PubMed: 16381880]
- (16)
- Galperin M. Y. The Molecular Biology Database Collection: 2006 update. Nucl. Acids Res. 2006;34:D3–D5. [PMC free article: PMC1347524] [PubMed: 16381871]
- (17)
- Ouzonis C. A. , Valencia A. Early bioinformatics: the birth of a discipline – a personal view. Bioinformatics. 2003;19:2176–2190. [PubMed: 14630646]
- (18)
- Benson D. A. , Karsch-Mizrachi I. , Lipman D. J. , Ostell J. , Wheeler D. L. GenBank. Nucl. Acids Res. 2006;34:D16–D20. [PMC free article: PMC1347519] [PubMed: 16381837]
- (19)
- Kouranov A. , Xie L. , de la Cruz J. , Chen L. , Westbrook J , Bourne P. E. , Berman H. M. The RCSB PDB information portal for structural genomics. Nucl. Acids Res. 2006;34:D302–D305. [PMC free article: PMC1347482] [PubMed: 16381872]
- (20)
- Altschul S. F. , Madden T. L. , Schaffer A. A. , Zhang J. , Zhang Z. , Miller W. , Lipman D. J . Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nuc. Acids Res. 1997;25:3389–3402. [PMC free article: PMC146917] [PubMed: 9254694]
- (21)
- Andreeva A. , Howorth D. , Brenner S. E. , Hubbard T. J. P. , Chothia C. G. , Murzin A. G. SCOP database in 2004: refinements integrate structure and sequence family data. Nuc. Acids. Res. 2004;32:D226–D229. [PMC free article: PMC308773] [PubMed: 14681400]
- (22)
- Murzin A. G. , Brenner S. E. , Hubbard T. , Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. [PubMed: 7723011]
- (23)
- Edwards Y. J. , Cottage A. Bioinformatics methods to predict protein structure and function. A practical approach. Mol. Biotechnol. 2003;23:139–166. [PubMed: 12632698]
- (24)
- Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 2005;15:285–289. [PubMed: 15939584]
- (25)
- Rajpal D. K. Understanding biology through bioinformatics. Int. J Toxicol. 2005;24:147–52. [PubMed: 16040566]
- (26)
- Martí-Renom M. A. , Stuart A. , Fiser A. , Sánchez R. , Melo F. , Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. [PubMed: 10940251]
- (27)
- Godzik A. Fold recognition methods. Methdos Biochem. Anal. 2003;44:525–546. [PubMed: 12647403]
- (28)
- Bonneau R. , Baker D.A. Ab initio protein structure prediction: progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001;30:173–189. [PubMed: 11340057]
- (29)
- Creighton, T. Proteins: Structures and Molecular Properties, 2nd Edition, W.H. Freeman, 1992.
- (30)
- Branden, C & Tooze, J. Introduction to protein Structure, 2nd Edition, Garland Publishing, New York, 1999.
- (31)
- Petsko, G. A. & Ringe, D. Protein Structure and Function, New Science Press, 2004.
- (32)
- Zhang Y , Baranov P.V. , Atkins J.F. , Gladyshev V.N. Selenocysteine and pyrrolysine use dissimilar decoding strategies. J. Biol. Chem. 2005;280:20740–20751. [PubMed: 15788401]
- (33)
- MacArthur M.W. , Thronton J.M. Influence of proline residues on protein conformation. J. Mol. Biol. 1991;218:397–412. [PubMed: 2010917]
- (34)
- Ramakrishna, C. , Ramachandran G.N. Stereochemical criteria for polypeptide and protein chain conformations. II. Allowed conformations for a pair of peptide units. Biophys. J. 1965;5:909–933. [PMC free article: PMC1367910] [PubMed: 5884016]
- (35)
- Karpen M.E. , Dehaseth P.L. , Neet K.E. Differences In The Amino-Acid Distributions Of 310-Helices And Alpha-Helices. Prot Sci. 1992;1:1333–1342. [PMC free article: PMC2142095] [PubMed: 1303752]
- (36)
- Fodje M,N , Al-Karadaghi S. Occurrence, conformational features and amino acid propensities for the pi-helix. Prot. Eng. 2002;15:353–358. [PubMed: 12034854]
- (37)
- Blundell T. , Barlow D. , Borkakoti N. , Thornton J. Solvent-induced distortions and the curvature of alpha-helices. Nature. 1983;306:281–283. [PubMed: 6646210]
- (38)
- Hol W.G.J. , Vanduijnen P.T. , Berendsen H.J.C. Alpha-Helix Dipole And Properties Of Proteins Nature. 1978;273:443–446. [PubMed: 661956]
- (39)
- Richardson J.S. , Richardson D.C. Amino acid preferences for specific locations at the ends of alpha helices. Science. 1988;240:1648–1652. [PubMed: 3381086]
- (40)
- Salemme F.R. Structural properties of protein beta-sheets. Prog. Biophys. Mol. Biol. 1983;42:95–133. [PubMed: 6359272]
- (41)
- Chou K-C. Prediction of tight turns and their types in proteins. Anal. Biochem. 2000;286:1–16. [PubMed: 11038267]
- (42)
- Sibanda B.L. , Blundell T.L. , Thornton J.M. Conformation of beta-hairpinin protein structures. J. Mol. Biol. 1989;206:759–777. [PubMed: 2500530]
- (43)
- Chothia C. , Levitt M. , Richardson D. Helix to helix packing in proteins. J. Mol. Biol. 1981;145:215–250. [PubMed: 7265198]
- (44)
- Chothia C. , Janin J. Relative orientation of close-packed beta-pleated sheets in proteins. Proc. Natl. Acad. Sci. 1981;78:4146–4150. [PMC free article: PMC319745] [PubMed: 16593054]
- (45)
- Janin J. , Chothia C. Packing of alpha-helices onto beta-pleated sheets and the anatomy of alpha/beta proteins. J. Mol. Biol. 1980;143:95–128. [PubMed: 7003165]
- (46)
- Orengo C.A. , Michie A.D. , Jones S. , Jones D.T. , Swindells M.B. , Thornton J.M. CATH - a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. [PubMed: 9309224]
- (47)
- Presnell S.R. , Cohen F.E. Topological distribution of four alpha-helix bundles. Proc. Natl. Acad. Sci. 1989;86:6592–6596. [PMC free article: PMC297890] [PubMed: 2771946]
- (48)
- Teichmann S.A. , Chothia C. , Gerstein M. Advances in Structural Genomics. Curr. Opin. Str. Biol. 1999;9:390–399. [PubMed: 10361097]
- (49)
- Orengo C.A. , Jones D.T. , Thornton J.M. Protein superfamilies and domain superfolds. Nature. 1994;372:631–634. [PubMed: 7990952]
- (50)
- Richardson J.S. Handedness of crossover connections in beta-sheets. Proc. Natl. Acad. Sci. 1976;73:2619–2623. [PMC free article: PMC430699] [PubMed: 183204]
- (51)
- Raetz C.R. , Roderick S.L. A left-handed parallel beta helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science. 1995;270:997–1000. [PubMed: 7481807]
- (52)
- Taylor W.R. A deeply knotted protein structure and how it might fold. Nature. 2000;406:916–919. [PubMed: 10972297]
- (53)
- Milton R.C.D. , Milton S.C.F. , Kent S. B. H. Total chemical synthesis of a d-enzyme: the enantiomers of HIV-1 protease show demonstration of reciprocal chiral substrate specificity. Science. 1992;256:1445–1448. [PubMed: 1604320]
- (54)
- Goodsell D.S. , Olson A.J. Structural Symmetry and Protein Function. Annu. Rev. Biophys. Biomol. Struct. 2000;29:105–153. [PubMed: 10940245]
- (55)
- Braig K. , Otwinowski Z. , Hegde R. , Boisvert D.C. , Joachimiak A. , Horwich A.L. , Sigler P.B. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature. 371:578–586. [PubMed: 7935790]
- (56)
- Seemuller E. , Lupas A. , Stock D. , Lowe J. , Huber R. , Baumeister W. Proteasome from Thermoplasma acidophilum: a threonine protease. Science. 1995;268:579–582. [PubMed: 7725107]
- (57)
- Hugo Kubinye - 3D QSAR in Drug Design: Theory Methods and Applications - Chapter: Molecular Superposition for Rational Design, page 200–201.
- (58)
- Pandi Veerapandian - Structure-Based drug design -1997.
- (59)
- Klaus Gubernator & Hans-Joachim Böhm - Structure-based Ligand Design -1998.
- (60)
- Myers S. , Baker A. Drug discovery - an operating model for a new era. Nature Biotechnology. 2001;19:727–730. [PubMed: 11479559]
- (61)
- Kuntz I.D. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. [PubMed: 1509259]
- (62)
- Verdonk M. L. , Cole J. C. , Hartshorn M. J. , Murray C. W. , Taylor R.D. Improved Protein–Ligand Docking Using GOLD. PROTEINS: Structure, Function, and Genetics. 2003;52:609–623. [PubMed: 12910460]
- (63)
- Vigers G. P. A , Rizzi J. P. Multiple Active Site Corrections for Docking and Virtual Screening. J. Med. Chem. 2004;47:80–89. [PubMed: 14695822]
- (64)
- Wang R. , Wang S. How Does Consensus Scoring Work for Virtual Library Screening? An Idealized Computer Experiment. J. Chem. Inf. Comput. Sci. 2001;41:1422–1426. [PubMed: 11604043]
- (65)
- Lyne, P. D. Structure-based virtual screening: an overview. Drug Discovery Today Vol. 7, No. 20. 2002. [PubMed: 12546894]
- (66)
- Wade, C. R., Henrich, S., Wang, t. Using 3D protein structures to derive 3D-QSARs. Drug Discovery Today, Vol.1, No 3. 2004. [PubMed: 24981491]
- (67)
- Anderson, A. C. Targeting DHFR in parasitic protozoa. DDT, Vol. 10, No 2. 2005. [PubMed: 15718161]
- (68)
- Shaw M.P. et al. High-resolution crystal structure of Trypanosoma brucei UDP-galactose 4_-epimerase: a potential target for structure-based development of novel trypanocides. Molecular & Biochemical Parasitology. 2003;126:173–180. [PubMed: 12615316]
- (69)
- Perbandt M. Structure of the Major Cytosolic Glutathione S-Transferase from the Parasitic Nematode Onchocerca volvulus. J.B.C. 2005;280(13):12630–12636. [PubMed: 15640152]
- (70)
- Nathan S. T. et al. Structure of glutathione S-transferase of the filarial parasite Wuchereria bancrofti: a target for drug development against adult worm. J. Mol. Model. 2005;11:194–199. [PubMed: 15864673]
- (71)
- Sánchez-Sixto C. et al. Structure-Based Design, Synthesis, and Biological Evaluation of Inhibitors of Mycobacterium tuberculosis Type II Dehydroquinase. J. Med. Chem. 2005;48:4871–4881. [PubMed: 16033267]
- (72)
- Olliaro P. L. , Yuthavong Y. An Overview of Chemotherapeutic Targets for Antimalarial Drug Discovery. Pharmacol. Ther. 1999;81(2):91–110. [PubMed: 10190581]
- (73)
- Schuttelkopff A. W. et al. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005;352:105–116. [PubMed: 16055151]
- (74)
- Buschiazzo A. et al. The Crystal Structure and Mode of Action of Trans-Sialidase, a Key Enzyme in Trypanosoma cruzi Pathogenesis. Molecular Cell. 2002;10:757–768. [PubMed: 12419220]
- (75)
- Jao, S. C. et al. (2005) Design of potent inhibitors for Schistosoma japonica glutathione S-transferase. Bioorg. Med. Chem. ARTICLE IN PRESS, 2005. [PubMed: 16275109]
- (76)
- Padmanabhan P.K. et al. Glyoxalase I from Leishmania donovani: A potential target for anti-parasite drug. Biochemical and Biophysical Research Communications. 2005;337:1237–1248. [PubMed: 16236261]
- (77)
- Guex N. , Peitsch M.C. SWISS-MODEL and The Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. [PubMed: 9504803]
- (78)
- Humphrey W. , Dalke A. , Schulten K. VMD - Visual Molecular Dynamics. J. Molec. Graphics. 1996;14:33–38. [PubMed: 8744570]
- 1. Why Is It Important to Study Proteins?
- 2. Explosion of Biological Sequence and Structure Data
- 3. Basic Principles of Protein Structure
- 4. The Use of Protein Structures in Inhibitor Discovery and Design
- 5. Practical Section: Computational Characterization of a Gene Product from a Partial Gene Sequence Only
- References
- Protein Structure, Modelling and Applications - Bioinformatics in Tropical Disea...Protein Structure, Modelling and Applications - Bioinformatics in Tropical Disease Research
- SLC35F4 solute carrier family 35 member F4 [Homo sapiens]SLC35F4 solute carrier family 35 member F4 [Homo sapiens]Gene ID:341880Gene
- Trappc11 trafficking protein particle complex 11 [Mus musculus]Trappc11 trafficking protein particle complex 11 [Mus musculus]Gene ID:320714Gene
- SIMC1P1 SIMC1 pseudogene 1 [Homo sapiens]SIMC1P1 SIMC1 pseudogene 1 [Homo sapiens]Gene ID:202181Gene
- LINC02870 long intergenic non-protein coding RNA 2870 [Homo sapiens]LINC02870 long intergenic non-protein coding RNA 2870 [Homo sapiens]Gene ID:170393Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...