

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Rao JN, Wang JY. Regulation of Gastrointestinal Mucosal Growth. San Rafael (CA): Morgan & Claypool Life Sciences; 2010.

GI hormones are chemical messengers that are implicated in many aspects of physiological functions of the gastrointestinal tract, including the regulation of secretion, absorption and digestion, and gut motility. GI hormones are a large family of peptides and are secreted by endocrine cells that are widely distributed throughout the GI mucosa and pancreas. Gastrin, secretin, and cholecystokinin (CCK) were the first discovered gut hormones, and as of today, there are more than 50 gut hormone genes and a multitude of bioactive peptides, which makes the gut as the largest endocrine organ of the body. GI hormones act through their specific receptors to activate particular signal pathways and ultimately provide functional signals for their physiological effects [81–84]. Initially, these hormones were described solely as endocrine products, but subsequent experiments have revealed that GI hormones also function as autocrine or paracrine to regulate GI functions. In addition, these hormones are thought to serve as transmitting agents for nerve impulses discharged into blood vessels after nervous stimulation in a true neurocrine fashion [85,86]. Besides their regulatory effects on secretion, absorption and digestion, and gut motility, GI hormones also modulate GI mucosal growth and are involved in the pathogenesis of gut mucosal atrophy, neoplasm, and cancers. The GI hormones that regulate gut mucosal growth positively or negatively include gastrin, CCK, secretin, somatostatin, ghrelin, bombesin, and gastrin-releasing peptide (GRP).
GASTRIN
Gastrin was first identified by Edkins in 1906 [87] when he discovered that extracts of antral mucosa stimulated acid secretion (gastric juice) from the gastric fundus, and then he named this new active agent within the antral extract as “gastrin.” Almost 55 years later, Gregory and Tracy [88] successfully isolated the pure form of gastrin from the antral mucosa of hogs. Since then, gastrin has been established as the major biological regulator of gut physiology and plays a critical role in the regulation of gastric acid secretion [89–91]. Gastrin is initially released from the G cells in the antral region of the stomach during a meal by vagal stimulation, distention and digested protein. Other organs and cells that also produce gastrin include pancreatic endocrine cells [92], pituitary [93], and extraantral G cells [94]. The cellular targets for gastrin in the stomach are the acid-secreting parietal cells and histamine producing enterochromaffin-like (ECL) cells. In addition to stimulating acid secretion from gastric parietal cells, gastrin is also considered to be a key growth regulator in the gut mucosa and is implicated in the development of various GI cancers [91,95–97].
The major biologically active forms of gastrin are G-17 and G-34 amino acid peptides containing tyrosine residues at carboxyl terminus [96]. The biosynthetic pathways, leading to the production of amidated gastrins from the precursor molecule, prograstrin, are well established [82,84,98]. In antral G cells progastrin is stored and processed into secretory granules; N- and C-terminal extensions are removed by prohormone convertases. The posttranslational modification of gastrin appears to have no functional significance, as all forms are equally potent at the receptor level. G-17 is cleared from the circulation faster than the G-34 form, therefore, the majority of gastrin in the circulation during fasting is G-34. In contrast, the major form of gastrin that is released after a meal is the G-17 [99] (Figure 6). The greatest proportion of gastrin in the circulation is fully processed. As mentioned above, gastrin secretion from antral G cells is tightly regulated by luminal, paracrine, endocrine, and different neuronal stimuli [99,100]. Small peptides, aromatic amino acids, and calcium in a meal are also key contributors for the stimulation of gastrin release (Figure 6). Negative regulation of gastrin release depends on the decreased pH levels following acid secretion, which is mediated by the paracrine effects of other hormones, such as somatostatin.

One of the most important functions of gastrin is to regulate gut mucosal growth and IEC proliferation, and it has been recognized as the single most important trophic hormone of the stomach [84]. The regulatory effect of gastrin on gut mucosal growth was initially identified more than three decades ago in two separate reports. Studies by Johnson et al. [101] and Crean et al. [102] revealed that administration of a synthetic gastrin analog, pentagastrin, increases protein synthesis and parietal cell mass in rats. These findings were quickly confirmed using the natural amidated gastrins, G-17 and G-34. G-17 and G-34 produced maximal stimulation on DNA synthesis in the oxyntic mucosa, duodenum, and colon at doses of 13.5 and 6.75 nmol/kg, respectively [84,103]. Removal of endogenous gastrin by antral resection causes mucosal atrophy that can be overcome by exogenous administration of gastrin. Subsequently, increased fundic mucosal proliferation was also demonstrated in rodent models following the administration of H2-receptor antagonists, resulting in hypergastrenemia [84]. These animals exhibit the gastric gland elongation and increased cell proliferation. In humans, infusion of gastrin at high doses was shown to increase gut epithelial cell proliferation [104]. Consistently, patients with Zollinger–Ellison syndrome are associated with an increase in gastric mucosal growth due to high levels of gastrin in the circulation [27].
A number of gastrin gene transgenic or knock-out mouse strains have been developed, in which gastrin concentrations in circulation is increased or decreased dramatically [99,105]. Overexpression of either unprocessed gastrins or the amidated gastrins, such as G-17 and G-34, in the transgenic mice increases 2-fold elevation in serum-amidated gastrin and induces gut mucosal growth, particularly induction in numbers of parietal cells that is associated with an elevated gastric acid secretion [99,100]. These observations are further supported by results in athymic nude mice bearing xenografts of a transplanted human gastrinoma demonstrating gastric and duodenal mucosal hyperplasia. Interestingly, with aging, there is a progressive loss of parietal cells and expansion of the mucous neck cell proliferation [84]. This condition resembles the human condition of atrophic gastritis, which is characterized by a loss of gastric glands, progressive loss of parietal cells and increased plasma gastrin levels [84] (Figure 7). On the other hand, gastrin-deficient mice exhibit a significant decrease in parietal cell mass (by 35%) compared with wild-type control animals [106,107]. Another study also show that in gastrin knock-out mice, ECL cells appear to be clustered toward the bottom of the gastric gland and there is a reduced rate of cell migration to the base of the gland (Figure 7). As opposed to the results using amidated gastrin, experiments using G-gly show colonic proliferation, sparking renewed interest in a role for gastrin in precursor products in colonic growth. Mice overexpressing progastrin truncated at glycine-72 (MTI/G-GLY) exhibit elevated serum and mucosal levels of G-gly compared with wild-type mice. MTI/G-GLY mice display increases in colonic mucosal thickness (by 43%) and in the percentage of goblet cells per crypt (by 41%) [108]. Furthermore, administration of G-gly increases colonic mucosal thickness by ∼10% and colonic proliferation by 81% in gastrin-deficient mice. These experiments using gastrin transgenic and knock-out models clearly show that gastrin is a potent stimulator of GI mucosal growth and epithelial cell proliferation.

In addition, gastrin receptor antagonists also have variable effects on gastrin-stimulated GI cancer growth [97]. For example, administration of proglumide (a potent gastrin-receptor blocker) inhibits the growth of MC-26 tumors in vivo and prolongs survival of tumor-bearing mice [99,109]. Hoosein et al. [110] demonstrate that gastrin stimulation is possibly attributable to an autocrine mechanism. Although discussion on the role of gastrin in the pathogenesis of GI cancers at detail is beyond the scope of this chapter, readers can be referred to understand most recent development of this area through several review articles [27,84,97,99,100,111].
CHOLECYSTOKININ
CCK is a member of the gut–brain family of peptide hormones, and it is produced by endocrine cells located predominantly in the proximal small intestine (duodenum and jejunum) as well as by the neurons in the myenteric plexus and brain. This gut peptide was first described in 1928 by Ivy and Oldberg [112] as a contaminant in impure secretin preparations. After complete purification and sequencing, CCK has been shown to be crucial for gallbladder contraction and pancreatic enzyme secretion. Other physiological functions of CCK in the GI tract include inhibition of gastric emptying, stimulation of bowel motility, potentiation of insulin secretion, and trophic effects on the pancreas and gut mucosa. Under biological conditions, CCK release is stimulated by fats, proteins, and amino acids.
Various forms of CCK are derived from the posttranslational modification of products of the pro-CCK gene which produces a cocktail of peptides with varying numbers of amino acids, each of which includes the minimal epitope for bioactivity [27,84]. In human embryonic development, CCK first appears in the duodenal and small intestinal mucosa at around 10 weeks of gestation, and the concentration progressively increases as gestation progresses and continue to present in endocrine cells of the GI tract. In the rat, maximal CCK concentration is present in the duodenum and jejunum. A high level of CCK is achieved in the suckling period, at around 14 days, and a decline is observed with weaning, and adult levels are reached at 28 days. CCK-58 is the largest form of the hormone, whereas CCK-8 is the smallest fragment containing 8-amino acids with complete biological activity [113]. The molecular forms of CCK are diverse and appear to be tissue-specific. For example, the most abundant form of CCK in the brain is CCK-8, although significant amounts of large carboxy-amidated forms such as CCK-33, CCK-58, and CCK-83 have been also isolated. CCK functions by directly interacting with the specific G-coupled CCK receptors (Figure 8). After binding to the receptor, a sequence of events collectively occurs, which culminates in an increase in concentration of intracellular calcium which in turn leads to degranulation of the pancreatic acinar cells and enzyme secretion. Additionally, CCK also stimulates the growth of the pancreas in experimental studies, although evidence for CCK regulating the growth of the GI mucosa is limited (Figure 8).
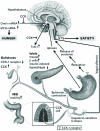
An increasing body of evidence demonstrates the trophic effects of CCK on the pancreas. Administration of CCK alone or combination with secretin causes marked increases in pancreatic weight, DNA, RNA, and protein content in rats [114]. The trophic effect of CCK on the pancreas is physiologically significant since amino acids infused into the duodenum leads to a marked increase in pancreatic growth, and this stimulatory effect is prevented by the CCK-receptor antagonist such as CR 1409. Chronic camostate feeding increases pancreatic growth, which is associated with increased CCK plasma levels. The administration of exogenous CCK produces similar increase in pancreatic growth, but the combination of camostate and CCK-8 induces an additive stimulatory effect on the pancreas. Inactivation of CCK-receptors by CR 1409 completely abolishes the trophic effects of exogenous CCK-8 and inhibits the effects of chronic camostate feeding. In addition, treatment with CR 1409 alone also decreases pancreatic weight, DNA, and protein content. Johnson and Guthrie [115] have reported that administration of CCK-8 at very low doses induced a significant increase in pancreatic DNA synthesis, although it did not stimulate the mucosal growth of the oxyntic gland area or duodenum. Taken together, these findings clearly show that CCK is a potent stimulant for pancreatic growth.
On the other hand, evidence has been reported, showing the role of CCK in the regulation GI mucosal growth. Treatment with CCK and secretin is shown to prevent atrophy in the jejunum and ileum of dogs given total parenteral nutrition (TPN) as a sole nutrient source [84,116]. Administration of this peptide also increases galactose absorption, suggesting that CCK is a positive enterotrophic factor for the gut. Fine et al. [117], using intestinal bypass models, found that trophic response induced by CCK and secretin in the small bowel is the indirect result of increased pancreatobiliary secretion, as opposed to a direct stimulatory effect of these peptides on the gut mucosa. In cultured rabbit jejunum and ileum preparations, Stange et al. [118] further confirmed the lack of a direct effect of CCK on small-bowel growth. Furthermore, CCK has also been proposed as a major mediator of the satiety response, which leads to the cessation of feeding when food is placed in the stomach or intestine [119]. Intravenous CCK inhibits food intake in rats, although the mechanisms by which CCK controls the appetite are yet to be clearly identified. In addition to the involvement of CCK in the induction of satiety, this hormone is also involved in several disease conditions, such as diabetes mellitus, gall stone disease, irritable bowel syndrome (IBS), and inflammation (Figure 8) [84,120].
Several studies examined the cellular mechanisms by which CCK regulates pancreatic and gut growth. It has been reported that CCK activates the MAPK cascade, leading to the activation of ERK, JNK, and p38 MAPK in the pancreas [84,121]. Other signaling pathways that are also involved in CCK-induced mitogenesis and cellular proliferation include the PI3K-mTOR (mammalian target of rapamycin)-p70S6K and eIF4A pathways [121–123]. CCK stimulates the phosphorylation and activation of p70S6K in rat pancreatic acini; this activation is blocked by inhibiting mTOR and PI3K. In addition, the PI3K-mTOR pathway activates protein synthesis by phosphorylating the binding protein eIF4E, the translation initiation factor that binds to the 7-methyl guanosine cap at the 5' end of most eukaryotic mRNA molecules.
SECRETIN
Secretin was initially identified by Bayliss and Starling in 1902 [124]. In the past century, the research of secretin has gone by many milestones, which includes isolation, purification, structural characterization, and chemical synthesis of secretin, establishment of its hormonal status, identification of the specific receptor, cloning of secretin and its receptor genes, and identification of secretin-releasing peptides. Secretin has been identified as a hormone-regulating pancreatic exocrine secretion of fluid and bicarbonate, gastric acid secretion, and gastric motility [125,126].
There are a few observations showing the involvement of secretin in the regulation of GI mucosal growth so far. Generally, secretin is shown to inhibit the trophic action of gastrin but it has no a direct antitrophic activity in the GI mucosa [27]. Secretin inhibits the gastrin-mediated stimulation of DNA synthesis in the gastric oxyntic gland region, duodenum, and colon [127,128]. This inhibitory effect of secretin is independent of its ability to inhibit gastrin-stimulated acid secretion. It is likely that secretin indirectly regulates GI mucosal growth by blocking the trophic effect of gastrin.
SOMATOSTATIN
Somatostatin (SST) is a natural peptide hormone secreted in various parts of the human body including the GI tract [129]. SST was first identified by Brazeau et al. in 1973 [130] and it was originally described as a growth hormone-releasing inhibitory factor, containing 14-amino acids. The single SST gene is expressed in numerous endocrine cells in the GI system, including gastric mucosa and pancreas. In the intestinal mucosa, a 92-amino acid precursor molecule is processed to release a 28-amino acid peptide, of which 14 amino acids occupy the N-terminal position [131]. SST acts via five different but related receptor molecules belonging to the superfamily of G-protein-coupled receptors [132,133].
SST is a regulatory–inhibitory peptide and functions as the universal endocrine off-switch. SST represses the release of growth hormones and all known GI hormones, and it also inhibits gastric acid secretion and motility, intestinal absorption, and pancreatic bicarbonate and enzyme secretion, and selectively reduces splanchnic and portal blood flow [129]. Importantly, SST also inhibits the growth of the GI mucosa and normal pancreas, and this effect is mediated through either an indirect mechanism such as inhibition of other trophic hormones or a direct mechanism via interaction with the SST receptor subtype 2 [134]. Experiments in vivo revealed that the SST administration decreases DNA synthesis and reduces the number of parietal cells in gastric mucosa and exocrine pancreatic cells [27,135]. Furthermore, SST given together with gastrin reduces gastrin-stimulated mucosal growth of the stomach. On the other hand, in the duodenum and jejunum, the effects of SST are less consistent, with nocturnal SST producing a slight decrease in DNA synthesis, suggesting that SST inhibits cell division in the mucosa of normal GI tract and, furthermore, antagonizes the trophic activity of gastrin. Similarly, there are inhibitory effects on rat mucosal growth of duodenum using the SST analog, sandostatin. The notion that SST represses GI mucosal growth is further supported by studies demonstrating that the normal adaptive hyperplasia noted in rats after 40% small-bowel resection is abolished by administration of SST.
In the stomach, SST is produced by fundic and antral D cells which are closely associated with parietal cells, enterochromaffin-like (ECL) cells and gastrin G cells either directly via cytoplasmic processes (paracrine secretion) or indirectly via the circulation (endocrine secretion) [136]. This close anatomical relationship provides the morphological base for the tonic inhibitory effect of SST on gastric acid secretion directly by inhibiting parietal cells and indirectly by inhibiting the release of histamine from the ECL cells and gastrin from G cells (Figure 7). Piqueras and Martinez [137] demonstrated that SST modulates the gastrin-ECL cell/parietal cell axis through SST receptor subtype-2. A synthetic SST analog peptide, octreotide, is clinically used, and its pharmacological actions in the GI tract include the inhibition of release of gastrin, motilin, secretin and vasoactive intestinal polypeptides, reduction in blood flow to the gut mucosa, and repression of intestinal motility and carbohydrate absorption. Octreotide treatment for patients with acromegaly prevents hormone hypersecretion from the tumor, normalizing circulating growth hormone and IGF-1 levels, thus altering tumor growth [133,138,139].
There are considerable amounts of results showing the effects of SST on the normal pancreatic growth [84,140]. The administration of SST reduces pancreatic weight, DNA, RNA, and protein content, whereas SST depletion with cysteamine stimulates pancreatic growth. In addition, treatment with cysteamine augments the trophic effect of bombesin on the pancreas. Blocking endogenous SST may release these inhibitory constraints and allow for increased proliferation of the normal pancreas. Although the exact mechanism underlying the inhibitory effects of SST remains largely unknown, the SST receptor subtype-2 associates and stimulates tyrosine phosphatase Src homology 2-containing tyrosine phosphatase-1 activity, which in turn arrests cells in the G0/G1 phase of the cell cycle associated with up-regulation of the cyclin-dependent kinase inhibitor p27kip1 and an increase in hypophosphorylated retinoblastoma protein levels.
GHRELIN
Ghrelin is a relatively new member of the gut hormones and was first isolated from the rat and the human stomach in 1999 [141]. Ghrelin is a 28-amino acid peptide and serves as an endogenous ligand for growth hormone secretagogue receptor (GHSR). Together with the recently discovered 23-amino acid obestatin, it is derived from a prohormone precursor (proghrelin) by the posttranslational processing. Cells immunoreactive to ghrelin are widely distributed in the gastric mucosa in domestic and laboratory animals and in humans. The greatest expression of ghrelin is in the stomach, particularly in endocrine A-type cells in the oxyntic mucosa, and smaller amounts were in the small intestine and colon. Ghrelin stimulates the release of growth hormone from the pituitary both in vitro and in vivo and plays an important role in the regulation of food intake, energy homeostasis, gastric emptying, and acid secretion [142,143]. In contrast, obestatin seems to induce the opposite effects. The active form of ghrelin known as acyl ghrelin binds to and activates its receptor, GHSR-1a, and crosses the blood–brain barrier [144]. Peripheral and central administration of ghrelin in rats stimulates acute food intake [145], whereas the circulating levels of ghrelin increase with fasting but fall in response to meal intake, which is proportional to the calorie load of the meal. Ghrelin secretion is increased by acetylcholine and gastric inhibitory peptide, but decreased by CCK, SST, insulin and infection with Helicobacter pylori [146,147]. Taheri et al. [148] reported that lack of sleep is also associated with an increase in ghrelin levels and a decrease in leptin levels.
Ghrelin and ghrelin receptor expression are found in developing GI fetal and neonatal tissues, and substantial amounts of ghrelin are also identified in colostrum, suggesting that it has a potential role in perinatal development. Subcutaneous administration of ghrelin to pregnant rats is shown to increase body weight of newborn animals, which may be due to its possible role in stimulating the gut development in the postnatal period [149]. In another study, exogenous administration of ghrelin reduces the gastric growth in suckling rats, as indicated by a decrease in levels of gastric mucosa weight, DNA synthesis, and total DNA content [149]. Furthermore, treatment with ghrelin produces a significant reduction in the pancreatic weight and pancreatic amylase enzyme activity, and the inhibitory effects of ghrelin result probably from the hypothalamus immaturity in postnatal animals. Several studies by Kotunia et al. [150,151] show that repetitive intragastric administration of ghrelin in neonatal pigs fed with milk formula results in a significant depletion in body weight and reduces small intestine length, and it also causes a remarkable reduction in villous length and thickness of the tunica mucosa and tunica muscularis in the jejunum and ileum. In humans, the expression of ghrelin in the stomach is increased during infancy which may be related to the increase in gastric acid output, growth hormone secretion, and meal intake [143,144,149,150,152–154]. Although intensive research has been carried out on ghrelin over the past decade, there remain many interesting questions regarding ghrelin-related biology. These include the identification of the pathways regulating ghrelin production and release from the GI tract, the enzyme that catalyzes its acyl-modification, and the continuing search for its physiological actions, especially its role and mechanism in modulating GI mucosal growth under various pathological condition.
BOMBESIN/GASTRIN-RELEASING PEPTIDE
Bombesin (BBS), a 14-amino acid peptide that was originally isolated from the extracts of skin from European amphibians in 1970, is analogous to mammalian gastrin-releasing peptide (GRP) [155]. BBS/GRP was found in a variety of species including rats, guinea pigs, dogs, and humans, and BBS/GRP-like immunoreactivity is widely distributed throughout the GI tract, predominantly in the neuronal populations of the gut and in the acid- and gastrin-secreting portions of the stomach [156]. In general, BBS/GRP functions as a universal on-switch and plays predominantly stimulatory effects. BBS/GRP stimulates the release of almost all GI hormones, intestinal and pancreatic secretions, and motility [157]. Although the most important functions of BBS/GRP are to regulate antral gastrin release and gastric acid secretion, this peptide also stimulates growth of the GI mucosa and pancreas.
Increasing evidence indicates that BBS/GRP is a potent trophic factor in the GI tract and pancreas [158]. Since BBS/GRP stimulates the release of gastrin, it is obvious that its effects are opposite to those of somatostatin. It has been shown that the administration of BBS/GRP for 7 days at 8-h intervals stimulates DNA synthesis and total DNA and RNA content in the gastric and colonic mucosa [157], but this stimulatory effect is attenuated by SST. Another experiment found that removal of the gastrin-secreting cells by antrectomy and subsequent administration of CCK receptor inhibitor prevents the proliferative effects of BBS/GRP, suggesting that the mucosal growth activities of BBS/GRP are mediated through gastrin and CCK stimulation [159]. Lehy et al. [160] observed that BBS/GRP administration twice daily for 1 week induces the gastrin-cell proliferation and increases the antral gastrin content. Gastric weight, fundic and antral mucosal weight, and the number of parietal cells are also increased in neonatal rats treated with BBS/GRP.
BBS/GRP is also shown to stimulate growth of the small-bowel mucosa in rats fed with liquid elemental diet [84]. When BBS/GRP was administered for 11 days, it not only prevents jejunal mucosal atrophy but also enhances ileal mucosal growth as determined by mucosal weight, RNA, DNA, and protein content. Furthermore, Chu et al. [161] showed that BBS/GRP-mediated stimulation of small intestinal mucosal growth is regulated by factors that are independent of luminal contents and pancreaticobiliary secretion. Several studies also revealed that BBS/GRP stimulates colonic mucosal growth [115] as well as the growth of the pancreas [160]. In addition, BBS/GRP improves integrity of the gut mucosal epithelium after exposure to severe burn injury by decreasing burn-induced gut mucosal atrophy and epithelial cell apoptosis [157,162]. These findings suggest that BBS/GRP plays an important role in intrinsic gastric mucosal defense system against various luminal noxious substances. Qiao et al. [163] recently reported that silencing BBS/GRP receptors suppresses tumor growth and reduces metastatic potential of neuroblastoma in vitro as well as in vivo.
OTHER GI HORMONES
As mentioned earlier, there are more than 50 gut hormones and peptides synthesized and released from the GI tract, of which only small proportions have been vigorously investigated for their potential roles in the regulation of GI mucosal growth. The glucagon-like peptides, GLP-1 and GLP-2, are peptide hormones released from the gut endocrine L-type cells in response to the products of a mixed carbohydrate and fat meal [164]. L-type cells are most abundant in the ileum and colon in human intestine, and these cells are the second most numerous populations of endocrine cells after ECL cells [165]. GLP-1 is shown to increase pancreatic islet mass by stimulating β-cell proliferation; it also promotes differentiation of exocrine cells or immature islet progenitors. On the other hand, GLP-2 stimulates cell proliferation in GI mucosa, leading to an expansion of the GI mucosal epithelium [166]. Mice treated with GLP-2 exhibit elongated villi by enhancing crypt cell proliferation and decreasing enterocyte apoptosis [167]. Furthermore, exogenous GLP-2 enhances mucosal regeneration in pathological conditions, such as colitis and small-bowel enteritis [84].
Vasoactive intestinal peptide (VIP) is a 28-amino acid peptide and also plays a role in regulating gut mucosal growth through a way similar to secretin [168]. Although VIP given alone shows no significant effect on DNA synthesis or total DNA content in the GI mucosa, VIP given together with pentagastrin prevents the gastrin-induced trophic effects on gastric or colonic mucosae [169].
In addition, neurotensin (NT) is produced by the endocrine N-type cells of the jejunum and ileal mucosas, its major functions in the GI tract are to stimulate pancreatic and biliary secretions and to suppress small-bowel gastric motility [170]. There are considerable amount of results showing that NT also promotes growth of the gastric antrum, small bowel, colon, and pancreas [84,170].
Gastric-inhibitory polypeptide (GIP) is a 42-amino acid peptide that is secreted by specific K-type endocrine cells. GIP is also implicated in the regulation of gut mucosal growth [171]. Additional information about the involvement of other gut peptides in controlling growth of gut mucosa and pancreas is available in several review articles [84,149,172,173].
- Role of GI Hormones on Gut Mucosal Growth - Regulation of Gastrointestinal Mucos...Role of GI Hormones on Gut Mucosal Growth - Regulation of Gastrointestinal Mucosal Growth
- Guide for the Care and Use of Laboratory AnimalsGuide for the Care and Use of Laboratory Animals
- Introduction - Screening for Hearing Loss in Adults Ages 50 Years and OlderIntroduction - Screening for Hearing Loss in Adults Ages 50 Years and Older
- Using Preferences - My NCBI HelpUsing Preferences - My NCBI Help
- Human Perceptions and Preferences for Fat-Rich Foods - Fat DetectionHuman Perceptions and Preferences for Fat-Rich Foods - Fat Detection
Your browsing activity is empty.
Activity recording is turned off.
See more...