

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Chronic inflammatory diseases, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and psoriasis and infections, such as periodontal disease and HIV, are associated with an increased risk of cardiovascular disease. Patients with these disorders also have an increase in coronary artery calcium measured by CT and carotid intima media thickness measured by ultrasound. Inflammation and infections induce a variety of alterations in lipid metabolism that may initially dampen inflammation or fight infection, but if chronic could contribute to the increased risk of atherosclerosis. The most common changes are decreases in serum HDL and increases in triglycerides. The increase in serum triglycerides is due to both an increase in hepatic VLDL production and secretion and a decrease in the clearance of triglyceride rich lipoproteins. The mechanisms by which inflammation and infection decrease HDL levels are uncertain. With inflammation there is also a consistent increase in lipoprotein (a) levels due to increased apolipoprotein (a) synthesis. LDL levels are frequently decreased but the prevalence of small dense LDL is increased due to exchange of triglycerides from triglyceride rich lipoproteins to LDL followed by triglyceride hydrolysis. In addition to affecting serum lipid levels, inflammation also adversely effects lipoprotein function. LDL is more easily oxidized as the ability of HDL to prevent the oxidation of LDL is diminished. Moreover, there are a number of steps in the reverse cholesterol transport pathway that are adversely affected during inflammation. The greater the severity of the underlying inflammatory disease, the more consistently these abnormalities in lipids and lipoproteins are observed. Treatment of the underlying disease leading to a reduction in inflammation results in the return of the lipid profile towards normal. The changes in lipids and lipoproteins that occur during inflammation and infection are part of the innate immune response and therefore are likely to play an important role in protecting the host. The standard risk calculators for predicting cardiovascular disease (ACC/AHA, Framingham, SCORE, etc.) underestimate the risk in patients with inflammation. It has been recommended to increase the calculated risk by approximately 50% in patients with severe inflammatory disorders. The treatment of lipid disorders in patients with inflammatory disorders is similar to patients without inflammatory disorders. Of note statins, fibrates, and fish oil have anti-inflammatory properties and have been reported to have beneficial effects on some of these inflammatory disorders. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
A number of chronic inflammatory diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ankylosing spondylitis, Sjögren's syndrome, polymyalgia rheumatica, inflammatory bowel disease, and psoriasis are associated with an increased risk of cardiovascular disease (1-9). For example, in a meta-analysis of twenty-four studies comprising 111,758 patients with 22,927 cardiovascular events it was observed that there was a 50% increased risk of CVD death in patients with RA (10). In some studies patients with RA have a similar risk for a cardiovascular event as patients with diabetes (11). Similarly, women with SLE in the 35- to 44-year age group were over 50 times more likely to have a myocardial infarction than were women of similar age in the Framingham Offspring Study (12). As a final example, a meta-analysis of 14 studies reported that in individuals with severe psoriasis the risk for cardiovascular mortality was 1.37, the risk for myocardial infarction was 3.04, and the risk for stroke was 1.59 times higher than the general population (13). It should be noted that the pathology in psoriasis is localized to the skin but nevertheless even this disorder, by inducing systemic inflammation, is associated with an increased risk of cardiovascular disease.
Further, supporting the link of RA, SLE, and psoriasis with atherosclerosis are studies showing that patients with these disorders have an increase in coronary artery calcium measured by CT and carotid intima media thickness measured by ultrasound (14-20). Finally, even children and adolescents with SLE have an increase in carotid intimal-medial thickness (21). Thus, it is clear that patients with a number of different chronic inflammatory diseases have an increased risk of atherosclerotic cardiovascular complications.
In addition, chronic infections are also associated with an increased risk of atherosclerosis (22-24). Since the development of effective anti-viral agents, it has been widely recognized that a major cause of morbidity and mortality in HIV infected patients is due to cardiovascular disease (25,26). Moreover, numerous studies have demonstrated an association of periodontal infections with an increased risk of atherosclerotic vascular disease (27). Additionally, carotid intima-media thickness is increased in patients with periodontal disease (28-31). The link between various chronic infections, such as HIV, dental infections, Helicobacter pylori, chronic bronchitis, and urinary tract infections with cardiovascular disease is presumably due to the chronic inflammation that accompanies these infections (32). For certain infections such as chlamydia pneumonia and cytomegalovirus it is possible that the association with cardiovascular disease is due to a direct role in the vessel wall.
To definitively link inflammation with cardiovascular disease studies determining the effect of anti-inflammatory drugs on cardiovascular events have been carried out. The Cantos study has provided data supporting a link between inflammation and cardiovascular disease (33). In this trial 10,061 patients with a previous myocardial infarction and a hsCRP level of 2 mg/L or more were randomized to canakinumab, a monoclonal antibody targeting interleukin-1β, or placebo. At 48 months canakinumab did not reduce lipid levels from baseline but did reduce hsCRP levels by approximately 30-40% indicating a decrease in inflammation. Most importantly, canakinumab administration led to a significantly lower rate of recurrent cardiovascular events than placebo. In addition, several randomized trials have demonstrated that colchicine reduces cardiovascular events in patients with chronic cardiovascular disease (34-36). These results support the hypothesis that inflammation increases the risk of cardiovascular events and that reducing inflammation will decrease events. In contrast to the positive trials described above, a trial using methotrexate to inhibit inflammation failed to reduce cardiovascular event (37). However, in this trial methotrexate did not reduce levels of interleukin-1β, interleukin-6, or C-reactive protein raising the possibility that methotrexate did not effectively inhibit inflammation and therefore did not reduce cardiovascular events. Clearly further studies determining the effect of drugs that reduce inflammation on cardiovascular events are required.
The mechanisms by which chronic inflammation and infection increase the risk of atherosclerotic cardiovascular disease are likely multifactorial. As will be discussed below inflammation and infection induce a variety of alterations in lipid and lipoprotein metabolism that could contribute to the increased risk of atherosclerosis.
LIPID AND LIPOPROTEIN ABNORMALITIES IN PATIENTS WITH INFLAMMATORY DISORDERS AND INFECTIONS
Rheumatoid Arthritis
The most consistent abnormality in patients with RA is a decrease in HDL-C and apolipoprotein A-I levels (9,38-41). In particular, small HDL particles are decreased in patients with RA (42). Patients with more severe RA have the greatest reductions in HDL-C levels (38-41,43). There is an inverse correlation of CRP levels with HDL-C levels (i.e., higher CRP levels are associated with lower HDL-C levels). With regards to total cholesterol and LDL-C, there is more variability with many studies showing a decrease, other studies showing no change, and some studies showing an increase in patients with RA (38-41,43). The more severe the RA the greater the likelihood that the LDL-C levels will be decreased. Small dense LDL levels are increased in RA (44,45). Serum triglyceride levels tend to be increased in patients with RA (38-41,43,46). Levels of lipoprotein (a) are characteristically elevated in patients with RA and correlate with CRP levels (47-49).
Systemic Lupus Erythematosus
The changes in serum lipids and lipoproteins seen in patients with SLE are very similar to those observed in patients with RA (50-52). Specifically, there is a decrease in HDL-C levels and an increase in serum triglyceride levels. LDL-C levels are variable and maybe increased, normal, or low but small dense LDL levels tend to be increased. Lipoprotein (a) levels are also increased (53). Similar to RA the more severe the disease state the greater the alterations in serum lipid levels.
Psoriasis
A large number of studies have compared serum lipid levels in controls and patients with psoriasis (54). However, many of these studies included only a small number of subjects and the results have therefore been extremely variable with some studies showing alterations in serum lipid levels in patients with psoriasis and other studies showing no changes. In general, there is a tendency for an increase in serum triglycerides and a decrease in HDL-C levels in patients with psoriasis (55-59). Additionally, a number of studies showed an increase in LDL-C and lipoprotein (a) levels in patients with psoriasis (55,56,58). Small dense LDL levels and oxidized Lp(a) are also increased in psoriasis (46) (60). This variability between studies is most likely due to differences in the severity of the psoriasis with more severe disease demonstrating more robust alterations in lipid levels. The prevalence of other abnormalities that affect lipid metabolism such as obesity and abnormalities in glucose metabolism could also account for the variability in results.
Other Inflammatory Disease
Decreased HDL-C levels have also been observed in patients with inflammatory bowel disease, Sjögren's syndrome, and ankylosing spondylitis (61-64). LDL-C and triglyceride levels varied but LDL-C levels tended to be decreased and triglyceride levels increased.
Periodontal Disease
Differences exist between studies but in general patients with periodontitis tend to have increased LDL-C and triglyceride levels and decreased HDL-C levels (65-69). Additionally, the prevalence of small dense LDL is increased in patients with periodontitis (68,70). The severity of the periodontitis correlated with the changes in the in the lipid profile with patients with increased periodontal disease having higher triglyceride levels, lower HDL-C levels, and smaller LDL particle size (71). Moreover, treatment of periodontitis improved the dyslipidemia, with the HDL-C levels increasing and the LDL-C levels decreasing (68,72,73).
Acute Infections
Patients with a variety of different infections (gram positive bacterial, gram negative bacterial, viral, tuberculosis, parasitic) have similar alterations in plasma lipid levels. Specifically, total cholesterol, LDL-C, and HDL-C levels are decreased while plasma triglyceride levels are elevated or inappropriately normal for the poor nutritional status (32,74-81). As expected apolipoprotein A-I, A-II, and B levels are reduced (74,79,80). While LDL-C levels were decreased, the concentration of small dense LDL has been found to be increased during infections (82-84).That plasma cholesterol levels decrease during infection has been known for many years as it was described by Denis in 1919 in the Journal of Biological Chemistry (JBC 29: 93, 1919). The alterations in lipids correlate with the severity of the underlying infection i.e., the more severe the infection the more severe the alterations in lipid and lipoprotein levels (85,86). The decreases in plasma cholesterol levels can be quite profound and a case report described HDL-C levels < 10mg/dl and LDL-C levels < 3mg/dl in sepsis (87).
Of note studies have demonstrated that the degree of reduction in total cholesterol, HDL-C, and apolipoprotein A-I are predictive of mortality in patients with severe sepsis (81,88-92). Moreover, epidemiologic studies have suggested that low cholesterol, LDL-C, and HDL levels increase the chance of developing an infection (93-96). Additionally, a genetic approach, which reduces the risk of confounding variables, has suggested a causal relationship between low HDL-C levels and an increased risk of infections (97,98). During recovery from the infection plasma lipid and lipoprotein abnormalities return towards normal. The changes in lipid and lipoproteins that occur during infection can be experimentally reproduced in humans and animals by the administration of endotoxin and lipoteichoic acid (32,99).
Summary
Thus, in these different inflammatory disorders and infectious diseases, the alterations in plasma lipid and lipoprotein levels are very similar with decreases in plasma HDL being consistently observed. Also of note is the consistent increase in small dense LDL and Lp(a) level (the increase in Lp(a) occurs in inflammatory diseases but not infections) (32,100). There is also a tendency for plasma triglyceride levels to be elevated and LDL-C levels decreased. The greater the severity of the underlying disease the more consistently these abnormalities in lipids are observed. Additionally, treatment of the underlying disease leading to a reduction in inflammation results in a return of the lipid profile towards normal. This is best illustrated in periodontal disease where intensive dental hygiene can reverse the abnormalities in the lipid profile (72,73).
Table 1.
Effect of Inflammation and Infection on Lipid and Lipoprotein Levels
| Triglycerides- Tend to be increased |
| HDL-C- Decreased |
| LDL-C- Variable but with more severe inflammation or infection they are decreased |
| Small dense LDL- Increased |
| Lp(a)- Increased with inflammation; may decrease with certain infections |
EFFECT OF ANTI-INFLAMMATORY DRUGS ON LIPID LEVELS
Treatments that reduce inflammation will return the lipid profile towards normal resulting in an increase in plasm HDL levels and a decrease in triglyceride levels. If LDL levels were reduced at baseline, treatment that reduces inflammation will also result in an increase in LDL levels (i.e., a return towards “normal” levels) (101-103). Many of the drugs used for the treatment of RA, SLE, and psoriasis decrease inflammation and have been shown to increase both HDL and LDL levels (9,101,102,104). The increase in HDL tends to be more robust. In a few instances, drugs used to treat inflammatory disorders have effects on lipid metabolism that are independent of the reduction in inflammation. For example, high dose glucocorticoid treatment results in an increase in serum triglyceride and LDL levels due to the increased production and secretion of VLDL by the liver (105-107) and hydroxychloroquine has been reported to lower total cholesterol, LDL, and triglycerides in patients with RA and SLE (108-110).
PATHOPHYSIOLOGY OF THE DYSLIPIDEMIA OF INFLAMMATION AND INFECTION
Inflammation and infections increase the production of a variety of cytokines, including TNF, IL-1, and IL-6, which have been shown to alter lipid metabolism (32). Many of the changes in plasma lipids and lipoproteins that are seen during chronic inflammation and infections are also observed following the acute administration of cytokines (32).
Increased Triglyceride Levels
Multiple cytokines increase serum triglyceride and VLDL levels (TNF, IL-1, IL-2, IL-6, etc.) (32). Following a single administration of a cytokine or LPS (a model of gram-negative infections), which stimulates cytokine production, an increase in serum triglyceride and VLDL levels can be seen within 2 hours and this effect is sustained for at least 24 hours. The increase in serum triglycerides is due to both an increase in hepatic VLDL production and secretion and a decrease in the clearance of triglyceride rich lipoproteins (figure 1) (32). The increase in VLDL production and secretion is a result of increased hepatic fatty acid synthesis, an increase in adipose tissue lipolysis with the increased transport of fatty acids to the liver, and a decrease in fatty acid oxidation in the liver. Together these changes provide an increased supply of fatty acids in the liver that stimulate an increase in hepatic triglyceride synthesis (32). The increased availability of triglycerides leads to the increased formation and secretion of VLDL. The decrease in the clearance of triglyceride rich lipoproteins is due to a decrease in lipoprotein lipase, the key enzyme that metabolizes triglycerides in the circulation (32). A variety of cytokines have been shown to decrease the synthesis of lipoprotein lipase in adipose and muscle tissue (32). Studies have also shown that inflammation also increases angiopoietin like protein 4, an inhibitor of lipoprotein lipase activity, which would further block the metabolism of triglyceride rich lipoproteins (111). In SLE, antibodies to lipoprotein lipase have been reported and are associated with increased triglyceride levels (112,113).
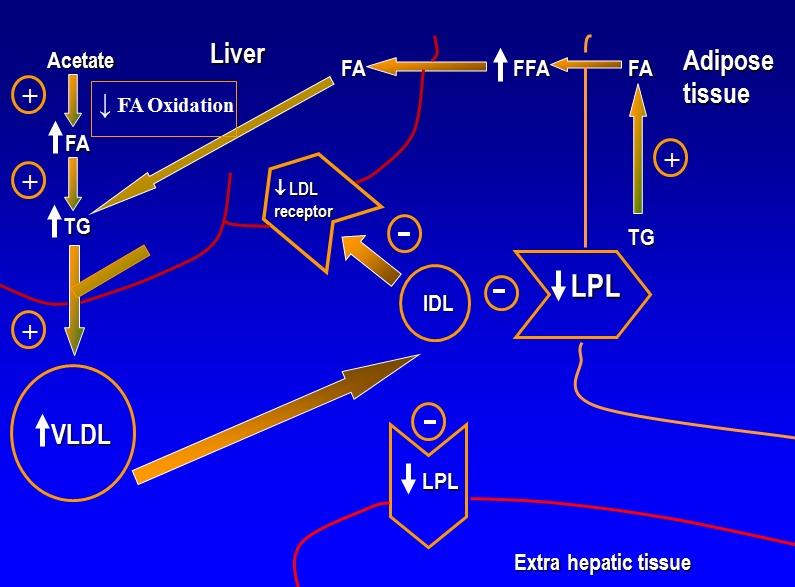
Figure 1.
Pathogenesis of Hypertriglyceridemia
Production of Small Dense LDL
The elevation in triglyceride rich lipoproteins in turn has effects on other lipoproteins (32). Specifically, cholesterol ester transfer protein (CETP) mediates the exchange of triglycerides from triglyceride rich VLDL and chylomicrons to LDL. The increase in triglyceride rich lipoproteins per se leads to an increase in CETP mediated exchange, increasing the triglyceride content of LDL. The triglyceride on LDL is then hydrolyzed by hepatic lipase leading to the increased production of small dense LDL.
Decreased HDL Levels
In addition to a decrease in HDL, inflammation can also lead to structural changes in this lipoprotein (32). During inflammation HDL particles tend to be larger with a decrease in cholesterol ester and an increase in free cholesterol, triglycerides, and free fatty acids. Furthermore, there are marked changes in HDL associated proteins and the enzymes and transfer proteins involved in HDL metabolism and function (figure 2 and 3).

Figure 2.
Changes in HDL Protein Composition During Inflammation

Figure 3.
Changes in Enzymes and Transfer Proteins During Inflammation
The precise mechanism by which inflammation and infection decrease HDL levels is uncertain and is likely to involve multiple mechanisms (32). Decreases in apolipoprotein A-I synthesis in the liver occur during inflammation and would result in the decreased formation of HDL. However, in acute infection and inflammation HDL decreases faster than would be predicted from decreased synthesis of apolipoprotein A-I. Increased serum amyloid A (SAA) production by the liver and other tissues occurs during inflammation and infection and the SAA binds to HDL displacing apolipoprotein A-I, which can accelerate the clearance of HDL. However, the overexpression in SAA in the absence of the acute phase response does not result in a decrease in HDL levels (114). Inflammation results in a decrease in LCAT leading to decreased cholesterol ester formation, which would prevent the formation of normal HDL, leading to decreased cholesterol carried in HDL. Elevations in triglyceride rich lipoproteins that accompany inflammation and infection can lead to the enrichment of HDL with triglycerides that can accelerate the clearance of HDL. Finally, cytokine induced increases in enzymes such as secretory phospholipase A2 (sPLA2) and endothelial cell lipase, which metabolize key constituents of HDL, could alter the stability and metabolism of HDL. Given the complexity of HDL metabolism it is not surprising that multiple pathways could be affected by inflammation, which together may account for the decrease in HDL levels.
Increased Lipoprotein (a)
The mechanism accounting for the increase in lipoprotein (a) (Lp(a)) during inflammation is likely due to increased apolipoprotein (a) synthesis, as apolipoprotein (a) is a positive acute phase protein whose expression is up-regulated during inflammation (32,115). The apolipoprotein (a) gene contains several IL-6 responsive elements that enhance transcription (116). Tocilizumab an antibody against IL-6, that is used to treat RA, has been shown to decrease Lp(a) levels (117) .
FUNCTIONAL CHANGES IN LIPOPROTEINS THAT INCREASE THE RISK OF ATHEROSCLEROSIS
LDL
While the levels of LDL do not consistently increase and may even decrease with inflammation and infection, many studies have indicated that inflammation and infection are associated with small dense LDL (32). These small dense LDL particles are believed to be more pro-atherogenic for a number of reasons (118). Small dense LDL particles have a decreased affinity for the LDL receptor resulting in a prolonged period of time in the circulation. Additionally, they more easily enter the arterial wall and bind more avidly to intra-arterial proteoglycans, which traps them in the arterial wall. Finally, small dense LDL particles are more susceptible to oxidation, which could result in an enhanced uptake by macrophages (119).
Several markers of lipid peroxidation, including conjugated dienes, thiobarbituric acid-reactive substances, malondialdehyde, and lipid hydroperoxides are increased in serum and/or circulating LDL during inflammation and infection (32,71,120-123). Moreover, LDL isolated from LPS-treated animals is more susceptible to oxidation in vitro (32). Oxidized LDL is taken up very efficiently by macrophages and is thought to play a major role in foam cell formation in the arterial wall (124). Additionally, antibodies to oxidized LDL are present in patients with SLE and could facilitate the uptake of an antibody LDL complex via the Fc-receptor in macrophages (120). Finally, studies have shown that LDL isolated from patients with periodontal disease leads to enhanced uptake of cholesterol esters by macrophages (71)
HDL
In addition to a decrease in serum HDL, inflammation and infection affects the anti-atherogenic properties of HDL (32,125,126). Reverse cholesterol transport plays a key role in preventing cholesterol accumulation in macrophages thereby reducing atherosclerosis. Many steps in the reverse cholesterol transport pathway are adversely affected during inflammation and infection (figure 4 and 5) (43,127). First, cytokines induced by inflammation and infection decrease the production of Apo A-I, the main protein constituent of HDL. Second, pro-inflammatory cytokines decrease the expression of ABCA1, ABCG1, SR-B1, and apolipoprotein E in macrophages, which will lead to a decrease in the efflux of phospholipids and cholesterol from the macrophage to HDL. Third, the structurally altered HDL formed during inflammation is a poor acceptor of cellular cholesterol and in fact may actually deliver cholesterol to the macrophage (43,61,127-134). HDL isolated from patients with RA, SLE, inflammatory bowel disease, psoriasis, ankylosing spondylitis, periodontal disease, and acute sepsis are poor facilitators of cholesterol efflux (61,128-133,135). Similarly, the experimental administration of endotoxin to humans also results in the formation of HDL that is a poor facilitator of the efflux of cholesterol from macrophages (136). Of note treatments that reduce inflammation in patients with RA, psoriasis, or periodontitis can restore towards normal the ability of HDL to remove cholesterol from cells (133,137-139). Fourth, pro-inflammatory cytokines decrease the production and activity of LCAT, which will limit the conversion of cholesterol to cholesteryl esters in HDL. This step is required for the formation of a normal spherical HDL particle and facilitates the ability of HDL to transport cholesterol. Fifth, pro-inflammatory cytokines decrease CETP levels, which will decrease the movement of cholesterol from HDL to Apo B containing lipoproteins, an important step in the delivery of cholesterol to the liver. Sixth, pro-inflammatory cytokines decrease the expression of SR-B1 in the liver. SR-B1 plays a key role in the uptake of cholesterol from HDL particles into hepatocytes. Finally, inflammation and infection decrease both the conversion of cholesterol to bile acids and the secretion of cholesterol into the bile, the two mechanisms by which cholesterol is disposed of by the liver.
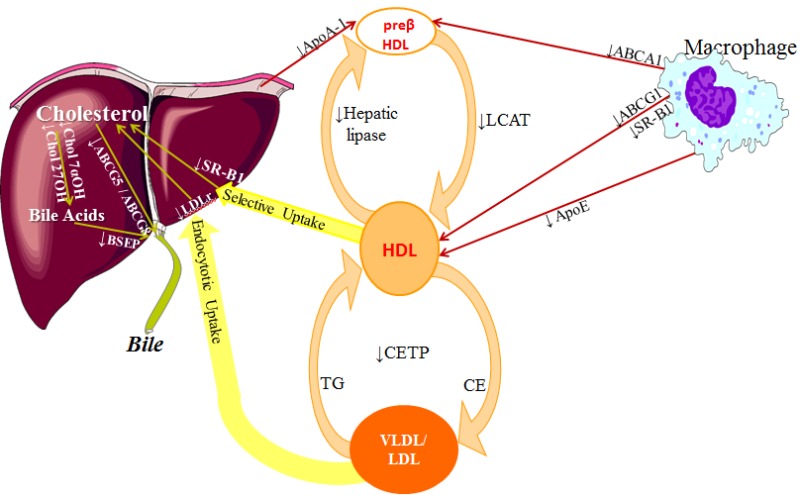

Figure 5.
Effect of Inflammation on the Factors Involved in Reverse Cholesterol Transport (from reference (127))
Another important function of HDL is to prevent the oxidation of LDL. Oxidized LDL is more easily taken up by macrophages and is pro-atherogenic (124). Paraoxonase is an enzyme that is associated with HDL and plays a key role in preventing the oxidation of LDL. Inflammation and infection decrease the expression of paraoxonase 1 in the liver resulting in a decrease in circulating paraoxonase activity (32). Plasma paraoxonase levels are decreased in patients with RA, SLE, psoriasis, and infections (140-148) Studies have shown that HDL isolated from patients with RA and SLE have a diminished ability to protect LDL from oxidation and in fact may facilitate LDL oxidation (125). Moreover, in patients with RA, reducing inflammation and disease activity with methotrexate treatment restored HDL function towards normal (149). Additionally, treatment with atorvastatin 80mg improved the function of HDL in patients with RA (150).
Thus, it should be recognized that in patients with inflammatory disorders and infections the absolute levels of lipids and lipoproteins may not be the only factor increasing the risk of atherosclerosis (32,54,121,125-127). Rather functional changes in LDL and HDL maybe pro-atherogenic and thereby contribute to the increased risk of atherosclerosis in inflammatory disorders and infections. Additionally, the increase in lipoprotein (a) may also play a role.
Table 2.
Pro-Atherogenic Changes During Inflammation
| Increased triglycerides |
| Decreased HDL |
| Increased small dense LDL |
| Increased Lp(a) |
| Oxidized LDL |
| Dysfunctional HDL |
BENEFICIAL EFFECTS OF LIPIDS DURING INFECTIONS AND INFLAMMATION
The changes in lipids and lipoproteins that occur during inflammation and infection are part of the innate immune response and therefore are likely to play an important role in protecting from the detrimental effects of infection and inflammatory stimuli (32,151-153). Some of the potential beneficial effects are listed in Table 3. Thus, the changes in lipid and lipoprotein metabolism that occur during inflammation may initially be protective but if chronic can increase the risk of atherosclerosis.
Table 3.
Beneficial Effects of Lipoproteins
| Redistribution of nutrients to immune cells that are important in host defense |
| Lipoproteins bind endotoxin, lipoteichoic acid, viruses and other biological agents and prevent their toxic effects |
| Lipoproteins bind urate crystals |
| Lipoproteins bind and target parasites for destruction |
| Apolipoproteins neutralize viruses |
| Apolipoproteins lyse parasites |
LIPID MANAGEMENT IN A PATIENT WITH AN INFLAMMATORY DISEASE
Deciding When to Treat
As noted earlier, patients with inflammatory disorders are at an increased risk for atherosclerosis and this is not totally accounted for by standard lipid profile measurements and other risk factors (1-3,9). Some authors have advocated considering inflammatory disorders as a cardiovascular risk equivalent similar to diabetes; risk calculators (ACC/AHA, Framingham, and SCORE) commonly used for deciding on lipid lowering therapy do not take into account this increased risk in patients with inflammatory disorders (3,154,155). It should be noted that the QRISK calculator (http://qrisk.org/) does factor in the presence of RA when calculating risk (156). Not surprisingly, the standard risk calculators for predicting cardiovascular disease (ACC/AHA and Framingham) underestimate the risk in this population (157-162). Even the Reynolds Risk Calculator (http://www.reynoldsriskscore.org/Default.aspx), which uses measurements of hsCRP levels, a marker of inflammation, underestimates the risk of cardiovascular events in patients with inflammatory disorders (157-161). Thus, using these calculators will underestimate cardiovascular risk in patients with inflammatory disorders. However, in both the 2018 American College of Cardiology/American Heart Association and 2019 European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) guideline recommendations, the presence of inflammatory disease is included as a risk factor, which can influence decisions on whether to initiate treatment (163,164).
A reasonable approach is to use the standard approach and calculators but increase the calculated risk by approximately 50% in patients with severe inflammatory disorders. For example, if a patient with severe RA has a 5% ten-year risk and 40% lifetime risk one might increase the ten-year risk to 7.5% and lifetime risk to 60%. This approach has been recommended by an expert committee who advocated introducing a 1.5 multiplication factor (i.e., 50% increase) in patients with RA (9). Alternatively, one could carry out imaging studies such as obtaining a coronary artery calcium score to better define risk. Whatever the approach taken, it is crucial to recognize that patients with inflammatory diseases have an increased risk of cardiovascular disease and therefore one needs to be more aggressive.
Guidelines from the American College of Cardiology (ACC)/American Heart Association (AHA) and European Society of Cardiology (ESC)/European Atherosclerosis Society (EAS) are briefly summarized in table 4, 5,and 6 (163,164) and are discussed in detail in the Endotext chapter “Guidelines for the Management of High Blood Cholesterol” (165).
Table 4.
ACC/AHA Guidelines
| In patients with clinical ASCVD initiate high intensity statin therapy or maximally tolerated statin therapy. High intensity statin therapy is atorvastatin 40-80mg per day or rosuvastatin 20-40mg per day. |
| In very high-risk ASCVD, use an LDL-C > 70 mg/dL (1.8 mmol/L) to consider addition of non-statins (ezetimibe or PCSK9 inhibitors). Very high-risk includes a history of multiple major ASCVD events or 1 major ASCVD event and multiple high-risk conditions. |
| In patients with LDL-C ≥190 mg/dL [≥4.9 mmol/L]) begin high-intensity statin therapy. If the LDL-C level remains ≥100 mg/dL (≥2.6 mmol/L), adding ezetimibe is reasonable. |
| In patients with diabetes aged 40-75 years with an LDL > 70mg/dL begin moderate intensity statin therapy. For patients > 50 year consider high intensity statin to achieve a 50% reduction in LDL-C. |
| In adults 40 to 75 years of age without diabetes mellitus and with LDL-C levels ≥70 mg/dL (≥1.8 mmol/L) start a moderate-intensity statin if the 10-year ASCVD risk is ≥7.5%. Moderate intensity therapy is atorvastatin 10-20mg, rosuvastatin 5-10mg, simvastatin 20-40mg, pravastatin 40mg. |
Table 5.
ESC/EAS Cardiovascular Risk Categories
| Very High-Risk |
| ASCVD, either clinical or unequivocal on imaging DM with target organ damage or at least three major risk factors or T1DM of long duration (>20 years) Severe CKD (eGFR <30 mL/min/1.73 m2) A calculated SCORE >10% for 10-year risk of fatal CVD. FH with ASCVD or with another major risk factor |
| High Risk |
| Markedly elevated single risk factors, in particular total cholesterol >8 mmol/L (>310mg/dL), LDL-C >4.9 mmol/L (>190 mg/dL), or BP >180/110 mmHg. Patients with FH without other major risk factors. Patients with DM without target organ damage, a with DM duration >_10 years or another additional risk factor. Moderate CKD (eGFR 30-59 mL/min/1.73 m2). A calculated SCORE >5% and <10% for 10-year risk of fatal CVD. |
| Moderate Risk |
| Young patients (T1DM <35 years; T2DM <50 years) with DM duration <10 years, without other risk factors. Calculated SCORE >1% and <5% for 10-year risk of fatal CVD. |
| Low Risk |
| Calculated SCORE <1% for 10-year risk of fatal CVD |
Table 6.
ESC/EAS LDL Cholesterol Goals
| Very High Risk | LDL-C reduction of >50% from baseline and an LDL-C goal of <1.4 mmol/L (<55 mg/dL) is recommended |
| High Risk | LDL-C reduction of >50% from baseline and an LDL-C goal of <1.8 mmol/L (<70 mg/dL) is recommended |
| Moderate Risk | LDL-C goal of <2.6 mmol/L (<100 mg/dL) should be considered |
| Low Risk | LDL-C goal <3.0 mmol/L (<116 mg/dL) may be considered. |
Treatment Approach
As in all patients with lipid abnormalities the initial approach is lifestyle changes. Dietary recommendations are not unique in patients with inflammatory disorders. Exercise is recommended but depending upon the clinical situation the ability of patients with certain inflammatory disorders to participate in an exercise regimen may be limited. Exercise programs will need to be tailored for each patient’s capabilities. Treatment of the underlying disease to decrease inflammation is likely to be beneficial (9,166). Studies have shown that increased disease activity is associated with a greater risk of cardiovascular disease while lower disease activity is associated with a lower risk (9,167-173). Moreover, treatments that reduce disease activity can decrease cardiovascular risk (9,166).
Drug Therapy
This section on drug therapy will focus solely on the studies that are unique to patients with inflammatory diseases. Detailed information on the use of these drugs can be found in the Endotext chapters on cholesterol lowering drugs and triglyceride lowering drugs (174,175).
STATIN THERAPY
As expected, studies have demonstrated that statins lower LDL-C levels in patients with inflammatory disorders to a similar degree as patients without inflammatory disorders. For example, in a randomized trial in 116 patients with RA with a mean LDL-C level of 125mg/dl, the effect of atorvastatin 40mg was compared to placebo (176). Atorvastatin reduced LDL-C by 54mgdl vs. 3mg/dl in the placebo group (176). Similarly in the IDEAL trial there was a small subgroup of patients with RA (177). The IDEAL trial compared the ability of atorvastatin 80mg vs. simvastatin 20-40mg to reduce cardiovascular events. The lowering of LDL-C with either simvastatin or atorvastatin was similar in the patients with and without RA (177). Finally, a combined analysis of the IDEAL, Treat to New Target (TNT), and CARDS trials reported that the decrease in LDL-C levels with statin therapy was similar in patients with or without psoriasis (178). Studies have shown similar reductions in LDL-C levels with statin therapy in patients with SLE (179-181). The effects of statin treatment on other lipid parameters were also similar in patients with and without inflammatory diseases. Thus, as expected statins improve the lipid profile in patients with inflammatory disorders. In some studies, the incidence of statin associated side effects have been increased in the patients with inflammatory disorders. Specifically, in the IDEAL trial RA patients reported myalgia more frequently than patients without RA (10.4% and 7.7% in RA patients vs 1.1% and 2.2% in non-RA patients receiving simvastatin and atorvastatin respectively) (177). Note that this does not necessarily indicate that statins induce myalgias more frequently in patients with RA as there was not a placebo group in the IDEAL trial. Rather it is likely that patients with RA have an increased prevalence of myalgias.
A key question is whether statin therapy will reduce cardiovascular events in patients with inflammatory diseases. A number of studies have looked at surrogate markers for events such as changes in carotid intima-media thickness or changes in cardiac calcium scores in patients treated with statins. The results have varied with some studies showing benefits and other studies showing no effects. Rollefstad et al measured changes in carotid plaque size in 86 patients with inflammatory joint disease treated with rosuvastatin for 18 months (182). The LDL-C levels decreased from 155mg/dl to 66mg/dl and plaque height was significantly reduced (182). Similarly, Mok et al treated 72 patients with SLE with rosuvastatin 10mg or placebo for 12 months and reported that carotid intima-media thickness appeared to decrease (179). Moreover, Plazak et al treated 60 patients with SLE with atorvastatin 40mg or placebo for 1 year and measured changes in coronary calcium score (180). They observed an increase in coronary calcium in the placebo group while there was no change in the patients treated with statin therapy (180). In contrast, Petri et al treated 200 patients with SLE with atorvastatin 40mg or placebo for 2 years and measured both carotid intima-media thickness and coronary calcium score (183). In this study no beneficial effects of statin therapy were observed (183). Similarly, Schanberg et al treated 221 children with SLE with atorvastatin 10-20mg or placebo for 36 months and did not observe a beneficial effect of statin treatment on carotid intima-media thickness (181). Additionally, Tam et al also failed to find a decrease in carotid intima-media thickness with rosuvastatin treatment in patients with RA (184). Thus, the effect of statin therapy in patients with inflammatory disorders on these surrogate markers of atherosclerosis is uncertain.
There are no large randomized controlled trials evaluating the impact of statin therapy on cardiovascular disease outcomes in patients with inflammatory disease. A subgroup analysis of a small number of patients with SLE in the ALERT study has been reported (185). The ALERT study was a randomized placebo-controlled trial examining the effect of fluvastatin 40-80mg on cardiovascular events after kidney transplantation. In this trial fluvastatin therapy reduced the risk of cardiovascular events by 74% in the patients with SLE (185). Additionally, a post hoc analysis of patients with inflammatory arthritis in the IDEAL and TNT trial has been reported (186). The IDEAL trial compared atorvastatin 80mg vs simvastatin 20-40mg and the TNT compared atorvastatin 80mg vs. atorvastatin 10mg. In these trials, statin therapy resulted in a decrease in lipid levels in the patients with inflammatory arthritis to a similar degree as patients without inflammatory arthritis (186). Moreover, there was an approximate 20% reduction in the risk of cardiovascular events in patients treated with atorvastatin 80mg compared to moderate dose statin therapy in patients with and without inflammatory arthritis (186). Similarly, a post hoc analysis of the IDEAL and TNT trials reported a similar reduction in cardiovascular events with high dose statin therapy compared to low dose statin therapy in patients with psoriasis (178). A trial that focused solely on patients with RA was initiated but stopped early due to a lower than expected event rate (187). In this trial 3,002 patients with RA were randomized to atorvastatin 40mg/day vs. placebo for a median of 2.51 years. As expected, the reduction in LDL-C levels was significantly greater in the atorvastatin group compared to placebo (-30mg/dL, p<0.001). There was a 34% risk reduction for major cardiovascular events in the atorvastatin group compared to placebo that was not statistically significant due to the small number of events. Of note, the decrease in events was actually greater than expected based on the Cholesterol Treatment Trialists’ Collaboration meta-analysis of the effect of statins in other populations (42% decrease per 39mg/dL in this trial whereas in the large collaboration meta-analysis there was a 21% decrease per 39mg/dL). The number and type of adverse events were similar in the atorvastatin and placebo groups. Taken together these results strongly suggest that patients with inflammatory diseases will have a reduction in cardiovascular events with statin therapy.
It is well recognized that statins have anti-inflammatory properties and studies have consistently demonstrated a decrease in CRP levels in patients treated with statins (175). Two meta-analyses have explored the effect of statin therapy on disease activity in patients with RA. A meta-analysis by Ly et al included 15 studies with 992 patients and reported that statin therapy decreased erythrocyte sedimentation rate, CRP, tender joint count, swollen joint count, and morning stiffness (188). Similarly, a meta-analysis by Xing et al included 13 studies with 737 patients (189). They reported that statin therapy decreased erythrocyte sedimentation rate, CRP, tender joint count, and swollen joint count (189). Additionally, the disease activity score 28 (DAS28), which focuses on joint pathology, decreased significantly in the patients treated with statin therapy and the patients with the most active disease benefited the most (189,190).
In contrast to the beneficial effects seen in patients with RA, in randomized placebo controlled trials in patients with SLE studies by Plazak et al and Petri et al failed to show a decrease in disease activity with statin therapy (180,183). In psoriasis treatment with statins has produced mixed results with some studies showing a decrease in skin abnormalities and others showing no significant effect or even an increase in disease activity (191). A meta-analysis of 5 randomized trials with 223 patients found that statins may improve psoriasis, particularly in patients with severe disease (192). Finally, treatment with statins has been shown to improve periodontal disease and reduce inflammation (193-195). Thus, statins can decrease the clinical manifestations of RA, periodontitis, and perhaps psoriasis but has no effect on the clinical manifestations of SLE. These differences could be due to the relative severity of the inflammatory response and/or the specific pathways that induce inflammation in these different disorders.
The effect of statins on outcomes in patients with sepsis has been extensively studied. Numerous observational studies have shown that patients treated with statins have a marked reduction in morbidity and mortality (196,197). For example, in a meta-analysis by Wan et al of 27 observational studies with 337,648 patients, statins were associated with a relative mortality risk of 0.65 (CI 0.57-0.75) (197). However, in randomized placebo controlled clinical trials statin administration has not been shown to reduce mortality or improve outcomes (196-198). For example in a meta-analysis by Wan et al of 5 randomized controlled trials with 867 patients the relative risk was 0.98 (197). Similarly, a meta-analysis by Pertzov et al of fourteen randomized trials evaluating 2628 patients also did not observe any benefits of statin therapy in patients with sepsis (199). Additionally, a recent study examining the effect of rosuvastatin on sepsis associated acute respiratory distress also failed to demonstrate a benefit of statin therapy (200). Finally, meta-analyses of observational studies have found that statins in patients with COVID-19 infections are beneficial (201,202) but a randomized trial failed to demonstrate that statin treatment was beneficial (203). Thus, while observational data suggested that statins may be beneficial the more rigorous randomized placebo-controlled trials have not provided evidence of benefit.
FIBRATE THERAPY
Fibrates, gemfibrozil and fenofibrate, are used to lower triglycerides and raise HDL-C levels. However, fibrates, by activating PPAR alpha, are well known to have anti-inflammatory effects. Several studies have shown that fibrate therapy improves the clinical manifestations in patients with RA. For example, Shirinsky et al treated 27 patients with RA with fenofibrate and reported a significant reduction in disease activity score (DAS28) (204). A recent review described 4 randomized trials and 2 observation trials of fibrates in patients with RA and in general these studies showed that fibrate therapy decreased disease activity in patients with RA (205). The authors are not aware of clinical trials of fibrate therapy in patients with sepsis, psoriasis, SLE, and periodontal disease. Thus, there is a suggestion that the anti-inflammatory properties of fibrates may beneficially impact disease activity, but clearly further studies are required.
BILE ACID SEQUESTRANT THERAPY
Bile acid binders are used to lower LDL-C levels. While there are no studies of the effect of bile acid binders in patients with either RA, SLE, or periodontal disease, there are two studies in patients with psoriasis. Both Roe and Skinner et al reported that the treatment of patients with psoriasis with bile acid binders improved the skin condition (206,207). The mechanism for this beneficial effect is unknown.
EZETIMIBE THERAPY
Ezetimibe is used to lower LDL-C levels. There is a single six-week trial in 20 patients with RA that demonstrated that ezetimibe treatment decreased total cholesterol, LDL-C, and CRP levels (208). Moreover, ezetimibe treatment reduced disease activity (208). The mechanism for this beneficial effect is unclear.
FISH OIL THERAPY
Fish oil (omega-3-fatty acids) is widely used to reduce serum triglyceride levels and is recognized to have anti-inflammatory properties. There are numerous studies examining the effect of fish oil therapy on inflammatory diseases. A meta-analysis of 17 randomized controlled trials by Goldberg and Katz of the effect of omega-3-fatty acids in patients with RA reported that treatment with omega-3-fatty acids reduced joint pain intensity, morning stiffness, number of painful and/or tender joints, and the use of non-steroidal anti-inflammatory medications (209). Similarly, a meta-analyses by Lee et al and Gioxari et al also demonstrated that fish oil had beneficial effects in patients with RA (210,211). In psoriasis, a recent review of 15 trials reported that overall, there was a moderate benefit of fish oil supplements with 12 trials showing clinical benefit and 3 trials showing no benefit (212). In contrast, Gamret et al evaluated fish oil treatment in patients with psoriasis in 20 studies (12 randomized controlled trials, 1 open-label nonrandomized controlled trial, and 7 uncontrolled studies) (213). They reported that most of the randomized controlled trials showed no significant improvement in psoriasis, whereas most of the uncontrolled studies showed benefit when fish oil was used daily. In a meta-analysis of eighteen randomized controlled trials involving 927 study participants reached the conclusion that fish oil as monotherapy for psoriasis had not affect but when combined with conventional treatments appeared to be beneficial (214). In SLE four randomized trials have demonstrated clinical benefit with fish oil therapy, while three trials failed to show disease improvement (215-221). Finally, there are data suggesting that treatment with fish oil reduces periodontal disease (222-224). A major limitation of the studies in patients with periodontal disease is that in these trials the experimental group treated with fish oil also was simultaneously treated with aspirin making it difficult to be sure that the beneficial effects were solely due to fish oil supplementation (222,223). A meta-analysis of 20 randomized trials involving 1514 patients with sepsis reported that parenteral or enteral omega-3 fatty acid supplementation was associated with a decrease in mortality and length of stay in the intensive care unit (225). Taken together these studies indicate that in addition to lowering serum triglyceride levels, fish oil therapy may have beneficial effects on the underlying inflammatory disorder in some instances.
NIACIN THERAPY
Niacin is used to lower LDL-C levels and triglycerides and raise HDL-C levels. The authors are not aware of clinical trials of niacin in patients with RA, SLE, psoriasis, or periodontal disease.
PCSK9 INHIBITORS
PCSK9 inhibitors are used to lower LDL-C level. In addition, PCSK9 inhibitors also lower Lp(a) levels. The authors are not aware of clinical trials of PCSK9 inhibitors in patients with RA, SLE, psoriasis, or periodontal disease.
BEMPEDOIC ACID
Bempedoic acid is used to lower LDL-C levels. The authors are not aware of clinical trials of bempedoic acid in patients with inflammatory diseases or infections.
Treatment Strategy
The first priority in treating lipid disorders is to lower the LDL-C levels to goal, unless triglycerides are markedly elevated (> 500-1000mg/dl), which increases the risk of pancreatitis. LDL-C is the first priority because the database linking lowering LDL-C with reducing cardiovascular disease is extremely strong and we now have the ability to markedly decrease LDL-C levels in the vast majority of patients. Dietary therapy is the initial step but, in many patients, will not be sufficient to achieve the LDL-C goals. If patients are willing and able to make major changes in their diet it is possible to achieve remarkable reductions in LDL-C levels but this seldom occurs in clinical practice (for details see the Endotext chapter on the effect of lifestyle changes on lipids and lipoproteins) (226).
Statins are the first-choice drugs to lower LDL-C levels and many patients with inflammatory disorders will require statin therapy. Statins are available as generic drugs and are relatively inexpensive. The choice of statin will depend on the magnitude of LDL-C lowering required and whether other drugs that the patient is taking might alter statin metabolism thereby increasing the risk of statin toxicity. For example, cyclosporine affects the metabolism of many of the statins and in patients taking cyclosporine fluvastatin appears to be the safest statin (227).
If a patient is unable to tolerate statins or statins as monotherapy are not sufficient to lower LDL-C to goal the second-choice drug is either ezetimibe or a PCSK9 inhibitor. Ezetimibe is a generic drug and relatively inexpensive and can be added to any statin. PCSK9 inhibitors can also be added to any statin and are the drugs of choice if a large decrease in LDL-C is required to reach goal (PCSK9 inhibitors will lower LDL-C levels by 50-60% when added to a statin, whereas ezetimibe will only lower LDL-C by approximately 20%). Bile acid sequestrants are an alternative particularly if a reduction in A1c level is also needed. Bempedoic acid also lowers LDL-C by approximately 20% and is another alternative. Ezetimibe, PCSK9 inhibitors, bempedoic acid, and bile acid sequestrants additively lower LDL-C levels when used in combination with a statin, because these drugs increase hepatic LDL receptor levels by different mechanisms, thereby resulting in a reduction in serum LDL-C levels. Niacin and the fibrates also lower LDL-C levels but are not usually employed to lower LDL-C levels
The second priority should be non-HDL-C (non-HDL-C = total cholesterol – HDL-C), which is particularly important in patients with elevated triglyceride levels (>150mg/dl). Non-HDL-C is a measure of all the pro-atherogenic apolipoprotein B containing particles. Numerous studies have shown that non-HDL-C is a strong risk factor for the development of cardiovascular disease. The non-HDL-C goals are 30mg/dl greater than the LDL-C goals. For example, if the LDL goal is <100mg/dl then the non-HDL-C goal would be <130mg/dl. Drugs that reduce either LDL-C or triglyceride levels will reduce non-HDL-C levels. If LDL-C is only slightly below goal increasing drug dose or adding drugs to further lower LDL-C is a reasonable approach. If the LDL-C is significantly below goal lowering TG levels is reasonable.
The third priority in treating lipid disorders is to decrease triglyceride levels. Initial therapy should focus on lifestyle changes including a decrease in simple sugars and ethanol intake and initiating and exercise program. Fibrates, niacin, statins, and omega-3-fatty acids all reduce serum triglyceride levels. Typically, one will target triglyceride levels when one is trying to lower non-HDL-C levels to goal. Patients with very high triglyceride levels (> 500-1000 mg/dl) are at risk of pancreatitis and therefore lifestyle and triglyceride lowering drug therapy should be initiated early. Note that there is limited evidence demonstrating that lowering triglyceride levels reduces cardiovascular events with fibrates, niacin, and most omega-3-fatty acid preparations. A study has shown that adding the omega-3-fatty acid icosapent ethyl (EPA) to statins in patients with elevated triglyceride levels reduces cardiovascular events (228). In addition, the potential beneficial effects of fish oil on disease activity in many patients with inflammatory diseases make the use of omega-3-fatty acids an attractive choice in patients with inflammatory diseases and elevated triglyceride levels/non-HDL-C levels.
The fourth priority in treating lipid disorders is to increase HDL-C levels. There is strong epidemiologic data linking low HDL-C levels with cardiovascular disease, but whether increasing HDL levels with drugs reduces cardiovascular disease is unknown and studies have not been encouraging (229). Life style changes are the initial step and include increased exercise, weight loss, and stopping cigarette smoking. The role of recommending ethanol, which increases HDL levels, is controversial but in patients who already drink moderately there is no reason to recommend that they stop. The most effective drug for increasing HDL levels is niacin, but studies have not demonstrated a reduction in cardiovascular events when niacin is added to statin therapy (230,231). Fibrates and statins also raise HDL-C levels but the increases are modest (usually less than 15%). Additionally, the ACCORD-LIPID trial failed to demonstrate that adding fenofibrate to statin therapy reduces cardiovascular disease (232). Unfortunately, given the currently available drugs, it is very difficult to significantly increase HDL-C levels and in many of our patients we are unable to achieve HDL-C levels in the recommended range. Furthermore, whether this will result in a reduction in cardiovascular events is unknown.
Note that there is very limited evidence that adding fibrates or niacin to lower triglyceride levels and/or increase HDL-C levels will reduce cardiovascular events. However, the studies of fibrates or niacin in combination with statins did not specifically target patients with high triglycerides, high non-HDL-C, and low HDL-C levels. The only drugs in combination with statin therapy that has been shown to further reduce cardiovascular events when added to statin therapy are ezetimibe, PCSK9 inhibitors, and icosapent ethyl (EPA), an omega-3-fatty acid (175).
In summary, modern therapy of patients with inflammatory diseases demands that we aggressively treat lipids to reduce the high risk of cardiovascular disease in this susceptible population. Furthermore, treatment with lipid lowering drugs in some instances may improve the underlying inflammatory disorder.
REFERENCES
- 1.
- Coumbe AG, Pritzker MR, Duprez DA. Cardiovascular risk and psoriasis: beyond the traditional risk factors. Am J Med. 2014;127:12–18. [PubMed: 24161194]
- 2.
- Haque S, Mirjafari H, Bruce IN. Atherosclerosis in rheumatoid arthritis and systemic lupus erythematosus. Curr Opin Lipidol. 2008;19:338–343. [PubMed: 18607179]
- 3.
- John H, Toms TE, Kitas GD. Rheumatoid arthritis: is it a coronary heart disease equivalent? Curr Opin Cardiol. 2011;26:327–333. [PubMed: 21499088]
- 4.
- Ogdie A, Yu Y, Haynes K, Love TJ, Maliha S, Jiang Y, Troxel AB, Hennessy S, Kimmel SE, Margolis DJ, Choi H, Mehta NN, Gelfand JM. Risk of major cardiovascular events in patients with psoriatic arthritis, psoriasis and rheumatoid arthritis: a population-based cohort study. Ann Rheum Dis. 2015;74:326–332. [PMC free article: PMC4341911] [PubMed: 25351522]
- 5.
- Eriksson JK, Jacobsson L, Bengtsson K, Askling J. Is ankylosing spondylitis a risk factor for cardiovascular disease, and how do these risks compare with those in rheumatoid arthritis? Ann Rheum Dis. 2017;76:364–370. [PubMed: 27283333]
- 6.
- Yong WC, Sanguankeo A, Upala S. Association between primary Sjogren's syndrome, cardiovascular and cerebrovascular disease: a systematic review and meta-analysis. Clin Exp Rheumatol. 2018;36 Suppl 112:190–197. [PubMed: 29600936]
- 7.
- Ungprasert P, Koster MJ, Warrington KJ, Matteson EL. Polymyalgia rheumatica and risk of coronary artery disease: a systematic review and meta-analysis of observational studies. Rheumatol Int. 2017;37:143–149. [PubMed: 27577940]
- 8.
- Feng W, Chen G, Cai D, Zhao S, Cheng J, Shen H. Inflammatory Bowel Disease and Risk of Ischemic Heart Disease: An Updated Meta-Analysis of Cohort Studies. J Am Heart Assoc. 2017:6. [PMC free article: PMC5586435] [PubMed: 28768646]
- 9.
- Agca R, Heslinga SC, Rollefstad S, Heslinga M, McInnes IB, Peters MJ, Kvien TK, Dougados M, Radner H, Atzeni F, Primdahl J, Sodergren A, Wallberg Jonsson S, van Rompay J, Zabalan C, Pedersen TR, Jacobsson L, de Vlam K, Gonzalez-Gay MA, Semb AG, Kitas GD, Smulders YM, Szekanecz Z, Sattar N, Symmons DP, Nurmohamed MT. EULAR recommendations for cardiovascular disease risk management in patients with rheumatoid arthritis and other forms of inflammatory joint disorders: 2015/2016 update. Ann Rheum Dis. 2017;76:17–28. [PubMed: 27697765]
- 10.
- Avina-Zubieta JA, Choi HK, Sadatsafavi M, Etminan M, Esdaile JM, Lacaille D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: a meta-analysis of observational studies. Arthritis Rheum. 2008;59:1690–1697. [PubMed: 19035419]
- 11.
- Lindhardsen J, Ahlehoff O, Gislason GH, Madsen OR, Olesen JB, Torp-Pedersen C, Hansen PR. The risk of myocardial infarction in rheumatoid arthritis and diabetes mellitus: a Danish nationwide cohort study. Ann Rheum Dis. 2011;70:929–934. [PubMed: 21389043]
- 12.
- Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA Jr, Jansen-McWilliams L, D'Agostino RB, Kuller LH. Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997;145:408–415. [PubMed: 9048514]
- 13.
- Samarasekera EJ, Neilson JM, Warren RB, Parnham J, Smith CH. Incidence of cardiovascular disease in individuals with psoriasis: a systematic review and meta-analysis. J Invest Dermatol. 2013;133:2340–2346. [PubMed: 23528816]
- 14.
- Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, Linton MF, Raggi P, Stein CM. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2407–2415. [PubMed: 14681506]
- 15.
- Chung CP, Oeser A, Raggi P, Gebretsadik T, Shintani AK, Sokka T, Pincus T, Avalos I, Stein CM. Increased coronary-artery atherosclerosis in rheumatoid arthritis: relationship to disease duration and cardiovascular risk factors. Arthritis Rheum. 2005;52:3045–3053. [PubMed: 16200609]
- 16.
- Giles JT, Szklo M, Post W, Petri M, Blumenthal RS, Lam G, Gelber AC, Detrano R, Scott WW Jr, Kronmal RA, Bathon JM. Coronary arterial calcification in rheumatoid arthritis: comparison with the Multi-Ethnic Study of Atherosclerosis. Arthritis Res Ther. 2009;11:R36. [PMC free article: PMC2688181] [PubMed: 19284547]
- 17.
- Ludwig RJ, Herzog C, Rostock A, Ochsendorf FR, Zollner TM, Thaci D, Kaufmann R, Vogl TJ, Boehncke WH. Psoriasis: a possible risk factor for development of coronary artery calcification. Br J Dermatol. 2007;156:271–276. [PubMed: 17223866]
- 18.
- Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, Crow MK, Schwartz JE, Paget SA, Devereux RB, Salmon JE. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003;349:2399–2406. [PubMed: 14681505]
- 19.
- Wang S, Yiu KH, Mok MY, Ooi GC, Khong PL, Mak KF, Lau CP, Lam KF, Lau CS, Tse HF. Prevalence and extent of calcification over aorta, coronary and carotid arteries in patients with rheumatoid arthritis. J Intern Med. 2009;266:445–452. [PubMed: 19549093]
- 20.
- Yiu KH, Yeung CK, Zhao CT, Chan JC, Siu CW, Tam S, Wong CS, Yan GH, Yue WS, Khong PL, Chan HH, Tse HF. Prevalence and extent of subclinical atherosclerosis in patients with psoriasis. J Intern Med. 2013;273:273–282. [PubMed: 23003220]
- 21.
- Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, Mieszkalski KL, Ilowite NT, Eberhard A, Levy DM, Kimura Y, von Scheven E, Silverman E, Bowyer SL, Punaro L, Singer NG, Sherry DD, McCurdy D, Klein-Gitelman M, Wallace C, Silver R, Wagner-Weiner L, Higgins GC, Brunner HI, Jung L, Soep JB, Reed A. Atherosclerosis Prevention in Pediatric Lupus Erythematosus I. Premature atherosclerosis in pediatric systemic lupus erythematosus: risk factors for increased carotid intima-media thickness in the atherosclerosis prevention in pediatric lupus erythematosus cohort. Arthritis Rheum. 2009;60:1496–1507. [PMC free article: PMC2770725] [PubMed: 19404953]
- 22.
- Becker AE, de Boer OJ, van Der Wal AC. The role of inflammation and infection in coronary artery disease. Annu Rev Med. 2001;52:289–297. [PubMed: 11160780]
- 23.
- Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999;100:e20–28. [PubMed: 10421626]
- 24.
- Leinonen M, Saikku P. Evidence for infectious agents in cardiovascular disease and atherosclerosis. Lancet Infect Dis. 2002;2:11–17. [PubMed: 11892489]
- 25.
- Triant VA, Grinspoon SK. Epidemiology of ischemic heart disease in HIV. Curr Opin HIV AIDS. 2017;12:540–547. [PMC free article: PMC5776691] [PubMed: 28799997]
- 26.
- Sarkar S, Brown TT. Lipid Disorders in People with HIV. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- 27.
- Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, Taubert KA, Newburger JW, Gornik HL, Gewitz MH, Wilson WR, Smith SC Jr, Baddour LM. American Heart Association Rheumatic Fever E, Kawasaki Disease Committee of the Council on Cardiovascular Disease in the Young CoE, Prevention CoPVD, Council on Clinical C. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: a scientific statement from the American Heart Association. Circulation. 2012;125:2520–2544. [PubMed: 22514251]
- 28.
- Beck JD, Elter JR, Heiss G, Couper D, Mauriello SM, Offenbacher S. Relationship of periodontal disease to carotid artery intima-media wall thickness: the atherosclerosis risk in communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2001;21:1816–1822. [PubMed: 11701471]
- 29.
- Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR Jr, Papapanou PN, Sacco RL, Oral I. Vascular Disease Epidemiology S. Relationship between periodontal disease, tooth loss, and carotid artery plaque: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Stroke. 2003;34:2120–2125. [PMC free article: PMC2677013] [PubMed: 12893951]
- 30.
- Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR Jr, Sacco RL, Papapanou PN. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST). Circulation. 2005;111:576–582. [PMC free article: PMC2812915] [PubMed: 15699278]
- 31.
- Soder PO, Soder B, Nowak J, Jogestrand T. Early carotid atherosclerosis in subjects with periodontal diseases. Stroke. 2005;36:1195–1200. [PubMed: 15879347]
- 32.
- Khovidhunkit W, Kim MS, Memon RA, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. [PubMed: 15102878]
- 33.
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ., Cantos Trial Group. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. [PubMed: 28845751]
- 34.
- Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ. The SHK, Xu XF, Ireland MA, Lenderink T, Latchem D, Hoogslag P, Jerzewski A, Nierop P, Whelan A, Hendriks R, Swart H, Schaap J, Kuijper AFM, van Hessen MWJ, Saklani P, Tan I, Thompson AG, Morton A, Judkins C, Bax WA, Dirksen M, Alings M, Hankey GJ, Budgeon CA, Tijssen JGP, Cornel JH, Thompson PL, LoDoCo2 Trial Investigators. Colchicine in Patients with Chronic Coronary Disease. N Engl J Med. 2020;383:1838–1847. [PubMed: 32865380]
- 35.
- Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, Berry C, Lopez-Sendon J, Ostadal P, Koenig W, Angoulvant D, Gregoire JC, Lavoie MA, Dube MP, Rhainds D, Provencher M, Blondeau L, Orfanos A, L'Allier PL, Guertin MC, Roubille F. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N Engl J Med. 2019;381:2497–2505. [PubMed: 31733140]
- 36.
- Tong DC, Bloom JE, Quinn S, Nasis A, Hiew C, Roberts-Thomson P, Adams H, Sriamareswaran R, Htun NM, Wilson W, Stub D, van Gaal W, Howes L, Yeap A, Yip B, Wu S, Perera P, Collins N, Yong A, Bhindi R, Whitbourn R, Lee A, Premaratne M, Asrress K, Freeman M, Amerena J, Layland J. Colchicine in Patients With Acute Coronary Syndrome: Two-Year Follow-Up of the Australian COPS Randomized Clinical Trial. Circulation. 2021;144:1584–1586. [PubMed: 34748393]
- 37.
- Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, Hasan A, Rosenberg Y, Iturriaga E, Gupta M, Tsigoulis M, Verma S, Clearfield M, Libby P, Goldhaber SZ, Seagle R, Ofori C, Saklayen M, Butman S, Singh N, Le May M, Bertrand O, Johnston J, Paynter NP, Glynn RJ. CIRT Investigators. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med. 2018 [PMC free article: PMC6587584] [PubMed: 30415610]
- 38.
- Choi HK, Seeger JD. Lipid profiles among US elderly with untreated rheumatoid arthritis--the Third National Health and Nutrition Examination Survey. J Rheumatol. 2005;32:2311–2316. [PubMed: 16331755]
- 39.
- Georgiadis AN, Papavasiliou EC, Lourida ES, Alamanos Y, Kostara C, Tselepis AD, Drosos AA. Atherogenic lipid profile is a feature characteristic of patients with early rheumatoid arthritis: effect of early treatment--a prospective, controlled study. Arthritis Res Ther. 2006;8:R82. [PMC free article: PMC1526648] [PubMed: 16646989]
- 40.
- Lazarevic MB, Vitic J, Mladenovic V, Myones BL, Skosey JL, Swedler WI. Dyslipoproteinemia in the course of active rheumatoid arthritis. Semin Arthritis Rheum. 1992;22:172–178. [PubMed: 1295090]
- 41.
- Steiner G, Urowitz MB. Lipid profiles in patients with rheumatoid arthritis: mechanisms and the impact of treatment. Semin Arthritis Rheum. 2009;38:372–381. [PubMed: 18395771]
- 42.
- Chung CP, Oeser A, Raggi P, Sokka T, Pincus T, Solus JF, Linton MF, Fazio S, Stein CM. Lipoprotein subclasses determined by nuclear magnetic resonance spectroscopy and coronary atherosclerosis in patients with rheumatoid arthritis. J Rheumatol. 2010;37:1633–1638. [PMC free article: PMC2914215] [PubMed: 20516025]
- 43.
- Knowlton N, Wages JA, Centola MB, Alaupovic P. Apolipoprotein-defined lipoprotein abnormalities in rheumatoid arthritis patients and their potential impact on cardiovascular disease. Scand J Rheumatol. 2012;41:165–169. [PubMed: 22401593]
- 44.
- Hurt-Camejo E, Paredes S, Masana L, Camejo G, Sartipy P, Rosengren B, Pedreno J, Vallve JC, Benito P, Wiklund O. Elevated levels of small, low-density lipoprotein with high affinity for arterial matrix components in patients with rheumatoid arthritis: possible contribution of phospholipase A2 to this atherogenic profile. Arthritis Rheum. 2001;44:2761–2767. [PubMed: 11762936]
- 45.
- Rizzo M, Spinas GA, Cesur M, Ozbalkan Z, Rini GB, Berneis K. Atherogenic lipoprotein phenotype and LDL size and subclasses in drug-naive patients with early rheumatoid arthritis. Atherosclerosis. 2009;207:502–506. [PubMed: 19643412]
- 46.
- Schulte DM, Paulsen K, Turk K, Brandt B, Freitag-Wolf S, Hagen I, Zeuner R, Schroder JO, Lieb W, Franke A, Nikolaus S, Mrowietz U, Gerdes S, Schreiber S, Laudes M. Small dense LDL cholesterol in human subjects with different chronic inflammatory diseases. Nutr Metab Cardiovasc Dis. 2018;28:1100–1105. [PubMed: 30143407]
- 47.
- Asanuma Y, Kawai S, Aoshima H, Kaburaki J, Mizushima Y. Serum lipoprotein(a) and apolipoprotein(a) phenotypes in patients with rheumatoid arthritis. Arthritis Rheum. 1999;42:443–447. [PubMed: 10088766]
- 48.
- Dursunoglu D, Evrengul H, Polat B, Tanriverdi H, Cobankara V, Kaftan A, Kilic M. Lp(a) lipoprotein and lipids in patients with rheumatoid arthritis: serum levels and relationship to inflammation. Rheumatol Int. 2005;25:241–245. [PubMed: 15290086]
- 49.
- Lee YH, Choi SJ, Ji JD, Seo HS, Song GG. Lipoprotein(a) and lipids in relation to inflammation in rheumatoid arthritis. Clin Rheumatol. 2000;19:324–325. [PubMed: 10941819]
- 50.
- Borba EF, Bonfa E. Dyslipoproteinemias in systemic lupus erythematosus: influence of disease, activity, and anticardiolipin antibodies. Lupus. 1997;6:533–539. [PubMed: 9256312]
- 51.
- Bruce IN, Urowitz MB, Gladman DD, Ibanez D, Steiner G. Risk factors for coronary heart disease in women with systemic lupus erythematosus: the Toronto Risk Factor Study. Arthritis Rheum. 2003;48:3159–3167. [PubMed: 14613278]
- 52.
- de Carvalho JF, Bonfa E, Borba EF. Systemic lupus erythematosus and "lupus dyslipoproteinemia". Autoimmun Rev. 2008;7:246–250. [PubMed: 18190886]
- 53.
- Borba EF, Santos RD, Bonfa E, Vinagre CG, Pileggi FJ, Cossermelli W, Maranhao RC. Lipoprotein(a) levels in systemic lupus erythematosus. J Rheumatol. 1994;21:220–223. [PubMed: 8182628]
- 54.
- Feingold KR, Grunfeld C. Psoriasis: it's more than just the skin. J Lipid Res. 2012;53:1427–1429. [PMC free article: PMC3540839] [PubMed: 22685321]
- 55.
- Friedewald VE, Cather JC, Gelfand JM, Gordon KB, Gibbons GH, Grundy SM, Jarratt MT, Krueger JG, Ridker PM, Stone N, Roberts WC. AJC editor's consensus: psoriasis and coronary artery disease. Am J Cardiol. 2008;102:1631–1643. [PubMed: 19064017]
- 56.
- Gottlieb AB, Dann F. Comorbidities in patients with psoriasis. Am J Med 2009; 122:1150 e1151-1159. [PubMed: 19958894]
- 57.
- Langan SM, Seminara NM, Shin DB, Troxel AB, Kimmel SE, Mehta NN, Margolis DJ, Gelfand JM. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol. 2012;132:556–562. [PMC free article: PMC3278499] [PubMed: 22113483]
- 58.
- Tobin AM, Veale DJ, Fitzgerald O, Rogers S, Collins P, O'Shea D, Kirby B. Cardiovascular disease and risk factors in patients with psoriasis and psoriatic arthritis. J Rheumatol. 2010;37:1386–1394. [PubMed: 20472927]
- 59.
- Fernandez-Armenteros JM, Gomez-Arbones X, Buti-Soler M, Betriu-Bars A, Sanmartin-Novell V, Ortega-Bravo M, Martinez-Alonso M, Gari E, Portero-Otin M, Santamaria-Babi L, Casanova-Seuma JM. Psoriasis, metabolic syndrome and cardiovascular risk factors. A population-based study. J Eur Acad Dermatol Venereol. 2018 [PubMed: 29953676]
- 60.
- Sorokin AV, Kotani K, Elnabawi YA, Dey AK, Sajja AP, Yamada S, Ueda M, Harrington CL, Baumer Y, Rodante JA, Gelfand JM, Chen MY, Joshi AA, Playford MP, Remaley AT, Mehta NN. Association Between Oxidation-Modified Lipoproteins and Coronary Plaque in Psoriasis. Circ Res. 2018;123:1244–1254. [PMC free article: PMC6345554] [PubMed: 30571459]
- 61.
- Ripolles Piquer B, Nazih H, Bourreille A, Segain JP, Huvelin JM, Galmiche JP, Bard JM. Altered lipid, apolipoprotein, and lipoprotein profiles in inflammatory bowel disease: consequences on the cholesterol efflux capacity of serum using Fu5AH cell system. Metabolism. 2006;55:980–988. [PubMed: 16784973]
- 62.
- Sappati Biyyani RS, Putka BS, Mullen KD. Dyslipidemia and lipoprotein profiles in patients with inflammatory bowel disease. J Clin Lipidol. 2010;4:478–482. [PubMed: 21122694]
- 63.
- Koutroumpakis E, Ramos-Rivers C, Regueiro M, Hashash JG, Barrie A, Swoger J, Baidoo L, Schwartz M, Dunn MA, Koutroubakis IE, Binion DG. Association Between Long-Term Lipid Profiles and Disease Severity in a Large Cohort of Patients with Inflammatory Bowel Disease. Dig Dis Sci. 2016;61:865–871. [PubMed: 26514677]
- 64.
- Papagoras C, Markatseli TE, Saougou I, Alamanos Y, Zikou AK, Voulgari PV, Kiortsis DN, Drosos AA. Cardiovascular risk profile in patients with spondyloarthritis. Joint Bone Spine. 2014;81:57–63. [PubMed: 23731637]
- 65.
- Bullon P, Morillo JM, Ramirez-Tortosa MC, Quiles JL, Newman HN, Battino M. Metabolic syndrome and periodontitis: is oxidative stress a common link? J Dent Res. 2009;88:503–518. [PubMed: 19587154]
- 66.
- Penumarthy S, Penmetsa GS, Mannem S. Assessment of serum levels of triglycerides, total cholesterol, high-density lipoprotein cholesterol, and low-density lipoprotein cholesterol in periodontitis patients. J Indian Soc Periodontol. 2013;17:30–35. [PMC free article: PMC3636940] [PubMed: 23633769]
- 67.
- Pussinen PJ, Mattila K. Periodontal infections and atherosclerosis: mere associations? Curr Opin Lipidol. 2004;15:583–588. [PubMed: 15361795]
- 68.
- Schenkein HA, Loos BG. Inflammatory mechanisms linking periodontal diseases to cardiovascular diseases. J Periodontol. 2013;84:S51–69. [PubMed: 23631584]
- 69.
- Nepomuceno R, Pigossi SC, Finoti LS, Orrico SRP, Cirelli JA, Barros SP, Offenbacher S, Scarel-Caminaga RM. Serum lipid levels in patients with periodontal disease: A meta-analysis and meta-regression. J Clin Periodontol. 2017;44:1192–1207. [PubMed: 28782128]
- 70.
- Rufail ML, Schenkein HA, Koertge TE, Best AM, Barbour SE, Tew JG, van Antwerpen R. Atherogenic lipoprotein parameters in patients with aggressive periodontitis. J Periodontal Res. 2007;42:495–502. [PubMed: 17956461]
- 71.
- Pussinen PJ, Vilkuna-Rautiainen T, Alfthan G, Palosuo T, Jauhiainen M, Sundvall J, Vesanen M, Mattila K, Asikainen S. Severe periodontitis enhances macrophage activation via increased serum lipopolysaccharide. Arterioscler Thromb Vasc Biol. 2004;24:2174–2180. [PubMed: 15388525]
- 72.
- Teeuw WJ, Slot DE, Susanto H, Gerdes VE, Abbas F, D'Aiuto F, Kastelein JJ, Loos BG. Treatment of periodontitis improves the atherosclerotic profile: a systematic review and meta-analysis. J Clin Periodontol. 2014;41:70–79. [PubMed: 24111886]
- 73.
- Buhlin K, Hultin M, Norderyd O, Persson L, Pockley AG, Pussinen PJ, Rabe P, Klinge B, Gustafsson A. Periodontal treatment influences risk markers for atherosclerosis in patients with severe periodontitis. Atherosclerosis. 2009;206:518–522. [PubMed: 19411077]
- 74.
- Alvarez C, Ramos A. Lipids, lipoproteins, and apoproteins in serum during infection. Clin Chem. 1986;32:142–145. [PubMed: 3940695]
- 75.
- Cappi SB, Noritomi DT, Velasco IT, Curi R, Loureiro TC, Soriano FG. Dyslipidemia: a prospective controlled randomized trial of intensive glycemic control in sepsis. Intensive Care Med. 2012;38:634–641. [PubMed: 22297666]
- 76.
- Gallin JI, Kaye D, O'Leary WM. Serum lipids in infection. N Engl J Med. 1969;281:1081–1086. [PubMed: 5824173]
- 77.
- Gordon BR, Parker TS, Levine DM, Saal SD, Wang JC, Sloan BJ, Barie PS, Rubin AL. Low lipid concentrations in critical illness: implications for preventing and treating endotoxemia. Crit Care Med. 1996;24:584–589. [PubMed: 8612407]
- 78.
- Kerttula Y, Weber TH. Serum lipids in viral and bacterial meningitis. Scand J Infect Dis. 1986;18:211–215. [PubMed: 3738432]
- 79.
- Sammalkorpi K, Valtonen V, Kerttula Y, Nikkila E, Taskinen MR. Changes in serum lipoprotein pattern induced by acute infections. Metabolism. 1988;37:859–865. [PubMed: 3419323]
- 80.
- van Leeuwen HJ, Heezius EC, Dallinga GM, van Strijp JA, Verhoef J, van Kessel KP. Lipoprotein metabolism in patients with severe sepsis. Crit Care Med. 2003;31:1359–1366. [PubMed: 12771603]
- 81.
- Feingold KR. The bidirectional link between HDL and COVID-19 infections. J Lipid Res. 2021;62:100067. [PMC free article: PMC7963524] [PubMed: 33741421]
- 82.
- Apostolou F, Gazi IF, Kostoula A, Tellis CC, Tselepis AD, Elisaf M, Liberopoulos EN. Persistence of an atherogenic lipid profile after treatment of acute infection with Brucella. J Lipid Res. 2009;50:2532–2539. [PMC free article: PMC2781324] [PubMed: 19535817]
- 83.
- Apostolou F, Gazi IF, Lagos K, Tellis CC, Tselepis AD, Liberopoulos EN, Elisaf M. Acute infection with Epstein-Barr virus is associated with atherogenic lipid changes. Atherosclerosis. 2010;212:607–613. [PubMed: 20594556]
- 84.
- Gazi IF, Apostolou FA, Liberopoulos EN, Filippatos TD, Tellis CC, Elisaf MS, Tselepis AD. Leptospirosis is associated with markedly increased triglycerides and small dense low-density lipoprotein and decreased high-density lipoprotein. Lipids. 2011;46:953–960. [PubMed: 21688175]
- 85.
- Deniz O, Gumus S, Yaman H, Ciftci F, Ors F, Cakir E, Tozkoparan E, Bilgic H, Ekiz K. Serum total cholesterol, HDL-C and LDL-C concentrations significantly correlate with the radiological extent of disease and the degree of smear positivity in patients with pulmonary tuberculosis. Clin Biochem. 2007;40:162–166. [PubMed: 17217941]
- 86.
- Deniz O, Tozkoparan E, Yaman H, Cakir E, Gumus S, Ozcan O, Bozlar U, Bilgi C, Bilgic H, Ekiz K. Serum HDL-C levels, log (TG/HDL-C) values and serum total cholesterol/HDL-C ratios significantly correlate with radiological extent of disease in patients with community-acquired pneumonia. Clin Biochem. 2006;39:287–292. [PubMed: 16487950]
- 87.
- Palacio C, Alexandraki I, Bertholf RL, Mooradian AD. Transient dyslipidemia mimicking the plasma lipid profile of Tangier disease in a diabetic patient with gram negative sepsis. Ann Clin Lab Sci. 2011;41:150–153. [PubMed: 21844573]
- 88.
- Barlage S, Gnewuch C, Liebisch G, Wolf Z, Audebert FX, Gluck T, Frohlich D, Kramer BK, Rothe G, Schmitz G. Changes in HDL-associated apolipoproteins relate to mortality in human sepsis and correlate to monocyte and platelet activation. Intensive Care Med. 2009;35:1877–1885. [PubMed: 19669126]
- 89.
- Chien JY, Jerng JS, Yu CJ, Yang PC. Low serum level of high-density lipoprotein cholesterol is a poor prognostic factor for severe sepsis. Crit Care Med. 2005;33:1688–1693. [PubMed: 16096442]
- 90.
- Gruber M, Christ-Crain M, Stolz D, Keller U, Muller C, Bingisser R, Tamm M, Mueller B, Schuetz P. Prognostic impact of plasma lipids in patients with lower respiratory tract infections - an observational study. Swiss Med Wkly. 2009;139:166–172. [PubMed: 19330560]
- 91.
- Lekkou A, Mouzaki A, Siagris D, Ravani I, Gogos CA. Serum lipid profile, cytokine production, and clinical outcome in patients with severe sepsis. J Crit Care. 2014;29:723–727. [PubMed: 24891152]
- 92.
- Cirstea M, Walley KR, Russell JA, Brunham LR, Genga KR, Boyd JH. Decreased high-density lipoprotein cholesterol level is an early prognostic marker for organ dysfunction and death in patients with suspected sepsis. J Crit Care. 2017;38:289–294. [PubMed: 28013095]
- 93.
- Grion CM, Cardoso LT, Perazolo TF, Garcia AS, Barbosa DS, Morimoto HK, Matsuo T, Carrilho AJ. Lipoproteins and CETP levels as risk factors for severe sepsis in hospitalized patients. Eur J Clin Invest. 2010;40:330–338. [PubMed: 20486994]
- 94.
- Iribarren C, Jacobs DR Jr, Sidney S, Claxton AJ, Feingold KR. Cohort study of serum total cholesterol and in-hospital incidence of infectious diseases. Epidemiol Infect. 1998;121:335–347. [PMC free article: PMC2809530] [PubMed: 9825784]
- 95.
- Guirgis FW, Donnelly JP, Dodani S, Howard G, Safford MM, Levitan EB, Wang HE. Cholesterol levels and long-term rates of community-acquired sepsis. Crit Care. 2016;20:408. [PMC free article: PMC5180408] [PubMed: 28010729]
- 96.
- Kaysen GA, Ye X, Raimann JG, Wang Y, Topping A, Usvyat LA, Stuard S, Canaud B, van der Sande FM, Kooman JP, Kotanko P. Monitoring Dialysis Outcomes I. Lipid levels are inversely associated with infectious and all-cause mortality: international MONDO study results. J Lipid Res. 2018;59:1519–1528. [PMC free article: PMC6071781] [PubMed: 29895699]
- 97.
- Madsen CM, Varbo A, Tybjaerg-Hansen A, Frikke-Schmidt R, Nordestgaard BG. U-shaped relationship of HDL and risk of infectious disease: two prospective population-based cohort studies. Eur Heart J. 2018;39:1181–1190. [PubMed: 29228167]
- 98.
- Trinder M, Walley KR, Boyd JH, Brunham LR. Causal Inference for Genetically Determined Levels of High-Density Lipoprotein Cholesterol and Risk of Infectious Disease. Arterioscler Thromb Vasc Biol. 2020;40:267–278. [PMC free article: PMC6946100] [PubMed: 31694394]
- 99.
- Patel PN, Shah RY, Ferguson JF, Reilly MP. Human experimental endotoxemia in modeling the pathophysiology, genomics, and therapeutics of innate immunity in complex cardiometabolic diseases. Arterioscler Thromb Vasc Biol. 2015;35:525–534. [PMC free article: PMC4344396] [PubMed: 25550206]
- 100.
- Missala I, Kassner U, Steinhagen-Thiessen E. A Systematic Literature Review of the Association of Lipoprotein(a) and Autoimmune Diseases and Atherosclerosis. Int J Rheumatol. 2012;2012:480784. [PMC free article: PMC3523136] [PubMed: 23304154]
- 101.
- Choy E, Sattar N. Interpreting lipid levels in the context of high-grade inflammatory states with a focus on rheumatoid arthritis: a challenge to conventional cardiovascular risk actions. Ann Rheum Dis. 2009;68:460–469. [PubMed: 19286905]
- 102.
- Robertson J, Peters MJ, McInnes IB, Sattar N. Changes in lipid levels with inflammation and therapy in RA: a maturing paradigm. Nat Rev Rheumatol. 2013;9:513–523. [PubMed: 23774906]
- 103.
- Heslinga SC, Peters MJ, Ter Wee MM, van der Horst-Bruinsma IE, van Sijl AM, Smulders YM, Nurmohamed MT. Reduction of Inflammation Drives Lipid Changes in Ankylosing Spondylitis. J Rheumatol. 2015;42:1842–1845. [PubMed: 26329334]
- 104.
- Park YB, Choi HK, Kim MY, Lee WK, Song J, Kim DK, Lee SK. Effects of antirheumatic therapy on serum lipid levels in patients with rheumatoid arthritis: a prospective study. Am J Med. 2002;113:188–193. [PubMed: 12208376]
- 105.
- Arnaldi G, Scandali VM, Trementino L, Cardinaletti M, Appolloni G, Boscaro M. Pathophysiology of dyslipidemia in Cushing's syndrome. Neuroendocrinology. 2010;92 Suppl 1:86–90. [PubMed: 20829625]
- 106.
- Ettinger WH Jr, Hazzard WR. Prednisone increases very low density lipoprotein and high density lipoprotein in healthy men. Metabolism. 1988;37:1055–1058. [PubMed: 3185288]
- 107.
- Mihailescu DV, Vora A, Mazzone T. Lipid effects of endocrine medications. Curr Atheroscler Rep. 2011;13:88–94. [PubMed: 21104166]
- 108.
- Cairoli E, Rebella M, Danese N, Garra V, Borba EF. Hydroxychloroquine reduces low-density lipoprotein cholesterol levels in systemic lupus erythematosus: a longitudinal evaluation of the lipid-lowering effect. Lupus. 2012;21:1178–1182. [PubMed: 22641182]
- 109.
- Munro R, Morrison E, McDonald AG, Hunter JA, Madhok R, Capell HA. Effect of disease modifying agents on the lipid profiles of patients with rheumatoid arthritis. Ann Rheum Dis. 1997;56:374–377. [PMC free article: PMC1752398] [PubMed: 9227167]
- 110.
- Tam LS, Gladman DD, Hallett DC, Rahman P, Urowitz MB. Effect of antimalarial agents on the fasting lipid profile in systemic lupus erythematosus. J Rheumatol. 2000;27:2142–2145. [PubMed: 10990225]
- 111.
- Lu B, Moser A, Shigenaga JK, Grunfeld C, Feingold KR. The acute phase response stimulates the expression of angiopoietin like protein 4. Biochem Biophys Res Commun. 2010;391:1737–1741. [PubMed: 20043872]
- 112.
- de Carvalho JF, Borba EF, Viana VS, Bueno C, Leon EP, Bonfa E. Anti-lipoprotein lipase antibodies: a new player in the complex atherosclerotic process in systemic lupus erythematosus? Arthritis Rheum. 2004;50:3610–3615. [PubMed: 15529371]
- 113.
- Reichlin M, Fesmire J, Quintero-Del-Rio AI, Wolfson-Reichlin M. Autoantibodies to lipoprotein lipase and dyslipidemia in systemic lupus erythematosus. Arthritis Rheum. 2002;46:2957–2963. [PubMed: 12428237]
- 114.
- Hosoai H, Webb NR, Glick JM, Tietge UJ, Purdom MS, de Beer FC, Rader DJ. Expression of serum amyloid A protein in the absence of the acute phase response does not reduce HDL cholesterol or apoA-I levels in human apoA-I transgenic mice. J Lipid Res. 1999;40:648–653. [PubMed: 10191288]
- 115.
- Ramharack R, Barkalow D, Spahr MA. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler Thromb Vasc Biol. 1998;18:984–990. [PubMed: 9633941]
- 116.
- Wade DP, Clarke JG, Lindahl GE, Liu AC, Zysow BR, Meer K, Schwartz K, Lawn RM. 5' control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc Natl Acad Sci U S A. 1993;90:1369–1373. [PMC free article: PMC45874] [PubMed: 7679504]
- 117.
- Schultz O, Oberhauser F, Saech J, Rubbert-Roth A, Hahn M, Krone W, Laudes M. Effects of inhibition of interleukin-6 signalling on insulin sensitivity and lipoprotein (a) levels in human subjects with rheumatoid diseases. PLoS One. 2010;5:e14328. [PMC free article: PMC3001451] [PubMed: 21179199]
- 118.
- Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43:1363–1379. [PubMed: 12235168]
- 119.
- Tribble DL, Rizzo M, Chait A, Lewis DM, Blanche PJ, Krauss RM. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low-density lipoproteins. Am J Med. 2001;110:103–110. [PubMed: 11165551]
- 120.
- Borba EF, Carvalho JF, Bonfa E. Mechanisms of dyslipoproteinemias in systemic lupus erythematosus. Clin Dev Immunol. 2006;13:203–208. [PMC free article: PMC2270754] [PubMed: 17162363]
- 121.
- Esteve E, Ricart W, Fernandez-Real JM. Dyslipidemia and inflammation: an evolutionary conserved mechanism. Clin Nutr. 2005;24:16–31. [PubMed: 15681098]
- 122.
- Frostegard J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, Horkko S, Witztum JL. Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum. 2005;52:192–200. [PubMed: 15641060]
- 123.
- Asha K, Singal A, Sharma SB, Arora VK, Aggarwal A. Dyslipidaemia & oxidative stress in patients of psoriasis: Emerging cardiovascular risk factors. Indian J Med Res. 2017;146:708–713. [PMC free article: PMC5926341] [PubMed: 29664028]
- 124.
- Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF, Vickers KC. The Role of Lipids and Lipoproteins in Atherosclerosis. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA) 2019.
- 125.
- Hahn BH, Grossman J, Chen W, McMahon M. The pathogenesis of atherosclerosis in autoimmune rheumatic diseases: roles of inflammation and dyslipidemia. J Autoimmun. 2007;28:69–75. [PubMed: 17433865]
- 126.
- Mehta NN, Gelfand JM. High-density lipoprotein cholesterol function improves after successful treatment of psoriasis: a step forward in the right direction. J Invest Dermatol. 2014;134:592–595. [PMC free article: PMC3929212] [PubMed: 24518110]
- 127.
- Feingold KR, Grunfeld C. The acute phase response inhibits reverse cholesterol transport. J Lipid Res. 2010;51:682–684. [PMC free article: PMC2842157] [PubMed: 20071695]
- 128.
- Charles-Schoeman C, Lee YY, Grijalva V, Amjadi S, FitzGerald J, Ranganath VK, Taylor M, McMahon M, Paulus HE, Reddy ST. Cholesterol efflux by high density lipoproteins is impaired in patients with active rheumatoid arthritis. Ann Rheum Dis. 2012;71:1157–1162. [PMC free article: PMC3428121] [PubMed: 22267330]
- 129.
- Holzer M, Wolf P, Curcic S, Birner-Gruenberger R, Weger W, Inzinger M, El-Gamal D, Wadsack C, Heinemann A, Marsche G. Psoriasis alters HDL composition and cholesterol efflux capacity. J Lipid Res. 2012;53:1618–1624. [PMC free article: PMC3540842] [PubMed: 22649206]
- 130.
- Mehta NN, Li R, Krishnamoorthy P, Yu Y, Farver W, Rodrigues A, Raper A, Wilcox M, Baer A, DerOhannesian S, Wolfe M, Reilly MP, Rader DJ, VanVoorhees A, Gelfand JM. Abnormal lipoprotein particles and cholesterol efflux capacity in patients with psoriasis. Atherosclerosis. 2012;224:218–221. [PMC free article: PMC3693845] [PubMed: 22858285]
- 131.
- Ronda N, Favari E, Borghi MO, Ingegnoli F, Gerosa M, Chighizola C, Zimetti F, Adorni MP, Bernini F, Meroni PL. Impaired serum cholesterol efflux capacity in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. 2014;73:609–615. [PubMed: 23562986]
- 132.
- Annema W, Nijstad N, Tolle M, de Boer JF, Buijs RV, Heeringa P, van der Giet M, Tietge UJ. Myeloperoxidase and serum amyloid A contribute to impaired in vivo reverse cholesterol transport during the acute phase response but not group IIA secretory phospholipase A(2). J Lipid Res. 2010;51:743–754. [PMC free article: PMC2842154] [PubMed: 20061576]
- 133.
- Pussinen PJ, Jauhiainen M, Vilkuna-Rautiainen T, Sundvall J, Vesanen M, Mattila K, Palosuo T, Alfthan G, Asikainen S. Periodontitis decreases the antiatherogenic potency of high density lipoprotein. J Lipid Res. 2004;45:139–147. [PubMed: 13130123]
- 134.
- Zimetti F, De Vuono S, Gomaraschi M, Adorni MP, Favari E, Ronda N, Ricci MA, Veglia F, Calabresi L, Lupattelli G. Plasma cholesterol homeostasis, HDL remodeling and function during the acute phase reaction. J Lipid Res. 2017;58:2051–2060. [PMC free article: PMC5625127] [PubMed: 28830907]
- 135.
- Gkolfinopoulou C, Stratikos E, Theofilatos D, Kardassis D, Voulgari PV, Drosos AA, Chroni A. Impaired Antiatherogenic Functions of High-density Lipoprotein in Patients with Ankylosing Spondylitis. J Rheumatol. 2015;42:1652–1660. [PubMed: 26233507]
- 136.
- McGillicuddy FC, de la Llera Moya M, Hinkle CC, Joshi MR, Chiquoine EH, Billheimer JT, Rothblat GH, Reilly MP. Inflammation impairs reverse cholesterol transport in vivo. Circulation. 2009;119:1135–1145. [PMC free article: PMC4937877] [PubMed: 19221221]
- 137.
- Holzer M, Wolf P, Inzinger M, Trieb M, Curcic S, Pasterk L, Weger W, Heinemann A, Marsche G. Anti-psoriatic therapy recovers high-density lipoprotein composition and function. J Invest Dermatol. 2014;134:635–642. [PMC free article: PMC4178282] [PubMed: 23985995]
- 138.
- Liao KP, Playford MP, Frits M, Coblyn JS, Iannaccone C, Weinblatt ME, Shadick NS, Mehta NN. The association between reduction in inflammation and changes in lipoprotein levels and HDL cholesterol efflux capacity in rheumatoid arthritis. J Am Heart Assoc. 2015:4. [PMC free article: PMC4345878] [PubMed: 25637346]
- 139.
- Ronda N, Greco D, Adorni MP, Zimetti F, Favari E, Hjeltnes G, Mikkelsen K, Borghi MO, Favalli EG, Gatti R, Hollan I, Meroni PL, Bernini F. New anti-atherosclerotic activity of methotrexate and adalimumab: Complementary effects on lipoprotein function and macrophage cholesterol metabolism. Arthritis Rheumatol. 2015 [PubMed: 25605003]
- 140.
- Cheng Y, Chen Y, Sun X, Li Y, Huang C, Deng H, Li Z. Identification of potential serum biomarkers for rheumatoid arthritis by high-resolution quantitative proteomic analysis. Inflammation. 2014;37:1459–1467. [PubMed: 24682873]
- 141.
- Ferretti G, Bacchetti T, Campanati A, Simonetti O, Liberati G, Offidani A. Correlation between lipoprotein(a) and lipid peroxidation in psoriasis: role of the enzyme paraoxonase-1. Br J Dermatol. 2012;166:204–207. [PubMed: 21790517]
- 142.
- He L, Qin S, Dang L, Song G, Yao S, Yang N, Li Y. Psoriasis decreases the anti-oxidation and anti-inflammation properties of high-density lipoprotein. Biochim Biophys Acta. 2014;1841:1709–1715. [PubMed: 25240836]
- 143.
- Isik A, Koca SS, Ustundag B, Celik H, Yildirim A. Paraoxonase and arylesterase levels in rheumatoid arthritis. Clin Rheumatol. 2007;26:342–348. [PubMed: 16642406]
- 144.
- Tanimoto N, Kumon Y, Suehiro T, Ohkubo S, Ikeda Y, Nishiya K, Hashimoto K. Serum paraoxonase activity decreases in rheumatoid arthritis. Life Sci. 2003;72:2877–2885. [PubMed: 12697270]
- 145.
- Tripi LM, Manzi S, Chen Q, Kenney M, Shaw P, Kao A, Bontempo F, Kammerer C, Kamboh MI. Relationship of serum paraoxonase 1 activity and paraoxonase 1 genotype to risk of systemic lupus erythematosus. Arthritis Rheum. 2006;54:1928–1939. [PubMed: 16729301]
- 146.
- Usta M, Turan E, Aral H, Inal BB, Gurel MS, Guvenen G. Serum paraoxonase-1 activities and oxidative status in patients with plaque-type psoriasis with/without metabolic syndrome. J Clin Lab Anal. 2011;25:289–295. [PMC free article: PMC6647565] [PubMed: 21786331]
- 147.
- Draganov D, Teiber J, Watson C, Bisgaier C, Nemzek J, Remick D, Standiford T, La Du B. PON1 and oxidative stress in human sepsis and an animal model of sepsis. Adv Exp Med Biol. 2010;660:89–97. [PubMed: 20221873]
- 148.
- Novak F, Vavrova L, Kodydkova J, Novak F Sr, Hynkova M, Zak A, Novakova O. Decreased paraoxonase activity in critically ill patients with sepsis. Clin Exp Med. 2010;10:21–25. [PubMed: 19760042]
- 149.
- Charles-Schoeman C, Watanabe J, Lee YY, Furst DE, Amjadi S, Elashoff D, Park G, McMahon M, Paulus HE, Fogelman AM, Reddy ST. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009;60:2870–2879. [PMC free article: PMC2828490] [PubMed: 19790070]
- 150.
- Charles-Schoeman C, Khanna D, Furst DE, McMahon M, Reddy ST, Fogelman AM, Paulus HE, Park GS, Gong T, Ansell BJ. Effects of high-dose atorvastatin on antiinflammatory properties of high density lipoprotein in patients with rheumatoid arthritis: a pilot study. J Rheumatol. 2007;34:1459–1464. [PubMed: 17552046]
- 151.
- Barcia AM, Harris HW. Triglyceride-rich lipoproteins as agents of innate immunity. Clin Infect Dis. 2005;41 Suppl 7:S498–503. [PubMed: 16237653]
- 152.
- Han R. Plasma lipoproteins are important components of the immune system. Microbiol Immunol. 2010;54:246–253. [PubMed: 20377753]
- 153.
- Pirillo A, Catapano AL, Norata GD. HDL in infectious diseases and sepsis. Handb Exp Pharmacol. 2015;224:483–508. [PubMed: 25522999]
- 154.
- Peters MJ, van Halm VP, Voskuyl AE, Smulders YM, Boers M, Lems WF, Visser M, Stehouwer CD, Dekker JM, Nijpels G, Heine R, Dijkmans BA, Nurmohamed MT. Does rheumatoid arthritis equal diabetes mellitus as an independent risk factor for cardiovascular disease? A prospective study. Arthritis Rheum. 2009;61:1571–1579. [PubMed: 19877093]
- 155.
- SCORE Working Group. E. S. C. Cardiovascular risk collaboration. SCORE2 risk prediction algorithms: new models to estimate 10-year risk of cardiovascular disease in Europe. Eur Heart J. 2021;42:2439–2454. [PMC free article: PMC8248998] [PubMed: 34120177]
- 156.
- Hippisley-Cox J, Coupland C, Robson J, Brindle P. Derivation, validation, and evaluation of a new QRISK model to estimate lifetime risk of cardiovascular disease: cohort study using QResearch database. BMJ. 2010;341:c6624. [PMC free article: PMC2999889] [PubMed: 21148212]
- 157.
- Arts EE, Popa C, Den Broeder AA, Semb AG, Toms T, Kitas GD, van Riel PL, Fransen J. Performance of four current risk algorithms in predicting cardiovascular events in patients with early rheumatoid arthritis. Ann Rheum Dis. 2015;74:668–674. [PubMed: 24389293]
- 158.
- Crowson CS, Matteson EL, Roger VL, Therneau TM, Gabriel SE. Usefulness of risk scores to estimate the risk of cardiovascular disease in patients with rheumatoid arthritis. Am J Cardiol. 2012;110:420–424. [PMC free article: PMC3398213] [PubMed: 22521305]
- 159.
- Kawai VK, Chung CP, Solus JF, Oeser A, Raggi P, Stein CM. The ability of the 2013 American College of Cardiology/American Heart Association cardiovascular risk score to identify rheumatoid arthritis patients with high coronary artery calcification scores. Arthritis Rheumatol. 2015;67:381–385. [PMC free article: PMC4369184] [PubMed: 25371313]
- 160.
- Kawai VK, Solus JF, Oeser A, Rho YH, Raggi P, Bian A, Gebretsadik T, Shintani A, Stein CM. Novel cardiovascular risk prediction models in patients with systemic lupus erythematosus. Lupus. 2011;20:1526–1534. [PMC free article: PMC3227752] [PubMed: 21976402]
- 161.
- Purcarea A, Sovaila S, Udrea G, Rezus E, Gheorghe A, Tiu C, Stoica V. Utility of different cardiovascular disease prediction models in rheumatoid arthritis. J Med Life. 2014;7:588–594. [PMC free article: PMC4316145] [PubMed: 25713628]
- 162.
- Eder L, Chandran V, Gladman DD. The Framingham Risk Score underestimates the extent of subclinical atherosclerosis in patients with psoriatic disease. Ann Rheum Dis. 2014;73:1990–1996. [PubMed: 23887287]
- 163.
- Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, Goldberg R, Heidenreich PA, Hlatky MA, Jones DW, Lloyd-Jones D, Lopez-Pajares N, Ndumele CE, Orringer CE, Peralta CA, Saseen JJ, Smith SC Jr, Sperling L, Virani SS, Yeboah J. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol. Circulation. 2018;2018:CIR0000000000000625.
- 164.
- Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, Chapman MJ, De Backer GG, Delgado V, Ference BA, Graham IM, Halliday A, Landmesser U, Mihaylova B, Pedersen TR, Riccardi G, Richter DJ, Sabatine MS, Taskinen MR, Tokgozoglu L, Wiklund O, Group ESCSD. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. [PubMed: 31504418]
- 165.
- Grundy SM, Feingold KR. Guidelines for the Management of High Blood Cholesterol. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2019.
- 166.
- Atzeni F, Rodriguez-Carrio J, Popa CD, Nurmohamed MT, Szucs G, Szekanecz Z. Cardiovascular effects of approved drugs for rheumatoid arthritis. Nat Rev Rheumatol. 2021;17:270–290. [PubMed: 33833437]
- 167.
- Ajeganova S, Andersson ML, Frostegard J, Hafstrom I. Disease factors in early rheumatoid arthritis are associated with differential risks for cardiovascular events and mortality depending on age at onset: a 10-year observational cohort study. J Rheumatol. 2013;40:1958–1966. [PubMed: 23950188]
- 168.
- Arts EE, Fransen J, Den Broeder AA, van Riel P, Popa CD. Low disease activity (DAS28</=3.2) reduces the risk of first cardiovascular event in rheumatoid arthritis: a time-dependent Cox regression analysis in a large cohort study. Ann Rheum Dis. 2017;76:1693–1699. [PubMed: 28606965]
- 169.
- Arts EE, Fransen J, den Broeder AA, Popa CD, van Riel PL. The effect of disease duration and disease activity on the risk of cardiovascular disease in rheumatoid arthritis patients. Ann Rheum Dis. 2015;74:998–1003. [PubMed: 24458537]
- 170.
- Myasoedova E, Chandran A, Ilhan B, Major BT, Michet CJ, Matteson EL, Crowson CS. The role of rheumatoid arthritis (RA) flare and cumulative burden of RA severity in the risk of cardiovascular disease. Ann Rheum Dis. 2016;75:560–565. [PMC free article: PMC4520792] [PubMed: 25637001]
- 171.
- Zhang J, Chen L, Delzell E, Muntner P, Hillegass WB, Safford MM, Millan IY, Crowson CS, Curtis JR. The association between inflammatory markers, serum lipids and the risk of cardiovascular events in patients with rheumatoid arthritis. Ann Rheum Dis. 2014;73:1301–1308. [PubMed: 24796336]
- 172.
- Juneblad K, Rantapaa-Dahlqvist S, Alenius GM. Disease Activity and Increased Risk of Cardiovascular Death among Patients with Psoriatic Arthritis. J Rheumatol. 2016;43:2155–2161. [PubMed: 27909142]
- 173.
- Ahlehoff O. Psoriasis and Cardiovascular Disease: epidemiological studies. Dan Med Bull. 2011;58:B4347. [PubMed: 22047936]
- 174.
- Feingold KR, Grunfeld C. Triglyceride Lowering Drugs. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA) 2021.
- 175.
- Feingold KR, Grunfeld C. Cholesterol Lowering Drugs. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA) 2021.
- 176.
- McCarey DW, McInnes IB, Madhok R, Hampson R, Scherbakov O, Ford I, Capell HA, Sattar N. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): double-blind, randomised placebo-controlled trial. Lancet. 2004;363:2015–2021. [PubMed: 15207950]
- 177.
- Semb AG, Holme I, Kvien TK, Pedersen TR. Intensive lipid lowering in patients with rheumatoid arthritis and previous myocardial infarction: an explorative analysis from the incremental decrease in endpoints through aggressive lipid lowering (IDEAL) trial. Rheumatology (Oxford). 2011;50:324–329. [PubMed: 20884656]
- 178.
- Ports WC, Fayyad R, DeMicco DA, Laskey R, Wolk R. Effectiveness of Lipid-Lowering Statin Therapy in Patients With and Without Psoriasis. Clin Drug Investig. 2017;37:775–785. [PubMed: 28573499]
- 179.
- Mok CC, Wong CK, To CH, Lai JP, Lam CS. Effects of rosuvastatin on vascular biomarkers and carotid atherosclerosis in lupus: a randomized, double-blind, placebo-controlled trial. Arthritis Care Res (Hoboken). 2011;63:875–883. [PubMed: 21309005]
- 180.
- Plazak W, Gryga K, Dziedzic H, Tomkiewicz-Pajak L, Konieczynska M, Podolec P, Musial J. Influence of atorvastatin on coronary calcifications and myocardial perfusion defects in systemic lupus erythematosus patients: a prospective, randomized, double-masked, placebo-controlled study. Arthritis Res Ther. 2011;13:R117. [PMC free article: PMC3239355] [PubMed: 21774822]
- 181.
- Schanberg LE, Sandborg C, Barnhart HX, Ardoin SP, Yow E, Evans GW, Mieszkalski KL, Ilowite NT, Eberhard A, Imundo LF, Kimura Y, von Scheven E, Silverman E, Bowyer SL, Punaro M, Singer NG, Sherry DD, McCurdy D, Klein-Gitelman M, Wallace C, Silver R, Wagner-Weiner L, Higgins GC, Brunner HI, Jung L, Soep JB, Reed AM, Provenzale J, Thompson SD. Atherosclerosis Prevention in Pediatric Lupus Erythematosus I. Use of atorvastatin in systemic lupus erythematosus in children and adolescents. Arthritis Rheum. 2012;64:285–296. [PMC free article: PMC4074430] [PubMed: 22031171]
- 182.
- Rollefstad S, Ikdahl E, Hisdal J, Olsen IC, Holme I, Hammer HB, Smerud KT, Kitas GD, Pedersen TR, Kvien TK, Semb AG. Rosuvastatin induced carotid plaque regression in patients with inflammatory joint diseases: The RORA-AS study. Arthritis Rheumatol. 2015 [PubMed: 25778850]
- 183.
- Petri MA, Kiani AN, Post W, Christopher-Stine L, Magder LS. Lupus Atherosclerosis Prevention Study (LAPS). Ann Rheum Dis. 2011;70:760–765. [PubMed: 21177297]
- 184.
- Tam LS, Li EK, Shang Q, Tomlinson B, Lee VW, Lee KK, Li M, Kuan WP, Li TK, Tseung L, Yip GW, Freedman B, Yu CM. Effects of rosuvastatin on subclinical atherosclerosis and arterial stiffness in rheumatoid arthritis: a randomized controlled pilot trial. Scand J Rheumatol. 2011;40:411–421. [PubMed: 21867445]
- 185.
- Norby GE, Holme I, Fellstrom B, Jardine A, Cole E, Abedini S, Holdaas H. Assessment of Lescol in Renal Transplantation Study G. Effect of fluvastatin on cardiac outcomes in kidney transplant patients with systemic lupus erythematosus: a randomized placebo-controlled study. Arthritis Rheum. 2009;60:1060–1064. [PubMed: 19333947]
- 186.
- Semb AG, Kvien TK, DeMicco DA, Fayyad R, Wun CC, LaRosa JC, Betteridge J, Pedersen TR, Holme I. Effect of intensive lipid-lowering therapy on cardiovascular outcome in patients with and those without inflammatory joint disease. Arthritis Rheum. 2012;64:2836–2846. [PubMed: 22576673]
- 187.
- Kitas GD, Nightingale P, Armitage J, Sattar N, Belch JJF, Symmons DPM, Consortium TR. A. Multicenter, Randomized, Placebo-Controlled Trial of Atorvastatin for the Primary Prevention of Cardiovascular Events in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2019;71:1437–1449. [PMC free article: PMC6771601] [PubMed: 30983166]
- 188.
- Lv S, Liu Y, Zou Z, Li F, Zhao S, Shi R, Bian R, Tian H. The impact of statins therapy on disease activity and inflammatory factor in patients with rheumatoid arthritis: a meta-analysis. Clin Exp Rheumatol. 2015;33:69–76. [PubMed: 25327393]
- 189.
- Xing B, Yin YF, Zhao LD, Wang L, Zheng WJ, Chen H, Wu QJ, Tang FL, Zhang FC, Shan G, Zhang X. Effect of 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitor on disease activity in patients with rheumatoid arthritis: a meta-analysis. Medicine (Baltimore). 2015;94:e572. [PMC free article: PMC4554148] [PubMed: 25715256]
- 190.
- Mowla K, Rajai E, Ghorbani A, Dargahi-Malamir M, Bahadoram M, Mohammadi S. Effect of Atorvastatin on the Disease Activity and Severity of Rheumatoid Arthritis: Double-Blind Randomized Controlled Trial. J Clin Diagn Res. 2016;10:OC32–36. [PMC free article: PMC4948444] [PubMed: 27437268]
- 191.
- Mosiewicz J, Pietrzak A, Chodorowska G, Trojnar M, Szepietowski J, Reich K, Rizzo M. Rational for statin use in psoriatic patients. Arch Dermatol Res. 2013;305:467–472. [PubMed: 23754638]
- 192.
- Socha M, Pietrzak A, Grywalska E, Pietrzak D, Matosiuk D, Kicinski P, Rolinski J. The effect of statins on psoriasis severity: a meta-analysis of randomized clinical trials. Arch Med Sci. 2020;16:1–7. [PMC free article: PMC6963135] [PubMed: 32051699]
- 193.
- Fajardo ME, Rocha ML, Sanchez-Marin FJ, Espinosa-Chavez EJ. Effect of atorvastatin on chronic periodontitis: a randomized pilot study. J Clin Periodontol. 2010;37:1016–1022. [PubMed: 20825523]
- 194.
- Norata GD, Catapano AL. Statins and periodontal inflammation: a pleiotropic effect of statins or a pleiotropic effect of LDL-cholesterol lowering? Atherosclerosis. 2014;234:381–382. [PubMed: 24747112]
- 195.
- Subramanian S, Emami H, Vucic E, Singh P, Vijayakumar J, Fifer KM, Alon A, Shankar SS, Farkouh M, Rudd JH, Fayad ZA, Van Dyke TE, Tawakol A. High-dose atorvastatin reduces periodontal inflammation: a novel pleiotropic effect of statins. J Am Coll Cardiol. 2013;62:2382–2391. [PMC free article: PMC6849694] [PubMed: 24070911]
- 196.
- Tralhao AF, Ces de Souza-Dantas V, Salluh JI, Povoa PM. Impact of statins in outcomes of septic patients: a systematic review. Postgrad Med. 2014;126:45–58. [PubMed: 25387213]
- 197.
- Wan YD, Sun TW, Kan QC, Guan FX, Zhang SG. Effect of statin therapy on mortality from infection and sepsis: a meta-analysis of randomized and observational studies. Crit Care. 2014;18:R71. [PMC free article: PMC4056771] [PubMed: 24725598]
- 198.
- Pasin L, Landoni G, Castro ML, Cabrini L, Belletti A, Feltracco P, Finco G, Carozzo A, Chiesa R, Zangrillo A. The effect of statins on mortality in septic patients: a meta-analysis of randomized controlled trials. PLoS One. 2013;8:e82775. [PMC free article: PMC3876996] [PubMed: 24391721]
- 199.
- Pertzov B, Eliakim-Raz N, Atamna H, Trestioreanu AZ, Yahav D, Leibovici L. Hydroxymethylglutaryl-CoA reductase inhibitors (statins) for the treatment of sepsis in adults - a systematic review and meta-analysis. Clin Microbiol Infect. 2018 [PubMed: 30472427]
- 200.
- National Heart. Lung Blood Institute, Ards Clinical Trials Network, Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, Brower RG, Shanholtz C, Rock P, Douglas IS, deBoisblanc BP, Hough CL, Hite RD, Thompson BT. Rosuvastatin for sepsis-associated acute respiratory distress syndrome. N Engl J Med. 2014;370:2191–2200. [PMC free article: PMC4241052] [PubMed: 24835849]
- 201.
- Diaz-Arocutipa C, Melgar-Talavera B, Alvarado-Yarasca A, Saravia-Bartra MM, Cazorla P, Belzusarri I, Hernandez AV. Statins reduce mortality in patients with COVID-19: an updated meta-analysis of 147 824 patients. Int J Infect Dis. 2021;110:374–381. [PMC free article: PMC8349445] [PubMed: 34375760]
- 202.
- Kollias A, Kyriakoulis KG, Kyriakoulis IG, Nitsotolis T, Poulakou G, Stergiou GS, Syrigos K. Statin use and mortality in COVID-19 patients: Updated systematic review and meta-analysis. Atherosclerosis. 2021;330:114–121. [PMC free article: PMC8233054] [PubMed: 34243953]
- 203.
- Inspiration- S. Investigators. Atorvastatin versus placebo in patients with covid-19 in intensive care: randomized controlled trial. BMJ. 2022;376:e068407. [PubMed: 34996756]
- 204.
- Shirinsky I, Polovnikova O, Kalinovskaya N, Shirinsky V. The effects of fenofibrate on inflammation and cardiovascular markers in patients with active rheumatoid arthritis: a pilot study. Rheumatol Int. 2013;33:3045–3048. [PubMed: 23263548]
- 205.
- van Eekeren IC, Clockaerts S, Bastiaansen-Jenniskens YM, Lubberts E, Verhaar JA, van Osch GJ, Bierma-Zeinstra SM. Fibrates as therapy for osteoarthritis and rheumatoid arthritis? A systematic review. Ther Adv Musculoskelet Dis. 2013;5:33–44. [PMC free article: PMC3582306] [PubMed: 23515070]
- 206.
- Roe DA. The clinical and biochemical significance of taurine excretion in psoriasis. J Invest Dermatol. 1962;39:537–542. [PubMed: 13982331]
- 207.
- Skinner RB, Rosenberg EW, Belew PW, Marley WM. Improvement of psoriasis with cholestyramine. Arch Dermatol. 1982;118:144. [PubMed: 7065658]
- 208.
- Maki-Petaja KM, Booth AD, Hall FC, Wallace SM, Brown J, McEniery CM, Wilkinson IB. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol. 2007;50:852–858. [PubMed: 17719471]
- 209.
- Goldberg RJ, Katz J. A meta-analysis of the analgesic effects of omega-3 polyunsaturated fatty acid supplementation for inflammatory joint pain. Pain. 2007;129:210–223. [PubMed: 17335973]
- 210.
- Lee YH, Bae SC, Song GG. Omega-3 polyunsaturated fatty acids and the treatment of rheumatoid arthritis: a meta-analysis. Arch Med Res. 2012;43:356–362. [PubMed: 22835600]
- 211.
- Gioxari A, Kaliora AC, Marantidou F, Panagiotakos DP. Intake of omega-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: A systematic review and meta-analysis. Nutrition. 2018;45:114–124 e114. [PubMed: 28965775]
- 212.
- Millsop JW, Bhatia BK, Debbaneh M, Koo J, Liao W. Diet and psoriasis, part III: role of nutritional supplements. J Am Acad Dermatol. 2014;71:561–569. [PMC free article: PMC4134971] [PubMed: 24780177]
- 213.
- Gamret AC, Price A, Fertig RM, Lev-Tov H, Nichols AJ. Complementary and Alternative Medicine Therapies for Psoriasis: A Systematic Review. JAMA Dermatol. 2018;154:1330–1337. [PubMed: 30193251]
- 214.
- Chen X, Hong S, Sun X, Xu W, Li H, Ma T, Zheng Q, Zhao H, Zhou Y, Qiang Y, Li B, Li X. Efficacy of fish oil and its components in the management of psoriasis: a systematic review of 18 randomized controlled trials. Nutr Rev. 2020;78:827–840. [PubMed: 31995220]
- 215.
- Bello KJ, Fang H, Fazeli P, Bolad W, Corretti M, Magder LS, Petri M. Omega-3 in SLE: a double-blind, placebo-controlled randomized clinical trial of endothelial dysfunction and disease activity in systemic lupus erythematosus. Rheumatol Int. 2013;33:2789–2796. [PMC free article: PMC3805738] [PubMed: 23817872]
- 216.
- Duffy EM, Meenagh GK, McMillan SA, Strain JJ, Hannigan BM, Bell AL. The clinical effect of dietary supplementation with omega-3 fish oils and/or copper in systemic lupus erythematosus. J Rheumatol. 2004;31:1551–1556. [PubMed: 15290734]
- 217.
- Wright SA, O'Prey FM, McHenry MT, Leahey WJ, Devine AB, Duffy EM, Johnston DG, Finch MB, Bell AL, McVeigh GE. A randomised interventional trial of omega-3-polyunsaturated fatty acids on endothelial function and disease activity in systemic lupus erythematosus. Ann Rheum Dis. 2008;67:841–848. [PubMed: 17875549]
- 218.
- Arriens C, Hynan LS, Lerman RH, Karp DR, Mohan C. Placebo-controlled randomized clinical trial of fish oil's impact on fatigue, quality of life, and disease activity in Systemic Lupus Erythematosus. Nutr J. 2015;14:82. [PMC free article: PMC4538741] [PubMed: 26283629]
- 219.
- Walton AJ, Snaith ML, Locniskar M, Cumberland AG, Morrow WJ, Isenberg DA. Dietary fish oil and the severity of symptoms in patients with systemic lupus erythematosus. Ann Rheum Dis. 1991;50:463–466. [PMC free article: PMC1004457] [PubMed: 1877851]
- 220.
- Westberg G, Tarkowski A. Effect of MaxEPA in patients with SLE. A double-blind, crossover study. Scand J Rheumatol. 1990;19:137–143. [PubMed: 2186476]
- 221.
- Clark WF, Parbtani A, Naylor CD, Levinton CM, Muirhead N, Spanner E, Huff MW, Philbrick DJ, Holub BJ. Fish oil in lupus nephritis: clinical findings and methodological implications. Kidney Int. 1993;44:75–86. [PubMed: 8355469]
- 222.
- Elkhouli AM. The efficacy of host response modulation therapy (omega-3 plus low-dose aspirin) as an adjunctive treatment of chronic periodontitis (clinical and biochemical study). J Periodontal Res. 2011;46:261–268. [PubMed: 21261621]
- 223.
- El-Sharkawy H, Aboelsaad N, Eliwa M, Darweesh M, Alshahat M, Kantarci A, Hasturk H, Van Dyke TE. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega-3 Fatty acids and low-dose aspirin. J Periodontol. 2010;81:1635–1643. [PubMed: 20572767]
- 224.
- Sculley DV. Periodontal disease: modulation of the inflammatory cascade by dietary n-3 polyunsaturated fatty acids. J Periodontal Res. 2014;49:277–281. [PubMed: 23889472]
- 225.
- Wang C, Han D, Feng X, Wu J. Omega-3 fatty acid supplementation is associated with favorable outcomes in patients with sepsis: an updated meta-analysis. J Int Med Res. 2020;48:300060520953684. [PMC free article: PMC7783898] [PubMed: 33373266]
- 226.
- Feingold KR. The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- 227.
- Launay-Vacher V, Izzedine H, Deray G. Statins' dosage in patients with renal failure and cyclosporine drug-drug interactions in transplant recipient patients. Int J Cardiol. 2005;101:9–17. [PubMed: 15860377]
- 228.
- Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr, Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM. REDUCE-IT Investigators. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med. 2018
- 229.
- Smit RA, Jukema JW, Trompet S. Increasing HDL-C levels with medication: current perspectives. Curr Opin Lipidol. 2017;28:361–366. [PubMed: 28426523]
- 230.
- Hps Thrive Collaborative Group. Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–212. [PubMed: 25014686]
- 231.
- AIM HIGH Investigators. Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–2267. [PubMed: 22085343]
- 232.
- ACCORD Study Group. Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr., Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–1574. [PMC free article: PMC2879499] [PubMed: 20228404]
- ABSTRACT
- INTRODUCTION
- LIPID AND LIPOPROTEIN ABNORMALITIES IN PATIENTS WITH INFLAMMATORY DISORDERS AND INFECTIONS
- EFFECT OF ANTI-INFLAMMATORY DRUGS ON LIPID LEVELS
- PATHOPHYSIOLOGY OF THE DYSLIPIDEMIA OF INFLAMMATION AND INFECTION
- FUNCTIONAL CHANGES IN LIPOPROTEINS THAT INCREASE THE RISK OF ATHEROSCLEROSIS
- BENEFICIAL EFFECTS OF LIPIDS DURING INFECTIONS AND INFLAMMATION
- LIPID MANAGEMENT IN A PATIENT WITH AN INFLAMMATORY DISEASE
- REFERENCES
- Review The Effect of Endocrine Disorders on Lipids and Lipoproteins.[Endotext. 2000]Review The Effect of Endocrine Disorders on Lipids and Lipoproteins.Feingold KR. Endotext. 2000
- Review Approach to the Patient with Dyslipidemia.[Endotext. 2000]Review Approach to the Patient with Dyslipidemia.Feingold KR. Endotext. 2000
- Review Lipid and Lipoprotein Levels in Patients with COVID-19 Infections.[Endotext. 2000]Review Lipid and Lipoprotein Levels in Patients with COVID-19 Infections.Feingold KR. Endotext. 2000
- Review Obesity and Dyslipidemia.[Endotext. 2000]Review Obesity and Dyslipidemia.Feingold KR. Endotext. 2000
- Review Diabetic dyslipidemia.[Am J Cardiol. 1998]Review Diabetic dyslipidemia.Kreisberg RA. Am J Cardiol. 1998 Dec 17; 82(12A):67U-73U; discussion 85U-86U.
- The Effect of Inflammation and Infection on Lipids and Lipoproteins - EndotextThe Effect of Inflammation and Infection on Lipids and Lipoproteins - Endotext
- HPN hepsin [Homo sapiens]HPN hepsin [Homo sapiens]Gene ID:3249Gene
- 3249[uid] AND (alive[prop]) (1)Gene
- Human papillomavirus type 65 complete genomeHuman papillomavirus type 65 complete genomegi|312100|emb|X70829.1|Nucleotide
- cwf21 [Schizosaccharomyces pombe]cwf21 [Schizosaccharomyces pombe]Gene ID:2543616Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...