By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 1994, Bruce Alberts, Dennis Bray, Julian
Lewis, Martin Raff, Keith Roberts, and James D Watson.
Bookshelf ID: NBK28336

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Alberts B, Bray D, Lewis J, et al. Molecular Biology of the Cell. 3rd edition. New York: Garland Science; 1994.


Having described some of the remarkable devices that cells make out of proteins, we now consider how these devices are produced and how they are destroyed. The mechanism of protein synthesis is discussed elsewhere. We begin by considering how a protein folds and assembles once it leaves the ribosome as a finished polypeptide chain.
Because many purified proteins will refold properly on their own after being unfolded in vitro, for many years it was thought that a protein will try out every conceivable conformation as it folds until it attains the one conformation with the lowest free energy, which was assumed to be its correctly folded state. We now know that this view is incorrect: despite the high speed of molecular motions in a protein (see p. 97), there are vastly more possible conformations for any large protein than can be explored in the few seconds that are typically required for folding. Moreover, the existence of mutant proteins that have specific defects in folding indicates that a protein's amino acid sequence has been selected during evolution, not only for the properties of its final structure, but also for the ability to fold rapidly into its native conformation.
The ability of pure, denatured proteins to reform their native structures on their own has made it possible to dissect the process of protein folding experimentally. Proteins appear to fold rapidly into a structure in which most (but not all) of the final secondary structure (α helices and β sheets) has formed and in which these elements of structure are aligned in roughly the right way (Figure 5-27). This unusually open and flexible conformation, which is called a molten globule (Figure 5-28), is the starting point for a relatively slow process in which many side-chain adjustments occur in order to form the correct tertiary structure. In the latter process a variety of pathways can be taken toward the final conformation. Some of these may be nonproductive dead ends without the help of a molecular chaperone, special proteins in cells whose function is to help other proteins fold and assemble into stable, active structures (see Figure 5-27).

A current view of protein folding. A newly synthesized protein rapidly attains a "molten globule" state (see Figure 5-28). Subsequent folding occurs more slowly and by multiple pathways, some of which reach dead ends without the help of a molecular chaperone. (more...)

The structure of a molten globule. (A) A molten globule form of cytochrome b562 is more open and less highly ordered than the native protein, shown in (B). Note that the molten globule contains most of the secondary structure of the native form, although (more...)
Molecular chaperones were first identified in bacteria when E. coli mutants that failed to allow bacteriophage lambda to replicate in them were studied. These mutants produce slightly altered versions of two components of the chaperone machinery, related to heat-shock proteins 60 and 70 (hsp60 and hsp70), and as a result are defective in specific steps in the assembly of the viral proteins.
Eucaryotic cells have families of hsp60 and hsp70 proteins, and different family members function in different organelles. Thus, as discussed in Chapter 12, mitochondria contain their own hsp60 and hsp70 molecules that are distinct from those that function in the cytosol, and a special hsp70 (called BIP) helps to fold proteins in the endoplasmic reticulum.
Both hsp60-like and hsp70 proteins work with a small set of associated proteins when they help other proteins to fold. They share an affinity for the exposed hydrophobic patches on incompletely folded proteins, and they hydrolyze ATP, possibly binding and releasing their protein with each cycle of ATP hydrolysis. Originally, molecular chaperones were thought to act only by preventing the promiscuous aggregation of still unfolded proteins (hence their name). It is now thought, however, that they also interact more intimately with their clients, producing effects that can be likened to a "protein massage." By binding to exposed hydrophobic regions, the chaperone massages those regions of a protein that are likely to have misfolded from the molten globule state, changing their structure in a way that gives the protein another chance to fold (see Figure 5-27).
In some other respects the two types of hsp proteins function differently. The hsp70 machinery is thought to act early in the life of a protein, binding to a string of about seven hydrophobic amino acids before the protein leaves the ribosome (Figure 5-29). In contrast, hsp60-like proteins form a large barrel-shaped structure (Figure 5-30) that acts later in a protein's life; this chaperone is thought to form an "isolation chamber" into which misfolded proteins are fed, providing them with a favorable environment in which to attempt to refold (see Figure 5-29).

Two families of molecular chaperones. The hsp70 proteins act early, recognizing small patches on a protein's surface. The hsp60-like proteins appear to act later and form a container into which proteins that have still failed to fold are transferred. (more...)

The structure of an hsp60-like chaperone, as determined by electron microscopy. A large number of negatively stained particles is shown in (A) and a 3-D model of a single particle, derived by computer-based image processing methods, is shown in (B). A (more...)
These molecular chaperones are called heat-shock proteins because they are synthesized in dramatically increased amounts following a brief exposure of cells to an elevated temperature (for example, 42°C). This seems to reflect the operation of a feedback system that responds to any increase in misfolded proteins (such as those produced by elevated temperatures) by boosting the synthesis of the chaperones that help the protein refold.
The folding of a newly synthesized protein often begins with the formation of a number of distinct structurally stable domains that correspond to functional units, which seem to have ancient evolutionary origins. Elsewhere we discuss the pathways by which proteins are thought to have evolved, emphasizing how new proteins have been created by the shuffling of exons that code for conserved domains with useful properties (see pp. 386-394). Evolution has preserved some of these domains as folding units that retain their structure even when cut out of the protein - either by selected proteolysis or, more efficiently, by genetic engineering techniques. Protein domains of this type that are very frequently involved in evolutionary exon shuffling are called modules; their importance has become clear now that DNA sequences are available for thousands of genes.
Protein modules are typically 40 to 100 amino acids in length. Their small size and ability to fold independently has made it possible to determine many of their three-dimensional structures in solution by high-resolution NMR techniques, which is a convenient alternative to x-ray crystallography. Some typical modules are illustrated in Figure 5-31. Each of these modules has a stable core structure formed from strands of β sheet, from which less-ordered loops of polypeptide chain protrude (shown in green). The loops are ideally situated to form binding sites for other molecules, as well demonstrated for the immunoglobulin fold, which was first recognized in antibody molecules (see Figure 23-35). The evolutionary success of β-sheet-based modules is likely to have been due to their forming a convenient framework for the generation of new binding sites for ligands through changes to these protruding loops.
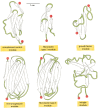
The three-dimensional structures of some protein modules. In these ribbon diagrams, beta-sheet strands are shown as arrows, and the N- and C-termini are marked with red balls. (Adapted from M. Baron, D.G. Norman, and I.D. Campbell, Trends Biochem. Sci. (more...)
A second feature of protein modules that explains their utility is the ease with which they can be integrated into other proteins. Five of the six modules illustrated in Figure 5-31 have their N- and C-terminal ends (marked with red balls) at opposite ends of the module. This "in-line" arrangement means that when the DNA encoding such a module undergoes tandem duplication, which is not unusual in the evolution of genomes (discussed in Chapter 8), the duplicated modules can be readily accommodated in the protein. In this way such modules can become linked in series to form extended structures, either with themselves (Figure 5-32) or with other in-line modules. Stiff extended structures composed of a series of modules are commonly found both in extracellular matrix molecules and in the extracellular portions of cell surface receptor proteins.
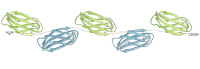
An extended structure formed from a series of in-line protein modules. Here, five fibronectin type 3 modules are shown forming a repeating array. Similar structures are found in several extracellular matrix molecules. Side-chain interactions between the ends (more...)
Other modules, like the kringle module in Figure 5-31, are of a "plug-in" type. After genomic rearrangements, they can be easily accommodated as an insertion into a loop region of a second protein. Some of these modules act as specific binding sites for other proteins or structures in the cell. An important example is the SH2 domain, which can bind tightly to a region of polypeptide chain that contains a phosphorylated tyrosine side chain. Because each SH2 domain also recognizes other features of the polypeptide, it binds only to a subset of proteins that contains phosphorylated tyrosines. The presence of an SH2 domain in a protein allows it to form complexes with proteins that become phosphorylated on tyrosines in response to cell-signaling events (Figure 5-33). Such protein complexes that form and break up as a result of changes in protein phosphorylation play a central part in transducing extracellular signals into intracellular ones, as described in Chapter 15.
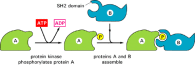
SH2 domains mediate protein assembly reactions that depend on protein phosphorylations. The structure of an SH2 domain, which has the form of a plug-in module, is illustrated in Figure 15-49.
Proteins can bind to other proteins in at least three ways. In many cases a portion of the surface of one protein contacts an extended loop of polypeptide chain (a "string") on a second protein (Figure 5-34A). Such a surface-string interaction, for example, allows the SH2 domain to recognize a phosphorylated loop of another protein, and it also enables a protein kinase to recognize the proteins that it will phosphorylate (see Figure 5-16B).

Three ways that two proteins can bind to each other. Only the interacting parts of the two proteins are shown. (A) A rigid surface on one protein can bind to an extended loop of polypeptide chain (a "string") on a second protein. (B) Two alpha helices (more...)
A second type of protein-protein interface is formed when two alpha helices, one from each protein, pair together to form a coiled-coil (Figure 5-34B). This type of protein interface is found in several families of gene regulatory proteins, as discussed in Chapter 9.
The most common way for proteins to interact, however, is by the precise matching of one rigid surface with that of another (Figure 5-34C). Such interactions can be very tight, since a large number of weak bonds can form between two surfaces that match well. For the same reason such surface-surface interactions can be extremely specific, allowing one protein to select a specific partner from the many thousands of different proteins found in a higher eucaryotic cell.
Many proteins are present in large complexes with other proteins. This requires that the protein bind to several other proteins at the same time. It is crucial for the cell that each protein complex form efficiently and that the formation of partial complexes, which can interfere with the function of complete complexes, be kept to a minimum. There must be mechanisms, therefore, for ensuring that assembly is an all-or-none process.
One important mechanism relies on the phenomenon of linkage, which we described earlier. Because of linkage, if a ligand changes the shape of an allo-steric protein so that the protein binds a second ligand more tightly, the second ligand must similarly increase the affinity of the protein for the first ligand (see Figure 5-5). The same principle applies to protein-protein interactions. When two proteins bind to each other, they often increase the affinity of one of the partners for a third protein. Because of linkage, the complex of all three proteins will be much more stable than a complex containing only two. A mechanism of this type can produce all-or-none assembly (Figure 5-35).
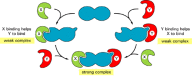
Linkage facilitates an efficient all-or-none assembly of protein complexes. As indicated, proteins X and Y each induce an allosteric shape change in a third protein (shown in blue) that helps the other protein to bind. As a result, the complex of all (more...)
Even if an all-or-none assembly mechanism drives the formation of complete protein complexes, unless the cell contains exactly the right proportions of each protein in the complex, unassembled proteins will be left over. In fact, cells do not always produce their components in precise amounts and are instead able to degrade selectively any protein component that is left unassembled (Figure 5-36). Cells therefore require a sophisticated system to identify abnormally assembled proteins and destroy them. Indeed, the eucaryotic cell contains an elaborate set of proteins that enables such incomplete assemblies to be selectively directed to its protein-degradation machinery, as we now discuss.
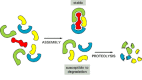
Proteolysis of the extra components of a protein complex prevents them from accumulating in a cell. The degradation shown here requires that an unassembled protein be recognized by enzymes that covalently add ubiquitin to it, as discussed in the text. (more...)
One function of intracellular proteolytic mechanisms is to recognize and eliminate unassembled proteins, as just described. Another is to dispose of damaged or misfolded proteins (see Figure 5-27). Yet another is to confer short half-lives on certain normal proteins whose concentrations must change promptly withalterations in the state of a cell; many of these short-lived proteins are degraded rapidly at all times, while others, most notably the cyclins, are stable until they are suddenly degraded at one particular point in the cell cycle. Although here we mainly discuss how proteins are degraded in the cytosol, important degradation pathways also operate in the endoplasmic reticulum (ER) and, as discussed in Chapter 13, in lysosomes.
Most of the proteins that are degraded in the cytosol are delivered to large protein complexes called proteasomes, which are present in many copies and are dispersed throughout the cell. Each proteasome consists of a central cylinder formed from multiple distinct proteases, whose active sites are thought to face an inner chamber. Each end of the cylinder is "stoppered" by a large protein complex formed from at least 10 types of polypeptides, some of which hydrolyze ATP (Figure 5-37). These protein stoppers are thought to select the proteins for destruction by binding to them and feeding them into the inner chamber of the cylinder, where multiple proteases degrade the proteins to short peptides that are then released.

A proteasome. A large number of negatively stained particles is shown in (A). A 3-D model of a single complete proteasome complex, derived by computer-based image processing of such images, is shown in (B). Many copies of this structure are present throughout (more...)
Proteasomes act on proteins that have been specifically marked for destruction by the covalent attachment of a small protein called ubiquitin (Figure 5-38). Ubiquitin exists in cells either free or covalently linked to proteins. Most ubiquinated proteins have been tagged for degradation. (Some long-lived proteins such as histones are also ubiquinated, but in these cases the function of ubiquitin is not understood.) Different ubiquitin-dependent proteolytic pathways employ structurally similar but distinct ubiquitin-conjugating enzymes that are associated with recognition subunits that direct them to proteins carrying a particular degradation signal. The conjugating enzyme adds ubiquitin to a lysine residue of a target protein and thereafter adds a series of additional ubiquitin moieties, forming a multiubiquitin chain (Figure 5-39) that is thought to be recognized by a specific receptor protein in the proteasome.

The three-dimensional structure of ubiquitin. This protein contains 76 amino acid residues. The addition of a chain of ubiquitin molecules to a protein results in the degradation of this protein by the proteasome (see Figure 5-39). (Based on S. Vijay-Kumar, (more...)

Ubiquitin-dependent protein degradation. In step 1 a target protein (containing a degradation signal) is recognized by the ubiquitinating enzyme complex. Then, in step 2 a repeated series of biochemical reactions joins ubiquitin molecules together to (more...)
Denatured or misfolded proteins, as well as proteins containing oxidized or otherwise abnormal amino acids, are recognized and degraded by ubiquitin-dependent proteolytic systems. The ubiquitin-conjugating enzymes presumably recognize signals that are exposed on these proteins as a result of their misfolding or chemical damage; such signals are likely to include amino acid sequences or conformational motifs that are buried and therefore inaccessible in the normal counterparts of these proteins.
A proteolytic pathway that recognizes and destroys abnormal proteins must be able to distinguish between completed proteins that have "wrong" conformations and the many growing polypeptides on ribosomes (as well as polypeptides just released from ribosomes) that have not yet achieved their normal folded conformation. That this is not a trivial problem can be demonstrated experimentally: if puromycin - an inhibitor of protein synthesis - is added to cells, the prematurely terminated proteins that are formed are rapidly degraded by a ubiquitin-dependent pathway. One possibility is that the normally forming proteins are temporarily protected by the translation machinery or by chaperone molecules. Another is that nascent and newly completed proteins are actually vulnerable to proteolysis but manage to fold up into their native conformations fast enough to escape being targeted for destruction by proteolysis.
One feature that has an important influence on the stability of a protein is the nature of the first (N-terminal) amino acid in the polypeptide chain. There is a strong relation, called the N-end rule, between the in vivo half-life of a protein and the identity of its N-terminal amino acid. Distinct versions of the N-end rule operate in all organisms examined, from bacteria to mammals. The amino acids Met, Ser, Thr, Ala, Val, Cys, Gly, or Pro, for example, protect proteins in the yeast S. cerevisiae when present at the N-terminus; these amino acids are not recognized by targeting components of the N-end rule pathway, while the remaining 12 amino acids attract a proteolytic attack. Most of the proteins that are rapidly degraded by the N-end rule pathway (which operates in both the cytosol and the nucleus) remain to be identified. Since destabilizing amino acids, however, are rare at the N-termini of cytosolic proteins but are frequently present at the N-terminus of proteins that have been transported to other compartments, one hypothetical function of the N-end rule pathway is to degrade proteins that normally function in the ER, the Golgi apparatus, or another membrane-bounded compartment but for some reason have leaked back into the cytosol.
It is not known how destabilizing amino acids become exposed at the N-terminus of a newly formed protein. As discussed in Chapter 6, all proteins are initially synthesized with methionine (or formyl-methionine in bacteria) as their N-terminal amino acid. This methionine, which is a stabilizing amino acid in the N-end rule, is often removed by a specific aminopeptidase. The presently known methionine aminopeptidases, however, will remove the N-terminal methionine if and only if the second amino acid is also stabilizing in the N-end rule. The proteases that produce physiological substrates of the N-end rule pathway, and the sequences they recognize as signals for cleavage, remain to be discovered.
Certain destabilizing N-terminal amino acids, such as aspartate and glutamate, are not recognized directly by the targeting component of the N-end rule pathway. Instead, they are modified by the enzyme arginyl-tRNA-protein transferase, which links arginine, one of the directly recognized destabilizing amino acids, to the N-terminus of proteins bearing N-terminal aspartate or glutamate. Arginine is thus one of the primary destabilizing amino acids in the N-end rule, while aspartate and glutamate are secondary destabilizing amino acids. In eucaryotes there are also tertiary destabilizing N-terminal amino acids - asparagine and glutamine - which are destabilizing through their conversion, by a specific amidase, into the secondary destabilizing amino acids aspartate and glutamate.
The N-terminal amino acid of a protein is often found to be resistant to hydrolysis by the reagents used in protein sequenators. Such proteins have a chemically modified ("blocked") N-terminus, the most frequent modification being acetylation. This modification was believed to play a role in protecting long-lived proteins from degradation. However, recent experiments with yeast mutants that lack the major species of N-terminal acetylase, so that the bulk of the normally acetylated proteins are unacetylated, show that most of these unacetylated proteins remain long-lived. The function of N-terminal acetylation in these proteins remains to be deciphered.
From the moment of its birth on a ribosome to its death by targeted proteolysis, a protein is accompanied by molecular chaperones and other surveying devices whose purpose is to massage it into shape, repair it, or eliminate it. Misfolded proteins are first induced to refold correctly by hsp70 or hsp60 chaperone molecules; if this fails, they are coupled to ubiquitin and thereby targeted for digestion in proteasomes.
Proteins are often composed of discrete modular domains that have been juxtaposed during evolution by duplication and shuffling of the DNA sequences that encode the modules. The modules often contain specific binding sites for other molecules, including other proteins, and they often enable proteins to assemble into large complexes. The principle of linkage explains how cells manage to use allosteric transitions to assemble such protein complexes in an all-or-none fashion.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...