By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 1999, American Society for
Neurochemistry.
Bookshelf ID: NBK28090

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Siegel GJ, Agranoff BW, Albers RW, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition. Philadelphia: Lippincott-Raven; 1999.

Because GABA is widely distributed and utilized throughout the CNS, early GABAergic drugs had very generalized effects on CNS function. The development of more selective agents has led to the identification of at least two distinct classes of GABA receptor, GABAA and GABAB. They differ in their pharmacological, electrophysiological and biochemical properties. Electrophysiological studies of the GABAA-receptor complex indicate that it mediates an increase in membrane conductance with an equilibrium potential near the resting level of −70 mV. This conductance increase often is accompanied by a membrane hyperpolarization, resulting in an increase in the firing threshold and, consequently, a reduction in the probability of action potential initiation, causing neuronal inhibition. This reduction in membrane resistance is accomplished by the GABA-dependent facilitation of Cl− ion influx through a receptor-associated channel. On the other hand, increased Cl− permeability can depolarize the target cell under some conditions of high intracellular Cl−. This in turn potentially can excite the cell to fire or to activate Ca2+ entry via voltage-gated channels and has been proposed as a physiologically relevant event, especially in embryonic neurons.
Electrophysiological data [8] suggest that there are two GABA-recognition sites per GABAA-receptor complex. An increase in the concentration of GABA results in an increase in the mean channel open time due to opening of doubly liganded receptor forms, which exhibit open states of long duration. It has been demonstrated, using a membrane preparation from rat brain, that the increase in the ionic permeability of the GABAA receptor complex is transient in the continuing presence of agonist [9]. This phenomenon is known as desensitization and is rapidly reversible. The molecular mechanism of desensitization is not understood, and various hypotheses remain under investigation. The existence of GABA-binding sites specific for the initiation of desensitization and distinct from sites mediating opening of the Cl− channel has been proposed [9].
Less is known about the GABAB receptor, primarily due to the limited number of pharmacological agents selective for this site. Originally, GABAB receptors were identified by their insensitivity to the GABAA antagonist bicuculline and certain GABAA-specific agonists [1,10]. The GABA analog ( − )baclofen (β-(4-chloro-phenyl)-γ-aminobutyric acid) was found to be a potent and selective GABAB agonist.
GABAB receptors are coupled indirectly to K+ channels. When activated, these receptors can decrease Ca2+ conductance and inhibit cAMP production via intracellular mechanisms mediated by G proteins. GABAB receptors can mediate both postsynaptic and presynaptic inhibition. Presynaptic inhibition may occur as a result of GABAB receptors on nerve terminals causing a decrease in the influx of Ca2+, thereby reducing the release of neurotransmitters. The cloning of the GABAB receptor [11] and its structural similarity to the metabotropic glutamate receptors should allow rapid progress in the pharmacological characterization of receptor subtypes and the development of new drugs of improved selectivity. Pharmacological responses to GABA that are insensitive to both bicuculline and baclofen have been termed GABAC receptors. Some, but not all, of these responses can be explained by a structural analog of GABAA receptors, the ρ subunit [10].
The complex includes five major binding domains (Fig. 16-2). These include binding sites localized in or near the Cl− channel for GABA, benzodiazepines, barbiturates and picrotoxin as well as binding sites for the anesthetic steroids. These binding domains modulate receptor response to GABA stimulation. In addition, other drugs, including volatile anesthetics, ethanol and penicillin, have been reported to have an effect on this receptor [3,8]. An integral part of this complex is the Cl− channel. The GABA-binding site is directly responsible for opening the Cl− channel. A variety of agonists bind to this site and elicit GABA-like responses. One of the most useful agonists is the compound muscimol, a naturally occurring GABA analog isolated from the psychoactive mushroom Amanita muscaria. It is a potent and specific agonist at GABAA receptors and has been a valuable tool for pharmacological and radioligand-binding studies [10,12]. Other GABA agonists include isoguvacine, 4,5,6,7-tetrahydroisoxazolo-[5,4-c]py-ridin-3-ol (THIP), 3-aminopropane-sulfonate and imidazoleacetic acid [12]. The classical GABAA-receptor antagonist is the convulsant bicuculline, which reduces current by decreasing the opening frequency and mean open time of the channel [8,10]. It is likely that bicuculline produces its antagonistic effects on GABAAreceptor currents by competing with GABA for binding to one or both sites on the GABAA receptor.
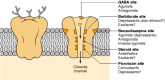
Structural model of the GABAA benzodiazepine receptor—chloride (Cl−) ionophore complex. The cut-away view demonstrates targets for a variety of compounds that influence the receptor complex. No specific drug receptor location is implied. (more...)
Among these are benzodiazepines, intravenous and volatile anesthetics and possibly ethanol. Benzodiazepine receptor-binding sites copurify with the GABA-binding sites [13]. In addition, benzodiazepine receptors are immunoprecipitated with antibodies that were developed to recognize the protein containing the GABA-binding site [14]. This indicates that the benzodiazepine receptor is an integral part of the GABAA receptor—Cl− channel complex.
Benzodiazepine agonists represent the newest group of agents in the general class of depressant drugs, which also includes barbiturates, that show anticonvulsant, anxiolytic and sedative—hypnotic activity. Well-known examples include diazepam and chlordiazepoxide, which often are prescribed for their anti-anxiety effects [1]. The mechanism of action of benzodiazepine agonists is to enhance GABAergic transmission. From electrophysiological studies, it is known that these benzodiazepines increase the frequency of channel opening in response to GABA, thus accounting for their pharmacological and therapeutic actions [8]. In addition, the benzodiazepine site is coupled allosterically to the barbiturate and picrotoxin sites [2]. Benzodiazepine receptors are heterogeneous with respect to affinity for certain ligands. A wide variety of nonbenzodiazepines, such as the β-carbolines, cyclopyrrolones and imidazopyridines, also bind to the benzodiazepine site.
Barbiturates comprise another class of drugs commonly used therapeutically for anesthesia and control of epilepsy. Phenobarbital and pentobarbital are two of the most commonly used barbiturates. Phenobarbital has been used to treat patients with epilepsy since 1912. Pentobarbital is also an anticonvulsant, but it has sedative side effects. Barbiturates at pharmacological concentrations allosterically increase binding of benzodiazepines and GABA to their respective binding sites [2]. Measurements of mean channel open times show that barbiturates act by increasing the proportion of channels opening to the longest open state (9 msec) while reducing the proportion opening to the shorter open states (1 and 3 msec), resulting in an overall increase in mean channel open time and Cl− flux [8].
Channel blockers, such as the convulsant compound picrotoxin, cause a decrease in mean channel open time. Picrotoxin works by preferentially shifting opening channels to the briefest open state (1 msec). Thus, both picrotoxin and barbiturates appear to act on the gating process of the GABAA receptor channel, but their effects on the open states are opposite to each other. Experimental convulsants like pentylenetetrazol and the cage convulsant t-butyl bicyclophosphorothionate (TBPS) act in a manner similar to picrotoxin, preventing Cl− channel permeability. The antibiotic penicillin is a channel blocker with a net negative charge. It blocks the channel by interacting with the positively charged amino acid residues within the channel pore, consequently occluding Cl− passage through the channel [8].
There have been numerous studies on the role of GABAA receptors in anesthesia. A considerable amount of evidence has been compiled to suggest that general anesthetics, including barbiturates, volatile gases, steroids and alcohols, enhance GABA-mediated Cl− conductance. A proper assessment of this phenomenon requires not only a behavioral assay of anesthesia but also in vitro models for the study of receptor function. In this regard, not only electrophysiological methods but also neurochemical measurements of Cl− flux and ligand binding have been useful. For example, a strong positive correlation exists between anesthetic potencies and the stimulation of GABA-mediated Cl− uptake. This is seen with barbiturates and anesthetics in other chemical classes [3].
Comparison of ligand-gated ion channels that vary in sensitivity to anesthetic modulation, using the chimera and site-directed mutagenesis approach, has identified two amino acids in the membrane-spanning domains that are critical for anesthetic sensitivity [14a]. Direct evidence of ethanol augmentation of GABAA receptor function, measured either by electrophysiological techniques or agonist-mediated Cl− flux, has been reported [3,15]. The similarity between the actions of ethanol and sedative drugs such as benzodiazepines and barbiturates that enhance GABA action suggests that ethanol may exert some of its effects by enhancing the function of GABAA receptors. Ethanol potentiation of GABAA receptor function appears to be dependent upon the cell type tested and the method of assay. This suggests that the ethanol interaction may be specific for certain receptor subtypes and/or that it may be an indirect action [3].
This enhancement by steroids involves direct action on the membrane receptor protein rather than through the classical genomic mechanism mediated by soluble high-affinity cytoplasmic steroid hormone receptors (see Chap. 49). Chemically reduced analogs of the hormones progesterone and corticosterone derivatives administered to animals and humans exert sedative—hypnotic and anti-anxiety effects. This led to the development of a synthetic steroid anesthetic, alphaxalone. These neuroactive steroids are potent modulators of GABAA-receptor function in vitro [2,3,16]. The neuroactive steroids can be produced in the brain endogenously and may influence CNS function under certain physiological or pathological conditions. Some observations suggesting that neurosteroids physiologically affect the CNS include the rapid behavioral effects of administered steroids; diurnal and estrous cycle effects on behavior; gender-specific pharmacology, especially of GABAergic drugs; and the development of withdrawal symptoms following cessation of chronically administered steroids. Neuroactive steroids have effects similar to those of barbiturates in that they enhance agonist binding to the GABA site and allosterically modulate benzodiazepine and TBPS binding [2,3]. Also, like barbiturates, high concentrations of neurosteroids directly activate the GABAA receptor Cl− channel. These observations led to the hypothesis that the neurosteroid-binding site may be similar to the barbiturate site, but the sites of action for the two classes of drugs are clearly not identical [2].
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...