

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Diabetic ketoacidosis (DKA) and hyperglycemic hyperosmolar state (HHS) are acute metabolic complications of diabetes mellitus that can occur in patients with both type 1 and 2 diabetes mellitus. Timely diagnosis, comprehensive clinical and biochemical evaluation, and effective management is key to the successful resolution of DKA and HHS. Critical components of the hyperglycemic crises’ management include coordinating fluid resuscitation, insulin therapy, and electrolyte replacement along with the continuous patient monitoring using available laboratory tools to predict the resolution of the hyperglycemic crisis. Understanding and prompt awareness of potential special situations such as DKA or HHS presentation in the comatose state, possibility of mixed acid-base disorders obscuring the diagnosis of DKA, and risk of brain edema during therapy are important to reduce the risks of complications without affecting recovery from hyperglycemic crisis. Identification of factors that precipitated DKA or HHS during the index hospitalization should help prevent subsequent episode of hyperglycemic crisis. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic state (HHS) represent two extremes in the spectrum of decompensated diabetes. DKA and HHS remain important causes of morbidity and mortality among diabetic patients despite well-developed diagnostic criteria and treatment protocols (1). The annual incidence of DKA from population-based studies in 1980s was estimated to range from 4 to 8 episodes per 1,000 patient admissions with diabetes (2); the annualized incidence remains stable based on the 2017 national inpatient sample analysis (3). Overall, the incidence of DKA admissions in the US continues to increase, accounting for about 140,000 hospitalizations in 2009 (Figure 1 a), 168,000 hospitalizations in 2014 (4,5), and most recently 220,340 admissions in 2017 (3) with similar trends observed in England (6) and Finland (7). The 2014 DKA hospitalization rates were the highest in persons aged <45 years (44.3 per 1,000) and lowest in persons aged ≥65 years (<2.0 per 1,000) (5); the age-related admission patterns remained the same in the 2017 analyses (3). The rate of hospital admissions for HHS is lower than of DKA and is less than 1% of all diabetic-related admissions (8,9). About 2/3 of adults presenting to the emergency department or admitted with DKA have a past history of type 1 diabetes (T1D), while almost 90% of the HHS patients have a known diagnosis of type 2 diabetes (T2D) (5). In 2014, there were reported 207,000 emergency department visits with a diagnosis of hyperglycemic crisis (10). Decompensated diabetes imposes a heavy burden in terms of economics and patient outcomes. DKA is responsible for more than 500,000 hospital days per year at an estimated annual direct medical expense and indirect cost of 2.4 billion USD in 1997 (CDC) (11). The cost of inpatient DKA care in the US has increased to 5.1 billion USD in 2014, corresponding to approximate charges related to DKA care varying between 20-26 thousand USD per admission (12,13) and continued to increase in 2017 when DKA admissions costed healthcare about 6.76 billion USD, corresponding to about 31 thousand USD per each admission (3). The mortality rate for DKA and hyperglycemic crises has been falling over the years (Figure 1b) (4) with estimates of fatality remaining under 1% for DKA (3); mortality can reach up to 20% in HHS (14). In 2010, among adults aged 20 years or older, hyperglycemic crisis caused 2,361 deaths (15). There was a decline in mortality from 2000 to 2014 across all age groups and both sexes with largest absolute decrease among persons aged ≥75 years (5). The mortality rate of HHS is higher, reaching 10-20% depending on associated comorbidities and severity of the initial presentation compared with DKA (14,16,17) and is highest in those with DKA+HHS (18). Severe dehydration, older age, and the presence of comorbid conditions in patients with HHS account for the higher mortality in these patients (17). Recent analyses suggested that patients who are black, female, and/or having Medicaid insurance had the highest risk of being admitted with DKA (3).

Figure 1.
Hyperglycemic Crises. A) Incidence of DKA 1980-2009 B) Crude and Age-Adjusted Death Rates for Hyperglycemic Crises as Underlying Cause per 100,000 Diabetic Population, United States, 1980–2009 C) Age-Adjusted DKA hospitalization rate per 1,000 persons with diabetes and in-hospital case-fatality rate, United States, 2000–2014 (5).
DEFINITIONS
DKA consists of the biochemical triad of hyperglycemia, ketonemia, and high anion gap metabolic acidosis (19) (Figure 2). The terms “hyperglycemic hyperosmolar nonketotic state” and “hyperglycemic hyperosmolar nonketotic coma” have been replaced with the term “hyperglycemic hyperosmolar state” (HHS) to highlight that 1) the hyperglycemic hyperosmolar state may consist of moderate to variable degrees of clinical ketosis detected by nitroprusside method, and 2) alterations in consciousness may often be present without coma.
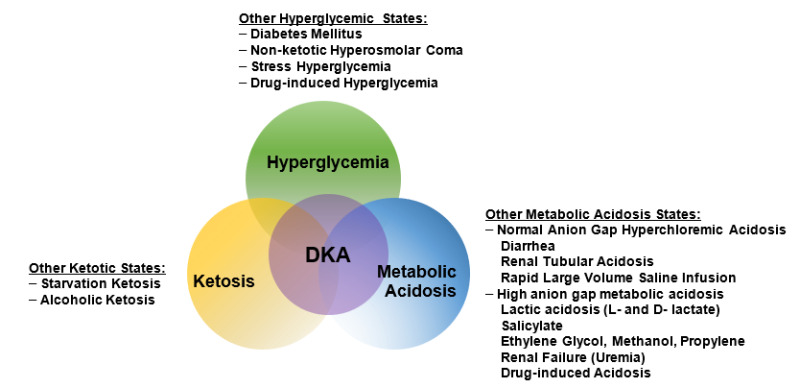
Figure 2.
The triad of DKA (hyperglycemia, acidemia, and ketonemia) and other conditions with which the individual components are associated. From Kitabchi and Wall (19).
Both DKA and HHS are characterized by hyperglycemia and absolute or relative insulinopenia. Clinically, they differ by the severity of dehydration, ketosis, and metabolic acidosis (17).
DKA most often occurs in patients with T1D. It also occurs in T2D under conditions of extreme stress, such as serious infection, trauma, cardiovascular or other emergencies, and, less often, as a presenting manifestation of T2D, a disorder called ketosis-prone T2D (16). Similarly, whereas HHS occurs most commonly in T2D, it can be seen in T1D in conjunction with DKA. Presentations with overlapping DKA and HHS accounted for 27% of admissions for hyperglycemic crises based on one report (18).
PATHOGENESIS
The underlying defects in DKA and HHS are 1) reduced net effective action of circulating insulin as a result of decreased insulin secretion (DKA) or ineffective action of insulin in HHS (20-22), 2) elevated levels of counter regulatory hormones: glucagon (23,24), catecholamines (23,25), cortisol (23), and growth hormone (26,27), resulting in increased hepatic glucose production and impaired glucose utilization in peripheral tissues, and 3) dehydration and electrolyte abnormalities, mainly due to osmotic diuresis caused by glycosuria (28) (Figure 3). Diabetic ketoacidosis is also characterized by increased gluconeogenesis, lipolysis, ketogenesis, and decreased glycolysis (16).
Diabetic Ketoacidosis
In DKA, there is a severe alteration of carbohydrate, protein, and lipid metabolism (8). In general, the body is shifted into a major catabolic state with breakdown of glycogen stores, hydrolysis of triglycerides from adipose tissues, and mobilization of amino acids from muscle (16). The released triglycerides and amino acids from the peripheral tissues become substrates for the production of glucose and ketone bodies by the liver (29). Hyperglycemia and ketone bodies production play central roles in developing this metabolic decompensation (30).
HYPERGLYCEMIA
The hyperglycemia in DKA is the result of three events: (a) increased gluconeogenesis; (b) increased glycogenolysis, and (c) decreased glucose utilization by liver, muscle, and fat. Insulinopenia and elevated cortisol levels also lead to a shift from protein synthesis to proteolysis with resultant increase in production of amino acids (alanine and glutamine), which further serve as substrates for gluconeogenesis (8,31). Furthermore, muscle glycogen is catabolized to lactic acid via glycogenolysis. The lactic acid is transported to the liver in the Cori cycle where it serves as a carbon skeleton for gluconeogenesis (32). Increased levels of glucagon, catecholamines, and cortisol with concurrent insulinopenia stimulate gluconeogenic enzymes, especially phosphoenol pyruvate carboxykinase (PEPCK) (26,33). Decreased glucose utilization is further exaggerated by increased levels of circulating catecholamines and FFA (34).
KETOGENESIS
Excess catecholamines coupled with insulinopenia promote triglyceride breakdown (lipolysis) to free fatty acids (FFA) and glycerol in adipose tissue. The latter provides a carbon skeleton for gluconeogenesis, while the former serves as a substrate for the formation of ketone bodies (35,36). The key regulatory site for fatty acid oxidation is known to be carnitine palmitoyl transferase 1(CPT1) which is inhibited by malonyl CoA in the normal non-fasted state but the increased ratio of glucagon and other counter regulatory hormones to insulin disinhibit fatty acid oxidation and incoming fatty acids from fat tissue can be converted to ketone bodies (37,38). Increased production of ketone bodies (β-hydroxybutyrate and acetoacetate) leads to ketonemia (39). Ketonemia is further maintained by the reduced liver clearance of ketone bodies in DKA. Extracellular and intracellular buffers neutralize hydrogen ions produced during hydrolysis of ketoacids. When overwhelming ketoacid production exceeds buffering capacity, a high anion gap metabolic acidosis develops. Studies in diabetic and pancreatectomized patients have demonstrated the cardinal role of hyperglucagonemia and insulinopenia in the genesis of DKA (40). In the absence of stressful situations, such as intravascular volume depletion or intercurrent illness, ketosis is usually mild (16,41).
Elevated levels of pro-inflammatory cytokines and lipid peroxidation markers, as well as procoagulant factors such as plasminogen activator inhibitor-1 (PAI-1) and C-reactive protein (CRP) have been demonstrated in DKA. The levels of these factors return to normal after insulin therapy and correction of hyperglycemia (42). This inflammatory and procoagulant state may explain the well-known association between hyperglycemic crisis and thrombotic state (43,44).
Hyperglycemic Hyperosmolar State
While DKA is a state of near absolute insulinopenia, there is sufficient amount of insulin present in HHS to prevent lipolysis and ketogenesis but not adequate to cause glucose utilization (as it takes 1/10 as much insulin to suppress lipolysis as it does to stimulate glucose utilization) (33,34). In addition, in HHS there is a smaller increase in counter regulatory hormones (20,45).
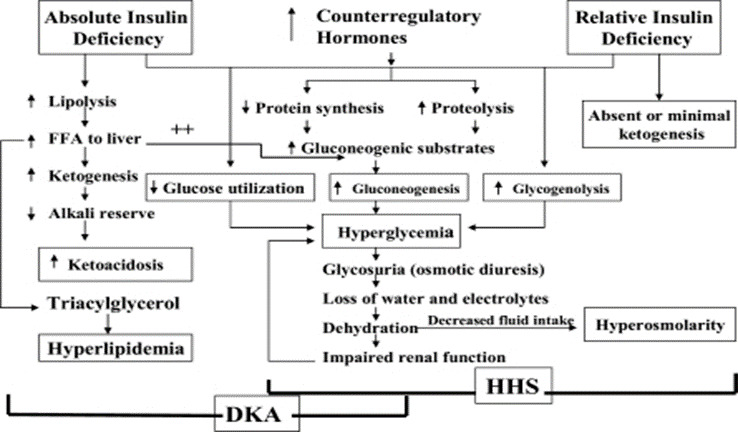
Figure 3.
Pathogenesis of DKA and HHS: stress, infection, or insufficient insulin. FFA, free fatty acid. Adapted from Kitabchi et al. (1).
PRECEPITATING FACTORS
The two most common precipitating factors in the development of DKA or HHS are inadequate insulin therapy (whether omitted or insufficient insulin regimen) or the presence of infection (46,47). Other provoking factors include myocardial infarction, cerebrovascular accidents, pulmonary embolism, pancreatitis, alcohol and illicit drug use (Figure 4). In addition, numerous underlying medical illness and medications that cause the release of counter regulatory hormones and/or compromise the access to water can result in severe volume depletion and HHS (46). Drugs such as corticosteroids, thiazide diuretics, sympathomimetic agents (e.g., dobutamine and terbutaline), and second generation antipsychotic agents may precipitate DKA or HHS (17). Most recently, two new classes of medications have emerged as triggers for DKA. Sodium-glucose cotransporter 2 (SGLT-2) inhibitors (canagliflozin, dapagliflozin, and empagliflozin) that are used for diabetes treatment have been implicated in the development of DKA in patients with both T1D and T2D (48). Though the absolute risk of DKA in patients treated with SGLT-2 inhibitors is small, this class of medications raises DKA risk by 2-4-fold in patients withT2D and its incidence can be up to 5% in patients with T1D (49,50). Also, anti-cancer medications that belong to classes of immune checkpoint inhibitors such as Ipilimumab, Nivolumab, Pembrolizumab, can cause new-onset diabetes mellitus in up to 1% of the patients receiving immune checkpoint inhibitors with about half of these patients presenting with DKA as the initial presentation of diabetes, particularly in those individuals who may have underlying beta-cell autoimmunity (51,52) (53-56). In young patients with T1D, insulin omission due to fear of hypoglycemia or weight gain, the stress of chronic disease, and eating disorders, may contribute in 20% of recurrent DKA (57). Cocaine use also is associated with recurrent DKA (58,59). Mechanical problems with continuous subcutaneous insulin infusion (CSII) devices can precipitate DKA (60); however, with an improvement in technology and better education of patients, the incidence of DKA have been declining in insulin pump users (61). There are also case reports of patients with DKA as the primary manifestation of acromegaly (62-64).
Increasing numbers of DKA cases have been reported in patients with Type 2 DM. Available evidence shows that almost 50% of newly diagnosed adult African American and Hispanic patients with DKA have T2D (65). These ketosis-prone type 2 diabetic patients develop sudden-onset impairment in insulin secretion and action, resulting in profound insulinopenia (66). Clinical and metabolic features of these patients include high rates of obesity, a strong family history of diabetes, a measurable pancreatic insulin reserve, and a low prevalence of autoimmune markers of β-cell destruction (67-69). Aggressive management with insulin improves β-cell function, leading to discontinuance of insulin therapy within a few months of follow-up and 40% of these patients remain non-insulin dependent for 10 years after the initial episode of DKA (68). The etiology of acute transient failure of β-cells leading to DKA in these patients is not known, however, the suggested mechanisms include glucotoxicity, lipotoxicity, and genetic predisposition (70,71). A genetic disease, glucose-6-phosphate dehydrogenase deficiency, has been also linked with ketosis-prone diabetes (72). In a most recent review of factors that can precipitate DKA, the authors emphasized that clinicians should consider factors such as socioeconomic disadvantage, adolescent age, female sex, prior DKA, and psychiatric comorbidities as potential DKA triggers in patients with T1D (50). Further, in US adults with T1D, HbA1c ≥ 9% was associated with 12-fold higher incidence of DKA (73). Finally, with recent accumulation of knowledge of health hazards related to the COVID-19 pandemic, there is early evidence that COVID-19 infection can trigger DKA in patients with diabetes who otherwise may not have risk factors to develop ketoacidosis (74). Particular attention should be provided to those DKA patients who are COVID-19-positive on admission as early evidence demonstrated a 6-fold increase in mortality in this group of patients compared with those admitted with DKA without COVID-19 (75).
With growing use of SGLT-2 inhibitors, it is worth elucidating potential risk factors that can mediate heightened DKA risk in patients with diabetes. It is now clear that T1D is an independent DKA risk factor regardless of whether other clinical circumstances known to trigger ketoacidosis are present or not. In people with T2D, low-carbohydrate diet, excessive ETOH intake, presence of autoimmunity, and/or exposure to stress situations such as infection, surgery, trauma, dehydration are now identified as DKA risk factors in those treated with SGLT-2 inhibitors (50,76).

Figure 4.
Common precipitating factors in DKA. Data are % of all cases except Nyenwe et al where new onset disease was not included in the percentage and complete data on these items were not given; therefore, the total is less than 100%. Adapted with modification from reference 1.
CLINICAL FEATURES
Symptoms and Signs
DKA usually evolves rapidly within a few hours of the precipitating event(s). On the other hand, development of HHS is insidious and may occur over days to weeks (16). The common clinical presentation of DKA and HHS is due to hyperglycemia and include polyuria, polyphagia, polydipsia, weight loss, weakness, and physical signs of intravascular volume depletion, such as dry buccal mucosa, sunken eye balls, poor skin turgor, tachycardia, hypotension and shock in severe cases. Of note, patients with euglycemic DKA including those treated with SGLT-2 inhibitors, may have less polydipsia and polyuria and may rather initially present with non-specific symptoms such as fatigue and malaise (77,78). Kussmaul respiration, acetone breath, nausea, vomiting, and abdominal pain may also occur primarily in DKA and are due to ketosis and acidosis. Abdominal pain, which correlates with the severity of acidosis (79), may be severe enough to be confused with acute abdomen in 50-75% of cases (80). Therefore, in the presence of acidosis, DKA as an etiology of abdominal pain should be considered. Patients usually have normal body temperature or mild hypothermia regardless of presence of infection (81). Therefore, a careful search for a source of infection should be performed even in the absence of fever. Neurological status in patients with DKA may vary from full alertness to a profound lethargy and coma, However, mental status changes in DKA are less frequent than HHS. The relationship of depressed consciousness and severity of hyperosmolality or DKA causes has been controversial (82,83). Some studies suggested that pH is the cause of mental status changes (84); while, others concluded that osmolality (85) is responsible for the comatose state. More recently, it has been proposed that consciousness level in adolescents with DKA was related to the severity of acidosis (pH) and not to a blood glucose levels (86). In our earlier studies of patients with DKA using low dose versus high dose insulin therapy, we evaluated the initial biochemical values of 48 patients with stupor/coma versus non comatose patients (87). Our study showed that glucose, bicarbonate, BUN and osmolality, and not pH were significantly different between non-comatose and comatose patients. Furthermore, in 3 separate studies in which 123 cases of DKA were evaluated, serum osmolality was also the most important determinant of mental status changes (19). However, in our recent retrospective study, it was shown that acidosis was independently associated with altered sensorium, but hyperosmolarity and serum “ketone” levels were not (88) (Figure 5). In that study, a combination of acidosis and hyperosmolarity at presentation may identify a subset of patients with severe DKA (7% in this study) who may benefit from more aggressive treatment and monitoring. Identifying this group of patients, who are at a higher risk for poorer prognosis, may be helpful in triaging them, thus further improving the outcome (88). Furthermore, according to one study, ICU-admitted patients with DKA are less ill, and have lower disease severity scores, mortality, and shorter length of ICU and hospital stay, than non-DKA patients. Disease severity scores are not, but precipitating cause is, predictive of prolonged hospital stays in patients with DKA (89).

Figure 5.
Admission clinical and biochemical profile in comatose vs non-comatose patients with DKA (88).
In patients with HHS, neurological symptoms include clouding of sensorium which can progress to mental obtundation and coma (90). Occasionally, patients with HHS may present with focal neurological deficits and seizures (91,92). Most of the patients with HHS and an effective serum osmolality of >320 mOsm/kg are obtunded or comatose; on the other hand, the altered mental status rarely exists in patients with serum osmolality of <320 mOsm/kg (8). Therefore, severe alteration in the level of consciousness in patients with serum osmolality of <320 mOsm/kg requires evaluation for other causes including CVA and other catastrophic events like myocardial and bowel infarctions.
LABORATORY ABNORMALITIES AND DIAGNOSIS OF HYPERGLYCEMIC CRISES
The initial laboratory evaluation of patients with suspected DKA or HHS should include determination of plasma glucose, blood urea nitrogen, serum creatinine, serum ketones, electrolytes (with calculated anion gap), osmolality, urinalysis, urine ketones by dipstick, arterial blood gases, and complete blood count with differential. An electrocardiogram, blood, urine or sputum cultures and chest X-ray should also be performed, if indicated. HbA1c may be useful in differentiating chronic hyperglycemia of uncontrolled diabetes from acute metabolic decompensation in a previously well-controlled diabetic patient (17). Figure 6 summarizes the biochemical criteria for DKA and HHS and electrolyte deficits in these two conditions. It also provides a simple method for calculating anion gap and serum osmolality.
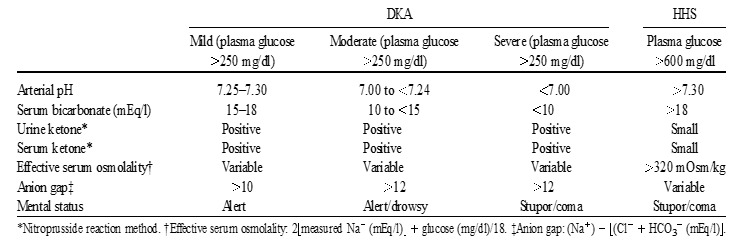
Figure 6.
Diagnostic Criteria and Typical Total Body Deficits of Water and Electrolytes in Diabetic Ketoacidosis (DKA) and Hyperglycemic Hyperosmolar Syndrome (HHS)
DKA can be classified as mild, moderate, or severe based on the severity of metabolic acidosis and the presence of altered mental status (17). Over 30% of patients have features of both DKA and HHS (16) with most recent evidence confirming that about 1 out of 4 patients will have both conditions at the time of presentation with hyperglycemic crisis (18). Patients with HHS typically have pH >7.30, bicarbonate level >20 mEq/L, and negative ketone bodies in plasma and urine. However, some of them may have ketonemia. Several studies on serum osmolarity and mental alteration have established a positive linear relationship between osmolarity, pH, and mental obtundation (87). Therefore, the occurrence of coma in the absence of definitive elevation of serum osmolality requires immediate consideration of other causes of mental status change. The levels of β-hydroxybutyrate (β-OHB) of ≥3.8mmol/L measured by a specific assay were shown to be highly sensitive and specific for DKA diagnosis (93). In patients with chronic kidney disease stage 4-5, the diagnosis of DKA could be challenging due to the presence of concomitant underlying chronic metabolic acidosis or mixed acid-base disorders. An anion gap of >20 mEq/L usually supports the diagnosis of DKA in these patients (94). Based on the 2009 American Diabetes Association publication, “euglycemic DKA” is characterized by metabolic acidosis, increased total body ketone concentration and blood glucose levels ≤250 mg/dL and is thought to occur in up to approximately 10% of patients with DKA and mostly associated with conditions associated with low glycogen reserves and/or increased rates of glucosuria such as pregnancy, liver disorders, and alcohol consumption (1). Since approval in 2013 of SGLT-2 inhibitors for therapy of T2D, multiple reports emerged demonstrating that the use of these medications can result in “euglycemic” DKA (48,78,95). Therefore, DKA must be excluded if high anion gap metabolic acidosis is present in a diabetic patient treated with SGLT-2 inhibitors irrespective if hyperglycemia is present or not. On the other hand, an SGLT-2 inhibitor can be also associated with hyperglycemic DKA in individuals who have sufficient glycogen storage to maintain hyperglycemia even in the setting of enhanced glucosuria (49,96).
The major cause of water deficit in DKA and HHS is glucose-mediated osmotic diuresis, which leads to loss of water in excess of electrolytes (97). Despite the excessive water loss, the admission serum sodium tends to be low. Because serum glucose in the presence of insulinopenia of DKA and HHS cannot penetrate to cells, in hyperglycemic crises, glucose becomes osmotically effective and causes water shifts from intracellular space to the extra cellular space resulting in dilution of sodium concentration – dilutional or hyperosmolar hyponatremia. Initially it has been thought that true sodium concentration (millimolar) can be obtained by multiplying excess glucose above 100 mg/dL by 1.6 /100 (98). It is, however, accepted now that true or corrected serum sodium concentration in patients experiencing hyperglycemic crisis should be calculated by adding 2.4 mmol/L to the measured serum sodium concentration for every 100 mg/dL incremental rise in serum glucose concentration above serum glucose concentration of 100 mg/dL (99). If the corrected sodium level remains low, hypertriglyceridemia (secondary to uncontrolled diabetes) should be also suspected. In this condition the plasma becomes milky and lipemia retinalis may be visible in physical examination (100). Osmotic diuresis and ketonuria also promote a total body sodium deficit via urinary losses, although concurrent conditions, such as diarrhea and vomiting, can further contribute to sodium losses. Total body sodium loss can result in contraction of extracellular fluid volume and signs of intravascular volume depletion. Serum potassium may be elevated on arrival due to insulin deficiency, volume depletion and a shift of potassium from intracellular to extra cellular compartments in response to acidosis (101). However, total body potassium deficit is usually present from urinary potassium losses due to osmotic diuresis and ketone excretion. More frequently, the initial serum potassium level is normal or low which is a danger sign. Initiation of insulin therapy, which leads to the transfer of potassium into cells, may cause fatal hypokalemia if potassium is not replaced early. Phosphate depletion in DKA is universal but on admission, like the potassium, it may be low, normal or high (102).
The differences and similarities in the admission biochemical data in patients with DKA or HHS are shown in Figure 7.

Leukocytosis is a common finding in patients with DKA or HHS, but leukocytosis greater than 25,000 /μL suggests ongoing infection requiring further work up (103). The exact etiology of this non-specific leukocytosis is not known. One study also showed nonspecific leukocytosis in subjects with hypoglycemia induced by insulin injection and suggested that this phenomenon may be due to the increased levels of catecholamines, cortisol, and proinflammatory cytokines such as TNF-α during acute stress (104). Hypertriglyceridemia may be present in HHS (105) and is almost always seen in DKA (79). Hyperamylasemia, which correlates with pH and serum osmolality and elevated level of lipase, may occur in 16 - 25% of patients with DKA (106). The origin of amylase in DKA is usually non-pancreatic tissue such as the parotid gland (107).
Pitfalls of Laboratory Tests and Diagnostic Considerations for Interpreting Acid Based Status in DKA
False positive values for lipase may be seen if plasma glycerol levels are very high due to rapid breakdown of adipose tissue triglycerides (glycerol is the product measured in most assays for plasma lipase). Therefore, elevated pancreatic enzymes may not be reliable for the diagnosis of pancreatitis in the DKA setting. Other pitfalls include artificial elevation of serum creatinine due to interference from ketone bodies when a colorimetric method is used (108). Most of the laboratory tests for ketone bodies use the nitroprusside method, which detects acetoacetate, but not β-hydroxybutyrate (β-OHB). Additionally, since β-OHB is converted to acetoacetate during treatment (109), the serum ketone test may remain positive for a prolonged period suggesting erroneously that ketonemia is deteriorating; therefore, the follow up measurement of ketones during the treatment by nitroprusside method is not recommended (16). Newer glucose meters have the capability to measure β-OHB, which overcomes this problem (110,111). Furthermore drugs that have sulfhydryl groups can interact with the reagent in the nitroprusside reaction, giving a false positive result (112). Particularly important in this regard is captopril, an angiotensin converting enzyme inhibitor prescribed for the treatment of hypertension and diabetic nephropathy. Therefore, for the diagnosis of DKA, clinical judgment and consideration of other biochemical data are required to interpret the value of positive nitroprusside reactions in patients on captopril. Most laboratories can now measure β-OHB levels.
The classical presentation of acid-base disorders in DKA consists of increased anion gap metabolic acidosis where the relation of plasma anion gap change and bicarbonate change (Δ-Δ, ratio of AG change over change in bicarbonate) equals to 1 due to parallel reduction in plasma bicarbonate with the addition of ketoacids into the extravascular fluid space. With frequent additional bicarbonate losses in urine in the form of ketoanions during DKA, the initiation of intravenous volume resuscitation with chloride-containing solutions can further lower plasma bicarbonate and unmask non-anion gap metabolic acidosis when Δ-Δ becomes less than 1 due to changes in plasma bicarbonate that exceed the expected changes in AG. Respiratory compensation will accompany metabolic acidosis with reduction in PCO2 in arterial blood gas. The expected changes in PCO2 can be calculated using Winter’s formula: PCO2 (mmHg) = 1.5 (Bicarbonate) + 8 ± 2 (113). Therefore, inappropriately high or low levels of PCO2, determined by ABG will suggest the presence of a mixed acid-based disorder. For example, DKA patients with concomitant fever or sepsis may have additional respiratory alkalosis manifesting by lower-than-expected PCO2. In contrast, a higher than calculated PCO2 level signifies additional respiratory acidosis and can be seen in patients with underlying chronic lung disease. Vomiting is a common clinical manifestation in DKA and leads to a loss of hydrogen ions in gastric content and the development of metabolic alkalosis. Patients with DKA and vomiting may have relatively normal plasma bicarbonate levels and close to normal pH. However, AG will remain elevated and be an important clue for DKA. In addition, Δ-Δ ratio will be over 2 suggesting that there is less than expected reduction in bicarbonate as compared with increase in AG and confirm the presence of a mixed acid-base disorder (combination of metabolic acidosis and metabolic alkalosis). We recommend measurement of β-OHB in instances when a mixed acid-base disorder is present in patients with hyperglycemic crisis and DKA is suspected.
DIFFERENTIAL DIAGNOSIS
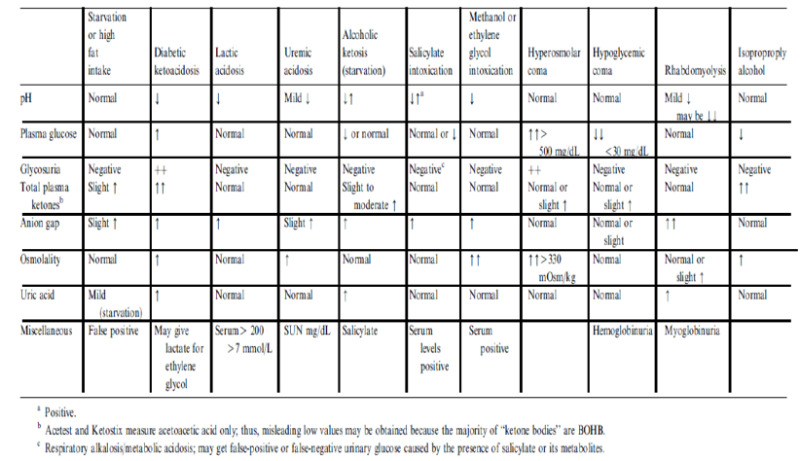
Patients may present with metabolic conditions resembling DKA or HHS. For example, in alcoholic ketoacidosis (AKA), total ketone bodies are much greater than in DKA with a higher β-OHB to acetoacetate ratio of 7:1 versus a ratio of 3:1 in DKA (8). The AKA patients seldom present with hyperglycemia (114). It is also possible that patients with a low food intake may present with mild ketoacidosis (starvation ketosis); however, serum bicarbonate concentration of less than 18 or hyperglycemia will be rarely present. Additionally, DKA has to be distinguished from other causes of high anion gap metabolic acidosis including lactic acidosis, advanced chronic renal failure, as well as ingestion of drugs such as salicylate, methanol, and ethylene glycol. Isopropyl alcohol, which is commonly available as rubbing alcohol, can cause considerable ketosis and high serum osmolar gap without metabolic acidosis. Moreover, there is a tendency to hypoglycemia rather than hyperglycemia with isopropyl alcohol injection (115,116). Finally, patients with diabetes insipidus presenting with severe polyuria and dehydration, who are subsequently treated with free water in a form of intravenous dextrose water, can have hyperglycemia- a clinical picture that can be confused with HHS (117) (Figure 8).
TREATMENT OF DKA
The goals of therapy in patients with hyperglycemic crises include: 1) improvement of circulatory volume and tissue perfusion, 2) gradual reduction of serum glucose and osmolality, 3) correction of electrolyte imbalance, and 4) identification and prompt treatment of co-morbid precipitating causes (8). It must be emphasized that successful treatment of DKA and HHS requires frequent monitoring of patients regarding the above goals by clinical and laboratory parameters. Suggested approaches for the management of patients with DKA and HHS are illustrated in Figures 9 and 10.
Fluid Therapy
DKA and HHS are volume-depleted states with total body water deficit of approximately 6 L in DKA and 9 L in HHS (16,118,119). Therefore, the initial fluid therapy is directed toward expansion of intravascular volume and securing adequate urine flow. The initial fluid of choice is isotonic saline at the rate of 15–20 ml /kg body weight per hour or 1–1.5 L during the first hour. The choice of fluid for further repletion depends on the hydration status, serum electrolyte levels, and urinary output. In patients who are hypernatremic or eunatremic, 0.45% NaCl infused at 4–14 ml/kg/hour is appropriate, and 0.9% NaCl at a similar rate is preferred in patients with hyponatremia. The goal is to replace half of the estimated water and sodium deficit over a period of 12-24 hours [161]. In patients with hypotension, aggressive fluid therapy with isotonic saline should continue until blood pressure is stabilized. The administration of insulin without fluid replacement in such patients may further aggravate hypotension (16). Furthermore, the use of hydrating fluid in the first hour of therapy before insulin administration provides time to obtain serum potassium value before insulin administration, prevents possible deterioration of hypotensive patients with the use of insulin without adequate hydration, and decreases serum osmolality (17). Hydration alone may also reduce the level of counter-regulatory hormones and hyperglycemia (28). Intravascular volume expansion reduces serum blood glucose, BUN, and potassium levels without significant changes in pH or HCO3.The mechanism for lowering glucose is believed to be due to osmotic diuresis and modulation of counter-regulatory hormone release (23,120). We recommend avoiding too rapid correction of hyperglycemia (which may be associated with cerebral edema especially in children) and also inhibiting hypoglycemia (23,120). In HHS, the reduction in insulin infusion rate and/or use of D5 ½ NS should be started when blood glucose reaches 300 mg/dL, because overzealous use of hypotonic fluids has been associated with the development of cerebral edema (121). In one recent review, authors suggested gradual reduction in osmolality not exceeding 3 mOsm/kg H2O per hour and a fall of serum sodium at a rate of less than 0.5 mmol/L per hour in order to prevent significant osmotic shifts of water to intracellular compartment during the management of hyperglycemic crises (122). It should be emphasized that urinary losses of water and electrolytes are also need to be considered.
Insulin Therapy
The cornerstone of DKA and HHS therapy is insulin in physiologic doses. Insulin should only be started after serum potassium value is > 3.3 mmol/L (8). In DKA, we recommend using intravenous (IV) bolus of regular insulin (0.1 u/kg body weight) followed by a continuous infusion of regular insulin at the dose of 0.1u/kg/hr. The insulin infusion rate in HHS should be lower as major pathophysiological process in these patients is severe dehydration. The optimal rate of glucose reduction is between 50-70 mg/hr. If desirable glucose reduction is not achieved in the first hour, an additional insulin bolus at 0.1 u/kg can be given. As mentioned earlier, when plasma glucose reaches 200-250 mg/dL in DKA or 300 in HHS, insulin rate should be decreased to 0.05 U/kg/hr, followed, as indicated, by the change in hydration fluid to D5 ½ NS. The rate of insulin infusion should be adjusted to maintain blood glucose between 150-200 mg/dL in DKA until it is resolved, and 250-300 mg/dL in HHS until mental obtundation and hyperosmolar state are corrected.
A study that investigated the optimum route of insulin therapy in DKA demonstrated that the time for resolution of DKA was identical in patients who received regular insulin via intravenous, intramuscular, or subcutaneous routes (123). However, patients who received intravenous insulin showed a more rapid decline in blood glucose and ketone bodies in the first 2 hours of treatment. Patients who received intravenous insulin attained an immediate pharmacologic level of insulin concentration. Thus, it was established that an intravenous loading dose of insulin would be beneficial regardless of the subsequent route of insulin administration during treatment. A follow up study demonstrated that a priming or loading dose given as one half by IV route and another half by intramuscular route was as effective as one dose given intravenously in lowering the level of ketone bodies in the first hour (124). A bolus or priming dose of insulin has been used in a number of studies. The need of such a method, when using intravenous infusion of insulin, is not clear, as there is no prospective randomized study to establish efficacy of bolus or priming dose before infusion of insulin. However, our study in children demonstrated the effectiveness of intravenous injection of insulin without a bolus dose (125). Therefore, it would appear that if intravenous insulin is used, priming or bolus dose insulin might not be necessary.
Several clinical studies have shown the potency and cost effectiveness of subcutaneous rapid-acting insulin analogs (lispro or aspart) in the management of patients with uncomplicated mild to moderate DKA (126,127). The patients received subcutaneous rapid-acting insulin doses of 0.2 U/kg initially, followed by 0.1 U/kg every 1 hour or an initial dose of 0.3 U/kg followed by 0.2 U/kg every 2 hours until blood glucose was < 250 mg/dL. Then the insulin dose was decreased by half to 0.05, or 0.1 U/kg respectively, and administered every 1 or 2 hours until resolution of DKA. There were no differences in length of hospital stay, total amount of insulin needed for resolution of hyperglycemia or ketoacidosis, or in the incidence of hypoglycemia among treatment groups. The use of insulin analogs allowed treatment of DKA in general wards or the emergency department and so reduced cost of hospitalization by 30% without any significant changes in hypoglycemic events (126). Similar results have been reported recently in pediatric patients with DKA (128). The administration of continuous IV infusion of regular insulin is the preferred route because of its short half-life and easy titration and the delayed onset of action and prolonged half-life of subcutaneous regular insulin. It is important to point out that the IV use of fast-acting insulin analogs is not recommended for patients with severe DKA or HHS, as there are no studies to support their use. Again, these agents may not be effective in patients with severe fluid depletion since they are given subcutaneously.
Potassium Therapy
Although total-body potassium is depleted (129,130), mild to moderate hyperkalemia frequently seen in patients with DKA is due to acidosis and insulinopenia. Insulin therapy, correction of acidosis, and volume expansion decrease serum potassium concentrations. To prevent hypokalemia, potassium replacement is initiated after serum levels fall below 5.3 mmol/L in patients with adequate urine output (50 ml/h). Adding 20–30 mmol potassium to each liter of infused fluid is sufficient to maintain a serum potassium concentration within the normal range of 4–5 mmol/L (8). Patients with DKA who had severe vomiting or had been on diuretics may present with significant hypokalemia. In such cases, potassium replacement should begin with fluid therapy, and insulin treatment should be postponed until potassium concentration becomes > 3.3 mmol/L in order to prevent arrhythmias and respiratory muscle weakness (131).

Bicarbonate Therapy
The use of bicarbonate in treatment of DKA remains controversial. In patients with pH >7.0, insulin therapy inhibits lipolysis and also corrects ketoacidosis without use of bicarbonate. Bicarbonate therapy has been associated with some adverse effects, such as hypokalemia (132), decreased tissue oxygen uptake and cerebral edema (133,134) and delay in the resolution of ketosis (135). However, patients with severe DKA (low bicarbonate <10 mEq/L, or Pco2 < 12) may experience deterioration of pH if not treated with bicarbonate. A prospective randomized study in patients with pH between 6.9 and 7.1 showed that bicarbonate therapy had no risk or benefit in DKA (136). Therefore, in patients with pH between 6.9 and 7.0, it may be beneficial to give 50 mmol of bicarbonate in 200 ml of sterile water with 10 mmol KCL over two hours to maintain the pH at > 7.0 (8,137,138). Considering the adverse effects of severe acidosis such as impaired myocardial contractility, adult patients with pH < 6.9 should be given 100 mmol sodium bicarbonate in 400 ml sterile water (an isotonic solution) with 20 mmol KCl administered at a rate of 200 ml/h for two hours until the venous pH becomes greater than 7.0. Venous pH should be assessed every 2 hours until the pH rises to 7.0; treatment can be repeated every 2 hours if necessary.
Phosphate Therapy
There is no evidence that phosphate therapy is necessary in treatment for better outcome of DKA (139-142). However, in patients with potential complications of hypophosphatemia, including cardiac and skeletal muscle weakness, the use of phosphate may be considered (143). Phosphate administration may result in hypocalcemia when used in high dose (139,142).
TREATMENT OF HHS
A similar therapeutic approach can be also recommended for treatment of HHS, but no bicarbonate therapy is needed for HHS, and changing to glucose-containing fluid is done when blood glucose reaches 300 mg/dL.

Figure 10.
Protocol for the management of adult patients with HHS.
Severe hyperosmolarity and dehydration associated with insulin resistance and presence of detectable plasma insulin level are the hallmarks of HHS pathophysiology. The main emphasis in the management of HHS is effective volume repletion and normalization of serum osmolality (14). There are no randomized controlled studies that evaluated safe and effective strategies in the treatment of HHS (121). It is important to start HHS therapy with the infusion of normal saline and monitor corrected serum sodium in order to determine appropriate timing of the change to hypotonic fluids. Insulin substitution approach should be very conservative as it is expected that insulin resistance will improve with rehydration. We recommend against rapid decreases in serum glucose and correction of serum sodium in order to avoid untoward effects of shifts in osmolarity on brain volume. This notion should particularly apply in the management of HHS in elderly and patients with multiple medical problems in whom it may not be clear how long these subjects experienced severe hyperglycemia prior to the admission to the hospital.
RESOLUTION OF DKA AND HHS
During follow up, blood should be drawn every 2-4 h for determination of serum electrolytes, glucose, blood urea nitrogen, creatinine, osmolality, and venous pH. After the initial arterial pH is drawn, venous pH can be used to assess the acid/base status. An equivalent arterial pH value is calculated by adding 0.03 to the venous pH value (144). The resolution of DKA is reached when the blood glucose is < 200 mg/dl, serum bicarbonate is ≥15 mEq/L, pH is >7.30 and anion gap is ≤12 mEq/L (17). HHS is resolved when serum osmolality is < 320 mOsm/kg with a gradual recovery to mental alertness. The latter may take twice as long as to achieve blood glucose control. Ketonemia typically takes longer to clear than hyperglycemia.
The proposed ADA criteria for DKA resolution include serum glucose level <200 mg/dL and two of the following: serum bicarbonate level ≥15 mEq/L, pH >7.3, and anion gap ≤12 mEq/L (1). Therefore, the treatment goal of DKA is to improve hyperglycemia and to stop ketosis with subsequent resolution of acidosis. In this regard, it is important to distinguish ketosis and acidosis, as the two terms are not always synonymous in DKA. Ketoacid production in DKA results in reduction in plasma bicarbonate (HCO3-) levels due to neutralization of hydrogen ion produced during dissociation of ketoacids in the extravascular fluid space. Concomitantly, ketoacid anion is added into extravascular space resulting in anion gap (AG) increase. The change in HCO3- concentration (Δ HCO3-/normal serum HCO3- – observed serum HCO3-) usually corresponds to equal changes in serum anion gap (Δ AG/observed AG – normal AG, both corrected for decreases and increases in plasma albumin concentration). Therefore, the ratio of AG excess to HCO3- deficit (delta-delta, or Δ-Δ) is close to 1 (143,145,146). In most patients with DKA bicarbonate deficit exceeds the addition of ketoanions, even though Δ-Δ ratio remains close to 1 (147). This is observed due to several reasons. First, hyperglycemia-induced osmotic diuresis leads to excretion of large amounts of sodium and potassium ions that is accompanied by the excretion of ketoanions. Ultimately, the amount of excreted ketoanions depends on degree of kidney function preservation with the largest amount of ketoanion loss in patients with relatively preserved glomerular filtration rate (145). Each ketoanion can be converted back to HCO3- during resolution of DKA and, therefore, ketoanion loss results in the loss of HCO3-. Additionally, extravascular fluid space contraction during DKA, leads to elevation of plasma HCO3-. Therefore, intravenous administration of sodium and chloride-containing fluids leads to further HCO3- reduction and hyperchloremic metabolic acidosis (143,145). This is an important point as persistent decrease in plasma HCO3- concentration should not be interpreted as a sign of continuous DKA if ketosis and hyperglycemia are resolving. Although not evaluated in prospective studies, measurement of serial levels of blood beta-hydroxybutyrate (β-OHB) can be useful adjunct to monitor the resolution of DKA (148). The expected fall in β-OHB with the adequate insulin dosing is 1mmol/L/hr; a lower decrease in blood β-OHB may suggest inadequate insulin provision.
Once DKA has resolved, patients who are able to eat can be started on a multiple dose insulin regimen with long-acting insulin and short/rapid acting insulin given before meals as needed to control plasma glucose. Intravenous insulin infusion should be continued for 2 hours after giving the subcutaneous insulin to maintain adequate plasma insulin levels. Immediate discontinuation of intravenous insulin may lead to hyperglycemia or recurrence of ketoacidosis. If the patient is unable to eat, it is preferable to continue the intravenous insulin infusion and fluid replacement. Patients with known diabetes may be given insulin at the dose they were receiving before the onset of hyperglycemic crises. In patients with new onset diabetes, a multi-dose insulin regimen should be started at a dose of 0.5-0.8 U/kg per day, including regular or rapid-acting and basal insulin until an optimal dose is established (17).
COMPLICATIONS
The most common complications of DKA and HHS include hypoglycemia and hypokalemia due to overzealous treatment with insulin and bicarbonate (hypokalemia), but these complications occur infrequently with current low dose insulin regimens. Nevertheless, in a recent retrospective study, both severe hypokalemia defined as K ≤ 2.5 mEq/L and severe hypoglycemia < 40 mg/dL were significantly and independently associated with increased risk of mortality in patients admitted to the tertiary care center for treatment of hypoglycemic crisis (18). During the recovery phase of DKA, patients commonly develop a short-lived hyperchloremic non-anion gap acidosis, which usually has few clinical consequences (149). Hyperchloremic acidosis is caused by the loss of large amounts of ketoanions, which are usually metabolized to bicarbonate during the evolution of DKA, and excess infusion of chloride containing fluids during treatment (150).
Cerebral edema, a frequently fatal complication of DKA, occurs in 0.7–1.0% of children, particularly those with newly diagnosed diabetes (120). It may also occur in patients with known diabetes and in very young adults usually under 20 years of age (151,152). Cerebral edema has also been reported in patients with HHS, with some cases of mortality (90). Clinically, cerebral edema is characterized by deterioration in the level of consciousness, lethargy, decreased arousal, and headache. Headache is the earliest clinical manifestation of cerebral edema. This is followed by altered level of consciousness and lethargy. Neurological deterioration may lead to seizures, incontinence, pupillary changes, bradycardia, and respiratory arrest. It may be so rapid in onset due to brain stem herniation that no papilledema is found. If deteriorating clinical symptoms occur, the mortality rate may become higher than 70%, with only 7–14% of patients recovering without permanent neurological deficit. Mannitol infusion and mechanical ventilation are used to combat cerebral edema. The cause of cerebral edema is not known with certainty. It may result from osmotically driven movement of water into the central nervous system when plasma osmolality declines too rapidly during treatment of DKA or HHS. As glucose concentration improves following insulin infusion and administration of the intravenous fluids, serum osmotic gradient previously contributed by hyperglycemia reduces which limits water shifts from the intracellular compartment. However, hyperglycemia treatment is associated with “recovery” in serum sodium that restores water transfer between extracellular and intracellular compartments and prevents water accumulation in cells (99). In cases when the serum glucose concentration improves to a greater extent than the serum sodium concentration rises, serum effective osmolality will decrease and may precipitate brain edema (153,154). Although the osmotically mediated mechanism seems most plausible, one study using magnetic resonance imaging (MRI) showed that cerebral edema was due to increased cerebral perfusion (135). Another postulated mechanism for cerebral edema in patients with DKA involves the cell membrane Na+/H+ exchangers, which are activated in DKA. The high H+ level allows more influx of Na+ thus increasing more influx of water to the cell with consequent edema (155). β-hydroxybutyrate and acetoacetate may also play a role in the pathogenesis of cerebral edema. These ketone bodies have been shown to affect vascular integrity and permeability, leading to edema formation (156). In summary, reasonable precautionary measures to decrease the risk of cerebral edema in high-risk patients include 1) avoidance of overenthusiastic hydration and rapid reduction of plasma osmolality and 2) close hemodynamic monitoring (157). Based on the recent reports, particular care should be offered to patients with end stage renal disease as these individuals are more likely to die, to have higher rates of hypoglycemia, or to be volume overloaded when admitted to the hospital with DKA (158).
Hypoxemia and rarely non-cardiogenic pulmonary edema may complicate the treatment of DKA [242]. Hypoxemia may be related to the reduction in colloid osmotic pressure that leads to accumulation of water in lungs and decreased lung compliance. The pathogenesis of pulmonary edema may be similar to that of cerebral edema suggesting that the sequestration of fluid in the tissues may be more widespread than is thought. Thrombotic conditions and disseminated intravascular coagulation may contribute to the morbidity and mortality of hyperglycemic emergencies (159-161). Prophylactic use of heparin, if there is no gastrointestinal hemorrhage, should be considered.
PREVENTION
About one in five patients with T1D admitted for DKA will be readmitted for DKA within 30 days (162). Several studies suggested that the omission of insulin is one of the most common precipitating factors of DKA, sometimes because patients are socio-economically underprivileged, and may not have access to or afford medical care (163-165). In addition, they may have a propensity to use illicit drugs such as cocaine, which has been associated with recurrent DKA (58), or live in areas with higher food deprivation risk (166). Therefore, it is important to continuously re-assess socio-economic status of patients who had at least one episode of DKA. The most recent data demonstrating a significant increase in DKA hospitalization rates in diabetic persons aged 45 years and younger (10) suggests that this group of patients may require particular attention to understand why they are more vulnerable than others to develop hyperglycemic crisis. Education of the patient about sick day management is very vital to prevent DKA, and should include information on when to contact the health care provider, blood glucose goals, use of insulin, and initiation of appropriate nutrition during illness and should be reviewed with patients periodically. Patients must be advised to continue insulin and to seek professional advice early in the course of the illness. COVID-19-positive patients with diabetes outside of the hospital environment should be particularly vigilant in point-of-care monitoring of home blood glucose and/or β-OHB until the resolution of infection. Close follow up is very important, as it has been shown that three-monthly visits to the endocrine clinic will reduce the number of ER admission for DKA (167). Close observation, early detection of symptoms and appropriate medical care would be helpful in preventing HHS in the elderly.
A study in adolescents with T1D suggests that some of the risk factors for DKA include higher HbA1c, uninsured children, and psychological problems (168). In other studies, education of primary care providers and school personnel in identifying the signs and symptoms of DKA has been shown to be effective in decreasing the incidence of DKA at the onset of diabetes (169). In another study outcome data of 556 patients with diabetes under continuing care over a 7-year period were examined. The hospitalization rates for DKA and amputation were decreased by 69% due to continuing care and education (170). There is early evidence that use of continuous glucose monitoring (CGM) can decrease DKA incidence (171,172). Contrary to the initial observations connecting DKA episodes with insulin pump malfunction, the newer pumps are associated with reduced DKA risk without or with concomitant CGM application in T1D youth (173). Given the increased DKA risks associated with HbA1c ≥ 9% in patients with T1D, all efforts should be applied to understand and potentially address reasons for poor chronic glycemic control as this may prevent DKA admission. Considering DKA and HHS as potentially fatal and economically burdensome complications of diabetes, every effort for diminishing the possible risk factors is worthwhile.
SGLT-2 inhibitor-induced DKA in patients with T2D is a potentially avoidable condition in light of accumulating knowledge of potential triggers prompting the development of this hyperglycemic emergency (174). A recent international consensus statement on the DKA risk management in patients with T1D treated with SGLT-2 inhibitors (76) can be effectively applied to the care of patients with T2D as well. Avoidance or temporary discontinuation of SGLT-2 inhibitors in clinical situations that independently increase risk of intravascular volume depletion and/or development of ketosis-prone conditions listed in the Figure 11 can mitigate the DKA risk. The DEEARAILS pneumonic can help recalling these clinical situations.

Figure 11.
Precipitating factors for DKA in patients taking SGLT2 inhibitors. LADA= latent autoimmune diabetes in adults
REFERENCES
- 1.
- Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN. Hyperglycemic crises in adult patients with diabetes. Diabetes care. 2009;32(7):1335–1343. [PMC free article: PMC2699725] [PubMed: 19564476]
- 2.
- Johnson DD, Palumbo PJ, Chu CP. Diabetic ketoacidosis in a community-based population. Mayo Clin Proc. 1980;55(2):83–88. [PubMed: 6766521]
- 3.
- Ramphul K, Joynauth J. An Update on the Incidence and Burden of Diabetic Ketoacidosis in the U.S. Diabetes care. 2020;43(12):e196–e197. [PubMed: 33051332]
- 4.
- CDC. 2014 national diabetes fact sheet (http://www
.cdc.gov/diabetes /statistics/dkafirst/index.htm). accessed 04.07.15. - 5.
- Benoit SR, Zhang Y, Geiss LS, Gregg EW, Albright A. Trends in Diabetic Ketoacidosis Hospitalizations and In-Hospital Mortality - United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2018;67(12):362–365. [PMC free article: PMC5877353] [PubMed: 29596400]
- 6.
- Zhong VW, Juhaeri J, Mayer-Davis EJ. Trends in Hospital Admission for Diabetic Ketoacidosis in Adults With Type 1 and Type 2 Diabetes in England, 1998-2013: A Retrospective Cohort Study. Diabetes care. 2018;41(9):1870–1877. [PubMed: 29386248]
- 7.
- Thomas M, Harjutsalo V, Feodoroff M, Forsblom C, Gordin D, Groop PH. The Long-Term Incidence of Hospitalization for Ketoacidosis in Adults with Established T1D-A Prospective Cohort Study. J Clin Endocrinol Metab. 2020;105(1) [PubMed: 31529090]
- 8.
- Kitabchi AE, Umpierrez GE, Murphy MB, Barrett EJ, Kreisberg RA, Malone JI, Wall BM. Management of hyperglycemic crises in patients with diabetes. Diabetes care. 2001;24(1):131–153. [PubMed: 11194218]
- 9.
- Matz R. Management of the hyperosmolar hyperglycemic syndrome. Am Fam Physician. 1999;60(5):1468–1476. [PubMed: 10524491]
- 10.
- CDC. 2017 national diabetes statistics (https://www
.cdc.gov/diabetes /pdfs/data/statistics /national-diabetes-statistics-report.pdf) accessed 05.05.18. - 11.
- Javor KA, Kotsanos JG, McDonald RC, Baron AD, Kesterson JG, Tierney WM. Diabetic ketoacidosis charges relative to medical charges of adult patients with type I diabetes. Diabetes care. 1997;20(3):349–354. [PubMed: 9051386]
- 12.
- Desai D, Mehta D, Mathias P, Menon G, Schubart UK. Health Care Utilization and Burden of Diabetic Ketoacidosis in the U.S. Over the Past Decade: A Nationwide Analysis. Diabetes care. 2018;41(8):1631–1638. [PubMed: 29773640]
- 13.
- Fernando SM, Bagshaw SM, Rochwerg B, McIsaac DI, Thavorn K, Forster AJ, Tran A, Reardon PM, Rosenberg E, Tanuseputro P, Kyeremanteng K. Comparison of outcomes and costs between adult diabetic ketoacidosis patients admitted to the ICU and step-down unit. Journal of critical care. 2019;50:257–261. [PubMed: 30640078]
- 14.
- Karslioglu French E, Donihi AC, Korytkowski MT. Diabetic ketoacidosis and hyperosmolar hyperglycemic syndrome: review of acute decompensated diabetes in adult patients. BMJ. 2019;365:l1114. [PubMed: 31142480]
- 15.
- Prevention. CfDCa. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: U.S. Department of Health and Human Services; 2014. 2014.
- 16.
- Kitabchi AE, Nyenwe EA. Hyperglycemic crises in diabetes mellitus: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Endocrinol Metab Clin North Am. 2006;35(4):725–751. viii. [PubMed: 17127143]
- 17.
- Kitabchi AE, Umpierrez GE, Murphy MB, Kreisberg RA. Hyperglycemic crises in adult patients with diabetes: a consensus statement from the American Diabetes Association. Diabetes care. 2006;29(12):2739–2748. [PubMed: 17130218]
- 18.
- Pasquel FJ, Tsegka K, Wang H, Cardona S, Galindo RJ, Fayfman M, Davis G, Vellanki P, Migdal A, Gujral U, Narayan KMV, Umpierrez GE. Clinical Outcomes in Patients With Isolated or Combined Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic State: A Retrospective, Hospital-Based Cohort Study. Diabetes care. 2020;43(2):349–357. [PMC free article: PMC6971788] [PubMed: 31704689]
- 19.
- Kitabchi AE, Wall BM. Diabetic ketoacidosis. Med Clin North Am. 1995;79(1):9–37. [PubMed: 7808097]
- 20.
- Chupin M, Charbonnel B, Chupin F. C-peptide blood levels in keto-acidosis and in hyperosmolar non-ketotic diabetic coma. Acta Diabetol Lat. 1981;18(2):123–128. [PubMed: 6787828]
- 21.
- Kipnis DM. Insulin secretion in diabetes mellitus. Annals of internal medicine. 1968;69(5):891–901. [PubMed: 5698485]
- 22.
- Kitabchi AE, Ayyagari V, Guerra SM. The efficacy of low-dose versus conventional therapy of insulin for treatment of diabetic ketoacidosis. Annals of internal medicine. 1976;84(6):633–638. [PubMed: 820228]
- 23.
- Kitabchi AE, Young R, Sacks H, Morris L. Diabetic ketoacidosis: reappraisal of therapeutic approach. Annu Rev Med. 1979;30:339–357. [PubMed: 122259]
- 24.
- Muller WA, Faloona GR, Unger RH. Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med. 1973;54(1):52–57. [PubMed: 4629972]
- 25.
- Christensen NJ. Plasma norepinephrine and epinephrine in untreated diabetics, during fasting and after insulin administration. Diabetes. 1974;23(1):1–8. [PubMed: 4203518]
- 26.
- Alberti KG. Role of glucagon and other hormones in development of diabetic ketoacidosis. Lancet. 1975;1(7920):1307–1311. [PubMed: 49515]
- 27.
- Unger RH. High Growth-Hormone Levels in Diabetic Ketoacidosis: A Possible Cause of Insulin Resistance. JAMA. 1965;191:945–947. [PubMed: 14260239]
- 28.
- Waldhausl W, Kleinberger G, Korn A, Dudczak R, Bratusch-Marrain P, Nowotny P. Severe hyperglycemia: effects of rehydration on endocrine derangements and blood glucose concentration. Diabetes. 1979;28(6):577–584. [PubMed: 109338]
- 29.
- Foster DW, McGarry JD. The metabolic derangements and treatment of diabetic ketoacidosis. N Engl J Med. 1983;309(3):159–169. [PubMed: 6408476]
- 30.
- Miles JM, Rizza RA, Haymond MW, Gerich JE. Effects of acute insulin deficiency on glucose and ketone body turnover in man: evidence for the primacy of overproduction of glucose and ketone bodies in the genesis of diabetic ketoacidosis. Diabetes. 1980;29(11):926–930. [PubMed: 6775996]
- 31.
- Felig P, Wahren J. Influence of endogenous insulin secretion on splanchnic glucose and amino acid metabolism in man. The Journal of clinical investigation. 1971;50(8):1702–1711. [PMC free article: PMC442070] [PubMed: 5097575]
- 32.
- Hue L. Gluconeogenesis and its regulation. Diabetes Metab Rev. 1987;3(1):111–126. [PubMed: 3552521]
- 33.
- Schade DS, Eaton RP. The temporal relationship between endogenously secreted stress hormones and metabolic decompensation in diabetic man. J Clin Endocrinol Metab. 1980;50(1):131–136. [PubMed: 7350176]
- 34.
- McGarry JD, Dobbins RL. Fatty acids, lipotoxicity and insulin secretion. Diabetologia. 1999;42(2):128–138. [PubMed: 10064091]
- 35.
- McGarry JD. Lilly Lecture 1978. New perspectives in the regulation of ketogenesis. Diabetes. 1979;28(5):517–523. [PubMed: 437381]
- 36.
- Nurjhan N, Consoli A, Gerich J. Increased lipolysis and its consequences on gluconeogenesis in non-insulin-dependent diabetes mellitus. The Journal of clinical investigation. 1992;89(1):169–175. [PMC free article: PMC442833] [PubMed: 1729269]
- 37.
- Balasse EO, Fery F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev. 1989;5(3):247–270. [PubMed: 2656155]
- 38.
- McGarry JD, Woeltje KF, Kuwajima M, Foster DW. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab Rev. 1989;5(3):271–284. [PubMed: 2656156]
- 39.
- Reichard GA Jr, Skutches CL, Hoeldtke RD, Owen OE. Acetone metabolism in humans during diabetic ketoacidosis. Diabetes. 1986;35(6):668–674. [PubMed: 3086164]
- 40.
- Barnes AJ, Bloom SR, Goerge K, Alberti GM, Smythe P, Alford FP, Chisholm DJ. Ketoacidosis in pancreatectomized man. N Engl J Med. 1977;296(22):1250–1253. [PubMed: 404553]
- 41.
- Cahill GF Jr. Starvation in man. N Engl J Med. 1970;282(12):668–675. [PubMed: 4915800]
- 42.
- Stentz FB, Umpierrez GE, Cuervo R, Kitabchi AE. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes. 2004;53(8):2079–2086. [PubMed: 15277389]
- 43.
- Cherian SV, Khara L, Das S, Hamarneh WA, Garcha AS, Frechette V. Diabetic ketoacidosis complicated by generalized venous thrombosis: a case report and review. Blood Coagul Fibrinolysis. 2012;23(3):238–240. [PubMed: 22356837]
- 44.
- Wakabayashi S, Tsujimoto T, Kishimoto M, Ikeda N, Inoue K, Ihana N, Hamasaki H, Noto H, Yamamoto-Honda R, Kajio H, Noda M. Acute Multiple Arteriovenous Thromboses in a Patient with Diabetic Ketoacidosis. Intern Med. 2015;54(16):2025–2028. [PubMed: 26278296]
- 45.
- Gerich JE, Martin MM, Recant L. Clinical and metabolic characteristics of hyperosmolar nonketotic coma. Diabetes. 1971;20(4):228–238. [PubMed: 4994561]
- 46.
- Wachtel TJ, Silliman RA, Lamberton P. Predisposing factors for the diabetic hyperosmolar state. Arch Intern Med. 1987;147(3):499–501. [PubMed: 3827427]
- 47.
- Wachtel TJ, Tetu-Mouradjian LM, Goldman DL, Ellis SE, O'Sullivan PS. Hyperosmolarity and acidosis in diabetes mellitus: a three-year experience in Rhode Island. J Gen Intern Med. 1991;6(6):495–502. [PubMed: 1765864]
- 48.
- Fadini GP, Bonora BM, Avogaro A. SGLT2 inhibitors and diabetic ketoacidosis: data from the FDA Adverse Event Reporting System. Diabetologia. 2017;60(8):1385–1389. [PubMed: 28500396]
- 49.
- Bonora BM, Avogaro A, Fadini GP. Euglycemic Ketoacidosis. Curr Diab Rep. 2020;20(7):25. [PubMed: 32424730]
- 50.
- Ehrmann D, Kulzer B, Roos T, Haak T, Al-Khatib M, Hermanns N. Risk factors and prevention strategies for diabetic ketoacidosis in people with established type 1 diabetes. Lancet Diabetes Endocrinol. 2020;8(5):436–446. [PubMed: 32333879]
- 51.
- Miyoshi Y, Ogawa O, Oyama Y. Nivolumab, an Anti-Programmed Cell Death-1 Antibody, Induces Fulminant Type 1 Diabetes. Tohoku J Exp Med. 2016;239(2):155–158. [PubMed: 27297738]
- 52.
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A. investigators K-. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–2532. [PubMed: 25891173]
- 53.
- Clotman K, Janssens K, Specenier P, Weets I, De Block CEM. Programmed Cell Death-1 Inhibitor-Induced Type 1 Diabetes Mellitus. J Clin Endocrinol Metab. 2018;103(9):3144–3154. [PubMed: 29955867]
- 54.
- Liu J, Zhou H, Zhang Y, Fang W, Yang Y, Huang Y, Zhang L. Reporting of Immune Checkpoint Inhibitor Therapy-Associated Diabetes, 2015-2019. Diabetes care. 2020;43(7):e79–e80. [PMC free article: PMC7305010] [PubMed: 32393586]
- 55.
- Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, Gettinger S, Sznol M, Young A, Rushakoff R, Lee J, Bluestone JA, Anderson M, Herold KC. Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes. 2018;67(8):1471–1480. [PMC free article: PMC6054443] [PubMed: 29937434]
- 56.
- Wright JJ, Salem JE, Johnson DB, Lebrun-Vignes B, Stamatouli A, Thomas JW, Herold KC, Moslehi J, Powers AC. Increased Reporting of Immune Checkpoint Inhibitor-Associated Diabetes. Diabetes care. 2018;41(12):e150–e151. [PMC free article: PMC7301161] [PubMed: 30305348]
- 57.
- Polonsky WH, Anderson BJ, Lohrer PA, Aponte JE, Jacobson AM, Cole CF. Insulin omission in women with IDDM. Diabetes care. 1994;17(10):1178–1185. [PubMed: 7821139]
- 58.
- Nyenwe EA, Loganathan RS, Blum S, Ezuteh DO, Erani DM, Wan JY, Palace MR, Kitabchi AE. Active use of cocaine: an independent risk factor for recurrent diabetic ketoacidosis in a city hospital. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2007;13(1):22-29. [PubMed: 17360297]
- 59.
- Warner EA, Greene GS, Buchsbaum MS, Cooper DS, Robinson BE. Diabetic ketoacidosis associated with cocaine use. Arch Intern Med. 1998;158(16):1799–1802. [PubMed: 9738609]
- 60.
- Peden NR, Braaten JT, McKendry JB. Diabetic ketoacidosis during long-term treatment with continuous subcutaneous insulin infusion. Diabetes care. 1984;7(1):1–5. [PubMed: 6423352]
- 61.
- Weissberg-Benchell J, Antisdel-Lomaglio J, Seshadri R. Insulin pump therapy: a meta-analysis. Diabetes care. 2003;26(4):1079–1087. [PubMed: 12663577]
- 62.
- Katz JR, Edwards R, Khan M, Conway GS. Acromegaly presenting with diabetic ketoacidosis. Postgrad Med J. 1996;72(853):682–683. [PMC free article: PMC2398638] [PubMed: 8944212]
- 63.
- Szeto CC, Li KY, Ko GT, Chow CC, Yeung VT, Chan JC, Cockram CS. Acromegaly in a woman presenting with diabetic ketoacidosis and insulin resistance. Int J Clin Pract. 1997;51(7):476–477. [PubMed: 9536592]
- 64.
- Vidal Cortada J, Conget Donlo JI, Navarro Tellez MP, Halperin Rabinovic I, Vilardell Latorre E. An Med Interna. 1995;12(2):76–8. [Diabetic ketoacidosis as the first manifestation of acromegaly] [PubMed: 7749014]
- 65.
- Umpierrez GE, Casals MM, Gebhart SP, Mixon PS, Clark WS, Phillips LS. Diabetic ketoacidosis in obese African-Americans. Diabetes. 1995;44(7):790–795. [PubMed: 7789647]
- 66.
- Umpierrez GE, Smiley D, Kitabchi AE. Narrative review: ketosis-prone type 2 diabetes mellitus. Annals of internal medicine. 2006;144(5):350–357. [PubMed: 16520476]
- 67.
- Kitabchi AE. Ketosis-prone diabetes--a new subgroup of patients with atypical type 1 and type 2 diabetes? J Clin Endocrinol Metab. 2003;88(11):5087–5089. [PubMed: 14602730]
- 68.
- Maldonado M, Hampe CS, Gaur LK, D'Amico S, Iyer D, Hammerle LP, Bolgiano D, Rodriguez L, Rajan A, Lernmark A, Balasubramanyam A. Ketosis-prone diabetes: dissection of a heterogeneous syndrome using an immunogenetic and beta-cell functional classification, prospective analysis, and clinical outcomes. J Clin Endocrinol Metab. 2003;88(11):5090–5098. [PubMed: 14602731]
- 69.
- Mauvais-Jarvis F, Sobngwi E, Porcher R, Riveline JP, Kevorkian JP, Vaisse C, Charpentier G, Guillausseau PJ, Vexiau P, Gautier JF. Ketosis-prone type 2 diabetes in patients of sub-Saharan African origin: clinical pathophysiology and natural history of beta-cell dysfunction and insulin resistance. Diabetes. 2004;53(3):645–653. [PubMed: 14988248]
- 70.
- Banerji MA, Chaiken RL, Huey H, Tuomi T, Norin AJ, Mackay IR, Rowley MJ, Zimmet PZ, Lebovitz HE. GAD antibody negative NIDDM in adult black subjects with diabetic ketoacidosis and increased frequency of human leukocyte antigen DR3 and DR4. Flatbush diabetes. Diabetes. 1994;43(6):741–745. [PubMed: 8194658]
- 71.
- Umpierrez GE, Woo W, Hagopian WA, Isaacs SD, Palmer JP, Gaur LK, Nepom GT, Clark WS, Mixon PS, Kitabchi AE. Immunogenetic analysis suggests different pathogenesis for obese and lean African-Americans with diabetic ketoacidosis. Diabetes care. 1999;22(9):1517–1523. [PubMed: 10480519]
- 72.
- Sobngwi E, Gautier JF, Kevorkian JP, Villette JM, Riveline JP, Zhang S, Vexiau P, Leal SM, Vaisse C, Mauvais-Jarvis F. High prevalence of glucose-6-phosphate dehydrogenase deficiency without gene mutation suggests a novel genetic mechanism predisposing to ketosis-prone diabetes. J Clin Endocrinol Metab. 2005;90(8):4446–4451. [PMC free article: PMC6143174] [PubMed: 15914531]
- 73.
- Pettus JH, Zhou FL, Shepherd L, Preblick R, Hunt PR, Paranjape S, Miller KM, Edelman SV. Incidences of Severe Hypoglycemia and Diabetic Ketoacidosis and Prevalence of Microvascular Complications Stratified by Age and Glycemic Control in U.S. Adult Patients With Type 1 Diabetes: A Real-World Study. Diabetes care. 2019;42(12):2220–2227. [PubMed: 31548241]
- 74.
- Li J, Wang X, Chen J, Zuo X, Zhang H, Deng A. COVID-19 infection may cause ketosis and ketoacidosis. Diabetes Obes Metab. 2020;22(10):1935–1941. [PMC free article: PMC7264681] [PubMed: 32314455]
- 75.
- Pasquel FJ, Messler J, Booth R, Kubacka B, Mumpower A, Umpierrez G, Aloi J. Characteristics of and Mortality Associated With Diabetic Ketoacidosis Among US Patients Hospitalized With or Without COVID-19. JAMA Netw Open. 2021;4(3):e211091. [PMC free article: PMC7948057] [PubMed: 33688962]
- 76.
- Danne T, Garg S, Peters AL, Buse JB, Mathieu C, Pettus JH, Alexander CM, Battelino T, Ampudia-Blasco FJ, Bode BW, Cariou B, Close KL, Dandona P, Dutta S, Ferrannini E, Fourlanos S, Grunberger G, Heller SR, Henry RR, Kurian MJ, Kushner JA, Oron T, Parkin CG, Pieber TR, Rodbard HW, Schatz D, Skyler JS, Tamborlane WV, Yokote K, Phillip M. International Consensus on Risk Management of Diabetic Ketoacidosis in Patients With Type 1 Diabetes Treated With Sodium-Glucose Cotransporter (SGLT) Inhibitors. Diabetes care. 2019;42(6):1147–1154. [PMC free article: PMC6973545] [PubMed: 30728224]
- 77.
- Ahmed M, McKenna MJ, Crowley RK. Diabetic Ketoacidosis in Patients with Type 2 Diabetes Recently Commenced on Sglt-2 Inhibitors: An Ongoing Concern. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2017;23(4):506-508. [PubMed: 28437153]
- 78.
- Peters AL, Buschur EO, Buse JB, Cohan P, Diner JC, Hirsch IB. Euglycemic Diabetic Ketoacidosis: A Potential Complication of Treatment With Sodium-Glucose Cotransporter 2 Inhibition. Diabetes care. 2015;38(9):1687–1693. [PMC free article: PMC4542270] [PubMed: 26078479]
- 79.
- Umpierrez G, Freire AX. Abdominal pain in patients with hyperglycemic crises. Journal of critical care. 2002;17(1):63–67. [PubMed: 12040551]
- 80.
- Campbell IW, Duncan LJ, Innes JA, MacCuish AC, Munro JF. Abdominal pain in diabetic metabolic decompensation. Clinical significance. JAMA. 1975;233(2):166–168. [PubMed: 805854]
- 81.
- Matz R. Hypothermia in diabetic acidosis. Hormones. 1972;3(1):36–41. [PubMed: 4631908]
- 82.
- Alberti KG, Nattrass M. Severe diabetic ketoacidosis. Med Clin North Am. 1978;62(4):799–814. [PubMed: 98677]
- 83.
- Ruderman NB, Goodman MN. Brain metabolism in diabetes. Horm Metab Res Suppl. 1980;9:1–8. [PubMed: 6769772]
- 84.
- Rosival V. The influence of blood hydrogen ion concentration on the level of consciousness in diabetic ketoacidosis. Ann Clin Res. 1987;19(1):23–25. [PubMed: 3107457]
- 85.
- Fulop M, Rosenblatt A, Kreitzer SM, Gerstenhaber B. Hyperosmolar nature of diabetic coma. Diabetes. 1975;24(6):594–599. [PubMed: 237799]
- 86.
- Edge JA, Roy Y, Bergomi A, Murphy NP, Ford-Adams ME, Ong KK, Dunger DB. Conscious level in children with diabetic ketoacidosis is related to severity of acidosis and not to blood glucose concentration. Pediatr Diabetes. 2006;7(1):11–15. [PubMed: 16489969]
- 87.
- Morris LR, Kitabchi AE. Efficacy of low-dose insulin therapy for severely obtunded patients in diabetic ketoacidosis. Diabetes care. 1980;3(1):53–56. [PubMed: 6773727]
- 88.
- Nyenwe EA, Razavi LN, Kitabchi AE, Khan AN, Wan JY. Acidosis: the prime determinant of depressed sensorium in diabetic ketoacidosis. Diabetes care. 2010;33(8):1837–1839. [PMC free article: PMC2909073] [PubMed: 20484127]
- 89.
- Freire AX, Umpierrez GE, Afessa B, Latif KA, Bridges L, Kitabchi AE. Predictors of intensive care unit and hospital length of stay in diabetic ketoacidosis. Journal of critical care. 2002;17(4):207–211. [PubMed: 12501147]
- 90.
- Arieff AI. Cerebral edema complicating nonketotic hyperosmolar coma. Miner Electrolyte Metab. 1986;12(5-6):383–389. [PubMed: 3543638]
- 91.
- Guisado R, Arieff AI. Neurologic manifestations of diabetic comas: correlation with biochemical alterations in the brain. Metabolism: clinical and experimental. 1975;24(5):665–679. [PubMed: 805337]
- 92.
- Harden CL, Rosenbaum DH, Daras M. Hyperglycemia presenting with occipital seizures. Epilepsia. 1991;32(2):215–220. [PubMed: 2004625]
- 93.
- Sheikh-Ali M, Karon BS, Basu A, Kudva YC, Muller LA, Xu J, Schwenk WF, Miles JM. Can serum beta-hydroxybutyrate be used to diagnose diabetic ketoacidosis? Diabetes care. 2008;31(4):643–647. [PubMed: 18184896]
- 94.
- Gosmanov AR, Gosmanova EO, Dillard-Cannon E. Management of adult diabetic ketoacidosis. Diabetes Metab Syndr Obes. 2014;7:255–264. [PMC free article: PMC4085289] [PubMed: 25061324]
- 95.
- Handelsman Y, Henry RR, Bloomgarden ZT, Dagogo-Jack S, DeFronzo RA, Einhorn D, Ferrannini E, Fonseca VA, Garber AJ, Grunberger G, LeRoith D, Umpierrez GE, Weir MR. American Association of Clinical Endocrinologists and American College of Endocrinology Position Statement on the Association of Sglt-2 Inhibitors and Diabetic Ketoacidosis. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2016;22(6):753-762. [PubMed: 27082665]
- 96.
- Kum-Nji JS, Gosmanov AR, Steinberg H, Dagogo-Jack S. Hyperglycemic, high anion-gap metabolic acidosis in patients receiving SGLT-2 inhibitors for diabetes management. Journal of diabetes and its complications. 2017;31(3):611–614. [PubMed: 27913012]
- 97.
- Umpierrez GE, Khajavi M, Kitabchi AE. Review: diabetic ketoacidosis and hyperglycemic hyperosmolar nonketotic syndrome. Am J Med Sci. 1996;311(5):225–233. [PubMed: 8615398]
- 98.
- Robin AP, Ing TS, Lancaster GA, Soung LS, Sparagana M, Geis WP, Hano JE. Hyperglycemia-induced hyponatremia: a fresh look. Clin Chem. 1979;25(3):496–497. [PubMed: 262202]
- 99.
- Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, Decaux G, Fenske W, Hoorn EJ, Ichai C, Joannidis M, Soupart A, Zietse R, Haller M, van der Veer S, Van Biesen W, Nagler E. Hyponatraemia Guideline Development G. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Nephrol Dial Transplant. 2014;29 Suppl 2:i1–i39. [PubMed: 24569496]
- 100.
- Kaminska ES, Pourmotabbed G. Spurious laboratory values in diabetic ketoacidosis and hyperlipidemia. The American journal of emergency medicine. 1993;11(1):77–80. [PubMed: 8447878]
- 101.
- Adrogue HJ, Lederer ED, Suki WN, Eknoyan G. Determinants of plasma potassium levels in diabetic ketoacidosis. Medicine (Baltimore). 1986;65(3):163–172. [PubMed: 3084904]
- 102.
- Wilson HK, Keuer SP, Lea AS, Boyd AE 3rd, Eknoyan G. Phosphate therapy in diabetic ketoacidosis. Arch Intern Med. 1982;142(3):517–520. [PubMed: 6802095]
- 103.
- Slovis CM, Mork VG, Slovis RJ, Bain RP. Diabetic ketoacidosis and infection: leukocyte count and differential as early predictors of serious infection. The American journal of emergency medicine. 1987;5(1):1–5. [PubMed: 3101715]
- 104.
- Razavi Nematollahi L, Kitabchi AE, Stentz FB, Wan JY, Larijani BA, Tehrani MM, Gozashti MH, Omidfar K, Taheri E. Proinflammatory cytokines in response to insulin-induced hypoglycemic stress in healthy subjects. Metabolism: clinical and experimental. 2009;58(4):443–448. [PubMed: 19303962]
- 105.
- Bewsher PD, Petrie JC, Worth HG. Serum lipid levels in hyperosmolar non-ketotic diabetic coma. Br Med J. 1970;3(5714):82–84. [PMC free article: PMC1701056] [PubMed: 5428783]
- 106.
- Vinicor F, Lehrner LM, Karn RC, Merritt AD. Hyperamylasemia in diabetic ketoacidosis: sources and significance. Annals of internal medicine. 1979;91(2):200–204. [PubMed: 111594]
- 107.
- Yadav D, Nair S, Norkus EP, Pitchumoni CS. Nonspecific hyperamylasemia and hyperlipasemia in diabetic ketoacidosis: incidence and correlation with biochemical abnormalities. Am J Gastroenterol. 2000;95(11):3123–3128. [PubMed: 11095328]
- 108.
- Gerard SK, Khayam-Bashi H. Characterization of creatinine error in ketotic patients. A prospective comparison of alkaline picrate methods with an enzymatic method. Am J Clin Pathol. 1985;84(5):659–664. [PubMed: 4061391]
- 109.
- Stephens JM, Sulway MJ, Watkins PJ. Relationship of blood acetoacetate and 3-hydroxybutyrate in diabetes. Diabetes. 1971;20(7):485–489. [PubMed: 4997333]
- 110.
- Fulop M, Murthy V, Michilli A, Nalamati J, Qian Q, Saitowitz A. Serum beta-hydroxybutyrate measurement in patients with uncontrolled diabetes mellitus. Arch Intern Med. 1999;159(4):381–384. [PubMed: 10030312]
- 111.
- Porter WH, Yao HH, Karounos DG. Laboratory and clinical evaluation of assays for beta-hydroxybutyrate. Am J Clin Pathol. 1997;107(3):353–358. [PubMed: 9052387]
- 112.
- Csako G, Elin RJ. Unrecognized false-positive ketones from drugs containing free-sulfhydryl group(s). JAMA. 1993;269(13):1634. [PubMed: 8455289]
- 113.
- Albert MS, Dell RB, Winters RW. Quantitative displacement of acid-base equilibrium in metabolic acidosis. Annals of internal medicine. 1967;66(2):312–322. [PubMed: 6016545]
- 114.
- Halperin ML, Hammeke M, Josse RG, Jungas RL. Metabolic acidosis in the alcoholic: a pathophysiologic approach. Metabolism: clinical and experimental. 1983;32(3):308–315. [PubMed: 6828000]
- 115.
- Bjellerup P, Kallner A, Kollind M. GLC determination of serum-ethylene glycol, interferences in ketotic patients. J Toxicol Clin Toxicol. 1994;32(1):85–87. [PubMed: 8308954]
- 116.
- Paulson WD, Gadallah MF. Diagnosis of mixed acid-base disorders in diabetic ketoacidosis. Am J Med Sci. 1993;306(5):295–300. [PubMed: 8238083]
- 117.
- Freidenberg GR, Kosnik EJ, Sotos JF. Hyperglycemic coma after suprasellar surgery. N Engl J Med. 1980;303(15):863–865. [PubMed: 7412803]
- 118.
- Hillman K. Fluid resuscitation in diabetic emergencies--a reappraisal. Intensive Care Med. 1987;13(1):4–8. [PubMed: 3104431]
- 119.
- Diabetes Canada Clinical Practice Guidelines Expert C. Goguen J, Gilbert J. Hyperglycemic Emergencies in Adults. Can J Diabetes. 2018;42 Suppl 1:S109–S114. [PubMed: 29650082]
- 120.
- Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes care. 1990;13(1):22–33. [PubMed: 2105195]
- 121.
- Pasquel FJ, Umpierrez GE. Hyperosmolar hyperglycemic state: a historic review of the clinical presentation, diagnosis, and treatment. Diabetes care. 2014;37(11):3124–3131. [PMC free article: PMC4207202] [PubMed: 25342831]
- 122.
- Cardoso L, Vicente N, Rodrigues D, Gomes L, Carrilho F. Controversies in the management of hyperglycaemic emergencies in adults with diabetes. Metabolism: clinical and experimental. 2017;68:43–54. [PubMed: 28183452]
- 123.
- Fisher JN, Shahshahani MN, Kitabchi AE. Diabetic ketoacidosis: low-dose insulin therapy by various routes. N Engl J Med. 1977;297(5):238–241. [PubMed: 406561]
- 124.
- Sacks HS, Shahshahani M, Kitabchi AE, Fisher JN, Young RT. Similar responsiveness of diabetic ketoacidosis to low-dose insulin by intramuscular injection and albumin-free infusion. Annals of internal medicine. 1979;90(1):36–42. [PubMed: 105656]
- 125.
- Burghen GA, Etteldorf JN, Fisher JN, Kitabchi AQ. Comparison of high-dose and low-dose insulin by continuous intravenous infusion in the treatment of diabetic ketoacidosis in children. Diabetes care. 1980;3(1):15–20. [PubMed: 6773725]
- 126.
- Umpierrez GE, Cuervo R, Karabell A, Latif K, Freire AX, Kitabchi AE. Treatment of diabetic ketoacidosis with subcutaneous insulin aspart. Diabetes care. 2004;27(8):1873–1878. [PubMed: 15277410]
- 127.
- Umpierrez GE, Latif K, Stoever J, Cuervo R, Park L, Freire AX. A EK. Efficacy of subcutaneous insulin lispro versus continuous intravenous regular insulin for the treatment of patients with diabetic ketoacidosis. Am J Med. 2004;117(5):291–296. [PubMed: 15336577]
- 128.
- Della Manna T, Steinmetz L, Campos PR, Farhat SC, Schvartsman C, Kuperman H, Setian N, Damiani D. Subcutaneous use of a fast-acting insulin analog: an alternative treatment for pediatric patients with diabetic ketoacidosis. Diabetes care. 2005;28(8):1856–1861. [PubMed: 16043723]
- 129.
- Atchley DW, Loeb RF, Richards DW, Benedict EM, Driscoll ME. ON. DIABETIC ACIDOSIS: A Detailed Study of Electrolyte Balances Following the Withdrawal and Reestablishment of Insulin Therapy. The Journal of clinical investigation. 1933;12(2):297–326. [PMC free article: PMC435909] [PubMed: 16694129]
- 130.
- Beigelman PM. Potassium in severe diabetic ketoacidosis. Am J Med. 1973;54(4):419–420. [PubMed: 4633105]
- 131.
- Abramson E, Arky R. Diabetic acidosis with initial hypokalemia. Therapeutic implications. JAMA. 1966;196(5):401–403. [PubMed: 5952215]
- 132.
- Viallon A, Zeni F, Lafond P, Venet C, Tardy B, Page Y, Bertrand JC. Does bicarbonate therapy improve the management of severe diabetic ketoacidosis? Crit Care Med. 1999;27(12):2690–2693. [PubMed: 10628611]
- 133.
- Glaser NS, Wootton-Gorges SL, Marcin JP, Buonocore MH, Dicarlo J, Neely EK, Barnes P, Bottomly J, Kuppermann N. Mechanism of cerebral edema in children with diabetic ketoacidosis. J Pediatr. 2004;145(2):164–171. [PubMed: 15289761]
- 134.
- Krane EJ, Rockoff MA, Wallman JK, Wolfsdorf JI. Subclinical brain swelling in children during treatment of diabetic ketoacidosis. N Engl J Med. 1985;312(18):1147–1151. [PubMed: 3920521]
- 135.
- Okuda Y, Adrogue HJ, Field JB, Nohara H, Yamashita K. Counterproductive effects of sodium bicarbonate in diabetic ketoacidosis. J Clin Endocrinol Metab. 1996;81(1):314–320. [PubMed: 8550770]
- 136.
- Morris LR, Murphy MB, Kitabchi AE. Bicarbonate therapy in severe diabetic ketoacidosis. Annals of internal medicine. 1986;105(6):836–840. [PubMed: 3096181]
- 137.
- Green SM, Rothrock SG, Ho JD, Gallant RD, Borger R, Thomas TL, Zimmerman GJ. Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998;31(1):41–48. [PubMed: 9437340]
- 138.
- Lever E, Jaspan JB. Sodium bicarbonate therapy in severe diabetic ketoacidosis. Am J Med. 1983;75(2):263–268. [PubMed: 6309004]
- 139.
- Fisher JN, Kitabchi AE. A randomized study of phosphate therapy in the treatment of diabetic ketoacidosis. J Clin Endocrinol Metab. 1983;57(1):177–180. [PubMed: 6406531]
- 140.
- Keller U, Berger W. Prevention of hypophosphatemia by phosphate infusion during treatment of diabetic ketoacidosis and hyperosmolar coma. Diabetes. 1980;29(2):87–95. [PubMed: 6766411]
- 141.
- Kreisberg RA. Phosphorus deficiency and hypophosphatemia. Hosp Pract. 1977;12(3):121–128. [PubMed: 402311]
- 142.
- Winter RJ, Harris CJ, Phillips LS, Green OC. Diabetic ketoacidosis. Induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67(5):897–900. [PubMed: 116547]
- 143.
- Adrogue HJ, Wilson H, Boyd AE 3rd, Suki WN, Eknoyan G. Plasma acid-base patterns in diabetic ketoacidosis. N Engl J Med. 1982;307(26):1603–1610. [PubMed: 6815530]
- 144.
- Kelly AM. The case for venous rather than arterial blood gases in diabetic ketoacidosis. Emerg Med Australas. 2006;18(1):64–67. [PubMed: 16454777]
- 145.
- Adrogue HJ, Eknoyan G, Suki WK. Diabetic ketoacidosis: role of the kidney in the acid-base homeostasis re-evaluated. Kidney Int. 1984;25(4):591–598. [PubMed: 6434787]
- 146.
- Oster JR, Epstein M. Acid-base aspects of ketoacidosis. Am J Nephrol. 1984;4(3):137–151. [PubMed: 6430087]
- 147.
- Kamel KS, Halperin ML. Acid-base problems in diabetic ketoacidosis. N Engl J Med. 2015;372(6):546–554. [PubMed: 25651248]
- 148.
- Wallace TM, Matthews DR. Recent advances in the monitoring and management of diabetic ketoacidosis. QJM. 2004;97(12):773–780. [PubMed: 15569808]
- 149.
- Fleckman AM. Diabetic ketoacidosis. Endocrinol Metab Clin North Am. 1993;22(2):181–207. [PubMed: 8325282]
- 150.
- Oh MS, Carroll HJ, Goldstein DA, Fein IA. Hyperchloremic acidosis during the recovery phase of diabetic ketosis. Annals of internal medicine. 1978;89(6):925–927. [PubMed: 102229]
- 151.
- Duck SC, Wyatt DT. Factors associated with brain herniation in the treatment of diabetic ketoacidosis. J Pediatr. 1988;113(1 Pt 1):10–14. [PubMed: 3133455]
- 152.
- Silver SM, Clark EC, Schroeder BM, Sterns RH. Pathogenesis of cerebral edema after treatment of diabetic ketoacidosis. Kidney Int. 1997;51(4):1237–1244. [PubMed: 9083292]
- 153.
- Haringhuizen A, Tjan DH, Grool A, van Vugt R, van Zante AR. Fatal cerebral oedema in adult diabetic ketoacidosis. Neth J Med. 2010;68(1):35–37. [PubMed: 20103820]
- 154.
- Hoorn EJ, Carlotti AP, Costa LA, MacMahon B, Bohn G, Zietse R, Halperin ML, Bohn D. Preventing a drop in effective plasma osmolality to minimize the likelihood of cerebral edema during treatment of children with diabetic ketoacidosis. J Pediatr. 2007;150(5):467–473. [PubMed: 17452217]
- 155.
- Smedman L, Escobar R, Hesser U, Persson B. Sub-clinical cerebral oedema does not occur regularly during treatment for diabetic ketoacidosis. Acta Paediatr. 1997;86(11):1172–1176. [PubMed: 9401508]
- 156.
- Isales CM, Min L, Hoffman WH. Acetoacetate and beta-hydroxybutyrate differentially regulate endothelin-1 and vascular endothelial growth factor in mouse brain microvascular endothelial cells. Journal of diabetes and its complications. 1999;13(2):91–97. [PubMed: 10432173]
- 157.
- Edge JA. Cerebral oedema during treatment of diabetic ketoacidosis: are we any nearer finding a cause? Diabetes Metab Res Rev. 2000;16(5):316–324. [PubMed: 11025556]
- 158.
- Galindo RJ, Pasquel FJ, Fayfman M, Tsegka K, Dhruv N, Cardona S, Wang H, Vellanki P, Umpierrez GE. Clinical characteristics and outcomes of patients with end-stage renal disease hospitalized with diabetes ketoacidosis. BMJ Open Diabetes Res Care. 2020;8(1) [PMC free article: PMC7050364] [PubMed: 32111715]
- 159.
- Buyukasik Y, Ileri NS, Haznedaroglu IC, Karaahmetoglu S, Muftuoglu O, Kirazli S, Dundar S. Enhanced subclinical coagulation activation during diabetic ketoacidosis. Diabetes care. 1998;21(5):868–870. [PubMed: 9589259]
- 160.
- McLaren EH, Cullen DR, Brown MJ. Coagulation abnormalities in diabetic coma before and 24 hours after treatment. Diabetologia. 1979;17(6):345–349. [PubMed: 535672]
- 161.
- Timperley WR, Preston FE, Ward JD. Cerebral intravascular coagulation in diabetic ketoacidosis. Lancet. 1974;1(7864):952–956. [PubMed: 4133643]
- 162.
- Everett E, Mathioudakis NN. Association of socioeconomic status and DKA readmission in adults with type 1 diabetes: analysis of the US National Readmission Database. BMJ Open Diabetes Res Care. 2019;7(1):e000621. [PMC free article: PMC6501850] [PubMed: 31114699]
- 163.
- Maldonado MR, Chong ER, Oehl MA, Balasubramanyam A. Economic impact of diabetic ketoacidosis in a multiethnic indigent population: analysis of costs based on the precipitating cause. Diabetes care. 2003;26(4):1265–1269. [PubMed: 12663608]
- 164.
- Musey VC, Lee JK, Crawford R, Klatka MA, McAdams D, Phillips LS. Diabetes in urban African-Americans. I. Cessation of insulin therapy is the major precipitating cause of diabetic ketoacidosis. Diabetes care. 1995;18(4):483–489. [PubMed: 7497857]
- 165.
- Randall L, Begovic J, Hudson M, Smiley D, Peng L, Pitre N, Umpierrez D, Umpierrez G. Recurrent diabetic ketoacidosis in inner-city minority patients: behavioral, socioeconomic, and psychosocial factors. Diabetes care. 2011;34(9):1891–1896. [PMC free article: PMC3161256] [PubMed: 21775761]
- 166.
- Lindner LME, Rathmann W, Rosenbauer J. Inequalities in glycaemic control, hypoglycaemia and diabetic ketoacidosis according to socio-economic status and area-level deprivation in Type 1 diabetes mellitus: a systematic review. Diabet Med. 2018;35(1):12–32. [PubMed: 28945942]
- 167.
- Laffel LM, Brackett J, Ho J, Anderson BJ. Changing the process of diabetes care improves metabolic outcomes and reduces hospitalizations. Qual Manag Health Care. 1998;6(4):53–62. [PubMed: 10339045]
- 168.
- Rewers A, Chase HP, Mackenzie T, Walravens P, Roback M, Rewers M, Hamman RF, Klingensmith G. Predictors of acute complications in children with type 1 diabetes. JAMA. 2002;287(19):2511–2518. [PubMed: 12020331]
- 169.
- Vanelli M, Chiari G, Ghizzoni L, Costi G, Giacalone T, Chiarelli F. Effectiveness of a prevention program for diabetic ketoacidosis in children. An 8-year study in schools and private practices. Diabetes care. 1999;22(1):7–9. [PubMed: 10333896]
- 170.
- Runyan JW Jr, Zwaag RV, Joyner MB, Miller ST. The Memphis diabetes continuing care program. Diabetes care. 1980;3(2):382–386. [PubMed: 7389550]
- 171.
- Charleer S, Mathieu C, Nobels F, De Block C, Radermecker RP, Hermans MP, Taes Y, Vercammen C. T'Sjoen G, Crenier L, Fieuws S, Keymeulen B, Gillard P, Investigators RT. Effect of Continuous Glucose Monitoring on Glycemic Control, Acute Admissions, and Quality of Life: A Real-World Study. J Clin Endocrinol Metab. 2018;103(3):1224–1232. [PubMed: 29342264]
- 172.
- Parkin CG, Graham C, Smolskis J. Continuous Glucose Monitoring Use in Type 1 Diabetes: Longitudinal Analysis Demonstrates Meaningful Improvements in HbA1c and Reductions in Health Care Utilization. J Diabetes Sci Technol. 2017;11(3):522–528. [PMC free article: PMC5505435] [PubMed: 28745091]
- 173.
- Cardona-Hernandez R, Schwandt A, Alkandari H, Bratke H, Chobot A, Coles N, Corathers S, Goksen D, Goss P, Imane Z, Nagl K, O'Riordan SMP, Jefferies C, Group SS. Glycemic Outcome Associated With Insulin Pump and Glucose Sensor Use in Children and Adolescents With Type 1 Diabetes. Data From the International Pediatric Registry SWEET. Diabetes care. 2021 [PubMed: 33653821]
- 174.
- Fleming N, Hamblin PS, Story D, Ekinci EI. Evolving Evidence of Diabetic Ketoacidosis in Patients Taking Sodium-Glucose Cotransporter 2 Inhibitors. J Clin Endocrinol Metab. 2020;105(8) [PubMed: 32302001]
- Pediatric Diabetic Ketoacidosis With Hyperosmolarity: Clinical Characteristics and Outcomes.[Endocr Pract. 2018]Pediatric Diabetic Ketoacidosis With Hyperosmolarity: Clinical Characteristics and Outcomes.Agrawal S, Baird GL, Quintos JB, Reinert SE, Gopalakrishnan G, Boney CM, Topor LS. Endocr Pract. 2018 Aug; 24(8):726-732. Epub 2018 Aug 7.
- Clinical Outcomes in Patients With Isolated or Combined Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic State: A Retrospective, Hospital-Based Cohort Study.[Diabetes Care. 2020]Clinical Outcomes in Patients With Isolated or Combined Diabetic Ketoacidosis and Hyperosmolar Hyperglycemic State: A Retrospective, Hospital-Based Cohort Study.Pasquel FJ, Tsegka K, Wang H, Cardona S, Galindo RJ, Fayfman M, Davis G, Vellanki P, Migdal A, Gujral U, et al. Diabetes Care. 2020 Feb; 43(2):349-357. Epub 2019 Nov 8.
- Review Treatment of Diabetic Ketoacidosis (DKA)/Hyperglycemic Hyperosmolar State (HHS): Novel Advances in the Management of Hyperglycemic Crises (UK Versus USA).[Curr Diab Rep. 2017]Review Treatment of Diabetic Ketoacidosis (DKA)/Hyperglycemic Hyperosmolar State (HHS): Novel Advances in the Management of Hyperglycemic Crises (UK Versus USA).Dhatariya KK, Vellanki P. Curr Diab Rep. 2017 May; 17(5):33.
- Clinical profiles, outcomes and risk factors among type 2 diabetic inpatients with diabetic ketoacidosis and hyperglycemic hyperosmolar state: a hospital-based analysis over a 6-year period.[BMC Endocr Disord. 2020]Clinical profiles, outcomes and risk factors among type 2 diabetic inpatients with diabetic ketoacidosis and hyperglycemic hyperosmolar state: a hospital-based analysis over a 6-year period.Wu XY, She DM, Wang F, Guo G, Li R, Fang P, Li L, Zhou Y, Zhang KQ, Xue Y. BMC Endocr Disord. 2020 Dec 14; 20(1):182. Epub 2020 Dec 14.
- Review Diabetic ketoacidosis and hyperglycemic hyperosmolar state.[Endocrinol Metab Clin North Am...]Review Diabetic ketoacidosis and hyperglycemic hyperosmolar state.Maletkovic J, Drexler A. Endocrinol Metab Clin North Am. 2013 Dec; 42(4):677-95.
- Hyperglycemic Crises: Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar State...Hyperglycemic Crises: Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar State - Endotext
- DVU3115 [Desulfovibrio vulgaris str. Hildenborough]DVU3115 [Desulfovibrio vulgaris str. Hildenborough]Gene ID:2796607Gene
- MARCHF11 membrane associated ring-CH-type finger 11 [Homo sapiens]MARCHF11 membrane associated ring-CH-type finger 11 [Homo sapiens]Gene ID:441061Gene
- Zdhhc5 zinc finger, DHHC domain containing 5 [Mus musculus]Zdhhc5 zinc finger, DHHC domain containing 5 [Mus musculus]Gene ID:228136Gene
- Kat7 K(lysine) acetyltransferase 7 [Mus musculus]Kat7 K(lysine) acetyltransferase 7 [Mus musculus]Gene ID:217127Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...