By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2001, Garland Science.
Bookshelf ID: NBK27112

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001.

Allergic reactions are triggered when allergens cross-link preformed IgE bound to the high-affinity receptor FcεRI on mast cells. Mast cells line the body surfaces and serve to alert the immune system to local infection. Once activated, they induce inflammatory reactions by secreting chemical mediators stored in preformed granules, and by synthesizing leukotrienes and cytokines after activation occurs. In allergy, they provoke very unpleasant reactions to innocuous antigens that are not associated with invading pathogens that need to be expelled. The consequences of IgE-mediated mast-cell activation depend on the dose of antigen and its route of entry; symptoms range from the irritating sniffles of hay fever when pollen is inhaled, to the life-threatening circulatory collapse that occurs in systemic anaphylaxis (Fig. 12.9). The immediate allergic reaction caused by mast-cell degranulation is followed by a more sustained inflammation, known as the late-phase response. This late response involves the recruitment of other effector cells, notably TH2 lymphocytes, eosinophils, and basophils, which contribute significantly to the immunopathology of an allergic response.
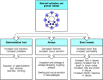
Mast-cell activation has different effects on different tissues.
Most antibodies are found in body fluids and engage effector cells, through receptors specific for the Fc constant regions, only after binding specific antigen through the antibody variable regions. IgE, however, is an exception as it is captured by the high-affinity Fcε receptor in the absence of bound antigen. This means that IgE is mostly found fixed in the tissues on mast cells that bear this receptor, as well as on circulating basophils and activated eosinophils. The ligation of cell-bound IgE antibody by specific antigen triggers activation of these cells at the site of antigen entry into the tissues. The release of inflammatory lipid mediators, cytokines, and chemokines at sites of IgE-triggered reactions results in the recruitment of eosinophils and basophils to augment the type I response.
There are two types of IgE-binding Fc receptor. The first, FcεRI, is a high-affinity receptor of the immunoglobulin superfamily that binds IgE on mast cells, basophils, and activated eosinophils (see Section 9-22). When the cell-bound IgE antibody is cross-linked by a specific antigen, FcεRI transduces an activating signal. High levels of IgE, such as those that exist in subjects with allergic diseases or parasite infections, can result in a marked increase in FcεRI on the surface of mast cells, enhanced sensitivity of such cells to activation by low concentrations of specific antigen, and markedly increased IgE-dependent release of chemical mediators and cytokines.
The second IgE receptor, FcεRII, usually known as CD23, is a C-type lectin and is structurally unrelated to FcεRI; it binds IgE with low affinity. CD23 is present on many different cell types, including B cells, activated T cells, monocytes, eosinophils, platelets, follicular dendritic cells, and some thymic epithelial cells. This receptor was thought to be crucial for the regulation of IgE antibody levels; however, knockout mouse strains lacking the CD23 gene show no major abnormality in the development of polyclonal IgE responses. However the CD23 knockout mice have demonstrated a role for CD23 in enhancing the antibody response to a specific antigen in the presence of that same antigen complexed with IgE. This antigen-specific, IgE-mediated enhancement of antibody responses fails to occur in mice lacking the CD23 gene. This demonstrates a role for CD23 on antigen-presenting cells in the capture of antigen by specific IgE.
Mast cells were described by Ehrlich in the mesentery of rabbits and named Mastzellen (‘fattened cells’). Like basophils, mast cells contain granules rich in acidic proteoglycans that take up basic dyes. However, in spite of this resemblance, and the similar range of mediators stored in these basophilic granules, mast cells are derived from a different myeloid lineage than basophils and eosinophils. Mast cells are highly specialized cells, and are prominent residents of mucosal and epithelial tissues in the vicinity of small blood vessels and postcapillary venules, where they are well placed to guard against invading pathogens (see Sections 9-20 and 9-21). Mast cells are also found in subendothelial connective tissue. They home to tissues as agranular cells; their final differentiation, accompanied by granule formation, occurs after they have arrived in the tissues. The major growth factor for mast cells is stem-cell factor (SCF), which acts on the cell-surface receptor c-Kit (see Section 7-2). Mice with defective c-Kit lack differentiated mast cells and cannot make IgE-mediated inflammatory responses. This shows that such responses depend almost exclusively on mast cells.
Mast cells express FcεRI constitutively on their surface and are activated when antigens cross-link IgE bound to these receptors (see Fig. 9.35). Degranulation occurs within seconds, releasing a variety of preformed inflammatory mediators (Fig. 12.10). Among these are histamine—a short-lived vasoactive amine that causes an immediate increase in local blood flow and vessel permeability—and enzymes such as mast-cell chymase, tryptase, and serine esterases. These enzymes can in turn activate matrix metalloproteinases, which break down tissue matrix proteins, causing tissue destruction. Large amounts of tumor necrosis factor (TNF)-α are also released by mast cells after activation. Some comes from stores in mast-cell granules; some is newly synthesized by the activated mast cells themselves. TNF-α activates endothelial cells, causing increased expression of adhesion molecules, which promotes the influx of inflammatory leukocytes and lymphocytes into tissues (see Section 2-22).

Molecules released by mast cells on activation. Mast cells produce a wide variety of biologically active proteins and other chemical mediators. The enzymes and toxic mediators listed in the first two rows are released from the preformed granules. The cytokines, (more...)
On activation, mast cells synthesize and release chemokines, lipid mediators such as leukotrienes and platelet-activating factor (PAF), and additional cytokines such as IL-4 and IL-13 which perpetuate the TH2 response. These mediators contribute to both the acute and the chronic inflammatory responses. The lipid mediators, in particular, act rapidly to cause smooth muscle contraction, increased vascular permeability, and mucus secretion, and also induce the influx and activation of leukocytes, which contribute to the late-phase response. The lipid mediators derive from membrane phospholipids, which are cleaved to release the precursor molecule arachidonic acid. This molecule can be modified by two pathways to give rise to prostaglandins, thromboxanes, and leukotrienes. The leukotrienes, especially C4, D4, and E4, are important in sustaining inflammatory responses in the tissues. Many anti-inflammatory drugs are inhibitors of arachidonic acid metabolism. Aspirin, for example, is an inhibitor of the enzyme cyclooxygenase and blocks the production of prostaglandins.
IgE-mediated activation of mast cells thus orchestrates an important inflammatory cascade that is amplified by the recruitment of eosinophils, basophils, and TH2 lymphocytes. The physiological importance of this reaction is as a defense mechanism against certain types of infection (see Section 9-23). In allergy, however, the acute and chronic inflammatory reactions triggered by mast-cell activation have important pathophysiological consequences, as seen in the diseases associated with allergic responses to environmental antigens.
Eosinophils are granulocytic leukocytes that originate in bone marrow. They are so called because their granules, which contain arginine-rich basic proteins, are colored bright orange by the acidic stain eosin (Fig. 12.11). Only very small numbers of these cells are normally present in the circulation; most eosinophils are found in tissues, especially in the connective tissue immediately underneath respiratory, gut, and urogenital epithelium, implying a likely role for these cells in defense against invading organisms. Eosinophils have two kinds of effector function. First, on activation they release highly toxic granule proteins and free radicals, which can kill microorganisms and parasites but can also cause significant tissue damage in allergic reactions. Second, activation induces the synthesis of chemical mediators such as prostaglandins, leukotrienes, and cytokines, which amplify the inflammatory response by activating epithelial cells, and recruiting and activating more eosinophils and leukocytes (Fig. 12.12).

Eosinophils can be detected easily in tissue sections by their bright refractile orange coloration. In this light micrograph, a large number of eosinophils are seen infiltrating a tumor of Langherhans' cells known as Langerhans' cell histiocytosis. The (more...)

Eosinophils secrete a range of highly toxic granule proteins and other inflammatory mediators.
The activation and degranulation of eosinophils is strictly regulated, as their inappropriate activation would be very harmful to the host. The first level of control acts on the production of eosinophils by the bone marrow. Few eosinophils are produced in the absence of infection or other immune stimulation. But when TH2 cells are activated, cytokines such as IL-5 are released that increase the production of eosinophils in the bone marrow and their release into the circulation. However, transgenic animals overexpressing IL-5 have increased numbers of eosinophils (eosinophilia) in the circulation but not in their tissues, indicating that migration of eosinophils from the circulation into tissues is regulated separately, by a second set of controls. The key molecules in this case are CC chemokines (see Section 2-20). Most of these cause chemotaxis of several types of leukocyte, but two are specific for eosinophils and have been named eotaxin 1 and eotaxin 2.
The eotaxin receptor on eosinophils, CCR3, is a member of the chemokine family of receptors (see Section 6-16). This receptor also binds the CC chemokines MCP-3, MCP-4, and RANTES, which also induce eosinophil chemotaxis. The eotaxins and these other CC chemokines also activate eosinophils. Identical or similar chemokines also stimulate mast cells and basophils. For example, eotaxin attracts basophils and causes their degranulation, and MCP-1, which binds to CCR2, similarly activates mast cells in both the presence or absence of antigen. MCP-1 can also promote the differentiation of naive TH0 cells to TH2 cells; TH2 cells also carry CCR3 and migrate toward eotaxin. These findings show that families of chemokines, as well as cytokines, can coordinate certain kinds of immune response.
A third set of controls regulates the state of eosinophil activation. In their nonactivated state, eosinophils do not express high-affinity IgE receptors and have a high threshold for release of their granule contents. After activation by cytokines and chemokines, this threshold drops, FcεRI is expressed, and the number of Fcγ receptors and complement receptors on the cell surface also increases. The eosinophil is now primed to carry out its effector activity, for example degranulation in response to antigen that cross-links specific IgE bound to FcεRI on the eosinophil surface.
The potential of eosinophils to cause tissue injury is illustrated by rare syndromes due to abnormally large numbers of eosinophils in the blood (hypereosinophilia). These syndromes are sometimes seen in association with T-cell lymphomas, in which unregulated IL-5 secretion drives a marked increase in the numbers of circulating eosinophils. The clinical manifestations of hypereosinophilia are damage to the endocardium (Fig. 12.13) and to nerves, leading to heart failure and neuropathy, both thought to be caused by the toxic effects of eosinophil granule proteins.

Hypereosinophilia can cause injury to the endocardium. The top panel shows a section of the endocardium from a patient with hyper-eosinophilic syndrome. There is an organized fibrous exudate and the underlying endocardium is thickened by fibrous tissue. Although (more...)
In a local allergic reaction, mast-cell degranulation and TH2 activation cause eosinophils to accumulate in large numbers and to become activated. Their continued presence is characteristic of chronic allergic inflammation and they are thought to be major contributors to tissue damage.
Basophils are also present at the site of an inflammatory reaction. Basophils share a common stem-cell precursor with eosinophils; growth factors for basophils are very similar to those for eosinophils and include IL-3, IL-5, and GM-CSF. There is evidence for reciprocal control of the maturation of the stem-cell population into basophils or eosinophils. For example, transforming growth factor (TGF)-β in the presence of IL-3 suppresses eosinophil differentiation and enhances that of basophils. Basophils are normally present in very low numbers in the circulation and seem to have a similar role to eosinophils in defense against pathogens. Like eosinophils, they are recruited to the sites of allergic reactions. Basophils express FcεRI on the cell surface and, on activation by cytokines or antigen, they release histamine and IL-4 from the basophilic granules after which they are named.
Eosinophils, mast cells, and basophils can interact with each other. Eosinophil degranulation releases major basic protein, which in turn causes degranulation of mast cells and basophils. This effect is augmented by any of the cytokines that affect eosinophil and basophil growth, differentiation, and activation, such as IL-3, IL-5, and GM-CSF.
The inflammatory response after IgE-mediated mast-cell activation occurs as an immediate reaction, starting within seconds, and a late reaction, which takes up to 8–12 hours to develop. These reactions can be distinguished clinically (Fig. 12.14). The immediate reaction is due to the activity of histamine, prostaglandins, and other preformed or rapidly synthesized mediators that cause a rapid increase in vascular permeability and the contraction of smooth muscle. The late-phase reaction is caused by the induced synthesis and release of mediators including leukotrienes, chemokines, and cytokines from the activated mast cells (see Fig. 12.10). These recruit other leukocytes, including eosinophils and TH2 lymphocytes, to the site of inflammation. Although the late-phase reaction is clinically less marked than the immediate response, it is associated with a second phase of smooth muscle contraction, sustained edema, and the development of one of the cardinal features of allergic asthma: airway hyperreactivity to nonspecific bronchoconstrictor stimuli such as histamine and methacholine.

Allergic reactions can be divided into an immediate response and a late-phase response. A wheal-and-flare allergic reaction develops within a minute or two of superficial injection of antigen into the epidermis and lasts for up to 30 minutes. The reaction (more...)
The late-phase reaction is an important cause of much serious long-term illness, as for example in chronic asthma. This is because the late reaction induces the recruitment of inflammatory leukocytes, especially eosinophils and TH2 lymphocytes, to the site of the allergen-triggered mast-cell response. This late response can easily convert into a chronic inflammatory response if antigen persists and stimulates allergen-specific TH2 cells, which in turn promote eosinophilia and further IgE production.
When reexposure to allergen triggers an allergic reaction, the effects are focused on the site at which mast-cell degranulation occurs. In the immediate response, the preformed mediators released are short-lived, and their potent effects on blood vessels and smooth muscles are therefore confined to the vicinity of the activated mast cell. The more sustained effects of the late-phase response are also focused on the site of initial allergen-triggered activation, and the particular anatomy of this site may determine how readily the inflammation can be resolved. Thus, the clinical syndrome produced by an allergic reaction depends critically on three variables: the amount of allergen-specific IgE present; the route by which the allergen is introduced; and the dose of allergen (Fig. 12.15).

The dose and route of allergen administration determine the type of IgE-mediated allergic reaction that results. There are two main anatomical distributions of mast cells: those associated with vascularized connective tissues, called connective tissue (more...)
If an allergen is introduced directly into the bloodstream or is rapidly absorbed
from the gut, the connective tissue mast cells associated with all blood vessels
can become activated. This activation causes a very dangerous syndrome called
systemic anaphylaxis ( Acute Systemic Anaphylaxis, in
Case Studies in Immunology, see Preface for details).
Disseminated mast-cell activation has a variety of potentially fatal effects:
the widespread increase in vascular permeability leads to a catastrophic loss of
blood pressure; airways constrict, causing difficulty in breathing; and swelling
of the epiglottis can cause suffocation. This potentially fatal syndrome is
called anaphylactic shock. It can
occur if drugs are administered to people who have IgE specific for that drug,
or after an insect bite in individuals allergic to insect venom. Some foods, for
example peanuts or brazil nuts, can cause systemic anaphylaxis in susceptible
individuals. This syndrome can be rapidly fatal but can usually be controlled by
the immediate injection of epinephrine, which relaxes the smooth muscle and
inhibits the cardiovascular effects of anaphylaxis.
Acute Systemic Anaphylaxis, in
Case Studies in Immunology, see Preface for details).
Disseminated mast-cell activation has a variety of potentially fatal effects:
the widespread increase in vascular permeability leads to a catastrophic loss of
blood pressure; airways constrict, causing difficulty in breathing; and swelling
of the epiglottis can cause suffocation. This potentially fatal syndrome is
called anaphylactic shock. It can
occur if drugs are administered to people who have IgE specific for that drug,
or after an insect bite in individuals allergic to insect venom. Some foods, for
example peanuts or brazil nuts, can cause systemic anaphylaxis in susceptible
individuals. This syndrome can be rapidly fatal but can usually be controlled by
the immediate injection of epinephrine, which relaxes the smooth muscle and
inhibits the cardiovascular effects of anaphylaxis.
The most frequent allergic reactions to drugs occur with penicillin and its relatives. In people with IgE antibodies against penicillin, administration of the drug by injection can cause anaphylaxis and even death. Great care should be taken to avoid giving a drug to patients with a past history of allergy to that drug or one that is closely related structurally. Penicillin acts as a hapten (see Section 9-2); it is a small molecule with a highly reactive β-lactam ring that is crucial for its antibacterial activity. This ring reacts with amino groups on host proteins to form covalent conjugates. When penicillin is ingested or injected, it forms conjugates with self proteins, and the penicillin-modified self peptides can provoke a TH2 response in some individuals. These TH2 cells then activate penicillin-binding B cells to produce IgE antibody to the penicillin hapten. Thus, penicillin acts both as the B-cell antigen and, by modifying self peptides, as the T-cell antigen. When penicillin is injected intravenously into an allergic individual, the penicillin-modified proteins can cross-link IgE molecules on the mast cells and cause anaphylaxis.
Inhalation is the most common route of allergen entry. Many people have mild allergies to inhaled antigens, manifesting as sneezing and a runny nose. This is called allergic rhinitis, and results from the activation of mucosal mast cells beneath the nasal epithelium by allergens such as pollens that release their protein contents, which can then diffuse across the mucus membranes of the nasal passages. Allergic rhinitis is characterized by intense itching and sneezing, local edema leading to blocked nasal passages, a nasal discharge, which is typically rich in eosinophils, and irritation of the nose as a result of histamine release. A similar reaction to airborne allergens deposited on the conjunctiva of the eye is called allergic conjunctivitis. Allergic rhinitis and conjunctivitis are commonly caused by environmental allergens that are only present during certain seasons of the year. For example, hay fever is caused by a variety of allergens, including certain grass and tree pollens. Autumnal symptoms may be caused by weed pollen, such as that of ragweed. These reactions are annoying but cause little lasting damage.
A more serious syndrome is allergic asthma, which is triggered by allergen-induced activation of submucosal mast cells in the lower airways (Fig. 12.16). This leads within seconds to bronchial constriction and increased secretion of fluid and mucus, making breathing more difficult by trapping inhaled air in the lungs. Patients with allergic asthma often need treatment, and asthmatic attacks can be life-threatening. An important feature of asthma is chronic inflammation of the airways, which is characterized by the continued presence of increased numbers of TH2 lymphocytes, eosinophils, neutrophils, and other leukocytes (Fig. 12.17).

The acute response in allergic asthma leads to TH2-mediated chronic inflammation of the airways. In sensitized individuals, cross-linking of specific IgE on the surface of mast cells by an inhaled allergen triggers them to secrete inflammatory mediators, (more...)
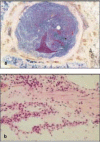
Morphological evidence of chronic inflammation in the airways of an asthmatic patient. Panel a shows a section through a bronchus of a patient who died of asthma; there is almost total occlusion of the airway by a mucus plug. In panel b, a close-up view (more...)
Although allergic asthma is initially driven by a response to a specific allergen, the subsequent chronic inflammation seems to be perpetuated even in the apparent absence of further exposure to allergen. The airways become characteristically hyperreactive and factors other than reexposure to antigen can trigger asthma attacks. For example, the airways of asthmatics characteristically show hyperresponsiveness to environmental chemical irritants such as cigarette smoke and sulfur dioxide; viral or, to a lesser extent, bacterial respiratory tract infections can exacerbate the disease by inducing a TH2-dominated local response.
The same dichotomy between immediate and delayed responses is seen in cutaneous allergic responses. The skin forms an effective barrier to the entry of most allergens but it can be breached by local injection of small amounts of allergen, for example by a stinging insect. The entry of allergen into the epidermis or dermis causes a localized allergic reaction. Local mast-cell activation in the skin leads immediately to a local increase in vascular permeability, which causes extravasation of fluid and swelling. Mast-cell activation also stimulates the release of chemicals from local nerve endings by a nerve axon reflex, causing the vasodilation of surrounding cutaneous blood vessels, which causes redness of the surrounding skin. The resulting skin lesion is called a wheal-and-flare reaction. About 8 hours later, a more widespread and sustained edematous response appears in some individuals as a consequence of the late-phase response (see Fig. 12.14). A disseminated form of the wheal-and-flare reaction, known as urticaria or hives, sometimes appears when ingested allergens enter the bloodstream and reach the skin. Histamine released by mast cells activated by allergen in the skin causes large, itchy, red swellings of the skin.
Allergists take advantage of the immediate response to test for allergy by injecting minute amounts of potential allergens into the epidermal layer of the skin. Although the reaction after the administration of antigen by intraepidermal injection is usually very localized, there is a small risk of inducing systemic anaphylaxis. Another standard test for allergy is to measure levels of IgE antibody specific for a particular allergen in a sandwich ELISA (see Appendix I, Section A-6).
Although acute urticaria is commonly caused by allergens, the causes of chronic urticaria, in which the urticarial rash can recur over long periods, are less well understood. In up to a third of cases, it seems likely that chronic urticaria is an autoimmune disease caused by autoantibodies against the α chain of FcεRI. This is an example of a type II hypersensitivity reaction in which an autoantibody against a cellular receptor triggers cellular activation, in this case causing mast-cell degranulation with resulting urticaria.
A more prolonged inflammatory response is sometimes seen in the skin, most often
in atopic children. They develop a persistent skin rash called eczema or atopic dermatitis (Atopic Dermatitis, in
Case Studies in Immunology, see Preface for details), due to a
chronic inflammatory response similar to that seen in the bronchial walls of
patients with asthma. The etiology of eczema is not well understood.
TH2 cells and IgE are involved, and it usually clears in
adolescence, unlike rhinitis and asthma, which can persist throughout life.
When an allergen is eaten, two types of allergic response are seen. Activation of mucosal mast cells associated with the gastrointestinal tract leads to transepithelial fluid loss and smooth muscle contraction, causing diarrhea and vomiting. For reasons that are not understood, connective tissue mast cells in the dermis and subcutaneous tissues can also be activated after ingestion of allergen, presumably by allergen that has been absorbed into the bloodstream, and this results in urticaria. Urticaria is a common reaction when penicillin is given orally to a patient who already has penicillin-specific IgE antibodies. Ingestion of food allergens can also lead to the development of generalized anaphylaxis, accompanied by cardiovascular collapse and acute asthmatic symptoms. Certain foods, most importantly peanuts, tree nuts, and shellfish, are particularly associated with this type of life-threatening response.
The approaches to the treatment and prevention of allergy are set out in Fig. 12.18. Two treatments are commonly used in clinical practice—one is desensitization and the other is blockade of the effector pathways. There are also several approaches still in the experimental stage. In desensitization the aim is to shift the antibody response away from one dominated by IgE toward one dominated by IgG; the latter can bind to the allergen and thus prevent it from activating IgE-mediated effector pathways. Patients are injected with escalating doses of allergen, starting with tiny amounts. This injection schedule gradually diverts the IgE-dominated response, driven by TH2 cells, to one driven by TH1 cells, with the consequent downregulation of IgE production. Recent evidence shows that desensitization is also associated with a reduction in the numbers of late-phase inflammatory cells at the site of the allergic reaction. A potential complication of the desensitization approach is the risk of inducing IgE-mediated allergic responses.

Approaches to the treatment of allergy. Possible methods of inhibiting allergic reactions are shown. Two approaches are in regular clinical use. The first is the injection of specific antigen in desensitization regimes, which are believed to divert the (more...)
An alternative, and still experimental, approach to desensitization is vaccination with peptides derived from common allergens. This procedure induces T-cell anergy (see Section 8-11), which is associated with multiple changes in the T-cell phenotype, including downregulation of cytokine production and reduced expression of the CD3:T-cell receptor complex. IgE-mediated responses are not induced by the peptides because IgE, in contrast to T cells, can only recognize the intact antigen. A major difficulty with this approach is that an individual's responses to peptides are restricted by their MHC class II alleles (see Section 5-12); therefore, patients with different MHC class II molecules respond to different allergen-derived peptides. As the human population is outbred and expresses a wide variety of MHC class II alleles, the number of peptides required to treat all allergic individuals might be very large.
Another vaccination strategy that shows promise in experimental models of allergy is the use of oligodeoxynucleotides rich in unmethylated cytosine guanine dinucleotides (CpG) as adjuvants (see Section 14-19) for desensitization regimes. These oligonucleotides mimic bacterial DNA sequences known as CpG motifs and strongly promote TH1 responses. Their mechanism of action is discussed in Sections 14-19 and 8-6 and Appendix I, Section A-4.
The signaling pathways that enhance the IgE response in allergic disease are also potential targets for therapy. Inhibitors of IL-4, IL-5, and IL-13 would be predicted to reduce IgE responses, but redundancy between some of the activities of these cytokines might make this approach difficult to implement in practice. A second approach to manipulating the response is to give cytokines that promote TH1-type responses. IFN-γ, IFN-α, IL-10, IL-12, and TGF-β have each been shown to reduce IL-4-stimulated IgE synthesis in vitro, and IFN-γ and IFN-α have been shown to reduce IgE synthesis in vivo.
Another target for therapeutic intervention might be the high-affinity IgE receptor. An effective competitor for IgE at this receptor could prevent the binding of IgE to the surfaces of mast cells, basophils, and eosinophils. Candidate competitors include humanized anti-IgE monoclonal antibodies, which bind to IgE and block its binding to the receptor, and modified IgE Fc constructs that bind to the receptor but lack variable regions and thus cannot bind antigen. Yet another approach would be to block the recruitment of eosinophils to sites of allergic inflammation. The eotaxin receptor CCR3 is a potential target for this type of therapy. The production of eosinophils in bone marrow and their exit into the circulation might also be reduced by a blockade of IL-5 action.
The mainstays of therapy at present, however, are drugs that treat the symptoms of allergic disease and limit the inflammatory response. Anaphylactic reactions are treated with epinephrine, which stimulates the reformation of endothelial tight junctions, promotes the relaxation of constricted bronchial smooth muscle, and also stimulates the heart. Inhaled bronchodilators that act on β-adrenergic receptors to relax constricted muscle are also used to relieve acute asthma attacks. Antihistamines that block the histamine H1 receptor reduce the urticaria that follows histamine release from mast cells and eosinophils. Relevant H1 receptors include those on blood vessels that cause increased permeability of the vessel wall, and those on unmyelinated nerve fibers that are thought to mediate the itching sensation. In chronic allergic disease it is extremely important to treat and prevent the chronic inflammatory tissue injury. Topical or systemic corticosteroids (see Section 14-1) are used to suppress the chronic inflammatory changes seen in asthma, rhinitis, and eczema. However, what is really needed is a means of converting the T-cell response to the allergenic peptide antigen from predominantly TH2 to predominantly TH1. This topic is also discussed in Chapter 14.
The allergic response to innocuous antigens reflects the pathophysiological aspects of a defensive immune response whose physiological role is to protect against helminthic parasites. It is triggered by antigen binding to IgE antibodies bound to the high-affinity IgE receptor FcεRI on mast cells. Mast cells are strategically distributed beneath the mucosal surfaces of the body and in connective tissue. Antigen cross-linking the IgE on their surface causes them to release large amounts of inflammatory mediators. The resulting inflammation can be divided into early events, characterized by short-lived mediators such as histamine, and later events that involve leukotrienes, cytokines, and chemokines, which recruit and activate eosinophils and basophils. The late phase of this response can evolve into chronic inflammation, characterized by the presence of effector T cells and eosinophils, which is most clearly seen in chronic allergic asthma.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...