Proteins perform most of the work of living cells. This versatile class of macromolecule is involved in virtually every cellular process: proteins replicate and transcribe DNA, and produce, process, and secrete other proteins. They control cell division, metabolism, and the flow of materials and information into and out of the cell. Understanding how cells work requires understanding how proteins function.
The question of what a protein does inside a living cell is not a simple one to answer. Imagine isolating an uncharacterized protein and discovering that its structure and amino acid sequence suggest that it acts as a protein kinase. Simply knowing that the protein can add a phosphate group to serine residues, for example, does not reveal how it functions in a living organism. Additional information is required to understand the context in which the biochemical activity is used. Where is this kinase located in the cell and what are its protein targets? In which tissues is it active? Which pathways does it influence? What role does it have in the growth or development of the organism?
In this section, we discuss the methods currently used to characterize protein structure and function. We begin with an examination of the techniques used to determine the three-dimensional structure of purified proteins. We then discuss methods that are used to predict how a protein functions, based on its homology to other known proteins and its location inside the cell. Finally, because most proteins act in concert with other proteins, we present techniques for detecting protein-protein interactions. But these approaches only begin to define how a protein might work inside a cell. In the last section of this chapter, we discuss how genetic approaches are used to dissect and analyze the biological processes in which a given protein functions.
The Diffraction of X-rays by Protein Crystals Can Reveal a Protein's Exact Structure
Starting with the amino acid sequence of a protein, one can often predict which secondary structural elements, such as membrane-spanning α helices, will be present in the protein. It is presently not possible, however, to deduce reliably the three-dimensional folded structure of a protein from its amino acid sequence unless its amino acid sequence is very similar to that of a protein whose three-dimensional structure is already known. The main technique that has been used to discover the three-dimensional structure of molecules, including proteins, at atomic resolution is x-ray crystallography.
X-rays, like light, are a form of electromagnetic radiation, but they have a much shorter wavelength, typically around 0.1 nm (the diameter of a hydrogen atom). If a narrow parallel beam of x-rays is directed at a sample of a pure protein, most of the x-rays pass straight through it. A small fraction, however, is scattered by the atoms in the sample. If the sample is a well-ordered crystal, the scattered waves reinforce one another at certain points and appear as diffraction spots when the x-rays are recorded by a suitable detector ().
X-ray crystallography. (A) A narrow parallel beam of x-rays is directed at a well-ordered crystal (B). Shown here is a protein crystal of ribulose bisphosphate carboxylase, an enzyme with a central role in CO2 fixation during photosynthesis. Some of the (more...)
The position and intensity of each spot in the x-ray diffraction pattern contain information about the locations of the atoms in the crystal that gave rise to it. Deducing the three-dimensional structure of a large molecule from the diffraction pattern of its crystal is a complex task and was not achieved for a protein molecule until 1960. But in recent years x-ray diffraction analysis has become increasingly automated, and now the slowest step is likely to be the generation of suitable protein crystals. This requires large amounts of very pure protein and often involves years of trial and error, searching for the proper crystallization conditions. There are still many proteins, especially membrane proteins, that have so far resisted all attempts to crystallize them.
Analysis of the resulting diffraction pattern produces a complex three-dimensional electron-density map. Interpreting this map—translating its contours into a three-dimensional structure—is a complicated procedure that requires knowledge of the amino acid sequence of the protein. Largely by trial and error, the sequence and the electron-density map are correlated by computer to give the best possible fit. The reliability of the final atomic model depends on the resolution of the original crystallographic data: 0.5 nm resolution might produce a low-resolution map of the polypeptide backbone, whereas a resolution of 0.15 nm allows all of the non-hydrogen atoms in the molecule to be reliably positioned.
A complete atomic model is often too complex to appreciate directly, but simplified versions that show a protein's essential structural features can be readily derived from it (see Panel 3-2, pp. 138–139). The three-dimensional structures of about 10,000 different proteins have now been determined by x-ray crystallography or by NMR spectroscopy (see below)—enough to begin to see families of common structures emerging. These structures or protein folds often seem to be more conserved in evolution than are the amino acid sequences that form them (see ).
X-ray crystallographic techniques can also be applied to the study of macromolecular complexes. In a recent triumph, the method was used to solve the structure of the ribosome, a large and complex cellular machine made of several RNAs and more than 50 proteins (see ). The determination required the use of a synchrotron, a radiation source that generates x-rays with the intensity needed to analyze the crystals of such large macromolecular complexes.
Molecular Structure Can Also Be Determined Using Nuclear Magnetic Resonance (NMR) Spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy has been widely used for many years to analyze the structure of small molecules. This technique is now also increasingly applied to the study of small proteins or protein domains. Unlike x-ray crystallography, NMR does not depend on having a crystalline sample; it simply requires a small volume of concentrated protein solution that is placed in a strong magnetic field.
Certain atomic nuclei, and in particular those of hydrogen, have a magnetic moment or spin: that is, they have an intrinsic magnetization, like a bar magnet. The spin aligns along the strong magnetic field, but it can be changed to a misaligned, excited state in response to applied radiofrequency (RF) pulses of electromagnetic radiation. When the excited hydrogen nuclei return to their aligned state, they emit RF radiation, which can be measured and displayed as a spectrum. The nature of the emitted radiation depends on the environment of each hydrogen nucleus, and if one nucleus is excited, it influences the absorption and emission of radiation by other nuclei that lie close to it. It is consequently possible, by an ingenious elaboration of the basic NMR technique known as two-dimensional NMR, to distinguish the signals from hydrogen nuclei in different amino acid residues and to identify and measure the small shifts in these signals that occur when these hydrogen nuclei lie close enough together to interact: the size of such a shift reveals the distance between the interacting pair of hydrogen atoms. In this way NMR can give information about the distances between the parts of the protein molecule. By combining this information with a knowledge of the amino acid sequence, it is possible in principle to compute the three-dimensional structure of the protein ().
NMR spectroscopy. (A) An example of the data from an NMR machine. This two-dimensional NMR spectrum is derived from the C-terminal domain of the enzyme cellulase. The spots represent interactions between hydrogen atoms that are near neighbors in the protein (more...)
For technical reasons the structure of small proteins of about 20,000 daltons or less can readily be determined by NMR spectroscopy. Resolution is lost as the size of a macromolecule increases. But recent technical advances have now pushed the limit to about 100,000 daltons, thereby making the majority of proteins accessible for structural analysis by NMR.
The NMR method is especially useful when a protein of interest has resisted attempts at crystallization, a common problem for many membrane proteins. Because NMR studies are performed in solution, this method also offers a convenient means of monitoring changes in protein structure, for example during protein folding or when a substrate binds to the protein. NMR is also used widely to investigate molecules other than proteins and is valuable, for example, as a method to determine the three-dimensional structures of RNA molecules and the complex carbohydrate side chains of glycoproteins.
Some landmarks in the development of x-ray crystallography and NMR are listed in .
Landmarks in the Development of X-ray Crystallography and NMR and Their Application to Biological Molecules.
Sequence Similarity Can Provide Clues About Protein Function
Thanks to the proliferation of protein and nucleic acid sequences that are catalogued in genome databases, the function of a gene—and its encoded protein—can often be predicted by simply comparing its sequence with those of previously characterized genes. Because amino acid sequence determines protein structure and structure dictates biochemical function, proteins that share a similar amino acid sequence usually perform similar biochemical functions, even when they are found in distantly related organisms. At present, determining what a newly discovered protein does therefore usually begins with a search for previously identified proteins that are similar in their amino acid sequences.
Searching a collection of known sequences for homologous genes or proteins is typically done over the World-Wide Web, and it simply involves selecting a database and entering the desired sequence. A sequence alignment program—the most popular are BLAST and FASTA—scans the database for similar sequences by sliding the submitted sequence along the archived sequences until a cluster of residues falls into full or partial alignment (). The results of even a complex search—which can be performed on either a nucleotide or an amino acid sequence—are returned within minutes. Such comparisons can be used to predict the functions of individual proteins, families of proteins, or even the entire protein complement of a newly sequenced organism.
Results of a BLAST search. Sequence databases can be searched to find similar amino acid or nucleic acid sequences. Here a search for proteins similar to the human cell-cycle regulatory protein cdc2 (Query) locates maize cdc2 (Subject), which is 68% identical (more...)
In the end, however, the predictions that emerge from sequence analysis are often only a tool to direct further experimental investigations.
Fusion Proteins Can Be Used to Analyze Protein Function and to Track Proteins in Living Cells
The location of a protein within the cell often suggests something about its function. Proteins that travel from the cytoplasm to the nucleus when a cell is exposed to a growth factor, for example, may have a role in regulating gene expression in response to that factor. A protein often contains short amino acid sequences that determine its location in a cell. Most nuclear proteins, for example, contain one or more specific short sequences of amino acids that serve as signals for their import into the nucleus after their synthesis in the cytosol (discussed in Chapter 12). These special regions of the protein can be identified by fusing them to an easily detectable protein that lacks such regions and then following the behavior of this surrogate protein in a cell. Such fusion proteins can be readily produced by the recombinant DNA techniques discussed previously.
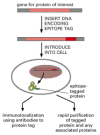
Another common strategy used both to follow proteins in cells and to purify them rapidly is epitope tagging. In this case, a fusion protein is produced that contains the entire protein being analyzed plus a short peptide of 8 to 12 amino acids (an “epitope”) that can be recognized by a commercially available antibody. The fusion protein can therefore be specifically detected, even in the presence of a large excess of the normal protein, using the anti-epitope antibody and a labeled secondary antibody that can be monitored by light or electron microscopy ().
Epitope tagging allows the localization or purification of proteins. Using standard genetic engineering techniques, a short epitope tag can be added to a protein of interest. The resulting protein contains the protein being analyzed plus a short peptide (more...)
Today large numbers of proteins are being tracked in living cells by using a fluorescent marker called green fluorescent protein (GFP). Tagging proteins with GFP is as simple as attaching the gene for GFP to one end of the gene that encodes a protein of interest. In most cases, the resulting GFP fusion protein behaves in the same way as the original protein, and its movement can be monitored by following its fluorescence inside the cell by fluorescence microscopy. The GFP fusion protein strategy has become a standard way to determine the distribution and dynamics of any protein of interest in living cells. We discuss its use further in Chapter 9.
GFP, and its derivatives of different color, can also be used to monitor protein-protein interactions. In this application, two proteins of interest are each labeled with a different fluorochrome, such that the emission spectrum of one fluorochrome overlaps the absorption spectrum of the second fluorochrome. If the two proteins—and their attached fluorochromes—come very close to each other (within about 1–10 nm), the energy of the absorbed light will be transferred from one fluorochrome to the other. The energy transfer, called fluorescence resonance energy transfer (FRET), is determined by illuminating the first fluorochrome and measuring emission from the second (). By using two different spectral variants of GFP as the fluorochromes in such studies, one can monitor the interaction of any two protein molecules inside a living cell.
Fluorescence resonance energy transfer (FRET). To determine whether (and when) two proteins interact inside the cell, the proteins are first produced as fusion proteins attached to different variants of GFP. (A) In this example, protein X is coupled to (more...)
Affinity Chromatography and Immunoprecipitation Allow Identification of Associated Proteins
Because most proteins in the cell function as part of a complex with other proteins, an important way to begin to characterize their biological roles is to identify their binding partners. If an uncharacterized protein binds to a protein whose role in the cell is understood, its function is likely to be related. For example, if a protein is found to be part of the proteasome complex, it is likely to be involved somehow in degrading damaged or misfolded proteins.
Protein affinity chromatography is one method that can be used to isolate and identify proteins that interact physically. To capture interacting proteins, a target protein is attached to polymer beads that are packed into a column. Cellular proteins are washed through the column and those proteins that interact with the target adhere to the affinity matrix (see ). These proteins can then be eluted and their identity determined by mass spectrometry or another suitable method.
Perhaps the simplest method for identifying proteins that bind to one another tightly is co-immunoprecipitation. In this case, an antibody is used to recognize a specific target protein; affinity reagents that bind to the antibody and are coupled to a solid matrix are then used to drag the complex out of solution to the bottom of a test tube. If this protein is associated tightly enough with another protein when it is captured by the antibody, the partner precipitates as well. This method is useful for identifying proteins that are part of a complex inside cells, including those that interact only transiently—for example when cells are stimulated by signal molecules (discussed in Chapter 15).
Co-immunoprecipitation techniques require having a highly specific antibody against a known cellular protein target, which is not always available. One way to overcome this requirement is to use recombinant DNA techniques to add an epitope tag (see ) or to fuse the target protein to a well-characterized marker protein, such as the small enzyme glutathione S-transferase (GST). Commercially available antibodies directed against the epitope tag or the marker protein can then be used to precipitate the whole fusion protein, including any cellular proteins associated with the protein of interest. If the protein is fused to GST, antibodies may not be needed at all: the hybrid and its binding partners can be readily selected on beads coated with glutathione ().
Purification of protein complexes using a GST-tagged fusion protein. GST fusion proteins, generated by standard recombinant DNA techniques, can be captured on an affinity column containing beads coated with glutathione. To look for proteins that bind (more...)
In addition to capturing protein complexes on columns or in test tubes, researchers are also developing high-density protein arrays for investigating protein function and protein interactions. These arrays, which contain thousands of different proteins or antibodies spotted onto glass slides or immobilized in tiny wells, allow one to examine the biochemical activities and binding profiles of a large number of proteins at once. To examine protein interactions with such an array, one incubates a labeled protein with each of the target proteins immobilized on the slide and then determines to which of the many proteins the labeled molecule binds.
Protein-Protein Interactions Can Be Identified by Use of the Two-Hybrid System
Methods such as co-immunoprecipitation and affinity chromatography allow the physical isolation of interacting proteins. A successful isolation yields a protein whose identity must then be ascertained by mass spectrometry, and whose gene must be retrieved and cloned before further studies characterizing its activity—or the nature of the protein-protein interaction—can be performed.
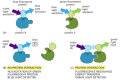
Other techniques allow the simultaneous isolation of interacting proteins along with the genes that encode them. The first method we discuss, called the two-hybrid system, uses a reporter gene to detect the physical interaction of a pair of proteins inside a yeast cell nucleus. This system has been designed so that when a target protein binds to another protein in the cell, their interaction brings together two halves of a transcriptional activator, which is then able to switch on the expression of the reporter gene.
The technique takes advantage of the modular nature of gene activator proteins (see ). These proteins both bind to DNA and activate transcription—activities that are often performed by two separate protein domains. Using recombinant DNA techniques, the DNA sequence that codes for a target protein is fused with DNA that encodes the DNA-binding domain of a gene activator protein. When this construct is introduced into yeast, the cells produce the target protein attached to this DNA-binding domain (). This protein binds to the regulatory region of a reporter gene, where it serves as “bait” to fish for proteins that interact with the target protein inside a yeast cell. To prepare a set of potential binding partners, DNA encoding the activation domain of a gene activator protein is ligated to a large mixture of DNA fragments from a cDNA library. Members of this collection of genes—the “prey”—are introduced individually into yeast cells containing the bait. If the yeast cell has received a DNA clone that expresses a prey partner for the bait protein, the two halves of a transcriptional activator are united, switching on the reporter gene (see ). Cells that express this reporter are selected and grown, and the gene (or gene fragment) encoding the prey protein is retrieved and identified through nucleotide sequencing.
The yeast two-hybrid system for detecting protein-protein interactions. The target protein is fused to a DNA-binding domain that localizes it to the regulatory region of a reporter gene as “bait.” When this target protein binds to another (more...)
Although it sounds complex, the two-hybrid system is relatively simple to use in the laboratory. Although the protein-protein interactions occur in the yeast cell nucleus, proteins from every part of the cell and from any organism can be studied in this way. Of the thousands of protein-protein interactions that have been catalogued in yeast, half have been discovered with such two-hybrid screens.
The two-hybrid system can be scaled up to map the interactions that occur among all of the proteins produced by an organism. In this case, a set of bait fusions is produced for every cellular protein, and each of these constructs is introduced into a separate yeast cell. These cells are then mated to yeast containing the prey library. Those rare cells that are positive for a protein-protein interaction are then characterized. In this way a protein linkage map has been generated for most of the 6,000 proteins in yeast (see ), and similar projects are underway to catalog the protein interactions in C. elegans and Drosophila.
A related technique, called a reverse two-hybrid system, can be used to identify mutations—or chemical compounds—that are able to disrupt specific protein-protein interactions. In this case the reporter gene can be replaced by a gene that kills cells in which the bait and prey proteins interact. Only those cells in which the proteins no longer bind—because an engineered mutation or a test compound prevents them from doing so—can survive. Like knocking out a gene (which we discuss shortly), eliminating a particular molecular interaction can reveal something about the role of the participating proteins in the cell. In addition, compounds that selectively interrupt protein interactions can be medically useful: a drug that prevents a virus from binding to its receptor protein on human cells could help people to avoid infections, for example.
Phage Display Methods Also Detect Protein Interactions
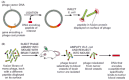
Another powerful method for detecting protein-protein interactions involves introducing genes into a virus that infects the E. coli bacterium (a bacteriophage, or “phage”). In this case the DNA encoding the protein of interest (or a smaller peptide fragment of this protein) is fused with a gene encoding one of the proteins that forms the viral coat. When this virus infects E. coli, it replicates, producing phage particles that display the hybrid protein on the outside of their coats (). This bacteriophage can then be used to fish for binding partners in a large pool of potential target proteins.
The phage display method for investigating protein interactions. (A) Preparation of the bacteriophage. DNA encoding the desired peptide is ligated into the phage vector, fused with the gene encoding the viral protein coat. The engineered phage are then (more...)
However, the most powerful use of this phage display method allows one to screen large collections of proteins or peptides for binding to selected targets. This approach requires first generating a library of fusion proteins, much like the prey library in the two-hybrid system. This collection of phage is then screened for binding to a purified protein of interest. For example, the phage library can be passed through an affinity column containing an immobilized target protein. Viruses that display a protein or peptide that binds tightly to the target are captured on the column and can be eluted with excess target protein. Those phage containing a DNA fragment that encodes an interacting protein or peptide are collected and allowed to replicate in E. coli. The DNA from each phage can then be recovered and its nucleotide sequence determined to identify the protein or peptide partner that bound to the target protein. A similar technique has been used to isolate peptides that bind specifically to the inside of the blood vessels associated with human tumors. These peptides are presently being tested as agents for delivering therapeutic anti-cancer compounds directly to such tumors ().
Phage display has also been used to generate monoclonal antibodies that recognize a specific target molecule or cell. In this case, a library of phage expressing the appropriate parts of antibody molecules is screened for those phage that bind to a target antigen.
Protein Interactions Can Be Monitored in Real Time Using Surface Plasmon Resonance
Once two proteins—or a protein and a small molecule—are known to associate, it becomes important to characterize their interaction in more detail. Proteins can bind to one another permanently, or engage in transient encounters in which proteins remain associated only temporarily. These dynamic interactions are often regulated through reversible modifications (such as phosphorylation), through ligand binding, or through the presence or absence of other proteins that compete for the same binding site.
To begin to understand these intricacies, one must determine how tightly two proteins associate, how slowly or rapidly molecular complexes assemble and break down over time, and how outside influences can affect these parameters. As we have seen in this chapter, there are many different techniques available to study protein-protein interactions, each with its individual advantages and disadvantages. One particularly useful method for monitoring the dynamics of protein association is called surface plasmon resonance (SPR). The SPR method has been used to characterize a wide variety of molecular interactions, including antibody-antigen binding, ligand-receptor coupling, and the binding of proteins to DNA, carbohydrates, small molecules, and other proteins.
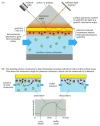
SPR detects binding interactions by monitoring the reflection of a beam of light off the interface between an aqueous solution of potential binding molecules and a biosensor surface carrying immobilized bait protein. The bait protein is attached to a very thin layer of metal that coats one side of a glass prism (). A light beam is passed through the prism; at a certain angle, called the resonance angle, some of the energy from the light interacts with the cloud of electrons in the metal film, generating a plasmon—an oscillation of the electrons at right angles to the plane of the film, bouncing up and down between its upper and lower surfaces like a weight on a spring. The plasmon, in turn, generates an electrical field that extends a short distance—about the wavelength of the light—above and below the metal surface. Any change in the composition of the environment within the range of the electrical field causes a measurable change in the resonance angle.
Surface plasmon resonance. (A) SPR can detect binding interactions by monitoring the reflection of a beam of light off the interface between an aqueous solution of potential binding molecules (green) and a biosensor surface coated with an immobilized (more...)
To measure binding, a solution containing proteins (or other molecules) that might interact with the immobilized bait protein is allowed to flow past the biosensor surface. When proteins bind to the bait, the composition of the molecular complexes on the metal surface change, causing a change in the resonance angle (see ). The changes in the resonance angle are monitored in real time and reflect the kinetics of the association—or dissociation—of molecules with the bait protein. The association rate (kon) is measured as the molecules interact, and the dissociation rate (koff) is determined as buffer washes the bound molecules from the sensor surface. A binding constant (K) is calculated by dividing koff by kon. In addition to determining the kinetics, SPR can be used to determine the number of molecules that are bound in each complex: the magnitude of the SPR signal change is proportional to the mass of the immobilized complex.
The SPR method is particularly useful because it requires only small amounts of proteins, the proteins do not have to be labeled in any way, and protein-protein interactions can be monitored in real time.
DNA Footprinting Reveals the Sites Where Proteins Bind on a DNA Molecule
So far we have concentrated on examining protein-protein interactions. But some proteins act by binding to DNA. Most of these proteins have a central role in determining which genes are active in a particular cell by binding to regulatory DNA sequences, which are usually located outside the coding regions of a gene.
In analyzing how such a protein functions, it is important to identify the specific nucleotide sequences to which it binds. A method used for this purpose is called DNA footprinting. First, a pure DNA fragment that is labeled at one end with 32P is isolated (see ); this molecule is then cleaved with a nuclease or a chemical that makes random single-stranded cuts in the DNA. After the DNA molecule is denatured to separate its two strands, the resultant fragments from the labeled strand are separated on a gel and detected by autoradiography. The pattern of bands from DNA cut in the presence of a DNA-binding protein is compared with that from DNA cut in its absence. When the protein is present, it covers the nucleotides at its binding site and protects their phosphodiester bonds from cleavage. As a result, the labeled fragments that terminate in the binding site are missing, leaving a gap in the gel pattern called a “footprint” (). Similar methods can be used to determine the binding sites of proteins on RNA.
The DNA footprinting technique. (A) This technique requires a DNA molecule that has been labeled at one end (see Figure 8-24B). The protein shown binds tightly to a specific DNA sequence that is seven nucleotides long, thereby protecting these seven nucleotides (more...)
Summary
Many powerful techniques are used to study the structure and function of a protein. To determine the three-dimensional structure of a protein at atomic resolution, large proteins have to be crystallized and studied by x-ray diffraction. The structure of small proteins in solution can be determined by nuclear magnetic resonance analysis. Because proteins with similar structures often have similar functions, the biochemical activity of a protein can sometimes be predicted by searching for known proteins that are similar in their amino acid sequences.
Further clues to the function of a protein can be derived from examining its subcellular distribution. Fusion of the protein with a molecular tag, such as the green fluorescent protein (GFP), allows one to track its movement inside the cell. Proteins that enter the nucleus and bind to DNA can be further characterized by footprint analysis, a technique used to determine which regulatory sequences the protein binds to as it controls gene transcription.
All proteins function by binding to other proteins or molecules, and many methods exist for studying protein-protein interactions and identifying potential protein partners. Either protein affinity chromatography or co-immunoprecipitation by antibodies directed against a target protein will allow physical isolation of interacting proteins. Other techniques, such as the two-hybrid system or phage display, permit the simultaneous isolation of interacting proteins and the genes that encode them. The identity of the proteins recovered from any of these approaches is then ascertained by determining the sequence of the protein or its corresponding gene.