

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition. Boston: Butterworths; 1990.

Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition.
Show detailsDefinition
Ketone bodies (acetoacetic acid, beta-hydroxybutyric acid, and acetone) are insignificant in the blood and urine of normal individuals in the postprandial or overnight-fasted state. However, these ketoacids become important sources of metabolic energy in circumstances in which the availability of glucose is restricted, as during prolonged fasting, or when the ability to use glucose is greatly diminished, as in decompensated diabetes mellitus. During prolonged starvation the arterial concentrations of these metabolically active strong organic acids increase approximately 70-fold to 10 to 12 mM and to significantly higher levels of 30 to 40 mM in diabetic ketoacidosis. The mechanisms responsible for the development of ketonemia are: (1) increased production by the liver; (2) decreased peripheral utilization in muscle; and (3) reduced volume of distribution. Since ketone bodies are not bound to plasma proteins, they are freely filterable solutes in the renal glomerulus and appear quantifiably in the tubular urine. At the very low plasma concentrations of ketone bodies that are encountered normally after an overnight fast, urinary excretion rates are negligible. When plasma levels increase beyond 0.1 to 0.2 mM, however, excretion increases and measurable amounts of ketone bodies appear in the urine.
Technique
Two semiquantitative tests are available for the rapid determination of primarily acetoacetate in urine and blood, although they have never been considered totally satisfactory for blood. The ferric chloride test is performed by adding 5 to 10 drops of freshly voided urine to a few drops of a 10% solution of ferric chloride. At first, a precipitate of ferric phosphate forms but with additional drops of urine the precipitate disappears and a burgundy red color appears. The intensity of the color is judged 1+ to 4+. The assay measures only acetoacetate and its sensitivity is less than 80 mg/dl. Unfortunately, drugs such as salicylates and antipyrines, as well as phenols, acetates, and cyanates, will cause false positive results. To overcome this the sample is boiled for 2 minutes to remove the unstable acetoacetate, which causes the red color to disappear. It is preferable to repeat the test on a previously boiled urine sample.
The sodium nitroprusside reaction measures both acetone and acetoacetate, but not hydroxybutyrate, in biological materials. There is no interference from most drugs and metabolic products. It is the most successful test devised. For the Rothera test A (named for the originator of the test), 1 gm ammonium sulfate is added to 5 ml urine and, after shaking to dissolve, 3 drops of freshly prepared sodium nitroprusside solution is added. After mixing, 1 to 2 ml of a saturated solution of ammonium hydroxide is layered onto the sample and after 2 minutes, the purple color which forms at the interface is judged for its intensity from 1 + to 4 +. The nitroprusside test will detect 1 to 2 mg/dl acetoacetate or 10 mg/dl acetone. In a less sensitive but perhaps more convenient assay (Rothera test B), a solution of ammonium sulfate is used and the reagents are mixed with the specimen so that the entire solution becomes purple. Ten mg/dl acetoacetate or 40 mg/dl acetone can be detected. To improve the convenience of the assay, the Joslin Clinic formulated a stable, dry preparation of sodium nitroprusside by combining it with ammonium sulfate (5 gm to 200 gm, respectively). Two to 3 ml of urine are saturated with this mixture, and the purple color at the interface is read 2 to 3 minutes after overlaying with ammonium hydroxide. The color intensity is from a purplish pink to deep purple and is scored 1 + to 4 +, but it deteriorates with standing. The ferric chloride and nitroprusside tests are still in clinical use in developing nations.
Nitroprusside is available as a test tablet (Acetest) and as a coated reagent strip (Ketostix), both manufactured by the Ames Division of Miles Laboratories, Inc., Elkhart, Indiana. With Acetest, after 30 seconds the color development is compared to a chart and judged negative, small, moderate or large. The tablet will detect 5 to 10 mg/dl of acetoacetate and 20 mg/dl of acetone. The quantitative range included in each category is 5 to 20 mg/dl for small, 20 to 40 mg/dl for moderate, and 40 mg/dl or greater for large. With Ketostix, the strip is momentarily dipped into the urine specimen or passed through in the urinary stream and compared to a color chart 1 minute later. The scale is negative, trace, small, moderate, and large. The strip is capable of detecting 5 mg/dl acetoacetate but is not reactive to acetone. The ranges are wider and shifted somewhat to the right in the higher zones compared to Acetest so that only 16% of samples containing 20 mg/dl acetoacetate are read as moderate while 24% of samples containing 80 mg/dl acetoacetate are still called moderate. Only 15% of the samples containing 40 mg/dl acetoacetate are judged to be large; 76% are large at 80 mg/dl and 100% at 160 mg/dl. The Ketostix test is most accurate when urines are tested with a high specific gravity (between 1.010 and 1.020) and low-pH. Highly pigmented urine specimens may yield false positive readings. Levodopa will also cause a false positive result. Ketostix strips are less sensitive than Acetest tablets and have a high degree of variability between lots. Acetest, with sensitivity in the 5 mg/dl range, is the preferable method.
Several other methods are available for measuring ketone bodies. With the isolation and purification of the enzyme 3-hydroxybutyrate dehydrogenase, it became possible to develop a sensitive and highly specific enzymatic assay for acetoacetate and hydroxybutyrate. The enzyme catalyzes the redox reaction:
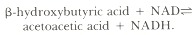
More recently, fluorometric techniques and an automated colorimetric method have been developed for this analysis, and other enzymatic methods have been described for the determination of acetoacetate. Finally, Kobayashi and co-workers (1983) have developed a gas chromatographic method for the determination of both acetone and acetoacetate in urine. They speculate that the method could be easily adapted to measure hydroxybutyrate as well, following its quantified oxidation to acetoacetate.
Basic Science
Ketones are most commonly detected when accelerated adipose tissue triglyceride hydrolysis and increased hepatic fatty acid permeability across the inner mitochondrial membrane (as produced by a decrease in circulating insulin and increased glucagon) cause increased glycogen breakdown, gluconeogenesis, lipolysis, fatty acid oxidation, and ketogenesis. In normal individuals plasma ketone levels are self-limited, because at 2 to 4 mM concentrations and greater, insulin release is stimulated, thereby preventing ketoacidosis by an insulin limitation of lipolysis and the availability of free fatty acids for hepatic oxidation. In the insulin-dependent diabetic, concentrations of free fatty acids continue to increase uncontrollably until ketoacidosis ensues. In addition to fasting, prolonged starvation, and uncontrolled diabetes, ketonuria may be observed in other physiologic conditions. Acute alcoholism and severe and prolonged exercise may result in ketonuria. Ketone bodies may also be detected in the urine during the third trimester of pregnancy, labor and delivery, the immediate postpartum period, and occasionally during lactation. Ketogenesis may also be increased in the neonate, resulting in significant ketonuria. These clinical conditions are all characterized by a temporary decrease in glucose availability, increased glucose utilization, and elevated levels of counterregulatory or stress-associated hormones such as cortisol and epinephrine.
To reduce the urinary loss of these important metabolic fuels, which furnish a significant amount of the caloric requirements during fasting, the kidney is capable of reabsorbing a portion of the increased filtered load of ketoacids. A linear correlation has been found between the urinary excretion of acetoacetate and the plasma concentration, once the renal threshold has been exceeded. Most reports have described a similar linear relationship for the excretion of hydroxybutyrate as the filtered load increases. At elevated plasma levels of ketone bodies, the mean fractional excretions are 0.15 to 0.19 for acetoacetate and hydroxybutyrate, respectively. Thus ketone bodies appear to be completely reabsorbed by the renal tubules at low plasma concentrations, but as plasma levels rise and the filtered load of ketone bodies increases, significant ketonuria appears. The net reabsorption rate remains directly proportional to the filtered load of ketone bodies so that, despite large increases in plasma concentrations, excretion rates of approximately 20% of the filtered load remain unchanged. Most recent evidence would support the conclusion that there is no tubular maximum for ketone bodies or that this exceeds the high filtered loads seen during the studies on starvation. Since the excretory rates are relatively unchanged, the reabsorption rates must therefore increase. The mechanisms that increase this reabsorption rate are unknown, but it has been shown that reabsorption rates are not altered by the reduction in extravascular volume and sodium balance that accompany starvation and decompensated diabetes.
Studies in the rat have demonstrated an initial decrease in the fractional reabsorption of hydroxybutyrate concentrations, but there was no further decline as the hydroxybutyrate concentration was raised to higher levels. These data may suggest both saturable and nonsaturable renal transport systems for hydroxybutyrate. The maximal transport rate of the saturable component is achieved at an arterial concentration of approximately 1.7 mM. Although the renal utilization of hydroxybutyrate rises with increasing arterial levels of this ketoacid, it does not affect the net reabsorption rate. The reabsorption rates of hydroxybutyrate always exceed utilization. As in humans, the excretion rate of acetoacetate in the rat is directly proportional to its filtration rate. No maximal rate was observed, indicating that a nonsaturable mechanism for the reabsorption of acetoacetate is also present in the kidney. The reabsorption of acetoacetate is decreased by increasing levels of hydroxybutyrate, indicating a common, competitive tubular transport mechanism. Acetoacetate was not released by the kidney into the blood with increasing hydroxybutyrate utilization, but there was simultaneous net utilization of acetoacetate by the kidney. This tended to increase with rising concentrations of arterial acetoacetate. There was evidence that with lower levels of acetoacetate reabsorption, higher rates of utilization were sustained by cellular uptake from the peritubular blood. Finally, membrane vesicles isolated from the brush border of the rat kidney have been known to contain a sodium-gradient-dependent carrier system that transports ketone bodies into an osmotically reactive space. The carrier demonstrates reciprocal inhibition between acetoacetate and hydroxybutyrate, accelerated exchange diffusion, saturation and competitive inhibition, and a lack of ouabain sensitivity.
Renal utilization of both ketoacids accounts for approximately 50% of the turnover of infused ketone bodies. Thus, oxidative metabolism accounts for a major fraction of the measured renal clearance of ketone bodies from the blood. In prolonged starvation it has been calculated that renal reabsorption of ketone bodies spares approximately 225 K cal/day, which would otherwise be lost in the urine. Up to 60% is consumed by the kidney, and the rest is released for utilization by the central nervous system during glucose deprivation. The ability of the kidney to reabsorb ketone bodies also preserves sodium, potassium, and ammonium ion, since urinary electroneutrality to equimolar excretion of cations is required during the loss of these anions. During the first few days of fasting or throughout the course of diabetic ketoacidosis, sodium and potassium are lost with the excretion of ketone bodies. If starvation continues, the obligate cation accompanying ketone bodies becomes ammonium ion. The reabsorption of ketone bodies therefore spares not only calories but also ammonium nitrogen, conserving at least 7g of nitrogen per day.
Clinical Significance
Poorly controlled or decompensated diabetes mellitus is the most common pathologic condition causing ketonuria. As a result, routine testing of urine for ketones in diabetics has been universally employed in hospitals and by outpatients to assess adequacy of diabetes control and determine insulin dosage during periods of loss of control. The widespread availability of self blood glucose monitoring and the superiority of this method over urine testing of glucose as a monitor of diabetic control have deemphasized the importance and hence the frequency of testing for ketone bodies in the urine. This is a serious omission, for there is no blood glucose level per se that is diagnostic of life-threatening ketoacidosis. Although it is not necessary to test every urine specimen for ketonuria, hyperglycemia at almost any level, when associated with an intercurrent illness or stress, demands urine ketone testing. Many cases of advanced ketoacidosis have gone undiagnosed and progressed to serious states because health care professionals as well as patients have abandoned urine testing not only for glucosuria but for ketonuria as well.
The presence or absence of ketonuria is also useful for clinical classifications of fasting hypoglycemia in infancy and childhood. The absence of ketone bodies is evidence for excess insulin secretion associated with beta cell neoplasia, whereas ketonuria suggests a lack of fuel availability. However, some caution is required for generalizations, as this discriminatory test may be inaccurate and unreliable. In cases of hyperinsulinism, the plasma levels of ketone bodies may be suppressed but not sufficiently low to prevent ketonuria. The same rationale, based on the action of insulin to suppress ketogenesis, has been used to serve as a rough indicator of the occurrence of nocturnal hypoglycemia in the insulin-requiring diabetic patient. Severe glucosuria with or without mild ketonuria upon arising is considered to be evidence of rebound hyperglycemia following insulin-induced hypoglycemia (Somogyi effect). Problems with a low renal threshold for ketone bodies, the high sensitivity of the nitroprusside test, and the problems with mixing of the urine within the bladder may make such a clinical judgment unreliable.
Excessive intake of ethanol is also a frequent cause of ketonuria. Often its use is associated with recent or chronic lack of sufficient nutrients, simulating a state of acute or chronic starvation. In addition, the metabolism of ethanol inhibits gluconeogenesis. Consequently, in most instances there is relative hypoglycemia and insulinopenia, although ethanol has been found to stimulate insulin secretion directly when combined with sucrose ingestion.
References
- Alkonyl I, Kerner J, Stabo D. A new enzymatic method for the determination of acetoacetate. Acta Biochem Biophys Acad Sci Hung. 1972;7:143–47. [PubMed: 4671865]
- Barac-Nieto M. Adrenal hydroxybutyrate and acetoacetate reabsorption and utilization in the rat. Am J Physiol. 1985;249:F40–48. [PubMed: 4014475]
- Behre JA. A modified salicylaldehyde method for determination of acetone bodies in blood and urine. J Biol Chem. 1940;136:25–34.
- Dawson WL. Levodopa and tests for ketonuria. N Engl J Med. 1969;281:1075. [PubMed: 5424737]
- Dobson HL, Shaffer R, Burns R. Accuracy of urine testing for sugar and acetone by hospital ward personnel. Diabetes. 1968;17:281–85. [PubMed: 5648371]
- Foster DW. From glycogen to ketones and back. Diabetes. 1984;33:1188–99. [PubMed: 6094292]
- Free HM, Smeby RR, Cook MH, Free AH. A comparative study of qualitative tests for ketones in urine and serum. Clin Chem. 1958;4:323–30. [PubMed: 13561546]
- Galvin RD, Harris JA, Johnson RE. Urinary excretion of beta-hydroxybutyrate and acetoacetate during experimenal ketosis. Quant J Exp Physiol. 1968;53:181–93. [PubMed: 5185570]
- Galvin RD, Johnson RE, Passmore R. Renal excretion of ketone bodies in post exercise ketosis. J Physiol (London). 1959;146:51P–52P.
- Garcia ML, Benavides J, Valdivieso F. Ketone body transport in renal brush border membrane vesicles. Biochem Biophys Acta. 1980;600:922–30. [PubMed: 7407151]
- Greenberg LA, Lester D. A micromethod for the determination of acetone and ketone bodies. J Biol Chem. 1944;154:177–90. [PubMed: 18871250]
- Ham TH. A syllabus of laboratory examination in clinical diagnosis. Cambridge, MA: Harvard Univ. Press, 1952;259.
- James RC, Chase GR. Evaluation of some commonly used semi-quantitative methods for urinary glucose and ketone determinations. Diabetes. 1974;23:474–79. [PubMed: 4830183]
- Kleiber M. The fire of life. New York: Wiley, 1961.
- Kobyashi K, Okada M, Yasuda, Kawai S. A gas chromatographic method for the determination of acetone acid in urine. Clin Chim Acta. 1983;133:223–26. [PubMed: 6627684]
- Levy LJ, Duqa J, Girgis M. et al. Ketoacidosis associated with alcoholism in non-diabetic subject. Ann Intern Med. 1973;78:213–25. [PubMed: 4683750]
- Marble A. Laboratory procedures useful in diagnosis and treatment. In: Marble EA, White P, Bradley RF, Krall LP, eds. Joslin's diabetes mellitus. 11th ed. Philadelphia: Lea & Febiger, 1971;196.
- Martin HE, Wick AN. Quantitative relationship between blood and urine ketone levels in diabetic acidosis. J Clin Invest. 1943;22:235–41. [PMC free article: PMC435232] [PubMed: 16694999]
- Miles J, Haymond M, Gerich J. Suppression of glucose production and stimulation of insulin secretion by physiological concentrations of ketone bodies in man. J Clin Endocrinol Metab. 1981;52:34–45. [PubMed: 7005257]
- Owen OE, Felig P, Morgan AP. et al. Liver and kidney metabolism during prolonged starvation. J Clin Invest. 1969;48:574–83. [PMC free article: PMC535723] [PubMed: 5773093]
- Owen OE, Morgan AP, Kemp HG. et al. Brain metabolism during fasting. J Clin Invest. 1967;46:1589–95. [PMC free article: PMC292907] [PubMed: 6061736]
- Owen OE, Reichard GA Jr,, Markus H. et al. Rapid intravenous sodium acetoacetate infusion in man. J Clin Invest. 1973;52:2606–16. [PMC free article: PMC302521] [PubMed: 4729054]
- Paterson P, Sheath J, Pincus T. et al. Maternal and foetal ketone concentrations in plasma and urine. Lancet. 1967;1:862–65. [PubMed: 4164365]
- Peters IP, Van Slyke DD. Acetone, acetoacetic acid, and beta-hydroxybutyric acid—principal of methods. In: Quantitative Clinical Chemistry, 2. Baltimore: Williams & Wilkins, 1932:623–46.
- Rapoport A, From GLA, Husdan H. Metabolic studies in prolonged fasting. I. Inorganic metabolism and kidney function. Metabolism. 1965;14:47–58. [PubMed: 14252341]
- Reichard GA Jr,, Owen OE, Haff AC. et al. Ketone-body production and oxidation in fasting humans. J Clin Invest. 1974;53:508–17. [PMC free article: PMC301493] [PubMed: 11344564]
- Ruderman NB, Goodman MN. Regulation of ketone body metabolism in skeletal muscle. Am J Physiol. 1973;224:1391–97. [PubMed: 4712152]
- Sansum WD, Koehler AB, Bowden RA. Manual for diabetic patients. New York: Macmillan, 1939;116.
- Sapir DG, Owen OE. Renal conservation of ketone bodies during starvation. Metabolism. 1975;24:23–33. [PubMed: 234169]
- Sapir DG, Owen OE, Cheng JT. et al. The effect of carbohydrates on ammonium and ketoacid exretion during starvation. J Clin Invest. 1972;51:2093–2102. [PMC free article: PMC292366] [PubMed: 5054466]
- Shipley RA, Long CNH. Studies on the ketogenic activity of an anterior pituitary. Biochem J. 1938;32:2242–56. [PMC free article: PMC1264319] [PubMed: 16746868]
- Todd JC, Sanford AH, Wells BB. Clinical diagnosis by laboratory methods. 12th ed. Philadelphia: W.B. Saunders, 1953:93.
- Wildenhoff KE. The renal excretion of acetoacetate and 3-hydroxybutyrate during absolute fasting. Acta Med Scand. 1972;192:475–79. [PubMed: 5083390]
- Williamson DH, Mellanby JD. Acetoacetate. In: Bergmeyer HB, ed. Methods of enzymatic analysis. New York: Academic Press, 1974;4:1840–43.
- Williamson DH, Mellanby JD. Hydroxybutyrate. In: Bergmeyer HB, ed. Methods of enzymatic analysis. New York: Academic Press, 1974;4:1836–39.
- Williamson DH, Mellanby J, Krebs HA. Enzymatic determination of D (−)-B- hydroxybutyric acid and acetoacetic acid in blood. Biochem J. 1962;82:90–96. [PMC free article: PMC1243411] [PubMed: 14007241]
- Wolfsdorf JL, Sadeghi-Nejad A, Senior B. Ketonuria does not exclude hyperinsulinemic hypoglycemia. Am J Dis Child. 1984;138:168–171. [PubMed: 6364785]
- Zivin JA, Snarr J. An automated colorimetric method for the measurement of 3-hydroxybutyrate concentrations. Anal Biochem. 1973;52:456–61. [PubMed: 4698839]
- Rapid intravenous sodium acetoacetate infusion in man. Metabolic and kinetic responses.[J Clin Invest. 1973]Rapid intravenous sodium acetoacetate infusion in man. Metabolic and kinetic responses.Owen OE, Reichard GA Jr, Markus H, Boden G, Mozzoli MA, Shuman CR. J Clin Invest. 1973 Oct; 52(10):2606-16.
- Ketoacidosis.[StatPearls. 2024]Ketoacidosis.Ghimire P, Dhamoon AS. StatPearls. 2024 Jan
- Ketoacidosis (Nursing).[StatPearls. 2024]Ketoacidosis (Nursing).Ghimire P, Dhamoon AS, Doerr C. StatPearls. 2024 Jan
- Review A focused review of the role of ketone bodies in health and disease.[J Med Food. 2013]Review A focused review of the role of ketone bodies in health and disease.Akram M. J Med Food. 2013 Nov; 16(11):965-7. Epub 2013 Oct 18.
- Review The influence of feeding and fasting on plasma metabolites in the dogfish shark (Squalus acanthias).[Comp Biochem Physiol A Mol Int...]Review The influence of feeding and fasting on plasma metabolites in the dogfish shark (Squalus acanthias).Wood CM, Walsh PJ, Kajimura M, McClelland GB, Chew SF. Comp Biochem Physiol A Mol Integr Physiol. 2010 Apr; 155(4):435-44. Epub 2009 Sep 24.
- Ketonuria - Clinical MethodsKetonuria - Clinical Methods
- The endocrine pancreas - EndocrinologyThe endocrine pancreas - Endocrinology
- The thyroid gland - EndocrinologyThe thyroid gland - Endocrinology
- The parathyroid glands and vitamin D - EndocrinologyThe parathyroid glands and vitamin D - Endocrinology
Your browsing activity is empty.
Activity recording is turned off.
See more...