

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Institute of Medicine (US) Committee on Military Nutrition Research; Marriott BM, editor. Food Components to Enhance Performance: An Evaluation of Potential Performance-Enhancing Food Components for Operational Rations. Washington (DC): National Academies Press (US); 1994.

Food Components to Enhance Performance: An Evaluation of Potential Performance-Enhancing Food Components for Operational Rations.
Show detailsRichard J.Wurtman 2
INTRODUCTION
Contrary to earlier expectations, it has now become well established that the amounts of neurotransmitter released when certain neurons fire normally vary over a broad range. One process that generates such variations involves receptors on the neurons' own presynaptic terminals: when activated by the neurotransmitter molecules that the neuron has released into the synapse, by concurrently released neuromodulators such as adenosine, or by other transmitters (e.g., the enkephalins) released at axoaxonal synapses, these receptors initiate intracellular events that diminish the number of neurotransmitter molecules released subsequently.
Another type of process that particularly affects the release of amine neurotransmitters depends on changes in the composition of the blood plasma induced by eating or by prolonged physical activity. Changes in plasma levels of choline or of certain amino acids lead to changes in brain levels of the precursors for these neurotransmitters—choline for acetylcholine, tryptophan for serotonin, and tyrosine for the catecholamines. These, in turn, regulate the rates at which the transmitters are synthesized, their concentrations within nerve terminals, and ultimately, the quantities released each time the neurons fire. For one transmitter—serotonin—the relevant variations in plasma composition probably affect most, if not all, of the neurons that release it. For other transmitters (e.g., the catecholamines), individual nerve cells can become more or less precursor dependent at any time, depending on the rates at which they happen to be firing.
Unlike the receptor-mediated presynaptic modulation of transmitter release, precursor-dependent modulation depends primarily on metabolic events occurring outside the brain and arising from a particular type of voluntary behavior, such as eating or exercise. Indeed, the primary physiological role of this dependency may be sensory (i.e., to provide the omnivore's brain with information about what has been eaten or about important changes in macronutrient requirements, so that the individual can better decide what to eat next). However, because precursor-dependent neurotransmitters are involved in a wide variety of normal (and pathological) brain mechanisms besides those controlling food intake, this relationship may have broad physiological and medical implications. It also provides benign ways of influencing neurotransmission, and thus mental and physical performance.
FOOD CONSUMPTION, TRYPTOPHAN AVAILABILITY, AND BRAIN SEROTONIN SYNTHESIS
The initial observation that physiological changes in precursor availability (i.e., after food consumption) could affect neurotransmitter synthesis was made in studies on rats performed in 1971 (Fernstrom and Wurtman, 1971). Animals were allowed to eat a test diet that contained carbohydrates and fat but that lacked protein. Soon after the start of the meal, brain levels of the essential (and scarce) amino acid tryptophan were found to have risen, thus increasing the substrate saturation of the enzyme that controls serotonin synthesis, tryptophan hydroxylase. The resulting increase in brain serotonin levels was associated with an increase in brain levels of serotonin's metabolite, 5-hydroxyindole acetic acid, thus suggesting that serotonin release had also been enhanced. (Direct evidence that physiological variations in brain tryptophan concentrations affect serotonin release was not obtained until 1987 [Schaechter and Wurtman, 1989].)
The rise in brain tryptophan levels after consumption of this test diet was accompanied by either a small increase (rats) or no change (humans) in plasma tryptophan levels. Both of these changes had been unanticipated, since the insulin secretion elicited by dietary carbohydrates was known to lower plasma levels of most of the other amino acids. However, the unusual response of plasma tryptophan to insulin was soon recognized as resulting from the amino acid's unusual propensity to bind loosely to circulating albumin. Insulin causes nonesterified fatty acid molecules to dissociate from albumin and to enter adipocytes. This dissociation increases the protein's capacity to bind circulating tryptophan; hence, whatever reduction insulin causes in free plasma tryptophan levels is compensated for by a rise in the tryptophan bound to albumin, yielding no net change in total plasma tryptophan levels in humans (Madras et al., 1974). Because this binding is of low affinity, the albumin-bound tryptophan is almost as able as free tryptophan to be taken up into the brain.
Considerably more difficult to explain were the data then obtained on what happens to brain tryptophan and serotonin levels after rats consume a meal rich in protein. Although plasma tryptophan levels were found to rise, reflecting the contribution of some of the tryptophan molecules in the protein, brain tryptophan and serotonin levels either failed to rise or, if the meal contained sufficient protein, actually fell (Fernstrom and Wurtman, 1972). The explanation for this paradox was found to lie in the transport systems that carry tryptophan across the blood-brain barrier (Pardridge, 1977) and into neurons. The endothelial cells that line central nervous system capillaries contain various macromolecules that shuttle specific nutrients or their metabolites between the blood and the brain's extracellular space. One such macromolecule mediates the transcapillary flux (by facilitated diffusion) of tryptophan and other large neutral amino acids (LNAAs) such as tyrosine; others move choline, basic or acidic amino acids, hexoses, monocarboxylic acids, adenosine, adenine, and various vitamins. The amount of any LNAA transported by the macromolecule depends on its ability to compete with the other circulating LNAAs for binding sites. Thus, the ability of circulating tryptophan molecules to enter the brain is increased when plasma levels of the other LNAAs fall (as occurs after insulin is secreted) and is diminished when the plasma levels of the other LNAAs rise, even if plasma tryptophan levels remain unchanged. Since all dietary proteins are considerably richer in the other LNAAs than in tryptophan (only 1.0–1.5 percent of most proteins), consumption of a protein-rich meal decreases the plasma/tryptophan ratio (the ratio of the plasma tryptophan concentration to the summed concentrations of its major circulating competitors for brain uptake, principally, tyrosine; phenylalanine; the branched-chain amino acids leucine, isoleucine, and valine; and methionine). This, in turn, decreases tryptophan's transport into the brain and slows its conversion to serotonin. (Similar plasma ratios predict brain levels of each of the other LNAAs—including drugs such as levodopa (L-dopa)—following meals or other treatments that modify plasma amino acid patterns (Wurtman et al., 1980). This is why a high-protein meal interferes with levodopa's therapeutic effect, whereas a high-carbohydrate, protein-free meal can lead to abnormal movements caused by too much levodopa suddenly entering the brain (Wurtman et al., 1988).
The fact that administration of pure tryptophan could increase brain serotonin synthesis, thereby affecting various serotonin-dependent brain functions (e.g., sleepiness and mood), has been known since at least 1968. What was novel and perhaps surprising about the above findings was their demonstration that brain tryptophan levels—and serotonin synthesis—normally undergo important variations in response, for example, to the decision to eat a carbohydrate-rich (as opposed to a protein-rich) breakfast or in response to the administration of a very low dose of tryptophan (Fernstrom and Wurtman, 1971).
It remained possible, however, that mechanisms external to the serotonin-releasing neuron might exist. These mechanisms kept such food-induced increases in serotonin's synthesis from causing parallel changes in the amounts released into synapses. Indeed, it was known that if rats were given very large doses of tryptophan that were sufficient to raise brain tryptophan levels well beyond their normal range, the firing frequencies of their serotonin-releasing raphe neurons decreased markedly; this was interpreted as reflecting the operation of a feedback system designed to keep serotonin release within a physiological range. Similar decreases in raphe firing had also been observed in animals given drugs, such as monoamine oxidase (MAO) inhibitors or serotonin-reuptake blockers, which cause persistent increases in intrasynaptic serotonin levels. Indeed, the administration of serotonin uptake inhibitors such as fluoxetine can cause the prolonged inhibition of serotonin release (Gardier and Wurtman, 1991). However, when rats were given small doses of tryptophan that were sufficient to raise brain tryptophan levels but not beyond their normal peaks or when they consumed a carbohydrate-rich meal, which raised brain tryptophan levels physiologically, no decreases in raphe firing occurred. Hence, food-induced changes in serotonin synthesis were found to affect the amounts of serotonin released per firing without slowing the neuron's firing frequencies, thus “allowing” modulation of the net output of information from serotonergic neurons.
BRAIN SEROTONIN, NUTRIENT CHOICE, AND CARBOHYDRATE CRAVING
If rats are allowed to pick from foods in two pans presented concurrently and containing differing proportions of protein and carbohydrate, they choose among the two so as to obtain fairly constant (for each animal) amounts of these macronutrients. However, if before “dinner” they receive either a carbohydrate-based snack or a drug that facilitates serotonergic neurotransmission, they quickly modify their food choice, selectively diminishing their intake of carbohydrates (Wurtman and Wurtman, 1979). These observations support the hypothesis that the responses of serotonergic neurons to food-induced changes in the relative concentrations of plasma amino acids allow these neurons to serve a special function as sensors in the brain's mechanisms governing nutrient choice (Wurtman, 1983, 1988). Perhaps these neurons participate in a feedback loop through which the composition of breakfast (i.e., its proportions of protein and carbohydrate) can, by increasing or decreasing brain serotonin levels, influence the choice of lunch. The ability of serotonin-containing neurons to distinguish between two foods (or the net compositions of two meals or snacks) depends upon the extent to which the foods produce significantly different plasma tryptophan/LNAA ratios. Thus, a food (e.g., berries for rats or popcorn for people) which contains carbohydrates but little or no protein is easily distinguished from one (e.g., meat or eggs) that is rich in protein. Less easily distinguished would be one containing, say, 10 percent protein from one containing 15 percent protein, unless one of the foods happens to lack carbohydrates entirely (Yokogoshi and Wurtman, 1986). Perhaps the food-plasma-serotonin connection evolved because certain carbohydrates taste too good; to maintain its muscle mass, the bear must eventually stop eating honey and go catch a fish.
A similar mechanism may operate in humans and may underlie the tendency of people in all known cultures to eat about 13 percent of their total calories as protein and about four to five times as much carbohydrate as protein. Subjects housed in a research hospital were allowed to choose from six different isocaloric foods (containing varying proportions of protein and carbohydrate but constant amounts of fat) at each meal, taking as many small portions as they liked; they also had continuous access to a computer-driven vending machine stocked with mixed carbohydrate-rich and protein-rich isocaloric snacks. It was observed (Wurtman and Wurtman, 1989) that the basic parameters of each person's food intake (total number of calories, grams of carbohydrate and protein, and number and composition of snacks) tended to vary only within a narrow range on a day-to-day basis and to be unaffected by placebo administration.
To assay the involvement of brain serotonin in maintaining this constancy of nutrient intake, pharmacological studies were undertaken in individuals in whom the feedback mechanism might be impaired. These were obese people who claimed to suffer from carbohydrate craving, manifested as their tendency to consume large quantities of carbohydrate-rich snacks, usually at a characteristic time of day or evening (Wurtman et al, 1985). (Too few protein-rich snacks were consumed by the subjects to allow assessment of drug effects on this source of calories.) Administration of dexfenfluramine, an antiobesity drug that increases intrasynaptic serotonin levels by releasing the transmitter and then blocking its reuptake, suppressed this carbohydrate craving. Other drugs thought to enhance serotonin-mediated neurotransmission selectively (e.g., the antidepressants zymelidine, fluvoxamine, and fluoxetine) have also been found to cause weight loss over the short term and may also selectively suppress carbohydrate intake. This contrasts with the weight gain (and carbohydrate craving) often associated with less chemically specific antidepressants such as amitriptyline.
Severe carbohydrate craving is also characteristic of patients suffering from seasonal affective disorder syndrome (SADS), a variant of bipolar clinical depression associated with a fall onset, a higher frequency in populations living far from the equator, and concurrent hypersomnia and weight gain (O'Rourke et al., 1989). A reciprocal tendency of many obese people to suffer from affective disorders (usually depression) has also been noted. Since serotonergic neurons apparently are involved in the actions of both appetite-reducing and antidepressant drugs, they might constitute the link between a patient's appetitive and affective symptoms. Some patients with disturbed serotonergic neurotransmission might present themselves to their physicians with problems of obesity, reflecting their overuse of dietary carbohydrates to treat their dysphoria. (The carbohydrates, by increasing intrasynaptic serotonin, would mimic the neurochemical actions of bona fide antidepressant drugs, such as the MAO inhibitors and tricyclic compounds [Wurtman, 1983].) Other patients might complain of depression, and their carbohydrate craving and weight gain would be perceived as secondary problems. Another group might include women suffering from premenstrual syndrome (PMS) who experience late-luteal-phase mood disturbances, weight gain, carbohydrate craving (Brzezinski et al., 1990), and sometimes bloating and fluid retention. Yet another group includes people attempting to withdraw from nicotine (Spring et al., 1991), a drug that releases serotonin (Ribeiro et al., submitted for publication). The participation of serotonergic neurons in a large number of brain functions besides nutrient choice regulation might have the effect of making such functions hostages to eating (seen in the sleepiness that can, for example, follow carbohydrate intake), just as it could cause mood-disturbed individuals to consume large amounts of carbohydrates for reasons related to neither the nutritional value nor the taste of these foods. In support of this view, it was observed that the serotonergic drug dexfenfluramine can be an effective treatment for both the affective and the appetitive symptoms of SADS (O'Rourke et al., 1989), PMS (Brzezinski et al., 1990), and smoking withdrawal (Spring et al., 1991).
UNDER WHAT CIRCUMSTANCES WILL NUTRIENT INTAKE AFFECT NEUROTRANSMISSION?
On the basis of the tryptophan-serotonin relationship, one can formulate a sequence of biochemical processes that would have to occur in order for any nutrient precursor to affect the synthesis and release of its neurotransmitter product.
First, plasma levels of the precursor (and of other circulating compounds, such as the LNAAs, that affect tryptophan's availability to the brain) must be allowed to increase after its administration (or after its consumption as a constituent of foods). In other words, plasma levels of tryptophan, the other LNAAs, or choline cannot be under tight homeostatic control comparable to, for example, that of plasma calcium or osmolarity. In actuality, plasma levels of tryptophan, tyrosine, and choline do vary severalfold after the consumption of normal foods, and those of the branched-chain amino acids may vary by as much as five- or sixfold.
Second, the brain level of the precursor must be dependent on its plasma level (i.e., there must not be an absolute blood-brain barrier for circulating tryptophan, tyrosine, or choline). In fact, such absolute barriers do not exist for these nutrients; rather, facilitated diffusion mechanisms that allow these compounds to enter the brain at rates that depend on the plasma levels of these ligands are in operation.
Third, the rate-limiting enzyme within presynaptic nerve terminals that initiates the conversion of the precursor to its neurotransmitter product must, similarly, be unsaturated with this substrate so that when presented with more tryptophan, tyrosine, or choline it can accelerate synthesis of the neurotransmitter. (Tryptophan hydroxylase and choline acetyltransferase [CAT] do indeed have very poor affinities for their substrates tryptophan and choline.) As discussed below, tyrosine hydroxylase activity becomes tyrosine-limited when neurons containing the enzyme have been activated and the enzyme has been phosphorylated (Wurtman, 1988; Wurtman et al., 1980).
Available evidence suggests that only some of the neurotransmitters present in the human brain are subject to such precursor control, principally, the monoamines mentioned above (serotonin; the catecholamines dopamine, norepinephrine, and epinephrine; and acetylcholine) and, possibly, histidine and glycine. Pharmacological doses of the amino acid histidine do elevate histamine levels within nerve terminals, and the administration of threonine, a substrate for the enzyme that normally forms glycine from serine, can elevate glycine levels within spinal cord neurons (and, probably, thereby ameliorate some of the clinical manifestations of spasticity [Growdon et al., 1991]). One large family of neurotransmitters, the peptides, is almost certainly not subject to precursor control. Brain levels of these compounds have never been shown to change with variations in brain amino acid levels; moreover, there are sound theoretical reasons why it is unlikely that brain peptide synthesis would respond. The immediate precursor for a brain protein or peptide is not an amino acid per se, as is the case for some of the monoamine neurotransmitters, but the amino acid molecule attached to its particular species of transfer RNA (tRNA). In brain tissue, the known enzymes that catalyze the coupling of an amino acid to its tRNA have very high affinities for their amino acid substrates, such that their ability to operate at full capacity in vivo is probably unaffected by amino acid levels (except possibly in pathological states that are associated with major disruptions in brain amino acid patterns, such as phenylketonuria).
Little information is available concerning the possible precursor control of the nonessential amino acids, such as glutamate, aspartate, and γ-aminobutyric acid (GABA), even though these are probably the most abundant neurotransmitters in the brain. It is difficult to do experiments on these relationships; the precise biochemical pathways that synthesize glutamate and aspartate within nerve terminals are not well established, and for GABA, although it is well established that its precursor is glutamate, brain levels of that amino acid cannot be raised experimentally without sorely disrupting normal brain functions. The macromolecule that transports acidic amino acids such as glutamate and aspartate across the blood-brain barrier is unidirectional and secretes these compounds from the brain into the blood by an active transport mechanism (Pardridge, 1977). Hence, administration of even an enormous dose of monosodium glutamate will not affect brain glutamate levels unless it elevates plasma osmolarity to the point of disrupting the blood-brain barrier.
TYROSINE EFFECT ON DOPAMINE AND NOREPINEPHRINE SYNTHESIS
Because tyrosine administration had not been shown to increase brain dopamine or norepinephrine levels in otherwise untreated animals, it was initially assumed that the catecholamine neurotransmitters were not under precursor control, even though (1) plasma tyrosine levels do increase severalfold after protein intake or tyrosine administration; (2) the LNAA transport system does ferry tyrosine, like tryptophan, across the blood-brain barrier; and (3) tyrosine hydroxylase, which catalyzes the rate-limiting step in catecholamine synthesis, is unsaturated in vivo (Wurtman et al., 1980). It did seem possible, however, that a pool of neuronal dopamine or norepinephrine might exist for which synthesis did depend on tyrosine levels, but which was of too small a size in relation to the total catecholamine mass to be detected.
Hence, studies were performed to determine whether catecholamine synthesis or release could be affected by changes in brain tyrosine concentrations. At first, catecholamine synthesis was estimated by following the rate at which dopa, the product of tyrosine's hydroxylation, accumulated in the brains of rats treated acutely with a drug that blocks the next enzyme in catecholamine formation (aromatic L-amino acid decarboxylase). Tyrosine administration did increase dopa accumulation, whereas other LNAAs decreased both dopa accumulation and brain tyrosine levels. Catecholamine release was then estimated by measuring the brain levels of metabolites of dopamine (homovanillic acid [HVA], dihydroxyphenylacetic acid [DOPAC]) or norepinephrine (methoxyhydroxyphenylglycol sulfate [MHPH-SO4]). Administration of even large doses of tyrosine had no consistent effect on these metabolites. However, if the experimental animals were given an additional treatment designed to accelerate the firing of dopaminergic or noradrenergic tracts (e.g., dopamine receptor blockers, cold exposure, partial lesions of dopaminergic tracts, and reserpine), the supplemental tyrosine caused a marked augmentation of catecholamine release (Wurtman, 1988; Wurtman et al., 1980). These initial observations formed the basis for the hypothesis that catecholaminergic neurons become tyrosine sensitive when they are physiologically active and lose this capacity when they are quiescent.
The biochemical mechanism that couples a neuron's firing frequency to its ability to respond to supplemental tyrosine involves phosphorylation of the tyrosine hydroxylase enzyme protein, a process that occurs when the neurons fire. This phosphorylation, which is short-lived, enhances the enzyme's affinity for its cofactor (tetrahydrobiopterin) and makes the enzyme insensitive to end product inhibition by catechols; these changes allow its net activity to depend on the extent to which it is saturated with tyrosine. An additional mechanism underlying this coupling may be an actual depletion of tyrosine within nerve terminals as a consequence of its accelerated conversion to catecholamines (Milner et al., 1987). If slices of rat caudate nucleus are superfused with a standard Krebs-Ringer solution (which lacks amino acids) and are depolarized repeatedly, they are unable to sustain their release of dopamine; concurrently, their contents of tyrosine, but not of other LNAAs, decline markedly. The addition of tyrosine to the superfusion solution enables the tissue to continue releasing dopamine at initial rates and also protects it against depletion of its tyrosine. The concentrations of tyrosine needed for these effects are proportional to the number of times the neurons are depolarized. (Of course, the intact brain is continuously perfused with tyrosine-containing blood, making it highly unlikely that tyrosine levels fall to a similar extent, even in continuously active brain neurons. However, they might decline somewhat, since tyrosine is poorly soluble in aqueous media and diffuses relatively slowly.)
More recently, in vivo dialysis techniques have been used to assess tyrosine's effects on brain dopamine release. When otherwise untreated animals receive the amino acid systemically, there is, after 20–40 min, a substantial increase in dopamine output from nigrostriatal neurons unaccompanied by detectable increases in dopamine's metabolites DOPAC or HVA. However, this effect is short-lived, and dopamine release returns to basal levels after 20–30 min. This latter response probably reflects receptor-mediated decreases in the firing frequencies of the striatal neurons (to compensate for the increase in dopamine release that occurs with each firing) and, perhaps, local presynaptic inhibition. If animals are given haloperidol, a dopamine receptor-blocking agent, before—or along with—the tyrosine, the supplemental tyrosine continues to amplify dopamine output for prolonged periods (During et al., 1989).
Tyrosine has now been shown to enhance the production and release of dopamine or norepinephrine in a variety of circumstances. This amino acid may ultimately have considerable utility in treating catecholamine-related diseases or conditions; it may also prove useful in promoting performance— particularly in high-stress situations.
EFFECTS OF CHOLINE ON SYNTHESIS OF ACETYLCHOLINE AND PHOSPHATIDYLCHOLINE
The amounts of acetylcholine released by physiologically active cholinergic neurons depend on the concentrations of choline available. In the absence of supplemental free choline, the neurons will continue to release constant quantities of the transmitter, especially when stimulated (Maire and Wurtman, 1985). However, when choline is available (in concentrations bracketing the physiological range), a clear dose relationship is observed between its concentration and acetylcholine release (Blusztajn and Wurtman, 1983; Marie and Wurtman, 1985). When no free choline is available, the source of the choline used for acetylcholine synthesis is the cells' own membranes (Blusztajn et al., 1987). Membranes are very rich in endogenous phosphatidylcholine (PC), and this phospholipid serves as a reservoir of free choline, much as bone and albumin serve as reservoirs for calcium and essential amino acids. It has been suggested that a prolonged imbalance between the amounts of free choline available to a cholinergic neuron and the amounts needed for acetylcholine synthesis might alter the dynamics of membrane phospholipids to the point of interfering with normal neuronal functioning (“autocannibalism”) (Blusztajn and Wurtman, 1983; Nitsch et al., 1992a), for example, in patients with Alzheimer's disease. In that event, providing the brain with supplemental choline would serve two purposes: it would enhance acetylcholine release from physiologically active neurons and it would replenish the choline-containing phospholipids in their membranes (Wurtman, 1985).
Neurons can draw on three sources of free choline for acetylcholine synthesis: that stored as PC in their own membranes, that formed intrasynaptically from the hydrolysis of acetylcholine (and taken back up into the presynaptic terminal by a high-affinity process estimated to be 30–50 percent efficient in the brain), and that present in the bloodstream (and taken into the brain by a specific blood-brain barrier transport system). The PC in foods (e.g., liver and eggs) is rapidly hydrolyzed to free choline in the intestinal mucosa (or is broken down more slowly after passage into the lymphatic circulation). Consumption of adequate quantities of PC can lead to severalfold elevations in plasma choline levels, thereby increasing brain choline levels and the substrate saturation of CAT.
The PC molecules consumed in the diet, as well as those formed endogenously in neuronal membranes, are very heterogeneous with respect to their fatty acid compositions. Some PCs (e.g., those in soybeans and nerve terminals) are relatively rich in polyunsaturated fatty acids; others (e.g., those in eggs) are highly saturated. PCs are also heterogeneous with reference to their mode of synthesis. Brain neurons produce PC by three distinct biochemical pathways: the sequential methylation of phosphatidylethanolamine (PE), the incorporation of preexisting free choline via the CDP-choline cycle, or the incorporation of free choline via the base exchange pathway (in which a choline molecule substitutes for the ethanolamine in PE or the serine in phosphatidylserine [PS]). Quite possibly, the different varieties of PC may subserve distinct functions; for example, one type of PC, distinguished by its fatty acid composition or its mode of synthesis, could be preferentially utilized to provide a choline source for acetylcholine synthesis or could be formed preferentially during the processes of cell division or synaptic remodeling. Similarly, one particular species might be especially involved in the pathogenesis of particular degenerative diseases afflicting cholinergic neurons (e.g., Alzheimer's disease).
Supplemental choline or PC has been used with some success in the treatment of tardive dyskinesia. A summary of related publications (Nasrallah et al., 1984) concluded that choline and the cholinesterase inhibitor physostigmine were about equally efficacious and that choline was less toxic. Most patients exhibited some reduction in the frequency of abnormal movement, but in only a few cases was there complete cessation of the movements. Choline sources have also been tried in the treatment of Alzheimer's disease. Most well-controlled studies have treated subjects for relatively short intervals (6–8 weeks) and have focused on younger subjects, with little or no success. A single double-blind study administered the PC for 6 months (Little et al., 1985). Improvement was noted in about one-third of the subjects; the average age of the responders was 83 years and that of nonresponders was 73 years, a relationship thought to be compatible with evidence that Alzheimer's disease may be more restricted to cholinergic neurons in subjects who become symptomatic at a later age. Occasional reports have also described the useful effects of choline or PC in treating mania, ataxia, myasthenic syndromes, and Tourette's syndrome. Very recently it has been observed (Nitsch et al., 1992a) that the brains of people dying of Alzheimer's disease (but not Down's Syndrome) contain reduced levels of PC and free choline (and PE and free ethanolamine) but major increases in those of the PC metabolite glycerophosphocholine and the PE metabolite glycerophosphoethanolamine. These changes were not restricted to regions containing plaques, tangles, or amyloid. Since low brain choline levels both impair acetylcholine synthesis and accelerate the breakdown of membrane PC and since adequate acetylcholine may be needed to prevent the formation of the amyloid protein of Alzheimer's disease (Nitsch et al., 1992b), supplemental choline and ethanolamine could have a role in the prevention of this disease.
CONCLUSIONS AND RECOMMENDATIONS
- The design of experiments to display the potentially useful effects of foods and nutrients on the ability to perform well, particularly under stressful circumstances, will require considerable sophistication. These chemicals are not nearly as potent as drugs and, in fact, lack intrinsic potency, having first to be converted to a neurotransmitter within a nerve terminal and then to be released from that terminal. (Of course, they are also likely to be significantly less toxic than drugs; this is perhaps their major advantage.) Such experimental design should be entrusted to people who are well trained in studying human behavior and who also fully understand the ground rules that determine when the food or nutrient is most likely to be effective (e.g., for tyrosine, when particular catecholamine-releasing neurons are firing frequently for long periods).
- At this point, too few adequate experiments have been done with human subjects to begin to assess the utilities of neurotransmitter precursors such as tyrosine or choline in increasing or sustaining performance; in fact, a number of poorly designed studies muddy the waters. Tyrosine's effect on performance must be examined in situations in which subjects are under real stress. Choline's effects on memory must be studied in experiments in which the nutrient is given for a sufficiently long period of time (i.e., one compatible with what is known about the dynamics of the choline-phosphatidylcholine interaction).
- The peripheral actions of the neurotransmitter precursors may turn out to be very useful (e.g., tyrosine's ability to normalize blood pressure when it is both too high and too low [Wurtman et al., 1980] and choline's ability to sustain exercise tolerance in subjects whose plasma choline levels have been reduced by, for example, long-distance running [Conlay et al., 1986; Sandage et al., 1992]).
- The development of foods or nutrients used to sustain performance—or otherwise to improve normal behaviors—requires guidance by the U.S. Food and Drug Administration, and perhaps other agencies as well, regarding how these compounds will be regulated. It is absolutely mandatory that all such preparations be safe and of adequate purity; it is also essential that they be adequately labeled, providing the user with full information about their indications, dosages, contraindications, and side effects. However, if and when it can be shown that their use is largely nutritional (i.e., to meet the body's needs for more of the particular nutrient because environmental circumstances have increased those needs), then perhaps they can be designated as foods.
- Considerable additional research should be done to identify special populations with unusual responses to foods or nutrients that affect neurotransmitters (e.g., the carbohydrate cravers who overconsume carbohydrate-rich snacks in order to relieve depressive symptoms). Heterogeneity of response will doubtless also exist among people in the military (e.g., those with mild seasonal depression or premenstrual syndrome and those giving up smoking).
ACKNOWLEDGMENTS
Some of these studies were supported by National Institute of Mental Health grant MH-28783, U.S. Air Force grant AFOSR-830366, National Aeronautics and Space Administration grant NAG-2–210, and National Institutes of Health grant RR00088–24 to the Clinical Research Center, Massachusetts Institute of Technology.
REFERENCES
- Blusztajn, J.K., and R.J.Wurtman 1983. Choline and cholinergic neurons. Science 221:614–620. [PubMed: 6867732]
- Blusztajn, J.K., M.Liscovitch, U.I.Richardson, and R.J.Wurtman 1987. Phosphatidylcholine as a precursor of choline for acetylcholine synthesis. Pp. 341–346 in Cellular and Molecular Basis of Cholinergic Function. M.J.Dowdall, editor; and J.N. Hawthorne, editor. , eds. Chichester (Sussex), U.K.: Ellis Harwood.
- Brzezinski, A.A., J.J.Wurtman, R.J.Wurtman, R.Gleason, T.Nader, and B.Laferrere 1990. D-fenfluramine suppresses the increased calorie and carbohydrate intakes and improves the mood of women with premenstrual syndrome. Obstet. Gynecol. 76:296–301. [PubMed: 2371034]
- Conlay, L., R.J.Wurtman, J.K.Blusztajn, G.Lopez-Coviella, T.J.Maher, and G.E.Evoniuk 1986. Decreased plasma choline concentrations in marathon runners. N. Eng. J. Med. 315:892. [PubMed: 3748109]
- During, M.J., I.N.Acworth, and R.J.Wurtman 1989. Dopamine release in rat striatum: Physiological coupling to tyrosine supply. J. Neurochem. 52:1449–1454. [PubMed: 2496199]
- Fernstrom, J.D., and R.J.Wurtman 1971. Brain serotonin content: Physiological dependence on plasma tryptophan levels. Science 173:149–152. [PubMed: 5581909]
- 1972. Brain serotonin content: Physiological regulation by plasma neutral amino acids. Science 178:414–416. [PubMed: 5077329]
- Gardier, A., and R.J.Wurtman 1991. Persistent blockade of potassium-evoked serotonin release from rat frontocortical terminals after fluoxetine administration. Brain Res. 540:325–330. [PubMed: 1711396]
- Growdon, J.H., T.M.A.Nader, D.Schoenfeld, and R.J.Wurtman 1991. L-Threonine in the treatment of spasticity. Clin. Neuropharm. 14(5):403–412. [PubMed: 1742749]
- Little, A., R.Levy, P.Chaqui-Kidd, and D.Hand 1985. A double-blind placebo controlled trial of high-dose lecithin in Alzheimer's disease. J Neurol. Neurosurg. Psychiatry 48:736–742. [PMC free article: PMC1028443] [PubMed: 3897460]
- Madras, B.K., E.L.Cohen, R.Messing, H.N.Munro, and R.J.Wurtman 1974. Relevance of serum-free tryptophan to tissue tryptophan concentrations. Metabolism 23:1107–1116. [PubMed: 4427559]
- Maire, J.-C., and R.J.Wurtman 1985. Effects of electrical stimulation and choline availability on release and contents of acetylcholine and choline in superfused slices from rat striatum. J. Physiol. ( Paris: ) 80:189–195. [PubMed: 3003350]
- Milner, J.D., D.K.Reinstein, and R.J.Wurtman 1987. Dopamine synthesis in rat striatum: Mobilization of tyrosine from non-dopaminergic cells. Experientia 43(10):1109. [PubMed: 3117583]
- Nasrallah, H.A., F.L.Dunner, R.E.Smith, M.McCalley-Whitters, and A.D.Sherman 1984. Variable clinical response to choline in tardive dyskinesia. Psychol. Med. 14:697–700. [PubMed: 6387757]
- Nitsch, R., J.K.Blusztajn, A.Pittas, B.E.Slack, J.H.Growdon, and R.J.Wurtman 1992. a Evidence for a membrane defect in Alzheimer's disease brain. Proc. Natl. Acad. Sci. USA: 89:1671–1675. [PMC free article: PMC48514] [PubMed: 1311847]
- Nitsch, R., B.Slack, R.Wurtman, and J.Growdon 1992. b Processing of Alzheimer amyloid precursor stimulated by activation of muscarinic acetylcholine receptors. Science 258(9):304–307. [PubMed: 1411529]
- O'Rourke, D., J.Wurtman, R.Wurtman, R.Chebli, and R.Gleason 1989. Treatment of seasonal depression with D-fenfluramine. J. Clin. Psychiatry 50:343–347. [PubMed: 2670915]
- Ribeiro, E., R.Bettiker, M.Bogdanov, and R.J.Wurtman 1993. Effects of systemic nicotine on serotonin release in rat brain. Brain Res. 621:311–318. [PubMed: 8242344]
- Pardridge, W.M. 1977. Regulation of amino acid availability to the brain. Pp. 141–204 in Nutrition and the Brain, vol. 7,R.J.Wurtman, editor; and J.J.Wurtman, editor. , eds. New York: Raven Press.
- Sandage, B.W., Jr., L.Sabounjian, R.White, and R.J.Wurtman 1992. Choline citrate may enhance athletic performance. Abstract. Fall meeting of the American Physiological Society.
- Schaechter, J., and R.J.Wurtman 1989. Tryptophan availability modulates serotonin release from rat hypothalamic slices. J Neurochem. 53:1925–1933. [PubMed: 2478666]
- Spring B., J.Wurtman, R.Gleason, R.Kessler, and R.J.Wurtman 1991. Weight gain and withdrawal symptoms after smoking cessation: A preventive intervention using D-fenfluramine. Health Psy. 10(3):216–223. [PubMed: 1879394]
- Wurtman, J.J., and R.J.Wurtman 1979. Drugs that enhance central serotonergic transmission diminish elective carbohydrate consumption by rats. Life Sci. 24:895–904. [PubMed: 221766]
- Wurtman, J.J., R.J.Wurtman, S.Mark, R.Tsay, W.Gilbert, and J.Growdon 1985. D-fenfluramine selectively suppresses carbohydrate snacking by obese subjects. Int. J. Eating Disord. 4:89–99. [PubMed: 11540865]
- Wurtman, R.J. 1983. Behavioral effects of nutrients. Lancet 1:145–147.
- 1988. Presynaptic control of release of amine neurotransmitters by precursor levels. NIPS (Am. Physiol. Soc.) 3:158–163.
- Wurtman, R.J., and J.J.Wurtman 1989. Carbohydrates and depression. Sci. Ame.: 68–75. [PubMed: 2642626]
- Wurtman, R.J., F.Hefti, and E.Melamed 1980. Precursor control of neurotransmitter synthesis. Pharmacol. Rev. 32:315. [PubMed: 6115400]
- Wurtman, R.J., B.Caballero, and E.Salzman 1988. Facilitation of levodopa-induced dyskinesias by dietary carbohydrates. N. Engl. J. Med. 319:1288–1289. [PubMed: 3185626]
- Yokogoshi, H., and R.J.Wurtman 1986. Meal composition and plasma amino acid ratios: Effects of various proteins or carbohydrates, and of various protein concentrations. Metabolism 35:837–842. [PubMed: 3747840]
DISCUSSION
WILLIAM WATERS: It occurred to me during various presentations that there is a difference between what happens to a nutrient when it is taken in a pure form and when it is taken in a natural food form. Does anybody have any information that they might share with us?
RICHARD WURTMAN: Take tryptophan ortyrosine as an example. If these are taken as constituents of dietary protein, much of what is taken in is just converted by the body to its own protein; very little of it enters the brain because of competition with other large neutral amino acids.
On the other hand, if tryptophan or tyrosine are taken alone, the body converts little or none of it to its own protein and much of it goes into the brain because of the lack of competition. That is the good news if you are looking for a drug effect; but not if you're not looking for a drug effect.
WILLIAM WATERS: The qualitative difference is: does that translate into behavior?
RICHARD WURTMAN: In the case of tryptophan versus carbohydrate, for instance, I think Harris Lieberman or Bonnie Spring would be better able to answer whether or not the effects of tryptophan are qualitative.
BONNIE SPRING: My comment was just that, qualitatively, they are very similar, but tryptophan is much more powerful.
WILLIAM WATERS: My comment was to agree with Bonnie Spring and to emphasize the fact that tryptophan produces clear effects that are quite easy to observe.
WILLIAM BEISEL: Should there be any concern for high-dose tryptophan, for the possibility that it may go to oxidative metabolism?
RICHARD WURTMAN: Timothy Maher was on the committee that reviewed that.
TIMOTHY MAHER: We have concerns that I will discuss tomorrow regarding the use of any amino acid in its pure form apart from protein. There were many concerns about the conversion of amino acids into other products that have never been studied. Therefore, your concern is one that is shared because the answer is not known.
ELDON ASKEW: This is directed to John Ivy: It seems like the provision of sugars during exercise elicits insulin responses to exercise, but the provision of simple sugar seems to be what would be provided during exercise. In the recovery phase you want something that stimulates insulin production to stimulate glycogen synthesis. Is it not appropriate to use polymers, glucose polymers, in the recovery phase? Should we be using a simpler sugar in the recovery phase?
JOHN IVY: Actually, some of the polymers that are used in drinks have a glycemic index—not much difference—so I do not think it is much of a problem.
IRWIN TAUB: For John Ivy also: You show that the fats were not very good as an additional energy source during exercise. That is, of course, short term. But what about high fat on a longer-term basis? Would that ultimately lead to some sparing with a glycogen, for example?
JOHN IVY: Let me back up. I think if you can get the fat in and get it converted out of the free fatty acids or somehow get large amounts of medium-chain triglycerides in, which seems to be difficult—I do not know all the ins and outs about possibilities for that—it may be beneficial. Taking a high-fat meal and then injecting heparin so that the triglycerides are broken down into free fatty acids does seem to be beneficial in sparing carbohydrate and enhancing endurance performance.
If you are talking about taking high-fat meals over a long period of time, we can go back a long ways. I think back during World War II they were looking at that with the Canadian Army, giving something called pemmican. That did not improve performance at all. In fact, it caused deterioration.
ROBERT NESHEIM: I think Ed Horton had a comment relative to that.
EDWARD HORTON: I think the pemmican studies were faulty because there was no adaptation. But a lot of people have looked at adaptation of high-fat, low-carbohydrate diets both in terms of intensity and duration of exercise.
I think the bottom line of that is that you can adapt: you lower your RQ [respiratory quotient], and you burn more fatty acid and less carbohydrate at moderate-intensity exercise. But everybody has shown that once you have to put out high-intensity exercise you still have an absolute requirement for carbohydrate oxidation. So they have not really panned out in terms of being able to enhance performance at high intensities.
IRWIN TAUB: But, as was pointed out, marching is equivalent to 40 percent 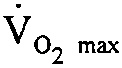 [maximum oxygen uptake]. So the question is, would it be useful in that context?
[maximum oxygen uptake]. So the question is, would it be useful in that context?
EDWARD HORTON: I think that the answer is maybe, when you are interested in trying to get high-caloric-density foods so that you can get more calories in with less weight. In fact, we reviewed this a couple of years ago in quite some detail. I think that the feeling was that you are okay at moderate-intensity exercise, but everybody has to put out a high-intensity exercise at some point. Under those circumstances, the high-fat diets do not stack up to having carbohydrates.
JOHN MILNER: I was really trying to understand what was going on. You presented information that the carbohydrate loading would improve performance. Then you turned right around and said that increasing free fatty acids would also do that.
I think that, mechanistically, when one is up, the other is down; so it should not work that way. Can you tell me mechanistically why you would think that an elevation in free fatty acids would be the same as ....
EDWARD HORTON: They function differently.
JOHN MILNER: So is it energy supply that really is a principal factor more than anything else?
EDWARD HORTON: If you have a substantial amount of free fatty acids in the blood, the rate of uptake of muscle free fatty acids is somewhat proportional to the amount available. You increase beta-oxidation. The high free fatty acid levels in the blood seem to block glucose transport as well as increase citrate levels in the muscle, which blocks lipolysis.
So you convert your reliance—you increase your reliance—on fats and spare carbohydrate, and therefore, you are able to work longer.
Typically, when you start exercising, you are not burning optimally the carbohydrates that are required for the exercise. You are actually burning more than what is required because there are plenty available. But if you can block that use initially above and beyond what is necessary, you can spare the carbohydrate and work longer. That is what the fats seem to be doing.
JOHN MILNER: So it is just this sparing, short-term effect in essence?
EDWARD HORTON: Short term from the standpoint that you can work 4 hours rather than 3 hours at 70 percent of maximum 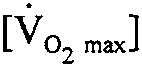 .
.
PEGGY BORUM: My question has to do with the different fuel sources that are available. This afternoon we heard that it makes a difference, depending upon what is eaten and also on the intensity and duration of exercise.
My assumption is that these studies like Alan Sherrington's in dogs, were with dogs that were well fed. What we heard this morning is that many individuals in the field are actually not maintaining their energy requirements and are actually not taking in enough fuel on a chronic basis, that is, they are losing weight. When you superimpose these experiments that did not have that element, how does that affect the fuel that is actually available to the muscles of these individuals when we give them stress such as sleep deprivation and then ask them to exercise at a fairly high intensity for an extended period of time?
EDWARD HORTON: That is a good point because you are right; most of the studies that have been done on dietary manipulation have been done on people who are on good caloric intakes.
PEGGY BORUM: Or dogs.
EDWARD HORTON: We have not really talked at all about protein turnover in these people out in the field in terms of what is happening to protein synthesis and protein degradation and the protein turnover rates that are going on when they are hypocaloric (losing weight). We know that they have a negative caloric balance and are losing weight. That certainly has to have an impact on muscle strength and for its immobilization of amino acids for gluconeogenesis, for example.
I think there is some real need for studies in that area that look at the effect of the stress hormone response, for example, on gluconeogenesis and glucose output in the liver when you may have a limitation of substrate in the form of amino acid substrates.
STEVEN ZEISEL: A number of people have described insulin growth factor [IGF] 1 and IGF 2 and shown that IGF 1, for instance, dropped very markedly during early malnutrition, very modest malnutrition.
Now they are using IGF 1 to increase anabolic metabolism and appetite in patients who have cachexia. Have any of you studied changes in IGF 1 at the same time you are studying the other parameters during exercise and starvation in these marching soldiers?
KARL FRIEDL: We looked at it in Rangers last year. It went down to about 50 percent of the normal level. I guess that is an adaptation of the semistarvation, the intense exercise, and the weight loss and sleep deprivation. It is a multistress environment.
DAVID SCHNAKENBERG: It is a cross between people who are doing nutrition work and people who are doing sleep work. Most people are engaged in doing sleep research. Do you know of any studies where they have tried to monitor what people are eating during the course of these 72 hours? Is there any change in terms of what they are eating during that period of time as to how much and when; or do you force people to eat?
GREGORY BELENKY: We keep track pretty much; we limit them. In our PET studies, which we are doing in collaboration with John Hopkins at the Gerontological Research Center, we are actually letting them ad lib it and we are keeping track of exactly what they eat and what they do not so we will have a much better notion of what their caloric intake is.
What we have done up to this time is simply state that this is the meal, this is what you get, here it is. During the sleep deprivation period, we provide snacks at around 2:00 in the morning. Of course, we see the nice regular decline in body temperature across the sleep deprivation period superimposed on the circadian cycle.
RICHARD WURTMAN: How many different foods can they choose from? Do they have a range of carbohydrate, protein, et cetera?
HARRIS LIEBERMAN: Yes, basically they have TV dinners, Healthy Choice, for example.
RICHARD WURTMAN: So they cannot decide they want to eat just the carbohydrates; they have to go with the whole thing?
HARRIS LIEBERMAN: No.
RICHARD WURTMAN: That is a shame because it does not really answer your question to see whether or not these people become carbohydrate cravers in the middle of the night, for instance. I would predict that they would.
DAVID SCHNAKENBERG: They are just looking at the possibility that there may be some opportunities for using diet as an augmentation to maintaining awareness.
HARRIS LIEBERMAN: First, I wanted to say that there is some evidence in the literature on both animals and humans that sleep deprivation produces hyperphagia. I do not know of anyone who actually studied whether that hyperphagia is specific or related to carbohydrate; one would certainly guess that it might be.
The other thing I wanted to mention was undernutrition. The Ranger study is really an extreme of undernutrition. There was a very important study that Eldon Askew did—the RLW30 study—where soldiers were deprived of a portion of their nutrition for a period of a month. They got about 2,000 calories per day but were burning something like 3,200 calories.
As part of that study, we measured both their physical and mental performances. There were only the subtlest changes in both as a function of a full month of undernutrition.
ROBERT NESHEIM: I remember that those studies were done for a month.
ELDON ASKEW: And they had plenty of sleep.
ROBERT NESHEIM: Yes, plenty of sleep.
MELVIN MATHIAS: John Ivy, you were alluding to this branched-chain amino acid cocktail and exercise enhancement. You were only using a neurotransmitter mechanism. Maybe Wayne Askew or Ed Horton can expand on the glucose-alanine cycle and the status of it. I do appreciate that there is not much energy in that cycle, but I think it still has some interest. I do not know how important that might be in the branched chain or if they used that in a hypothesis.
JOHN IVY: They did not use it in a hypothesis. I brought it up just because I saw that people were going to talk about it. I do not know much about the study.
ROBERT NESHEIM: There will be some more discussion of amino acids tomorrow.
JOHN MILNER: There are other studies also showing increases in protein synthesis or retention—the increase in protein degradation especially in the diaphragm. So I assume that some of that relates to that as well as anything else.
WILLIAM BEISEL: You have to remember there is a profound metabolic adaptation to the difference between simple starvation and the cachexia that results from the acute-phase reaction which causes hypermetabolism and gets most of the energy from muscle breakdown. So we have a profound difference here, and we have to determine what the soldiers are facing.
PATRICK DUNNE: As more of a follow-up on the pathways that we are looking at in muscle, in the branched-chain, I hear one strategy is to recognize that muscle will use branched-chain amino acids for energy much better than other tissues will. So, indeed, you might be sparing some of your other energy requirements and maybe feeding your alanine cycle. That is one of the strategies.
EDWARD HORTON: If you look at it quantitatively, the branched-chain amino acids oxidized by exercising muscle never contribute more than 1 or 2 percent of the total energy. It is true that the amino groups are basically converting to alanine with pyruvate, so you can feed that way.
Quantitatively, it is just a very small thing. I cannot believe that it has any major effect on exercise performance in the same way that giving a carbohydrate supplement would or from trying to get the body to use more fatty acids and spare carbohydrates. It is just quantitatively too small.
PATRICK DUNNE: With regard to the related issue of all proteins not being the same, which protein were you using in your supplement?
JOHN IVY: We were using milk isolate and whey.
PATRICK DUNNE: So it is basically a whey protein. Richard Wurtman showed that different proteins give some spectrum of ratio of the large neutral amino acids to the others. So one could be very leery when you say a universal response to protein. A casein may have one response.
PEGGY BORUM: I think part of the theory is that in exercise and in other conditions where free fatty acid levels increase in the plasma, the free fatty acid competes with the tryptophan, at least theoretically, and that you wind up getting more free tryptophan instead of it being bound to albumin because the free fatty acids and the tryptophan are competing.
Theoretically, that is very interesting, but I do not know whether anyone has any data to show that really takes place, where, if you increase the free fatty acid concentration in the plasma, you actually increase the tryptophan concentration in the plasma enough to increase serotonin production in the brain.
RICHARD WURTMAN: We spent about 2 years figuring it out. It does not matter because albumin-bound tryptophan is transported into the brain about 79 percent as effectively as free tryptophan. There is a slight retardation associated with binding of the tryptophan molecule to albumin, but it is so slight that unless you have a mega increase in free fatty acid levels, it is not going to matter or have much of an effect on brain tryptophan levels.
There used to be a great debate 15 or 18 years ago about the determinant of tryptophan's uptake into the brain: is it competition with large neutral amino acids, or is it the proportion that is bound to albumin?
Then definitive studies were done by Pardriole's and other people showing just what I have said.
PEGGY BORUM: But if you add free fatty acids into the mix, does the presence of free fatty acids have some effect?
RICHARD WURTMAN: Yes, it will increase free tryptophan in plasma, but it will not have much of an effect on the passage of plasma tryptophan into the brain.
PEGGY BORUM: That is the only effect?
RICHARD WURTMAN: That is the only effect.
EDWARD HORTON: I wanted to come back to the question that Peggy Borum asked earlier, and what Bill Beisel just said kind of triggered my thinking about this. If you look at people in negative caloric balance—simple starvation, with, say, anorexia nervosa, or people who have been starving or who are hypocaloric—hepatic glucose production actually decreases to very low baseline levels. Go back to the classic George Cahill-type studies. They decrease their gluconeogenic amino acids, they slow down hepatic glucose production to about half of normal, and basically reach an adapted state.
It is just the opposite of what is happening in these stress situations, and that is what I was trying to point out. With stress, it is very similar to exercise, where you increase peripheral utilization, and hepatic glucose production increases to match the peripheral utilization. In a stress situation, you are driving hepatic glucose production it seems, and it is very catabolic.
So if you take somebody out in the field and you are not giving them enough calories to meet their demands and they are under a lot of stress, they are going to be very, very catabolic under that circumstance.
ROBERT NESHEIM: I think that is true. Studies of moderate undernutrition in people show how they adapt. They cut down on their activities. If you are in a situation where you cannot cut down your activities—and as a matter of fact, you are forced to be active—then the picture is totally different.
JOHN IVY: Edward Horton, do you think that individuals under stress and exercise, for example, in a situation of combat where they are actually physically active and under stress, would require more carbohydrates?
EDWARD HORTON: I cannot tell you what the experimental data would show, but I would predict that they would. The real question to me is whether by giving them more carbohydrate in their diet can you affect hepatic glucose production and slow it down. You might be able to to some extent, but I think that some of the earlier studies looking at endotoxin responses show that you cannot shut it off. Also, some of the trauma studies from John Penny show that you cannot shut off hepatic glucose production even when giving glucose.
Footnotes
- 1
Portions of this manuscript have been adapted from Wurtman (1988).
- 2
Richard J.Wurtman, Massachusetts Institute of Technology, E25–604, Cambridge, MA 02139
- INTRODUCTION
- FOOD CONSUMPTION, TRYPTOPHAN AVAILABILITY, AND BRAIN SEROTONIN SYNTHESIS
- BRAIN SEROTONIN, NUTRIENT CHOICE, AND CARBOHYDRATE CRAVING
- UNDER WHAT CIRCUMSTANCES WILL NUTRIENT INTAKE AFFECT NEUROTRANSMISSION?
- TYROSINE EFFECT ON DOPAMINE AND NOREPINEPHRINE SYNTHESIS
- EFFECTS OF CHOLINE ON SYNTHESIS OF ACETYLCHOLINE AND PHOSPHATIDYLCHOLINE
- CONCLUSIONS AND RECOMMENDATIONS
- ACKNOWLEDGMENTS
- REFERENCES
- DISCUSSION
- Effects of Nutrients on Neurotransmitter Release - Food Components to Enhance Pe...Effects of Nutrients on Neurotransmitter Release - Food Components to Enhance Performance
- TRIM55 tripartite motif containing 55 [Homo sapiens]TRIM55 tripartite motif containing 55 [Homo sapiens]Gene ID:84675Gene
- 84675[uid] AND (alive[prop]) (1)Gene
- Systematic Review of the Literature Regarding the Diagnosis of Sleep ApneaSystematic Review of the Literature Regarding the Diagnosis of Sleep Apnea
- EEF1A1 eukaryotic translation elongation factor 1 alpha 1 [Homo sapiens]EEF1A1 eukaryotic translation elongation factor 1 alpha 1 [Homo sapiens]Gene ID:1915Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...