Polymicrogyria Overview – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY
Chloe A Stutterd, MBBS, FRACP, William B Dobyns, MD, Anna Jansen, MD, PhD, Ghayda Mirzaa, MD, and Richard J Leventer, MBBS, BMedSci, PhD, FRACP.
Author Information and AffiliationsInitial Posting: April 18, 2005; Last Update: August 16, 2018.
Estimated reading time: 18 minutes
Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
The following are the goals of this overview.
Goal 3.
Provide an evaluation strategy to identify the genetic cause of PMG in a proband (when possible).
1. Definition and Clinical Characteristics of Polymicrogyria
Polymicrogyria (PMG) is a malformation of the developing brain characterized by abnormal cortical lamination and an unusual folding pattern of the cerebral cortex such that all or part of the brain surface is taken up by an excessive number of small gyri (folds). PMG, one of the most common brain malformations, accounts for approximately 20% of all malformations of cortical development [Leventer et al 1999].
MRI. The diagnosis of PMG is typically made by MRI since computed tomography (CT) and other imaging methods do not have high enough resolution or adequate contrast to identify the small folds that define PMG. It is important that optimal imaging techniques (including thin sections) and age-specific protocols be used to provide the best contrast between gray and white matter with good spatial resolution and adequate signal-to-noise ratio [Barkovich 2010].
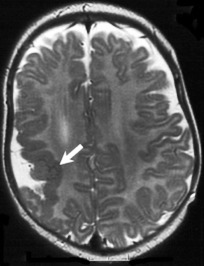
With high-quality MRI, microgyri and microsulci may be appreciated and stippling of the gray-white junction (a specific feature of PMG not seen in other malformations of cortical development) may be observed [Leventer et al 2010] (see Table 1). Of note, the Sylvian fissures (best seen on sagittal imaging) should be scrutinized in particular, as PMG often affects these areas preferentially.
While PMG is usually an isolated finding (i.e., occurring in the absence of other brain malformations), it can be seen in association with other brain malformations including gray matter heterotopia and ventriculomegaly, as well as abnormalities of the white matter, corpus callosum, brain stem, and cerebellum.
MRI – when interpreted by an expert – can reliably differentiate PMG from other malformations of cortical development (see Differential Diagnosis).
Prenatal ultrasound examination. In 5% of cases, PMG is detected on prenatal ultrasound examination by the presence of abnormalities – usually microcephaly and/or associated brain malformations. Note: PMG is difficult to detect by ultrasound examination until late in gestation [Leventer et al 2010].
Table 1.
Patterns of PMG: MRI and Clinical Findings
View in own window
| PMG Pattern | Affected Regions | MRI Findings |
|---|
Bilateral
perisylvian
PMG (BPP) |
| Bilateral PMG maximal in perisylvian regions (arrows), which may be symmetric or asymmetric & may extend beyond perisylvian regions; sylvian fissures often extended posteriorly & superiorly |
Unilateral
perisylvian
PMG (UPP) |
| Unilateral PMG maximal in perisylvian cortex (arrow), but may extend beyond perisylvian region; sylvian fissure often extended posteriorly & superiorly |
Bilateral
generalized
PMG (BGP) |
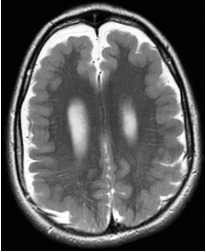
| Symmetric generalized PMG w/no obvious gradient or region of maximal severity; may have abnormal high signal in white matter |
Bilateral frontal
PMG (BFP) |
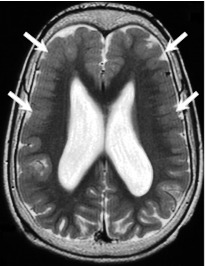
| Symmetric PMG w/anterior > posterior gradient extending from frontal poles (arrows) posteriorly to precentral gyrus & inferiorly to frontal operculum |
Bilateral
frontoparietal
PMG (BFPP) |
| Symmetric generalized PMG with decreasing anterior-posterior gradient, most prominent in frontoparietal cortex (arrows) |
Bilateral
parasagittal
parieto-occipital
PMG (BPPP) |
| Bilateral PMG in parasagittal & mesial aspects of parieto-occipital cortex (arrows) |
PMG associated
w/periventricular
nodular
heterotopia
(PNH-PMG) |
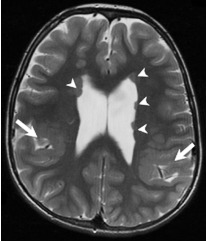
| PMG of variable patterns (mainly perisylvian or parieto-occipital) (large arrows) associated w/nodules of periventricular gray matter heterotopia (arrowheads) |
All MRI images are T2-weighted axial.
Bilateral perisylvian PMG (BPP), the most common pattern of PMG, is associated with oromotor dysfunction and a seizure disorder. Other findings typically include difficulties with tongue movement, expressive speech, and sucking and swallowing, as well as excessive drooling and facial diplegia as seen in the Worster-Drought syndrome. Mild-moderate intellectual disability is seen in up to 75%. Motor dysfunction may include limb spasticity, which – if present – is rarely severe [Kuzniecky et al 1993].
For information on neuropathology of PMG click here (pdf).
Differential Diagnosis
Other malformations of cortical development that need to be distinguished from PMG:
Pachygyria/lissencephaly, a distinct brain malformation in which reduced or absent cortical folding is combined with a thick cortex. Pachygyria and PMG may look similar on low-resolution neuroimaging (e.g., CT) because in both conditions the cortical thickness can appear to be increased and the gyri can appear to be broad and smooth. The cortex in PMG is overfolded but not thickened. The microgyri and microsulci of PMG and stippling of the gray-white junction that can be appreciated with high-quality MRI can distinguish PMG from pachygyria [
Leventer et al 2010].
Cobblestone lissencephaly, a brain surface that has a bumpy contour resulting from migration of neurons and glial cells through the basement membrane into the leptomeninges. Sometimes regions populated by these misplaced cells are incorrectly initially diagnosed as PMG based on the MRI appearance and are only distinguished from PMG by the presence of other brain abnormalities (e.g., white matter and cerebellar abnormalities) in combination with ocular anomalies and congenital muscular dystrophy. The cobblestone lissencephalies comprise brain malformations associated with congenital muscular dystrophy, including Walker-Warburg syndrome, muscle-eye-brain disease, and
Fukuyama congenital muscular dystrophy.
Schizencephaly, a specific pattern of PMG that describes a full-thickness cleft in the brain lined by PMG. While either CT or MRI is usually sufficient to diagnose schizencephaly, MRI is preferred when determining if the schizencephaly is open- or closed-lipped.
2. Genetic Causes of Polymicrogyria
Genetic causes of polymicrogyria (PMG) include both contiguous-gene and single-gene disorders.
Contiguous-Gene Disorders
PMG has been implicated in a number of chromosome copy number variants (CNVs) (summarized in Robin et al [2006], Dobyns et al [2008]).
The most common copy number variants associated with PMG:
22q11.2 deletion, characterized by cardiac defects, parathyroid hypoplasia, facial dysmorphism, and thymus hypoplasia, ± microcephaly and bilateral perisylvian PMG
1p36 deletion (OMIM
607872), characterized by developmental delay / intellectual disability and dysmorphic facial features, ± microcephaly and bilateral perisylvian PMG
Other copy number variants with which PMG may be associated are deletion of 4q21, 6q26, 13q3, 18p11, and 21q2 and duplication of 2p13 [Guerrini & Dobyns 2014].
Single-Gene Disorders
To date more than 40 genes have been associated with PMG. This GeneReview covers the following pathways and groups of related disorders implicated in PMG:
The mTORopathies (the PI3K-AKT-MTOR pathway)
The tubulinopathies
Cobblestone dysplasia – alpha dystroglycanopathies
Cobblestone dysplasia – other (including laminopathies and congenital disorders of glycosylation)
Other pathways/pathologies
Table 2 presents the genes associated with the above categories (in alphabetic order within each category). Several conditions with PMG are described in more detail following Table 2.
Additional disorders not included in Table 2 are Aicardi syndrome (in which PMG is almost uniformly seen) [Hetts et al 2006], and the monogenic syndromes Mowat-Wilson syndrome, Yunis Varon syndrome [Baulac et al 2014], Adams-Oliver syndrome [Amor et al 2000], and Sturge-Weber syndrome (in all of which PMG is seen at low frequency).
Table 2.
PMG-Associated Genes Grouped by Pathway/Pathology
View in own window
Pathway/
Pathology | Gene 1 | Head Size 2 | Brain Findings | Disorder Name | MOI | Reference 3 |
|---|
| MAC | MIC | N/MS |
|---|
|
mTORopathies
|
AKT3
| x | | | Bilateral perisylvian PMG | MPPH syndrome | AD |
MPPH Syndrome
|
|
CCND2
| x | | |
|
MTOR
| x | | | Smith-Kingsmore syndrome | AD | OMIM 616638 |
|
PI4KA
| | | x | Perisylvian PMG | | AR |
PI4KA-Related Disorder
|
|
PIK3CA
| x | | | Bilateral perisylvian PMG | MCAP syndrome | See footnote 5. |
PIK3CA-Related Segmental Overgrowth
|
|
PIK3R2
| x | | | MPPH syndrome | AD |
MPPH Syndrome
|
|
PTEN
| x | | | Diffuse, focal, or multifocal PMG | | AD | Elia et al [2012], Saletti et al [2017] |
|
Tubulinopathies
|
DYNC1H1
| | x | x | Frontal or diffuse PMG | | AD | OMIM 600112 |
|
KIF5C
| | | x | Perisylvian PMG | | AD | OMIM 615282 |
|
TUBA1A
| | x | x | Diffuse, focal, or multifocal PMG; bilateral, asymmetric, perisylvian PMG | Lissencephaly 3 | AD |
Tubulinopathies Overview
|
|
TUBB
| | x | | PMG | | AD |
|
TUBB2A
| | x | | Bilateral, asymmetric, anterior predominant PMG | | AD |
|
TUBB2B
| | x | x | |
|
TUBB3
| | x | x | Frontoparietal PMG | | AD |
Cobblestone
dysplasia – alpha
dystroglycan-
opathies
|
FKTN
| | | x | Diffuse (cerebral & cerebellar) PMG | | AR |
Fukuyama Congenital Muscular Dystrophy
|
|
POMGNT1
| | | x | PMG | | AR | OMIM 606822 |
|
POMT2
| | | x | PMG | | AR | OMIM 607439 |
Cobblestone
dysplasia –
other (incl
laminopathies
& congenital
disorders of
glycosylation)
| ADGRG1 (GPR56) | | | x | Bilateral frontoparietal PMG | | AR | OMIM 604110 |
|
COL3A1
| x | | x | Diffuse cobblestone cortex; PMG A > P | | AR |
Horn et al [2017]
|
|
ATP6V0A2
| | x | x | Frontoparietal PMG | Autosomal recessive cutis laxa type 2A | AR |
ATP6V0A2-Related Cutis Laxa
|
|
LAMA2
| | | x | Occipital PMG; white-matter signal abnormalities | Muscular dystrophy, congenital merosin-deficient, 1A | AR | OMIM 607855 |
|
LAMB1
| x | | x | Porencephaly; cobblestone lissencephaly P > A | Lissencephaly 5 | AR | OMIM 615191 |
|
LAMC3
| | | x | Occipital PMG | | AR | OMIM 614115 |
|
SNAP29
| | x | | Perisylvian or diffuse PMG | | AR | OMIM 609528 |
|
SRD5A3
| | | x | Frontal PMG | SRD5A3-CDG (CDG-Iq) | AR |
Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview
|
|
Other
|
BICD2
| | | x | Perisylvian PMG | | AD |
Ravenscroft et al [2016]
|
|
COL18A1
| | | x | Frontal PMG | Knobloch syndrome 1 | AR |
Keren et al [2007]
|
|
DDX3X
| | x | x | Frontoparietal or diffuse PMG | DDX3X-related neurodevelopmental disorder | XL |
DDX3X-Related Neurodevelopmental Disorder
|
|
EML1
| x | | | Ribbon-like heterotopia w/overlying PMG; ACC | | AR | OMIM 600348 |
| EOMES (TBR2) | | x | | Bilateral perisylvian or diffuse PMG | | AR | OMIM 604615 |
|
EZH2
| x | | | Bilateral perisylvian PMG | Weaver syndrome | AD |
EZH2-Related Overgrowth
|
|
FIG4
| | x | x | Bilateral or temporo-occipital PMG | | AR | OMIM 612691 |
|
GPSM2
| x | | | Parasagittal PMG; ACC | Chudley-McCollough syndrome | AR | OMIM 604213 |
|
GRIN1
| | x | | Extensive bilateral PMG | | AD |
GRIN1-Related Neurodevelopmental Disorder
|
|
GRIN2B
| | x | x | Diffuse PMG | | AD |
GRIN2B-Related Neurodevelopmental Disorder
|
| KIFBP (KIAA1279) | | x | | Diffuse PMG | Goldberg-Shprintzen syndrome | AR | OMIM 609460 |
|
MAP1B
| | | | Perisylvian PMG; PNH | | AD |
Heinzen et al [2018]
|
|
NDE1
| | x | | Diffuse PMG | | AR | OMIM 609449 |
|
NEDD4L
| | | x | Bilateral perisylvian PMG; PNH | | AD |
Kato et al [2017]
|
|
OCLN
| | x | | Band-like calcifications w/diffuse PMG | Pseudo-TORCH syndrome 1 | AR | OMIM 251290 |
|
OFD1
| x | | | Frontal and parietal PMG | Joubert syndrome (XLR); orofaciodigital syndrome 1 (XLD) | XL |
Joubert Syndrome
|
|
PAX6
| | x | | Variable temporal PMG | | AR | OMIM 607108 |
|
RAB18
| | x | x | Diffuse or frontal PMG | RAB18 deficiency 4 | AR |
RAB18 Deficiency
|
|
RAB3GAP1
| | x | x |
|
RAB3GAP2
| | x | x |
|
RTTN
| | x | | Variable diffuse, asymmetric PMG | | AR | OMIM 614833 |
|
TBC1D20
| | | x | Diffuse or bilateral frontal PMG | RAB18 deficiency 4 | AR |
RAB18 Deficiency
|
|
TCTN1
| | | x | Frontal PMG | Joubert syndrome | AR |
Joubert Syndrome
|
|
TMEM216
| | | x | Variable PMG | Meckel-Gruber syndrome, Joubert syndrome | AR |
|
WDR62
| | x | | ± diffuse or asymmetric PMG | | AR |
WDR62 Primary Microcephaly
|
A = anterior; ACC = absence of the corpus callosum; AD = autosomal dominant; AR = autosomal recessive; CDG = congenital disorder of glycosylation; MCAP = megalencephaly-capillary malformation-PMG; MOI = mode of inheritance; MPPH = megalencephaly-polymicrogyria-polydactyly-hydrocephalus; P = posterior; PNH = periventricular nodular heterotopia; XL = X-linked; XLD = X-linked dominant; XLR = X-linked recessive
- 1.
Genes are in alphabetic order.
- 2.
Head size:
N/MS = normal / mildly small (i.e., head circumference >3 SD and <97%)
MIC = severe microcephaly (i.e., birth head circumference <3 SD or earliest HC <4 SD)
MAC = macrocephaly (i.e., head circumference >97%)
- 3.
GeneReview, citation, or OMIM entry
- 4.
RAB18 deficiency is a spectrum that includes Warburg micro syndrome (at the severe end) and Martsolf syndrome (at the mild end). Additional findings are eye involvement (bilateral congenital cataracts, microphthalmia, and microcornea); severe-to-profound intellectual disability; and hypogonadism.
- 5.
Most affected individuals with MCAP reported to date (21/24) had somatic mosaicism for pathogenic variants in PIK3CA, suggesting that mutation occurred post fertilization in one cell of the multicellular embryo. Two of 24 affected individuals had a de novo germline pathogenic variant in PIK3CA.
Tubulinopathies (see Tubulinopathies Overview) are caused by a heterozygous pathogenic variant in TUBA1A (see Note), TUBB, TUBB2A, TUBB2B, or TUBB3 – genes encoding different isotypes of α-tubulin. Brain malformations include asymmetric perisylvian PMG with callosal defects, cerebellar vermis dysplasia, pontine hypoplasia, and dysmorphic basal ganglia. Because dysmorphic basal ganglia [Amrom et al 2014] and atypical microgyration (which may not be as evident as in typical PMG) are common in tubulin-associated PMG, it has been proposed that these complex cortical malformations be called "tubulin-associated dysgyria."
Note: While heterozygous pathogenic variants in TUBA1A can be associated with PMG, they are more frequently associated with pachygyria [Jansen et al 2011].
ADGRG1 (formerly GPR56). The spectrum of brain malformations includes the relatively rare phenotype of bilateral frontoparietal-predominant PMG [Piao et al 2005], a cobblestone-like lissencephaly (with no eye or muscle involvement), cerebellar dysplasia, and diffuse myelination abnormalities [Dobyns et al 1996, Bahi-Buisson et al 2010]. Although biallelic ADGRG1 pathogenic variants were initially reported as a cause of bilateral frontoparietal PMG (BFPP), subsequent reports and further delineation of the phenotype showed that the cortical malformation was most often cobblestone in nature or complex cortical dysgyria resembling PMG, rather than typical PMG [Bahi-Buisson et al 2010].
Metabolic disorders most commonly reported with PMG are summarized in Table 3.
3. Evaluation Strategy to Identify the Genetic Cause of PMG in a Proband
The approach outlined in this section can be used to determine if a specific genetic cause or mode of inheritance can be identified to aid in discussions of prognosis and genetic counseling.
Confirming the Clinical Diagnosis
Confirming PMG as the underlying cortical malformation and ruling out other brain malformations that may be confused with PMG (e.g., pachgyria and other forms of cortical dysplasia) requires consultation with an expert in interpretation of the MRI findings of PMG. Note that while the gold standard for the diagnosis of PMG is postmortem histopathologic examination, the clinical diagnosis relies on neuroimaging.
History
Pregnancy. A detailed pregnancy history with attention to infections, trauma, multiple gestations, and other documented problems that could provide insight into possible acquired causes of PMG. PMG is commonly associated with congenital cytomegalovirus [Kwak et al 2018].
Family. Parental consanguinity may suggest an autosomal recessive disorder (Table 2).
If no genetic cause has been identified and if familial PMG is suspected, it may be reasonable to perform a brain MRI (if not previously performed) on close relatives (i.e., parents or sibs) with the following:
Neurologic problems including seizures, cognitive delay, motor impairment, pseudobulbar signs, and/or focal weakness
What appear to be minor findings, such as a lisp or an isolated learning disability
Medical. Other specific tests targeting individual neurometabolic disorders can be obtained if clinically suggested (see Table 3).
Physical Examination
Head size is a useful first step in clinical assessment to aid in focusing genetic testing.
Macrocephaly is the group in which a genetic cause of PMG is most likely to be identified. The most common disorders with macrocephaly are the PI3K-AKT-MTOR-related disorders. Diagnostic possibilities:
Note that a subset of individuals have hydrocephalus (requiring shunting) and/or polydactyly of fingers/toes.
In the presence of other systemic features such as somatic overgrowth or pigmentary skin changes:
Microcephaly, a highly genetically heterogeneous group that includes numerous multisystem disorders, requires thorough physical assessment and detailed review of the brain MRI findings. Diagnostic possibilities in the presence of:
Normocephaly (or borderline microcephaly). The most commonly identified genetic factors are copy number variants resulting in disorders such as 22q11.2 deletion and 1p36 deletion (OMIM 607872), which involve multiple systems and have numerous additional features including (but not limited to) intellectual disability, dysmorphic facial features, and congenital heart defects.
A general physical examination should look for any associated craniofacial, musculoskeletal, and/or visceral findings that could indicate the mostly commonly considered syndromes (Table 2).
Molecular Genetic Testing
Testing approaches can include a combination of gene-targeted testing (single-gene testing or a multigene panel) and comprehensive genomic testing (exome sequencing, exome array, or chromosomal microarray analysis [CMA]). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not. Options include the following:
4. Genetic Counseling of Family Members of an Individual with PMG
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Hereditary polymicrogyria (PMG) may be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. Genetic counseling and risk assessment depend on determination of the specific genetic cause of PMG in an individual.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Some individuals diagnosed with PMG have an affected parent.
Many individuals diagnosed with PMG have the disorder as the result of a de novo pathogenic variant.
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant (i.e., the proband appears to be the only affected family member) include molecular genetic testing.
The family history may appear to be negative because of failure to recognize the disorder in a family member or reduced penetrance in a heterozygous parent. Therefore, an apparently negative family history cannot be confirmed unless appropriate molecular genetic testing and/or clinical evaluation has been performed.
Note: Due to reduced penetrance, there may be asymptomatic family members or several generations of individuals with the pathogenic variant who remain symptom-free.
Sibs of a proband. The risk to sibs depends on the genetic status of the proband's parents: if one of the proband's parents has a PMG-related pathogenic variant, the risk to the sibs of inheriting the pathogenic variant is 50%. Note that phenotypic variability can be observed within families.
Offspring of a proband. Each child of an individual with autosomal dominant PMG has a 50% chance of inheriting the pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the PMG-related pathogenic variant, the parent's family members may be at risk.
Autosomal Recessive Inheritance ‒ Risk to Family Members
Parents of a proband
The parents of an affected individual are obligate heterozygotes (i.e., carriers of one PMG-related pathogenic variant).
Heterozygotes are asymptomatic.
Sibs of a proband. At conception, each sib of a proband has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier (heterozygote), and a 25% chance of being unaffected and not a carrier.
Offspring of a proband. All offspring are obligate heterozygotes (carriers) for a pathogenic variant in a PMG-related gene.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a PMG-related pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the PMG-related pathogenic variants in the family.
X-Linked Inheritance ‒ Risk to Family Members
Parents of a male proband
The father of an affected male will not have the disorder nor will he be hemizygous for the PMG-related pathogenic variant; therefore, he does not require further evaluation/testing.
In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected child and no other affected relatives and if the PMG-related pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier) or the affected male may have a de novo pathogenic variant, in which case the mother is not a carrier.
Parents of a female proband. See Oral-Facial-Digital Syndrome Type I.
Sibs of a male proband. The risk to sibs depends on the genetic status of the proband's mother:
If the mother of the proband has the PMG-related pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the variant will be affected; females who inherit the variant will be heterozygotes (carriers) and will not typically be affected.
If the proband represents a simplex case (i.e., a single occurrence in a family) and if the PMG-related pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the possibility of maternal germline mosaicism.
Sibs of a female proband. See Oral-Facial-Digital Syndrome Type I.
Offspring of a proband. Affected males will transmit the pathogenic variant to all of their daughters and none of their sons.
Other family members. The proband's maternal aunts may be at risk of being heterozygotes (carriers) for the pathogenic variant, and the aunts' offspring, depending on their sex, may be at risk of being heterozygotes (carriers) for the pathogenic variant or of being affected.
Heterozygote detection. Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the PMG-related pathogenic variant has been identified in the proband.
Prenatal Testing and Preimplantation Genetic Testing
Once the PMG-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Chapter Notes
Author Notes
Dr Ghayda Mirzaa is a geneticist and researcher at Seattle Children's Hospital. The goal of her research is to study the underlying causes, natural history, and medical management of developmental brain disorders including brain malformations, autism, and epilepsy.
Acknowledgments
We acknowledge the assistance of Ms Stefanie Brock in the preparation of Table 2.
Author History
Kira Apse, ScM; Harvard Medical School (2004-2018)
Adria Bodell, MS, CGC; Harvard Medical School (2004-2018)
Bernard Chang, MD; Harvard Medical School (2004-2018)
William Dobyns, MD (2018-present)
Anna Jansen, MD, PhD (2018-present)
Richard J Leventer, MBBS, BMedSci, PhD, FRACP (2018-present)
Ghayda Mirzaa, MD (2018-present)
Chloe Stutterd, MBBS, FRACP (2018-present)
Christopher A Walsh, MD, PhD; Harvard Medical School (2004-2018)
Revision History
6 June 2024 (ma) Chapter retired: phenotype is too broad
16 August 2018 (bp) Comprehensive update posted live
6 August 2007 (me) Comprehensive update posted live
18 April 2005 (me) Overview posted live
24 August 2004 (bc) Original submission
References
Literature Cited
Amor
DJ, Leventer
RJ, Hayllar
S, Bankier
A. Polymicrogyria associated with scalp and limb defects: variant of Adams-Oliver syndrome.
Am J Med Genet.
2000;93:328-34.
[
PubMed: 10946361]
Amrom
D, Tanyalçin
I, Verhelst
H, Deconinck
N, Brouhard
GJ, Décarie
JC, Vanderhasselt
T, Das
S, Hamdan
FF, Lissens
W, Michaud
JL, Jansen
AC. Polymicrogyria with dysmorphic basal ganglia? Think tubulin!
Clin Genet.
2014;85:178-83.
[
PubMed: 23495813]
Bahi-Buisson
N, Poirier
K, Boddaert
N, Fallet-Bianco
C, Specchio
N, Bertini
E, Caglayan
O, Lascelles
K, Elie
C, Rambaud
J, Baulac
M, An
I, Dias
P, des Portes
V, Moutard
ML, Soufflet
C, El Maleh
M, Beldjord
C, Villard
L, Chelly
J. GPR56-related bilateral frontoparietal polymicrogyria: further evidence for an overlap with the cobblestone complex.
Brain.
2010;133:3194-209.
[
PubMed: 20929962]
Baulac
S, Lenk
GM, Dufresnois
B, Ouled Amar Bencheikh
B, Couarch
P, Renard
J, Larson
PA, Ferguson
CJ, Noé
E, Poirier
K, Hubans
C, Ferreira
S, Guerrini
R, Ouazzani
R, El Hachimi
KH, Meisler
MH, Leguern
E. Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria.
Neurology.
2014;82:1068-75
[
PMC free article: PMC3962989] [
PubMed: 24598713]
Dobyns
WB, Mirzaa
G, Christian
SL, Petras
K, Roseberry
J, Clark
GD, Curry
CJ, McDonald-McGinn
D, Medne
L, Zackai
E, Parsons
J, Zand
DJ, Hisama
FM, Walsh
CA, Leventer
RJ, Martin
CL, Gajecka
M, Shaffer
LG. Consistent chromosome abnormalities identify novel polymicrogyria loci in 1p36.3, 2p16.1-p23.1, 4q21.21-q22.1, 6q26-q27, and 21q2.
Am J Med Genet A.
2008;146A:1637-54.
[
PMC free article: PMC2801020] [
PubMed: 18536050]
Dobyns
WB, Patton
MA, Stratton
RF, Mastrobattista
JM, Blanton
SH, Northrup
H. Cobblestone lissencephaly with normal eyes and muscle.
Neuropediatrics.
1996;27:70-5.
[
PubMed: 8737821]
Elia
M, Amato
C, Bottitta
M, Grillo
L, Calabrese
G, Esposito
M, Carotenuto
M. An atypical patient with Cowden syndrome and PTEN gene mutation presenting with cortical malformation and focal epilepsy.
Brain Dev.
2012;34:873-6.
[
PubMed: 22469695]
Heinzen
EL, O'Neill
AC, Zhu
X, Allen
AS, Bahlo
M, Chelly
J, Chen
MH, Dobyns
WB, Freytag
S, Guerrini
R, Leventer
RJ, Poduri
A, Robertson
SP, Walsh
CA, Zhang
M, et al.
De novo and inherited private variants in MAP1B in periventricular nodular heterotopia.
PLoS Genet.
2018;14:e1007281.
[
PMC free article: PMC5965900] [
PubMed: 29738522]
Hetts
SW, Sherr
EH, Chao
S, Gobuty
S, Barkovich
AJ. Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations.
AJR Am J Roentgenol.
2006;187:1343-8.
[
PubMed: 17056927]
Horn
D, Siebert
E, Seidel
U, Rost
I, Mayer
K, Abou Jamra
R, Mitter
D, Kornak
U.
Biallelic COL3A1 mutations result in a clinical spectrum of specific structural brain anomalies and connective tissue abnormalities.
Am J Med Genet A.
2017;173:2534-8.
[
PubMed: 28742248]
Jansen
AC, Oostra
A, Desprechins
B, De Vlaeminck
Y, Verhelst
H, Régal
L, Verloo
P, Bockaert
N, Keymolen
K, Seneca
S, De Meirleir
L, Lissens
W. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria.
Neurology.
2011;76:988-92.
[
PubMed: 21403111]
Kato
K, Miya
F, Hori
I, Ieda
D, Ohashi
K, Negishi
Y, Hattori
A, Okamoto
N, Kato
M, Tsunoda
T, Yamasaki
M, Kanemura
Y, Kosaki
K, Saitoh
S.
A novel missense mutation in the HECT domain of NEDD4L identified in a girl with periventricular nodular heterotopia, polymicrogyria and cleft palate.
J Hum Genet.
2017;62:861-3.
[
PubMed: 28515470]
Keren
B, Suzuki
OT, Gérard-Blanluet
M, Brémond-Gignac
D, Elmaleh
M, Titomanlio
L, Delezoide
AL, Passos-Bueno
MR, Verloes
A. CNS malformations in Knobloch syndrome with splice mutation in COL18A1 gene.
Am J Med Genet A.
2007;143A:1514-8.
[
PubMed: 17546652]
Kuzniecky
R, Andermann
F, Guerrini
R. Congenital bilateral perisylvian syndrome: study of 31 patients. The CBPS Multicenter Collaborative Study.
Lancet.
1993;341:608-12.
[
PubMed: 8094839]
Kwak
M, Yum
MS, Yeh
HR, Kim
HJ, Ko
TS. Brain magnetic resonance imaging findings of congenital cytomegalovirus infection as a prognostic factor for neurological outcome.
Pediatr Neurol.
2018;83:14-18
[
PubMed: 29681488]
Leventer
RJ, Jansen
A, Pilz
DT, Stoodley
N, Marini
C, Dubeau
F, Malone
J, Mitchell
LA, Mandelstam
S, Scheffer
IE, Berkovic
SF, Andermann
F, Andermann
E, Guerrini
R, Dobyns
WB. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients.
Brain.
2010;133:1415-27.
[
PMC free article: PMC2859156] [
PubMed: 20403963]
Leventer
RJ, Phelan
EM, Coleman
LT, Kean
MJ, Jackson
GD, Harvey
AS. Clinical and imaging features of cortical malformations in childhood.
Neurology.
1999;53:715-22.
[
PubMed: 10489031]
Piao
X, Chang
BS, Bodell
A, Woods
K, Benzeev
B, Topcu
M, Guerrini
R, Goldberg-Stern
H, Sztriha
L, Dobyns
WB, Barkovich
AJ, Walsh
CA. Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes.
Ann Neurol.
2005;58:680-7.
[
PubMed: 16240336]
Ravenscroft
G, Di Donato
N, Hahn
G, Davis
MR, Craven
PD, Poke
G, Neas
KR, Neuhann
TM, Dobyns
WB, Laing
NG. Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria.
Neuromuscul Disord.
2016;26:744-8.
[
PubMed: 27751653]
Robin
NH, Taylor
CJ, McDonald-McGinn
DM, Zackai
EH, Bingham
P, Collins
KJ, Earl
D, Gill
D, Granata
T, Guerrini
R, Katz
N, Kimonis
V, Lin
JP, Lynch
DR, Mohammed
SN, Massey
RF, McDonald
M, Rogers
RC, Splitt
M, Stevens
CA, Tischkowitz
MD, Stoodley
N, Leventer
RJ, Pilz
DT, Dobyns
WB. Polymicrogyria and deletion 22q11.2 syndrome: window to the etiology of a common cortical malformation.
Am J Med Genet A.
2006;140:2416-25.
[
PubMed: 17036343]
Saletti
V, Esposito
S, Maccaro
A, Giglio
S, Valentini
LG, Chiapparini
L. Chiari I malformation in a child with PTEN hamartoma tumor syndrome: association or coincidence?
Eur J Med Genet.
2017;60:261-4.
[
PubMed: 28286253]