Summary
The purpose of this overview is to increase clinician awareness of the genetic basis of dilated cardiomyopathy (DCM) and the benefits of early diagnosis and management to individuals with genetic DCM.
The following are the goals of this overview.
Goal 4.
Provide a basic view of genetic risk assessment of at-risk asymptomatic relatives of a proband with DCM to inform cardiac surveillance and allow early detection and treatment of DCM to improve long-term outcome.
1. Dilated Cardiomyopathy (DCM): Definition
The diagnosis of DCM is established when both of the following are present:
Left ventricular enlargement. Enlargement is most commonly assessed in adults by either echocardiography or cardiac MRI. Because of rapid growth in children, expert cardiovascular assessment is recommended to assess left ventricular enlargement in the pediatric population.
Systolic dysfunction, a reduction in the myocardial force of contraction. An ejection fraction of less than 50% is considered systolic dysfunction. The left ventricular ejection fraction is the most commonly used clinical measure of systolic function, and is usually estimated from a two-dimensional echocardiogram or from cardiac MRI. Another noninvasive approach is a cardiac nuclear study. Ejection fractions can also be estimated from a left ventricular angiogram.
Note: Arrhythmogenic right ventricular cardiomyopathy (ARVC) with predominant left ventricular involvement may present as DCM [Sen-Chowdhry et al 2008].
DCM usually initially manifests in adults in the fourth to sixth decade, although it may present at any age (prenatally; in infancy, early or late childhood, or adolescence; or in the elderly). Extensive additional clinical and genetic information on DCM is available [Burkett & Hershberger 2005, Sivasankaran et al 2005, Judge 2009, Dellefave & McNally 2010, Hershberger et al 2010a, Jordan & Hershberger 2021].
Persons with DCM may be asymptomatic for a number of years. Manifestations usually occur late in the disease course with one or more of the following findings:
Heart failure. Symptoms include those of congestion (edema, orthopnea, paroxysmal nocturnal dyspnea) and/or reduced cardiac output (fatigue, dyspnea on exertion).
Arrhythmias and/or conduction system disease. These commonly accompany advanced cardiomyopathy and heart failure. Some genetic causes (e.g., pathogenic variants in DES, FLNC, LMNA, and SCN5A) may involve prominent conduction system disease or arrhythmias out of proportion to the degree of left ventricular dysfunction.
Thromboembolic disease. Stroke or systemic embolus secondary to left ventricular mural thrombus may also occur.
Pregnancy. Peripartum or pregnancy-associated cardiomyopathy (PPCM/PACM) that occurs during or soon after pregnancy was once considered distinct from DCM, but is now recognized as a part of the clinical spectrum of DCM.
2. Dilated Cardiomyopathy (DCM): Categories
DCM can be categorized as acquired, syndromic, or nonsyndromic ().
Categories of dilated cardiomyopathy
Acquired (Secondary) DCM
The most common cause of acquired DCM is ischemic injury, such as that caused by prior myocardial infarction from coronary artery disease.
Other less common causes include valvular and congenital heart disease, toxins (most commonly, anthracyclines or other chemotherapeutic agents; various drugs with idiosyncratic reactions), thyroid disease, inflammatory or infectious conditions, severe long-standing hypertension, and radiation. While emerging evidence suggests that DCM arising after chemotherapy exposure may also have a genetic background, the clinical relevance of this information is currently undefined [Garcia-Pavia et al 2019].
Note: Acquired DCM will not be discussed further in this overview.
Syndromic DCM
In GeneReviews, "syndromic" refers to a disorder characterized by a constellation of phenotypic features that either: (1) specifically suggests the diagnosis (which can be confirmed by molecular genetic testing) or (2) allows diagnosis of the disorder in the absence of confirmatory molecular genetic findings. A selected list of syndromic DCM is provided in Table 1 [Hershberger et al 2009, Hershberger et al 2013].
Note: Syndromic DCM will not be discussed further in this overview.
Table 1.
Selected List of Syndromic Dilated Cardiomyopathy
View in own window
AD = autosomal dominant; AR = autosomal recessive; Mat = maternal inheritance; MOI = mode of inheritance; XL = X-linked
- 1.
Disorders are in alphabetic order.
Nonsyndromic DCM
Individuals with DCM who do not have acquired (secondary) DCM or syndromic DCM (Table 1) have nonsyndromic dilated cardiomyopathy (defined for this GeneReview as DCM with no other systemic involvement). See Table 2 for a current list of known DCM-related genes organized by strength of ClinGen classification and alphabetically [Jordan et al 2021].
The Clinical Genome Resource (ClinGen) DCM Gene Curation Expert Panel has classified DCM-related genes using the ClinGen framework for the strength of their relationship with monogenic, nonsyndromic DCM. A summary of the data curated for each gene can be accessed at ClinGen Gene Validity Classification.
Note: Left ventricular non-compaction (LVNC) is a feature of the heart muscle that has been observed in the general population, and reported in conjunction with a DCM phenotype as well as numerous other cardiovascular phenotypes; its relationship (if any) to the presence or severity of the DCM phenotype remains unknown [Hershberger et al 2017, Ross & Semsarian 2018, Ross et al 2020].
Table 2.
Nonsyndromic Dilated Cardiomyopathy Genes
View in own window
AD = autosomal dominant; AR = autosomal recessive; CMT = Charcot-Marie-Tooth hereditary neuropathy; HCM = hypertrophic cardiomyopathy; LGMD = limb-girdle muscular dystrophy; LGMDR = limb-girdle muscular dystrophy autosomal recessive; MOI = mode of inheritance; RCM = restrictive cardiomyopathy; XL = X-linked
- 1.
Genes are organized first by strength of ClinGen classification, then frequency of causation of DCM, and then alphabetically.
- 2.
The percentages provided (based on ≥2 reports screening large numbers of probands with HNDCM) should be interpreted as preliminary estimates.
- 3.
Allelic disorders = other phenotypes caused by pathogenic variants in the same gene.
- 4.
Note: Although 10%-20% of DCM in three cohorts (with or without a family history of DCM) was attributed to TTN pathogenic truncating variants [Herman et al 2012], determining the role of pathogenic variants in TTN in DCM is difficult given that: (a) 3% of controls also have truncating variants; and (b) TTN pathogenic truncating variants have not segregated with DCM in all families with DCM [Norton et al 2013]. Truncating TTN variants found in individuals with DCM have been reported to cluster in the A-band region of titin, the protein encoded by TTN [Roberts et al 2015]. To date, TTN missense variants have not been associated with disease.
- 5.
The hot spot for pathogenic and likely pathogenic variants associated with DCM is located in exon 9 of RBM20; it is unclear if non-hot spot variants in RBM20 can be associated with DCM.
- 6.
3. Establishing (When Possible) the Specific Genetic Cause of DCM
Molecular genetic testing should be offered to every individual of any age with nonischemic DCM [Hershberger et al 2018] including those with peripartum or pregnancy-associated cardiomyopathy (PPCM/PACM) [Goli et al 2021] (). See Table 2 for a current list of known DCM-related genes. The purpose of establishing a molecular diagnosis of DCM is to inform risk assessment of relatives of a proband (see Section 4).
Variants (pathogenic, likely pathogenic, or of unknown significance) in more than 30 genes have been identified in up to 30%-35% of individuals with familial DCM (i.e., in ≥2 first-degree family members) [Hershberger et al 2013, Jordan & Hershberger 2021] or in simplex cases (i.e., in only 1 family member) [Hershberger et al 2008, Hershberger et al 2010b, Pugh et al 2014, Morales et al 2020]. The detection rate of pathogenic and likely pathogenic variants is about 27% [Pugh et al 2014].
A cardiomyopathy multigene panel that includes the genes with a ClinGen classification of definitive, strong, or moderate (as listed in Table 2) is most likely to identify the genetic cause of the condition while also limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype.
Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Health care providers ordering genetic testing should be familiar with the genetics of DCM [Burkett & Hershberger 2005, Judge 2009, Caleshu et al 2010, Hershberger et al 2010a, Hershberger & Siegfried 2011, Hershberger et al 2013, Jordan et al 2021]. Given the complexity of interpreting genetic test results and their implications for surveillance and management, health care providers should consider referral to a cardiovascular genetics center or a genetic counselor specializing in cardiac genetics (see NSGC – Find a Genetic Counselor).
4. Genetic Risk Assessment and Cardiac Surveillance of At-Risk Relatives for Detection of Early Treatable Manifestations of DCM
Cardiovascular screening of asymptomatic first-degree family members of an individual with DCM can allow early detection of DCM, prompt initiation of treatment, and improvement in long-term outcome [Morales & Hershberger 2015]. Clarification of the genetic status of first-degree family members of an individual with DCM can inform who is at risk and the recommended frequency of subsequent cardiovascular screening [Hershberger et al 2018].
A basic view of nonsyndromic dilated cardiomyopathy (DCM) genetic risk assessment and cardiac surveillance for at-risk relatives is presented in this section; issues that may be specific to a given family or genetic cause of nonsyndromic DCM are not comprehensively addressed.
Note: If a proband has a specific syndrome associated with DCM (e.g., Barth syndrome or Duchenne muscular dystrophy), counseling for that condition is indicated (see Table 1). Genetic risk assessment in families with syndromic DCM is not discussed further in this section.
Genetic Risk Assessment
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Nonsyndromic dilated cardiomyopathy (DCM) is typically inherited in an autosomal dominant manner. JPH2- and TNNI3-related nonsyndromic DCM can be inherited in an autosomal dominant or autosomal recessive manner.
DMD-related DCM – which can present as syndromic DCM in males or isolated DCM in carrier females (Table 1) – is inherited in an X-linked manner (see Dystrophinopathies).
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Some individuals diagnosed as having autosomal dominant DCM have an affected parent.
Some individuals diagnosed with autosomal dominant DCM have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with autosomal dominant DCM caused by a de novo pathogenic variant is unknown.
When evaluating for autosomal dominant inheritance, both the maternal and paternal lineages should be considered as possibly contributing to familial DCM. In an unknown proportion of cases, both parents may have evidence of DCM and/or DCM-related pathogenic variants, and thus the proband may have inherited pathogenic variants from one or both parents [
Liu et al 2015,
Cowan et al 2018].
Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status.
If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
The proband has a de novo pathogenic variant.
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
If a parent of the proband is affected and/or has a DCM-related pathogenic or likely pathogenic variant, the risk to the sibs of inheriting the variant is 50%. Because of variable expression and reduced penetrance, no predictions can be made regarding age of onset or severity of disease.
If both parents of a proband have a DCM-related pathogenic or likely pathogenic variant, sibs have a 75% chance of inheriting one or two DCM-related variants and a 25% chance of inheriting neither pathogenic variant.
If the parents are clinically unaffected but their genetic status is unknown, sibs are still presumed to be at increased risk for DCM because of the possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant DCM has a 50% chance of inheriting the DCM-related pathogenic variant.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected individual are obligate heterozygotes (i.e., presumed to have one JPH2 or TNNI3 pathogenic variant based on family history).
Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a JPH2 or TNNI3 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
The heterozygous parents of a child with biallelic pathogenic variants in JPH2 or TNNI3 may be at risk for DCM. JPH2-related DCM and TNNI3-related DCM can be inherited in an autosomal recessive or an autosomal dominant manner and the mechanisms to distinguish between pathogenic variants that solely incur risk in a dominant or recessive model are currently unknown. Therefore, it is reasonable for parents of the proband to undergo cardiac surveillance even if they have only one of the two pathogenic variants identified in the affected proband.
Sibs of a proband
If both parents are known to be heterozygous for a JPH2 or TNNI3 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting biallelic pathogenic variants, a 50% chance of inheriting one of the two pathogenic variants identified in the proband, and a 25% chance of inheriting neither pathogenic variant.
A sib who is heterozygous for one of the pathogenic variants identified in the proband may be at risk for DCM. JPH2 and TNNI3 pathogenic variants that solely incur risk in a dominant or recessive model are not distinguishable at this time. Therefore, it is reasonable for sibs of the proband to undergo cardiac surveillance even if they have only one of the two pathogenic variants identified in the affected proband.
Offspring of a proband. The offspring of an individual with autosomal recessive JPH2- or TNNI3-related DCM are obligate heterozygotes for a pathogenic variant in JPH2 or TNNI3 and may be at risk for DCM. Offspring should therefore undergo cardiac surveillance for DCM.
Cardiac Surveillance
It is appropriate to clarify the clinical and genetic status of asymptomatic family members at risk for DCM prior to the onset of manifestations to identify those with asymptomatic DCM and permit initiation of medical therapy aimed at preventing/delaying the morbidity of late-stage symptomatic disease [Morales & Hershberger 2015].
The following recommendations for surveillance of asymptomatic at-risk family members reflect the practice guidelines of the Heart Failure Society of America [Hershberger et al 2018].
If the Proband Has a Known Pathogenic Variant (or Pathogenic Variants) in a DCM-Related Gene
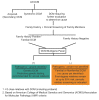
See , teal box ("Pathogenic variant identified"). Molecular genetic testing is recommended for parents, sibs, offspring, and other at-risk family members in order to clarify their genetic status.
Those identified as having a familial DCM-related pathogenic variant have an increased lifetime risk for DCM and, when asymptomatic, should undergo cardiovascular clinical screening at intervals based on the individual's age [Hershberger et al 2018].
Note: Asymptomatic at-risk relatives who do not meet criteria for DCM (with other causes ruled out) may represent early DCM when echocardiogram results are ambiguous (e.g., left ventricular enlargement with normal systolic function, decreased ejection fraction but normal-sized left ventricle) and/or echocardiogram results are normal but EKG results are abnormal (e.g., significant conduction system disease and/or arrhythmias).
In general, family members without the DCM-related pathogenic variant identified in the proband are no longer considered to be at increased risk for DCM and thus may be discharged from cardiac surveillance. However, because multiple variants in DCM-associated genes have been observed in individuals with nonsyndromic DCM [Morales et al 2020] and because families may segregate pathogenic variants in more than one DCM-related gene [Liu et al 2015, Cowan et al 2018], thorough individualized risk assessment through clinical, genetic, and family history analysis is warranted to determine if discharge from high-risk cardiac surveillance is appropriate.
If the Specific Genetic Cause of DCM in the Proband Has Not Been Identified
See , orange box ("Pathogenic variant not identified"). Perform cardiovascular screening on asymptomatic at-risk family members at intervals based on the individual's age [Hershberger et al 2018].
Note: Asymptomatic at-risk relatives who do not meet criteria for DCM (with other causes ruled out) may represent early DCM when echocardiogram results are ambiguous (e.g., left ventricular enlargement with normal systolic function, decreased ejection fraction but normal-sized left ventricle) and/or echocardiogram results are normal but EKG results are abnormal (e.g., significant conduction system disease and/or arrhythmias).
If a first-degree at-risk relative shows evidence of dilated cardiomyopathy, a diagnosis of familial DCM is made and the surveillance recommendations should extend to that person's first-degree relatives.
Future additional genetic testing for the proband (and other informative family members) may be considered when:
Multigene panels are expanded to include more genes and test sensitivity increases (e.g., resulting from better coverage of the genes included and improved detection of deletions/duplications); and
Genomic testing (exome sequencing and genome sequencing) becomes more suitable for clinical use.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Heart Association
Cardiomyopathy UK
United Kingdom
Phone: 0800 018 1024 (UK only)
Email: contact@cardiomyopathy.org
Children's Cardiomyopathy Foundation
DCM Foundation
Phone: 833-DCM-HOPE (833-326-4673)
Email: Info@DCMFoundation.org
MedlinePlus
Dilated Cardiomyopathy Research Project
Phone: 877-800-8430
Email: DCM.Research@osumc.edu
Chapter Notes
Author Notes
Web: Dilated Cardiomyopathy Research Project
The Dilated Cardiomyopathy Research Project, originally launched in 1993 by Dr Ray Hershberger while at the Oregon Health & Science University, aims to advance our understanding of dilated cardiomyopathy genetics. Multiple studies including data from more than 1500 families affected by DCM across the country have contributed genetic and clinical information to this research program. This multi-institutional effort, still led by Dr Hershberger and now housed at The Ohio State University, leverages the many sites collaborating in the DCM Consortium to identify families eligible for studies within the DCM Research Project, including the recently completed DCM Precision Medicine Study and the ongoing DCM Discovery Study. More information about the DCM Project, affiliated research studies, and other information and resources can be found on the website (www.dcmproject.com) or by emailing dcm.research@osumc.edu.
Acknowledgments
We are deeply grateful for the numerous families who have contributed to the research efforts in the DCM Research Project for nearly three decades. Without their gracious participation and engagement, the advances driven by the work of the DCM Research Project to the field would not be possible.
Author History
Ray E Hershberger, MD (2007-present)
Elizabeth Jordan, MS, LGC (2021-present)
Jessica D Kushner, MS, CGC; Oregon Health & Science University (2007-2013)
Ana Morales, MS, CGC; The Ohio State University (2013-2021)
Sharie Parks, PhD; Oregon Health & Science University (2007-2013)
Revision History
29 July 2021 (ha) Comprehensive update posted live
23 August 2018 (bp) Comprehensive update posted live
24 September 2015 (me) Comprehensive update posted live
9 May 2013 (me) Comprehensive update posted live
19 March 2009 (cd) Revision: sequence analysis and prenatal testing available clinically for TCAP, ABCC9, VCL, ACTN2, and CSRP3
10 July 2008 (cd) Revision: clinical testing available for TTN mutations as a cause of dilated cardiomyopathy
27 July 2007 (me) Review posted live
6 December 2006 (jdk) Original submission
References
Published Guidelines / Consensus Statements
Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899-909. [
PubMed]
Literature Cited
Burkett
EL, Hershberger
RE. Clinical and genetic issues in familial dilated cardiomyopathy.
J Am Coll Cardiol.
2005;45:969-81.
[
PubMed: 15808750]
Caleshu
C, Day
S, Rehm
HL, Baxter
S. Use and interpretation of genetic tests in cardiovascular genetics.
Heart.
2010;96:1669-75.
[
PubMed: 20937756]
Cowan
JR, Kinnamon
DD, Morales
A, Salyer
L, Nickerson
DA, Hershberger
RE. Multigenic disease and bilineal inheritance in dilated cardiomyopathy is illustrated in nonsegregating LMNA pedigrees.
Circ Genom Precis Med.
2018;11:e002038.
[
PMC free article: PMC6294440] [
PubMed: 30012837]
Garcia-Pavia
P, Kim
Y, Restrepo-Cordoba
MA, Lunde
IG, Wakimoto
H, Smith
AM, Toepfer
CN, Getz
K, Gorham
J, Patel
P, Ito
K, Willcox
JA, Arany
Z, Li
J, Owens
AT, Govind
R, Nuñez
B, Mazaika
E, Bayes-Genis
A, Walsh
R, Finkelman
B, Lupon
J, Whiffin
N, Serrano
I, Midwinter
W, Wilk
A, Bardaji
A, Ingold
N, Buchan
R, Tayal
U, Pascual-Figal
DA, de Marvao
A, Ahmad
M, Garcia-Pinilla
JM, Pantazis
A, Dominguez
F, John Baksi
A, O'Regan
DP, Rosen
SD, Prasad
SK, Lara-Pezzi
E, Provencio
M, Lyon
AR, Alonso-Pulpon
L, Cook
SA, DePalma
SR, Barton
PJR, Aplenc
R, Seidman
JG, Ky
B, Ware
JS, Seidman
CE. Genetic variants associated with cancer therapy-induced cardiomyopathy.
Circulation.
2019;140:31-41.
[
PMC free article: PMC6613726] [
PubMed: 30987448]
Goli
R, Li
J, Brandimarto
J, Levine
LD, Riis
V, McAfee
Q, DePalma
S, Haghighi
A, Seidman
JG, Seidman
CE, Jacoby
D, Macones
G, Judge
DP, Rana
S, Margulies
KB, Cappola
TP, Alharethi
R, Damp
J, Hsich
E, Elkayam
U, Sheppard
R, Alexis
JD, Boehmer
J, Kamiya
C, Gustafsson
F, Damm
P, Ersbøll
AS, Goland
S, Hilfiker-Kleiner
D, McNamara
DM, Arany
Z, et al.
Genetic and phenotypic landscape of peripartum cardiomyopathy.
Circulation.
2021;143:1852-62.
[
PMC free article: PMC8113098] [
PubMed: 33874732]
Herman
DS, Lam
L, Taylor
MR, Wang
L, Teekakirikul
P, Christodoulou
D, Conner
L, DePalma
SR, McDonough
B, Sparks
E, Teodorescu
DL, Cirino
AL, Banner
NR, Pennell
DJ, Graw
S, Merlo
M, Di Lenarda
A, Sinagra
G, Bos
JM, Ackerman
MJ, Mitchell
RN, Murry
CE, Lakdawala
NK, Ho
CY, Barton
PJ, Cook
SA, Mestroni
L, Seidman
JG, Seidman
CE. Truncations of titin causing dilated cardiomyopathy.
N Engl J Med.
2012;366:619-28.
[
PMC free article: PMC3660031] [
PubMed: 22335739]
Hershberger
RE, Cowan
J, Morales
A, Siegfried
JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy.
Circ Heart Fail.
2009;2:253-61.
[
PMC free article: PMC2927103] [
PubMed: 19808347]
Hershberger
RE, Givertz
MM, Ho
CY, Judge
DP, Kantor
PF, McBride
KL, Morales
A, Taylor
MRG, Vatta
M, Ware
SM. Genetic evaluation of cardiomyopathy-a Heart Failure Society of America Practice Guideline.
J Card Fail.
2018;24:281-302.
[
PMC free article: PMC9903357] [
PubMed: 29567486]
Hershberger
RE, Hedges
DJ, Morales
A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture.
Nat Rev Cardiol.
2013;10:531-47.
[
PubMed: 23900355]
Hershberger
RE, Morales
A, Cowan
J. Is left ventricular noncompaction a trait, phenotype, or disease? The evidence points to phenotype.
Circ Cardiovasc Genet.
2017;10:e001968.
[
PubMed: 29212902]
Hershberger
RE, Norton
N, Morales
A, Li
D, Siegfried
JD, Gonzalez-Quintana
J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy.
Circ Cardiovasc Genet.
2010b;3:155-61.
[
PMC free article: PMC2908892] [
PubMed: 20215591]
Hershberger
RE, Parks
SB, Kushner
JD, Li
D, Ludwigsen
S, Jakobs
P, Nauman
D, Burgess
D, Partain
J, Litt
M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy.
Clin Transl Sci.
2008;1:21-6.
[
PMC free article: PMC2633921] [
PubMed: 19412328]
Jónsson
H, Sulem
P, Kehr
B, Kristmundsdottir
S, Zink
F, Hjartarson
E, Hardarson
MT, Hjorleifsson
KE, Eggertsson
HP, Gudjonsson
SA, Ward
LD, Arnadottir
GA, Helgason
EA, Helgason
H, Gylfason
A, Jonasdottir
A, Jonasdottir
A, Rafnar
T, Frigge
M, Stacey
SN, Th Magnusson
O, Thorsteinsdottir
U, Masson
G, Kong
A, Halldorsson
BV, Helgason
A, Gudbjartsson
DF, Stefansson
K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature.
2017;549: 519-22.
[
PubMed: 28959963]
Jordan
E, Peterson
L, Ai
T, Asatryan
B, Bronicki
L, Brown
E, Celeghin
R, Edwards
M, Fan
J, Ingles
J, James
CA, Jarinova
O, Johnson
R, Judge
DP, Lahrouchi
N, Lekanne Deprez
RH, Lumbers
RT, Mazzarotto
F, Medeiros Domingo
A, Miller
RL, Morales
A, Murray
B, Peters
S, Pilichou
K, Protonotarios
A, Semsarian
C, Shah
P, Syrris
P, Thaxton
C, van Tintelen
JP, Walsh
R, Wang
J, Ware
J, Hershberger
RE. An evidence-based assessment of genes in dilated cardiomyopathy.
Circulation.
2021;144:7-19.
[
PMC free article: PMC8247549] [
PubMed: 33947203]
Judge
DP. Use of genetics in the clinical evaluation of cardiomyopathy.
JAMA. 2009;302:2471-6.
[
PubMed: 19996403]
Liu
GS, Morales
A, Vafiadaki
E, Lam
CK, Cai
WF, Haghighi
K, Adly
G, Hershberger
RE, Kranias
EG. A novel human R25C-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia.
Cardiovasc Res.
2015;107:164-74.
[
PMC free article: PMC4490203] [
PubMed: 25852082]
Miura
A, Kondo
H, Yamamoto
T, Okumura
Y, Nishio
H. Sudden unexpected death of infantile dilated cardiomyopathy with JPH2 and PKD1 gene variants.
Int Heart J.
2020;61:1079-83.
[
PubMed: 32879264]
Morales
A, Kinnamon
DK, Jordan
E, Platt
J, Vatta
M, Dorschner
MO, Starkey
CA, Mead
JO, Ai
T, Burke
W, Gastier-Foster
J, Jarvik
GP, Rehm
HL, Nickerson
DA, Hershberger
RE, et al.
Variant interpretation for dilated cardiomyopathy: refinement of the American College of Medical Genetics and Genomics/ClinGen guidelines for the DCM Precision Medicine Study.
Circ Genom Precis Med.
2020;13:e002480.
[
PMC free article: PMC8070981] [
PubMed: 32160020]
Norton
N, Li
D, Rampersaud
E, Morales
A, Martin
ER, Zuchner
S, Guo
S, Gonzalez
M, Hedges
DJ, Robertson
PD, Krumm
N, Nickerson
DA, Hershberger
RE, et al.
Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy.
Circ Cardiovasc Genet.
2013;6:144-53.
[
PMC free article: PMC3815606] [
PubMed: 23418287]
Pugh
TJ, Kelly
MA, Gowrisankar
S, Hynes
E, Seidman
MA, Baxter
SM, Bowser
M, Harrison
B, Aaron
D, Mahanta
LM, Lakdawala
NK, McDermott
G, White
ET, Rehm
HL, Lebo
M, Funke
BH. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing.
Genet Med.
2014;16:601-8.
[
PubMed: 24503780]
Roberts
AM, Ware
JS, Herman
DS, Schafer
S, Baksi
J, Bick
AG, Buchan
RJ, Walsh
R, John
S, Wilkinson
S, Mazzarotto
F, Felkin
LE, Gong
S, MacArthur
JA, Cunningham
F, Flannick
J, Gabriel
SB, Altshuler
DM, Macdonald
PS, Heinig
M, Keogh
AM, Hayward
CS, Banner
NR, Pennell
DJ, O'Regan
DP, San
TR, de Marvao
A, Dawes
TJ, Gulati
A, Birks
EJ, Yacoub
MH, Radke
M, Gotthardt
M, Wilson
JG, O'Donnell
CJ, Prasad
SK, Barton
PJ, Fatkin
D, Hubner
N, Seidman
JG, Seidman
CE, Cook
SA. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease.
Sci Transl Med.
2015;7:270ra6.
[
PMC free article: PMC4560092] [
PubMed: 25589632]
Ross
SB, Jones
K, Blanch
B, Puranik
R, McGeechan
K, Barratt
A, Semsarian
C. A systematic review and meta-analysis of the prevalence of left ventricular non-compaction in adults.
Eur Heart J.
2020;41:1428-36.
[
PubMed: 31143950]
Ross
SB, Semsarian
C. Clinical and genetic complexities of left ventricular noncompaction: preventing overdiagnosis in a disease we do not understand.
JAMA Cardiol.
2018;3:1033-4.
[
PubMed: 30140926]
Sabater-Molina
M, Navarro
M, García-Molina Sáez
E, Garrido
I, Pascual-Figal
D, González Carrillo
J, Gimeno Blanes
JR. Mutation in JPH2 cause dilated cardiomyopathy.
Clin Genet.
2016;90:468-9.
[
PubMed: 27471098]
Sen-Chowdhry
S, Syrris
P, Prasad
SK, Hughes
SE, Merrifield
R, Ward
D, Pennell
DJ, McKenna
WJ. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity.
J Am Coll Cardiol.
2008;52:2175-87.
[
PubMed: 19095136]
Sivasankaran
S, Sharland
GK, Simpson
JM. Dilated cardiomyopathy presenting during fetal life.
Cardiol Young.
2005;15:409-16
[
PubMed: 16014190]