By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2003, BC Decker Inc.
Bookshelf ID: NBK12722

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Kufe DW, Pollock RE, Weichselbaum RR, et al., editors. Holland-Frei Cancer Medicine. 6th edition. Hamilton (ON): BC Decker; 2003.

Benign tumors of the lid, conjunctiva, iris, and choroid are common, whereas those of the retina and cornea are rare. Benign tumors of the lens and vitreous do not occur. The most important benign tumors of the eye are the choroidal nevi.
Choroidal nevi are never present at birth. Pigmentation that looks like a choroidal nevus in infancy is usually from choroidal neurofibroma, seen as a part of systemic neurofibromatosis. Nevi of the choroid can be seen before puberty but they are unusual. With puberty they become visible. In the United States, 10% to 13% of the adult population have choroidal nevi. They are racially related; choroidal nevi in blacks are very rare.
Choroidal nevi are flat, pigmented benign lesions with edges that can be feathered and irregular or rounded (Figure 85-6). They are usually slate-gray to light chocolate in color. With time (months to years), there may be associated findings with the nevi. Many demonstrate changes on their surface such as drusen or subretinal fluid and may cause overlying visual field defects and can even be associated with overlying neovascular membranes. Because 10% of the adult population has choroidal nevi and there are only 1,500 choroidal melanomas in the United States yearly, it is assumed that the chance of a choroidal nevus becoming a melanoma is less than 1 in 1,000. A number of studies have demonstrated which nevi are likely to develop into melanomas. Table 85-8 lists the predictive factors.

Choroidal nevus. (Four-color version of figure on CD-ROM)
Predictive Factors: Nevus to Melanoma Transformation.
Photocoagulation with lasers of the leaking areas over the nevi has been done. Although the fluid usually resorbs and leakage stops within days to weeks, it has been suggested that photocoagulation itself somehow weakens the layer between the retina and choroid (Bruch membrane) and in some way may stimulate transformation to malignancy.
A special type of nevus that is not flat has been described in the choroid. It is a melanocytoma, or pathologically, a magnocellular nevus with jet-black pigmentation. This consists of large cells in darker-skinned whites, who are frequently of Mediterranean origin. These lesions originate in cells within the optic nerve itself and may obscure a view of the nerve. The lesions may be several millimeters high, grow slowly, and can affect the visual field or visual acuity. Although these lesions are benign, rare cases of transformation to malignancy have been recorded. Even less common are melanocytomas originating in the choroid, ciliary body, or iris.
The differential diagnosis of a nevus is straightforward. Other flat lesions in the choroid that have been confused with nevi and melanoma include hyperplasia of the retinal pigment epithelium, hamartomas of the retinal pigment epithelium, and hemorrhages within the retina, especially hemorrhages beneath the retinal pigment epithelium (as part of macular degeneration). Hyperplasia of the retinal pigment epithelium is a flat, dark, round, circular lesion with sharp edges and bare spots devoid of pigment. Distinguishing these lesions from melanoma is assisted with fluorescein patterns and ultrasonography, as well as judgment about size and thickness.
Iris nevi are, by definition, pigmented and flat. They are common, may be multiple, and occur more often in blue-eyed patients. Iris nevi are also rarely present at birth and like all other ocular nevi become apparent around puberty. They are always benign and of no real consequence for the eye or for life. Confusion exists, however, in the use of the term iris melanoma. Ophthalmologists have traditionally described elevated pigmented lesions of the iris as iris melanomas to differentiate them from the flat nevi. This has been further confused by pathologic interpretation of elevated pigmented iris masses that were excised (or enucleated) which were described as malignant. Elevated iris pigmented masses may grow and shed cells into the angle, clogging the trabecular meshwork and causing a severe secondary glaucoma that can blind the eye, but iris melanomas metastasize extremely rarely, if ever. Lack of metastasis may be a function of size. An iris melanoma that filled the anterior chamber would, if in the back of the eye (in the choroid), be small enough never to cause metastasis. Management of iris melanoma is based on the presence or absence of glaucoma, and is never guided by a need to prevent metastasis. Ciliary body melanomas that present as iris lesions do metastasize, however.
The most common primary ocular malignant tumor in adults is malignant melanoma, previously referred to as melanosarcoma of the choroid.80 There are approximately 1,500 new cases per year of choroidal malignant melanoma in the United States. The average age at diagnosis is 55 to 65 years, with men and women equally affected and the two eyes equally susceptible. In the United States, 99% of choroidal melanomas occur in whites. The most common way in which the tumor is detected is on routine exam (41%).81 Men more often present with symptoms and when there are symptoms the right eye is more often found to have the tumor. The most common symptom is a decrease in the peripheral visual field followed by decreased vision. The lesion is not painful, unlike metastatic tumors to the eye in which pain is not unusual.
The visual field defect is characteristic. There is an absolute scotoma overlying the tumor associated with a surrounding relative field defect that does not obey the horizontal meridian (as most ocular defects do) and does not observe the vertical meridian (as many CNS defects do).82
Melanomas of the choroid originate in melanocytes that normally lie within the choroid. The choroid, the layer between the sclera and retina is a rich, high-flow syncytium of vascular lobules that not only supply blood to the photoreceptors (rods and cones) of the retina but also serve as a heat sink to dissipate heat energy liberated by absorbed visible light.
Whether melanomas of the choroid originate only in nevi is not known, but patients with flat pigmented, untreated nevi followed for more than 20 years have developed melanomas arising from the previously dormant lesion. The cause of choroidal melanomas is unknown but predisposing medical conditions include melanosis oculi (nevus of Ota), dysplastic nevus syndrome, and possibly human immunodeficiency virus (HIV), pregnancy/estrogen replacement therapy, and levodopa therapy for Parkinson disease. Occupational associations include agriculture and farming work and several industrial operations.83 We have also seen an increased risk in World War II holocaust survivors.
The diagnosis of choroidal melanoma can usually be made on ophthalmoscopic grounds alone. With the direct ophthalmoscope it may be difficult to appreciate the three-dimensional shape, but with dilated pupils and the indirect ophthalmoscope, the tumor is easily identified. The tumor can have many shapes. A flat, diffuse type may be difficult to detect ophthalmoscopically, but most tumors are elevated, frequently dome shaped (the height of the tumor being half the base diameter), and occasionally multilobed. The tumor is held back by Bruch's membrane but when it ruptures this taut, transparent layer, it develops a rounded top on the surface of a dome-shaped mass. These tumors are referred to as mushroom shaped or collar button.
The diagnosis of melanoma is also aided by fundus photography (Figure 85-7) and sometimes fluorescein angiography. Of great value in confirming the diagnosis is ocular ultrasonography. Typically, the B-scan ultrasonogram demonstrates an elevated solid tumor84; the A-scan ultrasonogram demonstrates medium to low reflectivity.85 With the use of rigorous and standardized ophthalmic and systemic examinations, a diagnostic accuracy of 99.7% was reached in the Collaborative Ocular Melanoma Study (COMS).86 Because clinical accuracy is so high, needle biopsy is rarely needed.
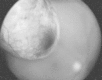
Malignant melanoma of the choroid. (Four-color version of figure on CD-ROM)
Pigmentation of choroidal melanomas varies from patient to patient, and frequently from area to area within the tumor. As many as 40% of the tumors have no pigment clinically. When pigmented, the tumors are frequently a dusky gray to charcoal in color, but occasionally they are deep brown. Black lesions within the eye are rarely melanomas.
All choroidal melanomas have associated retinal detachments. In some cases, it may be difficult to detect the detachments ophthalmoscopically, whereas in others, the retinal detachment may be so extensive that the melanoma is not seen clinically.
Because there are no lymphatics in the eye or within the orbit, melanomas of the choroid metastasize through vascular channels. More than 75% of such metastases are first identified within the liver. Fewer than 1% of patients with metastasis survive 5 years.
A number of clinical and pathologic features correlate with patient survival.87 First, larger tumors, measured clinically using the height, and/or greatest base diameter to volume, are more likely to metastasize. Second, location of the melanoma affects prognosis. Patients with melanomas in the iris have the best outcome. Ciliary body melanomas have a threefold mortality as compared to choroidal melanomas. Third, patients younger than age 60 years have better survival than those older than age 60 years. Fourth, patients with extraocular extension have higher mortality rates than those who do not. The greater the amount of local extension into the orbit, the poorer is the prognosis. Finally, many pathologic features (only available in cases where the eye has been removed) correlate with survival and are discussed extensively elsewhere.88 The best known of these is cell type. Ocular melanomas that contain epithelioid cells, which are larger and more pleomorphic cell types, are more likely to be contained in large tumors, which carry a worse prognosis.
The COMS Group, which included 44 institutions across the United States, has now completed multiple prospective, randomized clinical trials and other reports on the prognosis and treatment of small, medium, and large choroidal melanomas. Table 85-9 shows the COMS size classification scheme and Table 85-10 details the results of the studies.
Classification of Choroidal Melanomas.
Results of the Collaborative Ocular Melanoma Study (COMS).
The COMS demonstrated that of choroidal melanomas initially managed by observation, 21% demonstrated growth to medium or large tumors by 2 years and 31% by 5 years.89 Factors significantly associated with growth were greater initial tumor thickness and diameter, presence of orange pigment, absence of drusen, and absence of areas of retinal pigment epithelial changes adjacent to the tumor. Overall, patients with small melanomas were shown to have a 5-year survival rate of 96%.90 Clinically, most patients with small melanomas are observed until tumor growth is documented by fundus photography or ultrasonography.
In the past, patients with tumors in this group were treated with local excision, enucleation, external beam radiation, proton-beam therapy, or brachytherapy. The COMS prospective, randomized clinical trial enrolling 1,317 patients with medium-sized melanomas was recently published.91 Patients were randomly assigned to enucleation or brachytherapy with 125I plaques at a tumor dose of 10,000 cGy. The study concluded mortality rates following brachytherapy (81% 5-year survival) did not differ from mortality rates following enucleation (82% 5-year survival) for up to 12 years after treatment. Given the findings of this study, patients with medium-sized melanomas are now offered both enucleation and brachytherapy as potential treatment options. Brachytherapy with 125I plaques is usually administered at doses of 7,500 to 10,000 cGy. The fractionation schemes vary markedly without an apparent effect on local control, metastasis or complications. Complications include radiation retinopathy and optic neuropathy.92
In the past, patients with large tumors were generally treated with enucleation, with or without preoperative radiation. The COMS prospective, randomized clinical trial for patients with large tumors found that there was no survival difference between patients treated with enucleation alone (5-year survival 57%) and patients treated with preenucleation radiation (5-year survival 62%).93 The patients assigned to the preenucleation radiation group in this study were treated with 5 fractions of 200-cGy external beam radiation and enucleation within 72 h. Patients with large melanomas are now generally treated with enucleation alone.
The most common malignant neoplasm in the eye or orbit, in children or adults, is metastatic carcinoma to the choroid. Although there are only 350 cases of retinoblastoma and 1,500 cases of choroidal melanoma yearly in the United States, it is estimated that 30,000 to 100,000 patients with cancer develop metastases to the eye each year.94
Metastasis to the eye most commonly occurs in adults aged 55 to 65 years, the same age distribution as that for ocular melanomas. Metastases in males most commonly originate from a primary lung cancer whereas metastases in females most commonly arise from a primary breast cancer. In the past, ocular metastases in men often presented as the first sign of disease, whereas in women, the primary tumor and even other metastases were typically already detected. As lung cancer deaths in women are now more common than deaths from breast cancer, it is anticipated that the patterns of presentation in women will become more similar to those in men. Many other cancers metastasize to the eye, including gastrointestinal, prostate (although more commonly to orbital bones), thyroid, ovarian, cutaneous melanoma, and sweat gland. Virtually all cancers are capable of metastasizing to the eye.
Most cancers metastasize to the uveal tract, but metastasis to the lids, conjunctiva, optic nerve, orbit, extraocular muscles, and orbital bones are also reported. Metastasis to the retina is rare. Although metastases to the iris and ciliary body are not unusual, metastases to the choroid are the most common.
Choroidal metastases are usually amelanotic, multiple, bilateral, minimally elevated, and painful when situated around the optic nerve or invading the sclera. In contrast, ocular melanomas are typically pigmented, solitary, unilateral, significantly elevated, and painless. Metastatic tumors, like ocular melanomas, always have an associated serous detachment, but the amount of detachment is proportionally greater with metastases. Ultrasonographically, most metastases have high reflectivity on ultrasonogram, in contrast to melanomas, which usually have low to medium reflectivity. Most ocular melanomas are detected on routine exams, whereas ocular metastases are typically identified because of symptoms of decreased visual field and diminished visual acuity caused by serous retinal detachments. Metastases are sometimes detected on routine exams in asymptomatic patients.
The most striking feature of metastatic ocular lesions is their association with concurrent CNS metastases. Although the true concordance of these two lesions is unknown, in our experience more than 75% of cases of ocular metastasis have concurrent CNS disease, although frequently the CNS disease is initially undetectable with imaging. Consequently, it has been speculated that some ocular metastases do not arrive through blood-borne routes, but that CNS metastases may actually seed the choroid via the subarachnoid space as they do in childhood leukemia.
Treatment for ocular metastasis is considered when symptoms of diminished vision, pain, or diplopia are present. Treating the ocular lesion rarely has an impact on survival, except in carcinoid metastases, but may significantly alter the quality of life. Many ocular metastases respond to chemotherapy the way other systemic metastases respond. Chemotherapy and/or hormonal manipulation may cause rapid regression of the tumor and of subretinal fluid. Patients with undiagnosed ocular metastases occasionally demonstrate coarse brown pigmentation on the surface of a regressed choroidal metastasis following successful chemotherapy. External beam irradiation is also used to palliate symptoms from ocular metastases. Choroidal metastases are treated with lateral photon fields, similar to the technique used for bilateral retinoblastoma described above, to a dose of 3,000 cGy in 10 fractions over 2 weeks, sparing the lens and anterior segment. Complications other than transient skin erythema are rare. When metastases involve the anterior segment, anterior irradiation is necessary and can cause dry eye, cataract, and red, uncomfortable eyes. Except for carcinoid and breast cancer, however, median survival in patients with metastatic choroidal lesions is just over 6 months.
Paraneoplastic visual loss is an autoimmune disorder characterized by visual loss in patients with cancer. It has been shown that autoantibodies cross-react with retinal cell antigens giving rise to loss of vision and optic atrophy. The most frequently described retinal autoantigen in cancer-associated retinopathies (CAR) is the 23-kDa antigen recoverin, a photoreceptor and bipolar cell-specific calcium-binding protein.95 Lung, endometrial sarcoma, lymphoma, and prostate cancer are associated with the syndrome. Steroids have been tried without much success. Plasmapheresis and intravenous immunoglobulin have been reported to work in a few patients.96
Intraocular lymphomas are increasing in incidence because of their association with HIV/AIDS. Both benign and malignant types occur. Benign reactive hyperplasia may diffusely involve the uvea and be difficult to diagnosis without biopsy. It is not associated with systemic findings.
Primary malignant lymphomas of the eye, also called reticulum cell sarcoma or microgliomatosis, usually present with cells in the vitreous and may have associated retinal and optic nerve involvement. These patients frequently have CNS disease, but rarely have systemic disease. Diagnosis is made by vitrectomy or spinal tap. Treatment is accomplished with systemic steroids and radiation therapy of 2,400 cGy to the affected eye and/or chemotherapy as if for intermediate-grade systemic lymphoma. There is controversy about whether to treat the brain in these cases when diagnostic spinal tap and MRI studies demonstrate no disease. Eventual CNS involvement is very common, as is bilateral ocular disease. Median survival is 3.5 years and is usually determined by the extent of brain involvement. Cure is rare.
Malignant lymphomas of the uvea present as diffuse uveal involvement and are usually associated with systemic disease with involvement of lymph nodes and viscera, but rarely with involvement of the CNS.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...