Summary
The goals of this overview on heritable thoracic aortic aneurysms and dissections (shortened in this GeneReview to heritable thoracic aortic disease [HTAD]) are the following.
Goal 2.
Review the causes of HTAD.
Goal 3.
Provide a strategy for evaluation and genetic risk assessment for HTAD in a proband.
Goal 4.
Review management recommendations for individuals with HTAD.
Goal 5.
Inform genetic counseling and risk assessment in family members of a proband with HTAD.
1. Clinical Characteristics of Thoracic Aortic Disease
Thoracic aortic disease refers to thoracic aortic aneurysms and aortic dissections for the purpose of this GeneReview.
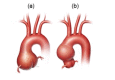
A thoracic aortic aneurysm is a permanent, localized enlargement of the thoracic aorta. Thoracic aortic aneurysms may involve different thoracic aortic segments; this review focuses on aneurysms involving the aortic root and/or ascending aorta (see ).
Thoracic aortic aneurysms involving the aortic root (a) and the ascending aorta (b) Printed with permission from Baylor College of Medicine, Copyright 2016
To evaluate for a thoracic aortic aneurysm, the aortic diameter is measured (perpendicular to the axis of blood flow) by echocardiography, CT, or MRI at reproducible anatomic locations. Measurements of aortic diameters obtained from transthoracic echocardiography tend to be smaller than measurements obtained from CT or MRI [Asch et al 2016]. The echocardiography convention for assessment of aortic root and ascending aortic diameters has been to measure leading edge to leading edge in end-diastole [Evangelista et al 2010]. Recent data indicate that measurements (using this convention) assessed by two-dimensional transthoracic echocardiography accurately correlate with internal diameters assessed by multidetector CT or MRI [Rodríguez-Palomares et al 2016].
Nomograms and z scores based on reference values for aortic root and ascending aortic diameters that account for biologic sex normalized to body surface area (aortic size index) are commonly used in the pediatric population to interpret clinically significant aortic dilatation in a growing child, but can also be applied to adults [Devereux et al 2012, Campens et al 2014]. Caution should be used when interpreting absolute aortic diameters in individuals with body size that differs significantly from the population mean. In these instances, normalizing aortic diameters to body surface area (aortic size index) and/or height may be more appropriate [Davies et al 2006, Zafar et al 2018].
An aortic dissection is defined as a tear in the intimal (innermost) layer of the aorta; blood flows from the lumen of the aorta and enters the inner wall separating the intima and media layers of the aortic wall.
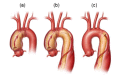
Aortic dissections are most commonly classified using Stanford and Debakey criteria. The Stanford system categorizes dissections based on the involvement of the ascending aorta. A newer classification system was proposed in 2020 to define aortic dissections in greater detail based on the location of entry tear and extent of dissection [Isselbacher et al 2022].
Stanford type A dissections (, ) involve the ascending aorta and typically originate at the junction between the root and ascending aorta. Type A dissections may or may not extend into the arch and descending thoracic aorta. In type A dissections blood within the aortic wall can either flow distally, up the ascending aorta (anterograde dissection), or back toward the aortic root (retrograde dissection).
Stanford type B dissections () do not involve the ascending aorta. Most often tears originate at the beginning of the descending thoracic aorta, just distal to the left subclavian artery, and propagate variable distances to the descending thoracic and abdominal aorta.
Thoracic aortic dissections: Type A (a and b) and Type B (c) Printed with permission from Baylor College of Medicine, Copyright 2016
Natural history
2. Causes of Heritable Thoracic Aortic Disease
A diagnosis of heritable thoracic aortic disease (HTAD) can be established in an individual with any of the following: (1) a highly penetrant pathogenic variant(s) in a known HTAD gene (see Table 1); (2) thoracic aortic disease and one or more additional family member(s) with thoracic aortic disease; or (3) thoracic aortic disease with specific clinical features of a syndrome associated with thoracic aortic disease (e.g., Marfan syndrome, Loeys-Dietz syndrome).
A genetic cause of HTAD is identified in the majority of individuals with a clinical diagnosis of Marfan syndrome or Loeys-Dietz syndrome. However, a genetic cause of HTAD is identified in only approximately 20%-30% of individuals with a family history of thoracic aortic disease who do not have syndromic manifestations [Robertson et al 2016, Hicks et al 2018, Raunsø et al 2020].
Known causes of HTAD. The Clinical Genome Resource (ClinGen) HTAD Gene Curation Expert Panel identified 11 genes with a definitive or strong HTAD association (see Table 1) [Renard et al 2018]. Additional genes were determined to have moderate or limited HTAD association (see Table 1), or uncertain associations with HTAD (see Table 2); classification may change as new evidence emerges. Variable expression is common with respect to age of disease onset and presentation (e.g., type A vs type B dissection, aneurysms confined to aortic root vs involvement of ascending aorta). Penetrance of thoracic aortic disease is reduced, particularly in women. In addition, other cardiovascular manifestations have been reported in individuals and families with various genetic causes of HTAD (see Table 1).
Table 1.
Genes Associated with Heritable Thoracic Aortic Disease
View in own window
| Gene 1 | MOI | ClinGen Classification 2 | Phenotype(s) | Other Cardiovascular Features |
|---|
|
ACTA2
| AD | Definitive | Nonsyndromic HTAD 3 Smooth muscle dysfunction syndrome (OMIM 613834)
| PDA, moyamoya-like cerebrovascular disease, early-onset coronary artery disease |
|
COL3A1
| AD | Definitive |
Vascular Ehlers-Danlos syndrome
| Dissection, rupture, & aneurysms throughout arterial tree |
|
FBN1
| AD | Definitive |
| Mitral valve disease, arterial tortuosity |
|
MYH11
| AD | Definitive | Nonsyndromic HTAD 3 | PDA 4 |
|
MYLK
| AD | Definitive | Nonsyndromic HTAD 3 | |
SMAD3
TGFB2
TGFBR1
TGFBR2
| AD | Definitive |
| Aneurysms & dissections throughout arterial tree, cardiac valve disease, arterial tortuosity |
|
LOX
| AD | Strong | Nonsyndromic HTAD 3 | Aneurysms & dissections reported in other arteries 5 |
|
PRKG1
| AD | Strong | Nonsyndromic HTAD 3 | Coronary artery aneurysm & dissection reported 6 |
|
EFEMP2
| AR | Moderate |
EFEMP2-related cutis laxa
| Arterial tortuosity, stenosis of pulmonary arteries & aortic isthmus |
|
FOXE3
| AD | Moderate | Nonsyndromic HTAD 7 | |
|
MFAP5
| AD | Moderate | Undefined 7 | Atrial fibrillation, mitral valve prolapse |
|
SMAD2
| AD | Moderate |
| Aneurysms & dissections throughout arterial tree, cardiac valve disease, arterial tortuosity |
|
BGN
| XL | Limited | Meester-Loeys syndrome (OMIM 300989) | Cardiac valve disease |
|
CBS
| AR | Limited |
Homocystinuria
| Thromboembolism; diagnosis primarily based on nonvascular features |
|
COL4A5
| XL | Limited |
Alport syndrome
| Abdominal aortic aneurysms; diagnosis primarily based on nonvascular features |
|
ELN
| AD | Limited |
ELN-related cutis laxa
| |
|
FBN2
| AD | Limited |
Congenital contractural arachnodactyly
| |
|
FLNA
| XL | Limited |
FLNA deficiency
9
| PDA, other congenital cardiac defects, thoracic aortic disease |
|
HCN4
| AD | Limited | Sick sinus syndrome (OMIM 163800) | Arrhythmia, cardiomyopathy |
|
NOTCH1
| AD | Limited | Aortic valve disease (OMIM 109730) | BAV |
|
MAT2A
| AD | Limited | Nonsyndromic HTAD 4 | BAV 4 |
PKD1
PKD2
| AD | Limited |
Polycystic kidney disease
| Intracranial aneurysms, thoracic aortic disease; diagnosis primarily based on nonvascular features |
|
SKI
| AD | Limited |
Shprintzen-Goldberg syndrome
| Mitral valve disease |
|
SLC2A10
| AR | Limited |
Arterial tortuosity syndrome
| Arterial tortuosity, stenosis of pulmonary artery, aorta, & other arteries |
|
SMAD4
| AD | Limited |
Juvenile polyposis syndrome
| Arteriovenous malformations, thoracic aortic disease, mitral valve disease |
|
TGFB3
| AD | Limited |
| Aneurysms & dissections throughout arterial tree |
BAV = bicuspid aortic valve; HTAD = heritable thoracic aortic disease; PDA = patent ductus arteriosus
- 1.
Genes are ordered first by strength of gene-disease validity classification and then alphabetically.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
FLNA was not classified as definitively associated with heritable thoracic aortic disease by Renard et al [2018] because FLNA pathogenic variants had not been identified in individuals with aortic dissection/rupture. In a more recent study, more than 20 individuals with FLNA pathogenic variants had thoracic aortic aneurysms and two had aortic rupture [Chen et al 2018].
Table 2.
Other Genes in which Thoracic Aortic Disease Is Reported
View in own window
| Gene 1 | MOI | Classification 2 | Phenotype(s) | Cardiovascular Features |
|---|
|
ARIH1
| AD | Not curated | Nonsyndromic HTAD 3 | |
|
LTBP3
| AR | Not curated | Dental anomalies & short stature (OMIM 601216) | Mitral valve disease, aneurysms throughout arterial tree |
|
THSD4
| AD | Not curated | Familial thoracic aortic aneurysm 12 (OMIM 619825) Nonsyndromic HTAD 4
| Dilatation of aortic root, ascending aorta, abdominal aorta, aortic root aneurysm & dissection |
BAV = bicuspid aortic valve; HTAD = heritable thoracic aortic disease; PDA = patent ductus arteriosus
- 1.
Genes are ordered first by validity classification and then alphabetically.
- 2.
Gene classification regarding association with heritable thoracic aortic disease is primarily from Renard et al [2018] and ClinGen. More recent information supporting classification is available for some genes as noted.
- 3.
- 4.
3. Evaluation and Genetic Risk Assessment for Heritable Thoracic Aortic Disease in a Proband
Risk assessment for heritable thoracic aortic disease (HTAD) should incorporate data from the proband's medical history, physical examination, family history, and aortic and arterial imaging studies. Molecular genetic testing is recommended in all individuals with suspected HTAD.
Medical history should include assessment of additional cardiovascular and thoracic aortic disease including poorly controlled hypertension, hyperlipidemia, sleep apnea, excessive isometric exercise, and smoking.
Physical examination should be directed at identifying syndromic features observed in a subset of individuals with HTAD with specific HTAD-related syndromes (e.g., Loeys-Dietz syndrome, Marfan syndrome, vascular Ehlers-Danlos syndrome).
Family history should include collection and interpretation of a three-generation family history with attention to aortic or other cardiovascular disease including sudden cardiac death in relatives, and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing, cardiovascular and physical examinations, and postmortem examination including aortic histopathology.
Aortic and arterial imaging studies (echocardiogram, CT, MRI) can provide information on aneurysm morphology, growth rate, and/or findings of extra-aortic aneurysms or occlusive disease to inform the likelihood of an underlying genetic cause and/or risk of aortic dissection (particularly in those with uninformative molecular genetic testing).
Molecular Genetic Testing
Establishing a molecular genetic cause of HTAD is useful for tailoring management recommendations based on the causative gene, and to identify and counsel at-risk relatives. When an HTAD-related pathogenic variant(s) is not identified, clinical and family history information also inform risk of aortic dissection and can be used to guide medical and surgical management (see Management).
The likelihood of identifying the genetic cause of HTAD in an individual with thoracic aortic disease varies based on clinical presentation and family history. Clinical and family history findings that increase the likelihood of identifying a pathogenic variant in an HTAD-related gene include:
Thoracic aortic aneurysm or dissection at age <60 years;
Family history of thoracic aortic disease, unexplained sudden death, or aneurysms/dissections in other arteries;
Aneurysms and dissections/ruptures of other arteries in an individual with thoracic aortic disease.
Note: Although these clinical and family history findings are useful for identifying individuals most likely to have a molecular cause identified, individuals without these findings can still benefit from molecular genetic testing and risk assessment [Cecchi et al 2022].
A multigene panel that includes genes associated with HTAD is recommended (see Table 1). The panel should include genes with a definitive or strong HTAD association; a panel that also includes genes with moderate or limited association with HTAD and/or emerging data regarding association with HTAD may be considered to increase the likelihood of identifying the genetic cause. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive
genomic testing (exome sequencing, genome sequencing) may be considered when evaluating an individual with thoracic aortic disease and clinical manifestations that overlap with other syndromes in which thoracic aortic disease is a rare manifestation (e.g., Noonan syndrome, neurofibromatosis type 1, Alagille syndrome). Genome sequencing may be useful to identify deep intronic variants not detected on a multigene panel. However, exome or genome sequencing are unlikely to yield a higher rate of clinically actionable findings in most individuals with thoracic aortic disease.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Karyotype or chromosomal microarray. In individuals with thoracic aortic aneurysm, aortic dissection, and/or bicuspid aortic valve and clinical features of Turner syndrome (short stature, ovarian insufficiency), a peripheral blood karyotype should be performed to identify an X chromosome deletion. FISH analysis or testing of additional tissue may be needed in those with suspected low-level mosaicism [Gravholt et al 2017].
4. Management
This section provides information regarding recommendations for imaging surveillance, medical management, and surgical management in individuals with heritable thoracic aortic disease (HTAD) and imaging recommendations for family members of individuals with thoracic aortic disease of unknown genetic cause. Management should be based on the genetic cause (when possible) or based on the clinical and family history when a molecular genetic diagnosis cannot be established [Isselbacher et al 2022]. Early detection of asymptomatic aneurysms through imaging surveillance allows for monitoring of aortic growth, pharmacotherapy to slow aortic root growth, and timely prophylactic aneurysm repair, which reduces the high morbidity and mortality associated with thoracic aortic dissections.
Imaging Surveillance
Thoracic Aortic Imaging Recommended for Individuals with HTAD
A baseline echocardiogram should be performed to assess aortic diameters (i.e., the aortic root and ascending aorta) and evaluate aortic valve anatomy and function. If the entire ascending aorta cannot be visualized on echocardiogram, CT or MRI may be indicated. Note that echocardiography may be needed to visualize the aortic root and cardiac valve structure and function unless a gated CT/MRI is performed.
Individuals diagnosed with a dilated aorta should undergo repeat imaging via echocardiogram, CT, or MRI (depending on baseline findings and aortic anatomy) in six to 12 months to assess the rate of aortic growth.
If the aortic diameter is stable on repeat imaging, continue imaging surveillance every six to 24 months based on aortic diameter and other dissection risk factors.
If the rate of increase in aortic diameter exceeds 0.5 cm per year, more frequent imaging should be considered. The frequency of imaging may be modified based on the individual's age and family history [
Isselbacher et al 2022].
Imaging of Other Arteries in Individuals with HTAD
The risk of aneurysms in arteries other than the aorta and/or other cardiovascular disease is variable and depends on the underlying cause of HTAD.
Imaging to assess for aneurysms in other arteries, moyamoya-like cerebrovascular disease, coronary artery disease, and other cardiovascular manifestations should be guided by family history and molecular genetic cause of HTAD, if established.
Imaging Recommendations for Family Members of Individuals with HTAD of Unknown Genetic Cause
When the genetic cause of HTAD in a family is unknown, thoracic aortic imaging is recommended for first-degree relatives (i.e., parents, sibs, offspring) of all individuals with thoracic aortic disease. This recommendation applies to all at-risk relatives regardless of the proband's age of thoracic aortic disease diagnosis and presentation.
Screening via echocardiogram is recommended if the aortic root and ascending aorta are adequately visualized; otherwise, a CT or MRI may be indicated.
Additional head and neck imaging may be indicated if there is also a family history of aneurysms/dissections in the intracranial and cervical arteries.
Note: Family members with HTAD-related pathogenic variant(s) have, by definition, HTAD, and implementation of gene-based management is recommended.
Pharmacotherapy
Beta-adrenergic blocking agents (beta-blockers) have been shown to slow the rate of aortic root growth in individuals with Marfan syndrome by reducing hemodynamic stress on the aortic wall [Ladouceur et al 2007, Forteza et al 2016]. Angiotensin receptor blockers have also been shown to slow aortic root dilation in children with Marfan syndrome [Lacro et al 2014]. Based on these data and studies investigating the effect of combination therapy (beta-blocker with angiotensin receptor blocker), American Heart Association (AHA) guidelines recommend treatment with a beta-blocker, angiotensin receptor blocker, or both to reduce the rate of aortic dilation. Randomized trials have not been conducted in individuals with Loeys-Dietz syndrome, but based on data from mouse models and individuals with Marfan syndrome, AHA guidelines note that treatment with a beta-blocker, angiotensin receptor blocker, or both is also reasonable [Gallo et al 2014, Forteza et al 2016, Isselbacher et al 2022].
Studies investigating the use of celiprolol, a beta-blocker with vasodilatory properties, in individuals with vascular Ehlers-Danlos syndrome have shown benefit [Ong et al 2010, Frank et al 2019]. However, the efficacy of celiprolol versus other more commonly prescribed beta-blockers has not been investigated. Use of angiotensin receptor blockers has not been investigated in individuals with vascular Ehlers-Danlos syndrome.
The efficacy of beta-blockers and angiotensin receptor blockers on slowing progressive aortic dilation has not been investigated in other known molecular causes of HTAD, but expert consensus generally supports the use of beta-blockers in individuals with other molecular causes of HTAD or no known molecular cause of HTAD [Isselbacher et al 2022].
Risk Factor Modification
Hypertension should be aggressively treated and controlled in all individuals with thoracic aortic disease (regardless of cause) and in at-risk family members (including those without aortic aneurysm or dissection). AHA treatment guidelines recommend beta-blockers as a first-line therapy to achieve optimal blood pressure in individuals with thoracic aortic disease, with adjunct use of an angiotensin receptor blocker if needed [Isselbacher et al 2022].
Exercise and physical activity. Data on the safety of specific types of exercise and physical activity in individuals with thoracic aortic disease is limited, but it is recommended that individuals with thoracic aortic disease avoid high-intensity isometric exercise (e.g., heavy weightlifting, activities requiring the Valsalva maneuver) and contact sports [Thijssen et al 2019, Isselbacher et al 2022]. See physical activity recommendations for Marfan syndrome, Loeys-Dietz syndrome, and vascular Ehlers-Danlos syndrome.
Other risk factors. Counseling on the contribution of other cardiovascular risk factors to thoracic aortic disease, including smoking and hyperlipidemia, should be provided. Approaches to mitigate associated risk should be discussed.
Surgical Management of Aneurysms of the Aortic Root and Ascending Aorta
Although pharmacotherapy and risk factor modification can slow progressive dilation of the aortic root and/or ascending aorta, the primary treatment to prevent premature death due to type A dissection is prophylactic surgical repair/replacement.
2022 AHA treatment guidelines proposed thresholds for prophylactic aortic root and ascending aortic repairs based on the causative HTAD gene. It is important to note that the presence of additional risk factors and family history should be considered when determining the timing of surgical repair; these are detailed in the AHA treatment guidelines [Isselbacher et al 2022] (see Table 3). Surgical thresholds refer to the maximum diameter of either the aortic root or ascending aorta.
Aneurysms in individuals with a pathogenic variant in certain genes (FBN1, SMAD3, TGFBR1, TGFBR2, TGFB2) almost always involve the aortic root and may also involve the ascending aorta; therefore, prophylactic replacement of both the aortic root and ascending aorta is recommended.
Table 3.
Surgical Thresholds for Prophylactic Replacement of the Aortic Root and Ascending Aorta by HTAD Gene
View in own window
| Gene | Diameter of Aortic Root or Ascending Aorta |
|---|
|
ACTA2
| ≥4.2 to ≥4.5 cm 1 |
|
FBN1
| ≥4.5 to ≥5.0 cm 1 |
|
PRKG1
| Normal aortic diameter to ≥4.2 cm 1 |
|
SMAD3
| ≥4.5 cm |
|
TGFB2
| ≥4.5 cm |
|
TGFB3
| ≥5.0 cm |
|
TGFBR1
| ≥4.0 to ≥4.5 cm 1 |
|
TGFBR2
| ≥4.0 to ≥4.5 cm 1 |
LOX
MYH11
MYLK
| There is limited data to establish surgical thresholds; however, type A dissections may present at aortic diameters <5.0 cm & differ by gene. Timing of aortic repair should account for aortic diameter, age, family history, other clinical risk factors, & studies reporting risk of dissection & aortic diameters at the time of dissection or repair in individuals w/pathogenic variant(s) in a specific HTAD-related gene. |
- 1.
Depending on presence of other risk factors and surgical expertise
In individuals with HTAD of unknown cause. Factors that increase the risk of aortic dissection in individuals with aneurysms of the aortic root or ascending aorta include: (1) family history of aortic dissection; (2) family history of sudden unexplained death at younger ages (usually <60 years); and (3) rapid aortic growth (≥0.5 cm in one year or ≥0.3 cm/year for two consecutive years) [Isselbacher et al 2022].
Timing of prophylactic surgical repair should be based on aortic diameter at the time of dissection or age at aneurysm repair in affected family members.
If aortic diameters are not known for affected family members and the affected individual does not have other dissection risk factors, prophylactic repair is recommended when the maximal aortic diameter reaches ≥5.0 cm.
In individuals with other dissection risk factors, prophylactic repair is reasonable when the maximal aortic diameter reaches ≥4.5 cm if performed by an experienced surgeon who is part of a multidisciplinary aortic team.
Pregnancy Management
The risk of peripartum aortic dissection and other pregnancy-related complications in individuals with HTAD varies based on the cause of HTAD, medical history, and family history. The majority of peripartum dissections occur in the third trimester and up to 12 weeks postpartum.
In the preconception period, individuals with HTAD should be counseled on recurrence risk (see Genetic Risk Assessment in Family Members of a Proband), molecular genetic testing options (see Molecular Genetic Testing), and pregnancy-related complications of HTAD. Prior to conception, thoracic aortic imaging by echocardiogram and/or MRI/CT is recommended for individuals with HTAD to evaluate aortic diameters and assess dissection risk. The recommended surgical threshold for prophylactic aortic repair prior to conception in individuals with HTAD-related pathogenic variant FBN1 is before the aortic root or ascending diameter exceeds 4.5 cm. However, surgical repair prior to pregnancy may be considered when the aortic diameter is 4.0-4.5 cm in individuals with other dissection risk factors (rapid aortic growth, family history of HTAD). Surgical repair before pregnancy may be considered when the aortic diameter reaches 4.0 cm for individuals with pathogenic variants in TGFBR1, TGFBR2, or SMAD3. In individuals with pathogenic variants in other HTAD-associated genes or in individuals with no known genetic cause, surgery is recommended when the aortic diameter reaches 4.5 cm, unless other risk factors are present.
Pregnant individuals with HTAD and those at risk for HTAD should be managed by a multidisciplinary team including a cardiologist and maternal-fetal medicine specialist [Isselbacher et al 2022]. During pregnancy, aortic imaging is recommended for individuals with aortic dilatation in each trimester and in the postpartum period. Echocardiogram should be used to evaluate the aortic root and ascending aorta or MRI (without contrast) if other portions of the aorta need to be imaged (arch, descending, abdominal aorta) based on indication [Isselbacher et al 2022].
In some instances, cesarean delivery may be recommended over vaginal delivery, but studies investigating indications and outcomes are limited. Factors that influence the method of delivery include aortic diameter and prior history of aortic dissection (e.g., chronic dissection or portion of aorta with residual dissection). In general, vaginal delivery is acceptable for individuals with HTAD who have an aortic diameter <4.0 cm. Cesarean delivery can be considered for individuals with aortic diameters between 4.0 cm and 4.5 cm and is recommended when the aortic diameter reaches 4.5 cm. Cesarean delivery is recommended when there is a history of aortic dissection (residually dissected aorta).
After delivery, aortic imaging by echocardiogram and/or MRI/CT is recommended.
See MotherToBaby for information on medication use during pregnancy.
5. Genetic Risk Assessment and Cascade Testing in Family Members of a Proband
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Genetic counseling regarding risk to family members of a proband with heritable thoracic aortic disease (HTAD) depends on accurate molecular diagnosis (i.e., identification of the HTAD-causing pathogenic variant[s] in an affected family member) and confirmation of the mode of inheritance in each family. When a molecular diagnosis is established in a proband, genetic counseling should be tailored to the molecular diagnosis and family history.
HTAD is typically inherited in an autosomal dominant manner. Less commonly, HTAD is inherited in an X-linked (e.g., FLNA deficiency) or autosomal recessive (e.g., LTBP3-related HTAD) manner (see Tables 1 and 2).
Note: A basic view of autosomal dominant nonsyndromic HTAD recurrence risk assessment is presented in this section; genetic counseling issues that may be specific to a given family or genetic cause of nonsyndromic HTAD and issues related to syndromic HTAD are not addressed.
Risk to Family Members of a Proband with a Pathogenic Variant in an Autosomal Dominant HTAD-Related Gene
Parents of a proband
Some individuals diagnosed with autosomal dominant HTAD have the disorder as the result of an HTAD-related pathogenic variant inherited from a parent. Because the penetrance of HTAD is reduced, the transmitting parent may or may not have a history of thoracic aortic or other associated vascular disease.
Some individuals diagnosed with autosomal dominant HTAD have the disorder as the result of a de novo pathogenic variant.
Molecular genetic testing for the HTAD-related pathogenic variant identified in the proband is recommended for the parents of the proband to evaluate their genetic status, inform recurrence risk assessment, and – if the HTAD-related pathogenic variant is identified in a parent – implement gene-based management recommendations for the heterozygous parent.
If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
The proband has a de novo pathogenic variant.
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the proband has the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
Sibs who inherit a pathogenic variant should be counseled and managed based on the causative HTAD gene, family history, and other clinical or lifestyle risk factors (see
Management).
If the HTAD-related pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If a molecular genetic diagnosis has been established in the proband but the genetic status of the parents is unknown (e.g., the parents have not been tested for the familial HTAD-related pathogenic variant), genetic testing should be offered to all sibs regardless of their clinical history due to the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant HTAD has a 50% chance of inheriting the HTAD-related pathogenic variant.
Cascade Testing of Relatives at Risk
If the HTAD-related pathogenic variant(s) in a family are known, cascade genetic testing is recommended for parents, sibs, offspring, and other at-risk family members in order to confirm their genetic status.
If the HTAD-related pathogenic variant(s) in a family are not known, parents, sibs, offspring, and other at-risk family members should be offered appropriate aortic imaging studies to screen for asymptomatic thoracic aortic disease (see Imaging Recommendations for Family Members of Individuals with HTAD of Unknown Genetic Cause and Isselbacher et al [2022]).
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Heart Association
MedlinePlus
The John Ritter Foundation for Aortic Health
Phone: 213-218-3329
The Marfan Foundation
Genetic Aortic Disorders Association (GADA) Canada
Canada
Phone: 905-826-3223; 866-722-1722
Email: info@gadacanada.ca
Chapter Notes
Acknowledgments
The authors are grateful to the patients and families participating in the research studies supported by the National Institutes of Health (R01HL109942), American Heart Association Merit Award, John Ritter Foundation for Aortic Health, Remembrin' Benjamin Foundation, Olivia Petrera-Cohen Research Fund, Family Fund, Genetic Aortic Disorders Association Canada (GADA Canada), and the Temerty Foundation.
Author History
Alana C Cecchi, MS, CGC (2023-present)
Dianna M Milewicz, MD, PhD (2003-present)
Ellen Regalado, MS, CGC; University of Texas Medical School (2011-2023)
Van Tran-Fadulu, MS, CGC; University of Texas Medical School (2003-2011)
Revision History
4 May 2023 (sw) Comprehensive update posted live
1 December 2016 (bp) Comprehensive update posted live; scope changed to overview
11 January 2011 (me) Comprehensive update posted live
10 May 2006 (me) Comprehensive update posted live
28 April 2005 (me) Comprehensive update posted live
13 February 2003 (me) Review posted live
11 July 2002 (dm) Original submission
References
Literature Cited
Asch
FM, Yuriditsky
E, Prakash
SK, Roman
MJ, Weinsaft
JW, Weissman
G, Weigold
WG, Morris
SA, Ravekes
WJ, Holmes
KW, Silberbach
M, Milewski
RK, Kroner
BL, Whitworth
R, Eagle
KA, Devereux
RB, Weissman
NJ, et al.
The need for standardized methods for measuring the aorta: multimodality core lab experience from the GenTAC Registry.
JACC Cardiovasc Imaging.
2016;9:219-26.
[
PMC free article: PMC4788536] [
PubMed: 26897684]
Boerio
ML, Engelhardt
NM, Cuddapah
S, Gold
JI, Marin
IC, Pinard
A, Guo
D, Prakash
SK, Milewicz
DM. Further evidence that ARIH1 rare variants predispose to thoracic aortic disease.
Circ Genom Precis Med.
2022;15:e003707.
[
PMC free article: PMC9771952] [
PubMed: 36350761]
Campens
L, Demulier
L, De Groote
K, Vandekerckhove
K, De Wolf
D, Roman
MJ, Devereux
RB, De Paepe
A, De Backer
J. Reference values for echocardiographic assessment of the diameter of the aortic root and ascending aorta spanning all age categories.
Am J Cardiol.
2014;114:914-20.
[
PubMed: 25092193]
Cannaerts
E, Kempers
M, Maugeri
A, Marcelis
C, Gardeitchik
T, Richer
J, Micha
D, Beauchesne
L, Timmermans
J, Vermeersch
P, Meyten
N, Chénier
S, van de Beek
G, Peeters
N, Alaerts
M, Schepers
D, Van Laer
L, Verstraeten
A, Loeys
B.
Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: further delineation of the phenotype.
J Med Genet.
2019;56:220-7.
[
PubMed: 29967133]
Cecchi
AC, Drake
M, Campos
C, Howitt
J, Medina
J, Damrauer
SM, Shalhub
S, Milewicz
DM, et al.
Current state and future directions of genomic medicine in aortic dissection: a path to prevention and personalized care.
Semin Vasc Surg.
2022;35:51-9.
[
PMC free article: PMC9258522] [
PubMed: 35501041]
Chen
MH, Choudhury
S, Hirata
M, Khalsa
S, Chang
B, Walsh
CA. Thoracic aortic aneurysm in patients with loss of function filamin A mutations: clinical characterization, genetics, and recommendations.
Am J Med Genet A.
2018;176:337-50.
[
PMC free article: PMC7534149] [
PubMed: 29334594]
Davies
RR, Gallo
A, Coady
MA, Tellides
G, Botta
DM, Burke
B, Coe
MP, Kopf
GS, Elefteriades
JA. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms.
Ann Thorac Surg.
2006;81:169-77.
[
PubMed: 16368358]
Devereux
RB, de Simone
G, Arnett
DK, Best
LG, Boerwinkle
E, Howard
BV, Kitzman
D, Lee
ET, Mosley
TH
Jr, Weder
A, Roman
MJ. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons ≥15 years of age.
Am J Cardiol.
2012;110:1189-94.
[
PMC free article: PMC3462295] [
PubMed: 22770936]
Elbitar
S, Renard
M, Arnaud
P, Hanna
N, Jacob
MP, Guo
DC, Tsutsui
K, Gross
MS, Kessler
K, Tosolini
L, Dattilo
V, Dupont
S, Jonquet
J, Langeois
M, Benarroch
L, Aubart
M, Ghaleb
Y, Abou Khalil
Y, Varret
M, El Khoury
P, Ho-Tin-Noé
B, Alembik
Y, Gaertner
S, Isidor
B, Gouya
L, Milleron
O, Sekiguchi
K, Milewicz
D, De Backer
J, Le Goff
C, Michel
JB, Jondeau
G, Sakai
LY, Boileau
C, Abifadel
M. Pathogenic variants in THSD4, encoding the ADAMTS-like 6 protein, predispose to inherited thoracic aortic aneurysm.
Genet Med.
2021;23:111-22.
[
PMC free article: PMC8559271] [
PubMed: 32855533]
Evangelista
A, Flachskampf
FA, Erbel
R, Antonini-Canterin
F, Vlachopoulos
C, Rocchi
G, Sicari
R, Nihoyannopoulos
P, Zamorano
J, Pepi
M, Breithardt
OA, Plonska-Gosciniak
E, et al.
Echocardiography in aortic diseases: EAE recommendations for clinical practice.
Eur J Echocardiogr.
2010;11:645-58.
[
PubMed: 20823280]
Forteza
A, Evangelista
A, Sánchez
V, Teixidó-Turà
G, Sanz
P, Gutiérrez
L, Gracia
T, Centeno
J, Rodríguez-Palomares
J, Rufilanchas
JJ, Cortina
J, Ferreira-González
I, García-Dorado
D. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial.
Eur Heart J.
2016;37:978-85.
[
PubMed: 26518245]
Frank
M, Adham
S, Seigle
S, Legrand
A, Mirault
T, Henneton
P, Albuisson
J, Denarié
N, Mazzella
JM, Mousseaux
E, Messas
E, Boutouyrie
P, Jeunemaitre
X. Vascular Ehlers-Danlos syndrome: long-term observational study.
J Am Coll Cardiol.
2019;73:1948-57.
[
PubMed: 30999998]
Gallo
EM, Loch
DC, Habashi
JP, Calderon
JF, Chen
Y, Bedja
D, van Erp
C, Gerber
EE, Parker
SJ, Sauls
K, Judge
DP, Cooke
SK, Lindsay
ME, Rouf
R, Myers
L, ap Rhys
CM, Kent
KC, Norris
RA, Huso
DL, Dietz
HC. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis.
J Clin Invest.
2014;124:448-60.
[
PMC free article: PMC3871227] [
PubMed: 24355923]
Gravholt
CH, Andersen
NH, Conway
GS, Dekkers
OM, Geffner
ME, Klein
KO, Lin
AE, Mauras
N, Quigley
CA, Rubin
K, Sandberg
DE, Sas
TCJ, Silberbach
M, Söderström-Anttila
V, Stochholm
K, van Alfen-van derVelden
JA, Woelfle
J, Backeljauw
PF, et al.
Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting.
Eur J Endocrinol.
2017;177:G1-G70.
[
PubMed: 28705803]
Guo
DC, Regalado
E, Casteel
DE, Santos-Cortez
RL, Gong
L, Kim
JJ, Dyack
S, Horne
SG, Chang
G, Jondeau
G, Boileau
C, Coselli
JS, Li
Z, Leal
SM, Shendure
J, Rieder
MJ, Bamshad
MJ, Nickerson
DA, Kim
C, Milewicz
DM, et al.
Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections.
Am J Hum Genet.
2013;93:398-404.
[
PMC free article: PMC3738837] [
PubMed: 23910461]
Guo
DC, Regalado
ES, Gong
L, Duan
X, Santos-Cortez
RL, Arnaud
P, Ren
Z, Cai
B, Hostetler
EM, Moran
R, Liang
D, Estrera
A, Safi
HJ, University of Washington Center for Mendelian Genomics, Leal SM, Bamshad MJ, Shendure J, Nickerson DA, Jondeau G, Boileau C, Milewicz DM. LOX mutations predispose to thoracic aortic aneurysms and dissections.
Circ Res.
2016;118:928-34.
[
PMC free article: PMC4839295] [
PubMed: 26838787]
Harris
KM, Nienaber
CA, Peterson
MD, Woznicki
EM, Braverman
AC, Trimarchi
S, Myrmel
T, Pyeritz
R, Hutchison
S, Strauss
C, Ehrlich
MP, Gleason
TG, Korach
A, Montgomery
DG, Isselbacher
EM, Eagle
KA. Early mortality in type A acute aortic dissection: insights from the International Registry of Acute Aortic Dissection.
JAMA Cardiol.
2022;7:1009-15.
[
PMC free article: PMC9403853] [
PubMed: 36001309]
Hicks
KL, Byers
PH, Quiroga
E, Pepin
MG, Shalhub
S. Testing patterns for genetically triggered aortic and arterial aneurysms and dissections at an academic center.
J Vasc Surg.
2018;68:701-11.
[
PubMed: 29510914]
Huynh
N, Thordsen
S, Thomas
T, Mackey-Bojack
SM, Duncanson
ER, Nwuado
D, Garberich
RF, Harris
KM. Clinical and pathologic findings of aortic dissection at autopsy: review of 336 cases over nearly 6 decades.
Am Heart J.
2019;209:108-15.
[
PubMed: 30660330]
Hysa
L, Khor
S, Starnes
BW, Chow
WB, Sweet
MP, Nguyen
J, Shalhub
S. Cause-specific mortality of type B aortic dissection and assessment of competing risks of mortality.
J Vasc Surg.
2021;73:48-60.e1.
[
PubMed: 32437949]
Isselbacher
EM, Preventza
O, Black
JH
3rd, Augoustides
JG, Beck
AW, Bolen
MA, Braverman
AC, Bray
BE, Brown-Zimmerman
MM, Chen
EP, Collins
TJ, DeAnda
A
Jr, Fanola
CL, Girardi
LN, ugtrde 4rytrewytrewd erytrytrertyttdsaRDS CW, Hui DS, Jones WS, Kalahasti V, Kim KM, Milewicz DM, Oderich GS, Ogbechie L, Promes SB, Ross EG, Schermerhorn ML, Times SS, Tseng EE, Wang GJ, Woo YJ. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines.
Circulation.
2022;146:e334-e482.
[
PMC free article: PMC9876736] [
PubMed: 36322642]
Lacro
RV, Dietz
HC, Sleeper
LA, Yetman
AT, Bradley
TJ, Colan
SD, Pearson
GD, Selamet Tierney
ES, Levine
JC, Atz
AM, Benson
DW, Braverman
AC, Chen
S, De Backer
J, Gelb
BD, Grossfeld
PD, Klein
GL, Lai
WW, Liou
A, Loeys
BL, Markham
LW, Olson
AK, Paridon
SM, Pemberton
VL, Pierpont
ME, Pyeritz
RE, Radojewski
E, Roman
MJ, Sharkey
AM, Stylianou
MP, Wechsler
SB, Young
LT, Mahony
L, et al.
Atenolol versus losartan in children and young adults with Marfan's syndrome.
N Engl J Med.
2014;371:2061-71.
[
PMC free article: PMC4386623] [
PubMed: 25405392]
Ladouceur
M, Fermanian
C, Lupoglazoff
JM, Edouard
T, Dulac
Y, Acar
P, Magnier
S, Jondeau
G. Effect of beta-blockade on ascending aortic dilatation in children with the Marfan syndrome.
Am J Cardiol.
2007;99:406-9.
[
PubMed: 17261408]
Milewicz
DM, Guo
D, Hostetler
E, Marin
I, Pinard
AC, Cecchi
AC. Update on the genetic risk for thoracic aortic aneurysms and acute aortic dissections: implications for clinical care.
J Cardiovasc Surg (Torino). 2021;62:203-10.
[
PMC free article: PMC8513124] [
PubMed: 33736427]
Olsson
C, Eriksson
N, Ståhle
E, Thelin
S. Surgical and long-term mortality in 2634 consecutive patients operated on the proximal thoracic aorta.
Eur J Cardiothorac Surg.
2007;31:963-9.
[
PubMed: 17336538]
Ong
KT, Perdu
J, De Backer
J, Bozec
E, Collignon
P, Emmerich
J, Fauret
AL, Fiessinger
JN, Germain
DP, Georgesco
G, Hulot
JS, De Paepe
A, Plauchu
H, Jeunemaitre
X, Laurent
S, Boutouyrie
P. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial.
Lancet.
2010;376:1476-84.
[
PubMed: 20825986]
Pannu
H, Tran-Fadulu
V, Papke
CL, Scherer
S, Liu
Y, Presley
C, Guo
D, Estrera
AL, Safi
HJ, Brasier
AR, Vick
GW, Marian
AJ, Raman
CS, Buja
LM, Milewicz
DM. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II.
Hum Mol Genet.
2007;16:2453-62.
[
PMC free article: PMC2905218] [
PubMed: 17666408]
Pape
LA, Tsai
TT, Isselbacher
EM, Oh
JK, O'gara
PT, Evangelista
A, Fattori
R, Meinhardt
G, Trimarchi
S, Bossone
E, Suzuki
T, Cooper
JV, Froehlich
JB, Nienaber
CA, Eagle
KA, et al.
Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD).
Circulation.
2007;116:1120-7.
[
PubMed: 17709637]
Prakash
SK, Haden-Pinneri
K, Milewicz
DM. Susceptibility to acute thoracic aortic dissections in patients dying outside the hospital: an autopsy study.
Am Heart J.
2011;162:474-9.
[
PubMed: 21884863]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016;48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Raunsø
J, Song
RJ, Vasan
RS, Bourdillon
MT, Nørager
B, Torp-Pedersen
C, Gislason
GH, Xanthakis
V, Andersson
C. Familial clustering of aortic size, aneurysms, and dissections in the community.
Circulation.
2020;142:920-8.
[
PubMed: 32580567]
Renard
M, Francis
C, Ghosh
R, Scott
AF, Witmer
PD, Adès
LC, Andelfinger
GU, Arnaud
P, Boileau
C, Callewaert
BL, Guo
D, Hanna
N, Lindsay
ME, Morisaki
H, Morisaki
T, Pachter
N, Robert
L, Van Laer
L, Dietz
HC, Loeys
BL, Milewicz
DM, De Backer
J. Clinical validity of genes for heritable thoracic aortic aneurysm and dissection.
J Am Coll Cardiol.
2018;72:605-15.
[
PMC free article: PMC6378369] [
PubMed: 30071989]
Robertson
EN, van der Linde
D, Sherrah
AG, Vallely
MP, Wilson
M, Bannon
PG, Jeremy
RW. Familial non-syndromal thoracic aortic aneurysms and dissections - Incidence and family screening outcomes.
Int J Cardiol.
2016;220:43-51.
[
PubMed: 27372041]
Rodríguez-Palomares
JF, Teixidó-Tura
G, Galuppo
V, Cuéllar
H, Laynez
A, Gutiérrez
L, González-Alujas
MT, García-Dorado
D, Evangelista
A. Multimodality assessment of ascending aortic diameters: comparison of different measurement methods.
J Am Soc Echocardiogr.
2016;29:819-26.e4.
[
PubMed: 27288090]
Thijssen
CGE, Bons
LR, Gökalp
AL, Van Kimmenade
RRJ, Mokhles
MM, Pelliccia
A, Takkenberg
JJM, Roos-Hesselink
JW. Exercise and sports participation in patients with thoracic aortic disease: a review.
Expert Rev Cardiovasc Ther.
2019;17:251-66.
[
PubMed: 30887852]
Van Gucht
I, Krebsova
A, Diness
BR, Laga
S, Adlam
D, Kempers
M, Samani
NJ, Webb
TR, Baranowska
AA, Van Den Heuvel
L, Perik
M, Luyckx
I, Peeters
N, Votypka
P, Macek
M, Meester
J, Van Laer
L, Verstraeten
A, Loeys
BL. Novel LOX variants in five families with aortic/arterial aneurysm and dissection with variable connective tissue findings.
Int J Mol Sci.
2021;22:7111.
[
PMC free article: PMC8269155] [
PubMed: 34281165]
Zafar
MA, Li
Y, Rizzo
JA, Charilaou
P, Saeyeldin
A, Velasquez
CA, Mansour
AM, Bin Mahmood
SU, Ma
WG, Brownstein
AJ, Tranquilli
M, Dumfarth
J, Theodoropoulos
P, Thombre
K, Tanweer
M, Erben
Y, Peterss
S, Ziganshin
BA, Elefteriades
JA. Height alone, rather than body surface area, suffices for risk estimation in ascending aortic aneurysm.
J Thorac Cardiovasc Surg.
2018;155:1938-50.
[
PubMed: 29395211]