

Variation Viewer: Help
Variation Viewer allows you to view, search, and navigate variations housed in dbSNP, dbVar, and ClinVar in genomic context. You can search based on chromosomal location, gene, variant IDs from dbSNP and dbVar, or phenotype; and explore results in the dynamic sequence viewer and annotated tables of variations.
For a brief overview about using Variation Viewer, please watch the introductory video tutorial . There you will learn how to select the assembly, focus on your region of interest, navigate by exons, review HGVS expressions, filter results, and go to linked databases.
An abbreviated version of this documentation, suitable for handouts, is available as the Variation Viewer Factsheet .
Table of contents
Application layout

How to search
You can locate regions of interest through the control panel at the upper left of the page. Select the assembly to query in the Pick Assembly section. Variation Viewer supports both the current and previous versions of the reference human genome assembly -- GRCh38 and GRCh37.p13.

Enter your query in the input box in the Search section. When you move your cursor to the area of the search box, note that the option 'Search examples' is displayed. Here you will note that Variation Viewer supports queries by location in multiple formats, by cytogenetic band, gene symbol, or dbSNP ID (rs number) or dbVar ID (esv and nsv number). An asterisk (*) can be used as a wild card for gene symbols only. To limit the scope of the display by size, indicate the size after a slash (/) as in the "chr1:1,500,000 / 200". Note that base pairs (bp) can be writen out, or abbreviated (K for kb, M for Mb)
By location:
- chr1:1,500,000-2,000,000
- chr2: 1.5M - 2M
- chr2: 1.5M-2,540.2K
- 21.33M - 22.01M
- 21.335M..21.337M
- chr1:1,500,000 / 200
- chr1:101,500,200
- 1,500K/0.5K
- chr5
- chr 10
- 5p15
By gene:
By dbSNP ID:
By dbVar ID:
If multiple results are returned by your query, select one for display by clicking on its row in the results under the search box. Note that the results section has two tabs, one for genes and one for all other features.
Gene search history
The "History" section displays your recent gene searches. Select a gene from the drop down menu to navigate to it.

Navigation in the sequence display
There are multiple tools provided to support navigation on the sequence being displayed. These include navigation by cytogenetic band supported by the horizontal ideogram display at the top of the page; by gene in the blue bar; by exon in the selected gene in the blue bar, modifying zoom level or panning in Sequence Viewer, clicking on a feature in Sequence Viewer, etc.
Chromosome overview

The chromosome overview -- or ideogram -- atop the page supports both changing the range of the display, and moving the range of the display. Locate the blue-colored section indicating the cytogenetic band(s) being displayed. If you click on the center, the cross-shaped icon will display indicating you can drag the view left or right. If you click on the edge of the blue-colored section, a double-headed arrow will indicate that you can expand or contract the range by dragging that edge left or right as appropriate.
Gene and exon navigator
Genes

When a region of sequence is displayed that includes one or more genes, the symbols for those genes are provided in the menu labeled 'Gene' in the blue bar under the ideogram. You can navigate quickly to a gene by clicking on its symbol. When a single gene is selected, you can move up and down the chromosome, one gene at a time, by clicking on the double arrows (circled red above) to the previous gene (left) or next gene (right) of the Gene selector region. Clicking on 'Region' will bring up a menu (Figure below) with options for limiting the view range for the gene with or without padding.

10% padding is selected by default.
Exons
When a gene is selected there will be one or more open circles at the right representing the exons of the gene. Navigate to a particular exon by clicking on the appropriate circle; move to the upstream or downstream exon by clicking on the appropriate arrow flanking the exon selector.
Sequence Viewer
There are multiple tools to support navigation within the graphical display, known as Sequence Viewer. Full documentation for Sequence Viewer is available as Sequence Viewer documentation in the Graphical Panel section. Sequence Viewer is used in several resources at NCBI, so you may already be familiar with its use.
Configuring Sequence Viewer

Custom Tracks
Your Data
The 'Your Data' widget allows you to add custom tracks for display in the Sequence Viewer alongside NCBI-provided tracks, by either uploading files or streaming data from remotely-hosted files. To start, click the "+" button. Uploaded tracks will expire 60 days after they are last touched in the browser; streamed tracks will persist until you remove them. If you are logged in with your My NCBI account and uploading/streaming human data, your custom tracks will also be available to you in other NCBI browsers displaying the same assembly version as your data (e.g. GDV or 1000 Genomes Browser).
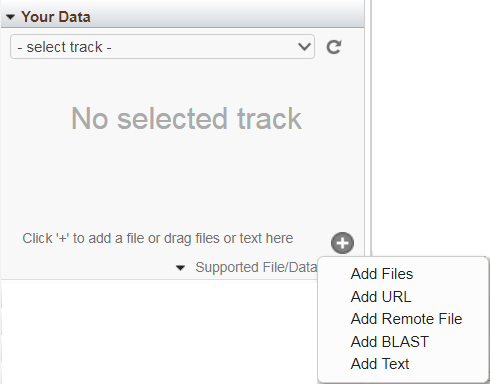
Uploading files: You can upload files by selecting one of the following menu options: "Add Files", "Add URL" or "Add Text", or by dragging the file into the widget. The following file types are supported: BED, GFF3, GTF, GVF, VCF, HGVS, ASN.1 (text and binary). Concurrent upload of multiple files is supported. The per-file upload limit is currently 4 GB. The file name will be used as the track's display name, unless an (optional) alternate name is provided. Once uploaded, the files will appear in the 'select tracks' drop-down menu in this widget.
Remote streaming files: BAM files hosted on HTTP can be streamed for display in Variation Viewer. To add these data as tracks, select “Add Remote Track” from supported files menu, and enter the corresponding URL in the display. Note that an index file with the .bai extension must be located at the same location as the BAM file. The file name will be used as the track’s display name, unless an (optional) alternate name is provided. A progress bar will indicate the status of the connection and validation processes. Once connected, remotely hosted files will appear in the graphical display and be listed in the 'select tracks' drop-down menu of the “Your Data” widget, designated by “(R)”. Other file types and BAM files hosted on FTP are not yet supported. To request streaming support for additional file types, click the "Support Center" link located at the lower right of the browser page.
BLAST: You can also enter the RID from a BLAST search into the Your Data widget. The results will appear as an alignment track in the display.
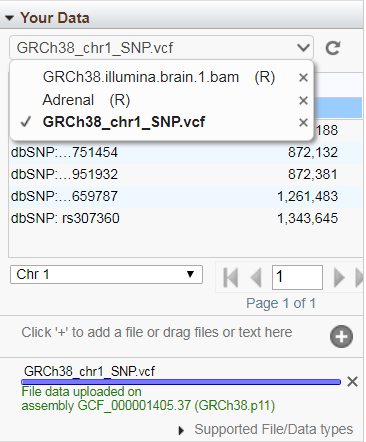
If your track has discrete features (e.g. SNPs or gene annotations, rather than graphs or alignments) on the currently displayed sequence, you can display those features in a paginated table if you select that track from the "Your Data" drop-down menu. Hovering the mouse over a table row opens a tool-tip with feature details. Click on a row to go to the location of that that feature in Sequence Viewer. Use the menus below the table for additional table naviation. To remove uploaded or remote tracks, select the track from this drop-down menu and then click on the 'minus' icon next to the track name. Note: A selected track will be marked by a check-mark in this menu, regardless of whether it has data suitable for tabular display.
The progress bar at the bottom of the window indicates the status of the upload or initial connection to the data file. If validation detects the presence of target sequences in your uploaded or streamed remote file that are not part of the assembly displayed in Variation Viewer, those sequences will be reported as errors, but you will still be connected and able to view tracks for all target sequences in the file that are part of the assembly. To see any warning or error messages associated with uploading or streaming your custom track data, click on the "Details" option that will be a part of the status message.
File uploads, streaming of remotely hosted files and BLAST results can also be managed via the track configuration menu found under the 'Tracks' button at the top right of the Sequence Viewer display. Files uploaded or connected by this mechanism will also appear in the 'Your Data' widget.
Variation data
The Variation Data section in the bottom half of the page has two panels. The panel on the left contains filters for narrowing variant sets by features of interest. The panel on the right is a table of variants.
Filtering variants
The left panel has filters you can use to restrict your display to variants of interest:

|
There are eight groups of filters. Selections between filter groups are handled as a boolean AND , e.g. variant type "single nucleotide variant" AND most severe clinical signficance "pathogenic". Selections within filter groups are handled as a boolean OR , e.g. molecular consequence "missense" OR "nonsense". The filter groups are listed in the following table.
| Name | Description and examples |
| Source database | dbSNP or dbVar |
| In ClinVar | A variation with medically important data in ClinVar |
| Most severe clinical significance | Most severe clinical significance annotated in ClinVar. The terms are definedhere. |
| Variant type | Type of variation, e.g. single nucleotide variant |
| Molecular consequence | Consequences of the variation that can be computed from genes and other features annotated on the genome, e.g. nonsense, missense |
| 1000 Genomes MAF | Minor allele frequency from the 1000 Genomes Project |
| GO-ESP MAF | Minor allele frequency from the GO-ESP Project |
| ExAC MAF | Minor allele frequency from the Exome Aggregation Consortium. "Singleton" and "2 - 10 alleles" are based on allele count and not frequency. The four frequencies only include variants with minor allele counts over 10. |
| Has publications | Whether the variant has associated publications in biomedical literature |
Variant table
The table on the right in the Variation Data section provides a paginated list of filtered results. It shows an overview of all variants in the current region displayed in Variation Viewer.
Allele-level data for each variant is available upon toggling table rows. Clicking the "Toggle all" arrow to the left of the Variant ID column (circled in red below) will toggle the allele tables for all variants in the current page of the table. Clicking the arrow to the left of each row (circled in green) will toggle the allele table for only that variant.

|
Column
|
Description
|
|---|---|
| (Toggle alleles) | Click this arrow to show the allele table for this variant (circled in green above). See also "Toggle all" arrow in table header (circled in red). |
| Variant ID | Identifier from an archival database (rs from dbSNP, nsv/esv from dbVar) |
| Location | Top level genomic location of the variant |
| Variant type | The type of variant using terms defined in Sequence Ontology (SO) athttp://www.sequenceontology.org. |
|
Gene |
For variations in dbVar (accession starting with nsv or esv), the field shows how the variation relates to nearby genes; for short variations, the value report the gene in which the variation is found. |
| Molecular consequence | Calculated molecular consequence(s) for the variation. Values are linked to their corresponding entries on the Sequence Ontology sitehttp://www.sequenceontology.org, where the consequence is defined. At this time, there is no molecular consequence reported for structural variations (nsv/esv). |
| The most severe clinical significance annotated for the variant in ClinVar. Values are linked to their corresponding record in ClinVar. | |
| 1000G MAF | Minor allele frequency from all 1000 Genomes Project (1000G) samples. If the minor allele is the reference allele, the value is bolded. |
| GO-ESP MAF | Minor allele frequency from the GO-ESP project. If the minor allele is the reference allele, the value is bolded. |
| ExAC MAF | Minor allele frequency from the Exome Aggregation Consortium project. If the minor allele is the reference allele, the value is bolded. |
| Publications | Number of publications associated with the variant. Click on the linked value to view the publications in PubMed. |
Resource links

- Reset All - resets Variation Viewer to its default settings
- Share this page - generates a temporary URL for 90 days to Variation Viewer display with current settings with "E-mail URL" option
- FAQ - frequently asked questions
- Help - Variation Viewer documentation
- Version # - release notes
Linking to Variation Viewer
URL links to Variation Viewer can be constructed to customize the display. For constructing URLs, the base URL is http://www.ncbi.nlm.nih.gov/variation/view/?<List-of-parameters>
| Parameter | Description | Comments and examples |
|---|---|---|
| assm | assembly |
Specifies the genome assembly to use, as identified by RefSeq accession. Defaults to GRCh38, but can be helpful for linking to features on GRCh37 -- a.k.a. hg19. Note that only GRCh38.p2 (RefSeq: GCF_000001405.28) and GRCh37.p13 (RefSeq: GCF_000001405.25) are currently supported.
|
| chr | chromosome oralternate locus | Can specify any chromosome name or top-level accession, e.g. 'chr=1', 'chr=X', 'chr=MT'. Examples: |
| from, to | range start, end (both 1-based) |
Used to specify a genomic range of interest. Example: |
| q | search term (gene, RS ID, etc.) |
Searches and selects the location of the first search result. Examples:
|
| filters | filter selections for the Variant Table | The 'filters' parameter can be used to load a table of variants that meet specific criteria. To put the data table in view without needing to scroll down past the sequence graphics, append "#data" to the end of the URL. Examples to load only variants that are...
|
| mk | markers to highlight regions of interest |
Markers in Sequence Viewer allow attributes of position and labels. The position may be a range (separated by :). The label is separated from the position by |. Multiple markers are separated by commas. Examples:
|
'filters' URL parameter mapping
Source database:
- In URL: source
- Filters:
|
In display
|
In URL
|
|---|---|
| dbSNP | dbsnp |
| dbVar | dbvar |
Most severe clinical significance:
- In URL: clinsig
- Definitions: Documentation
- Filters:
|
In display
|
In URL
|
|---|---|
| Pathogenic | pathogenic |
| Likely pathogenic | likely-pathogenic |
| drug response | drug-response |
| other | other |
| confers sensitivity | confers-sensitivity |
| risk factor | risk-factor |
| association | association |
| protective | protective |
| Uncertain significance | uncertain-significance |
| not provided | not-provided |
| Likely benign | likely-benign |
| Benign | benign |
| not specified | not-specified |
In ClinVar:
- In URL: inclinvar
- Filters:
|
In display
|
In URL
|
|---|---|
| Yes | 1 |
| No | 0 |
Variant type
- In URL: vartype
- Filters:
|
In display
|
In URL
|
|---|---|
| single nucleotide variant | single-nucleotide-variant |
| copy number variation | copy-number-variation |
| deletion | deletion |
| insertion | insertion |
| microsatellite | microsatellite |
| multiple nucleotide polymorphism | multiple-nucleotide-polymorphism |
| indel | indel |
| inversion | inversion |
| complex structural alteration | complex-structural-alteration |
| novel sequence insertion | novel-sequence-insertion |
| sequence alteration | sequence-alteration |
Molecular consequence
- In URL: molcons
- Filters
|
In display
|
In URL
|
|---|---|
| missense variant | missense-variant |
| nonsense | nonsense |
| stop lost | stop-lost |
| inframe variant | inframe-variant |
| frameshift variant | frameshift-variant |
| synonymous variant | synonymous-variant |
| splice acceptor variant | splice-acceptor-variant |
| splice donor variant | splice-donor-variant |
| intron variant | intron-variant |
| 5 prime UTR variant | 5-prime-utr-variant |
| 3 prime UTR variant | 3-prime-utr-variant |
| nc transcript variant | nc-transcript-variant |
| 500B downstream variant | 500b-downstream-variant |
| 2KB upstream variant | 2kb-upstream-variant |
| intergenic variant | intergenic-variant |
| not specified | not-specified |
1000 Genomes MAF
- In URL: 1000gmaf
- Filters:
|
In display
|
In URL
|
|---|---|
| < 0.005 | lt-05 |
| 0.005 - 0.01 | gte-05-lt-1 |
| 0.01 - 0.05 | gte-1-lt-5 |
| >= 0.05 | gte-5 |
| not specified | na |
GO-ESP MAF
- In URL: goespmaf
- Filters:
|
In display
|
In URL
|
|---|---|
| < 0.005 | lt-05 |
| 0.005 - 0.01 | gte-05-lt-1 |
| 0.01 - 0.05 | gte-1-lt-5 |
| >= 0.05 | gte-5 |
| not specified | na |
ExAC MAF
- In URL: exacmaf
- Filters:
|
In display
|
In URL
|
|---|---|
| Singleton | singleton |
| 2 - 10 alleles | 2-10-alleles |
| < 0. 001 | lt-01 |
| 0.001 - 0.01 | gte-01-lt-1 |
| 0.01 - 0.1 | gte-1-lt-10 |
| >= 0.1 | gte-10 |
| not specified | na |
Has Publication
- In URL: haspub
- Filters:
|
In display
|
In URL
|
|---|---|
| Yes | 1 |
| No | 0 |