

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.
This publication is provided for historical reference only and the information may be out of date.

INTRODUCTION
Growth in childhood, one of the most fascinating, complex and dynamic biological processes, is tightly controlled and regulated. Growth evaluation, height and weight determinations, remains one of the most useful of all indices of public health and economic well being, specifically in developing countries and countries in transition. This chapter does not intend to reduce to a brief summary the complex interactions involved in the processes of human growth and great scientific advances of the last decades. Rather, it aims to highlight two key themes. Firstly, the regulation and interaction of the main hormonal axis involved in human growth, the growth hormone (GH)-insulin-like growth factor axis (IGF-I) including the biochemical characteristics of the key members, the ontogeny and main modulators. An updated revision of the "somatomedin" hypothesis is presented. Secondly, current studies revisiting the organization of normal growth from infancy into puberty are briefly discussed. GH is important in promoting somatic growth and in regulating body composition, intermediary muscle and bone metabolism. Some of these GH effects are direct actions whereas others are mediated via IGF-I. IGF-I is the main effector of GH action in linear growth in man; in addition IGF-I and IGF-II are important growth factors involved in diverse cellular actions such as proliferation, differentiation and apoptosis. This chapter should be read in conjunction with chapter 5c dealing with GH in the adult (link)
THE GROWTH HORMONE-INSULIN-LIKE GROWTH FACTOR AXIS
GH is synthesized in the anterior pituitary, where it is store in secretory granules. It is the most abundant hormone in the pituitary accounting for 25% of the gland"s hormones (1). GH is a single polypeptide chain of 191 amino acids with 2 disulphide bridges between amino acids 53-165 and 282-189, respectively. The GH variant 20-kDa accounts for 10-20% of the total content of pituitary GH, results from a different mRNA by alternative splicing of intron B that encodes amino acids in position 32-46. There is another form 17-kDa, which is produced by the same mechanism (2). In man two GH genes are localized on chromosome 17. GH-N gene is expressed in the pituitary and encodes GH. It contains 5 exons and 4 introns. The GH-V gene is expressed in the placenta. There is considerable structural homology between GH, prolactin and hCG (3). In the fetus GH is produced by the pituitary gland from the end of the 1st trimester of gestation and circulating concentrations of GH can reach relatively high levels (4). The biological role of GH during fetal life has been the source of many studies. Early studies with anencephalic fetuses and neonates suggested that GH was not primarily involved in human fetal growth (5). However, the identification of the GH receptor (GHR) and its widespread localization in fetal tissues support a functional role for both GH and its receptor (6-9). There is growing evidence for a role of GH in fetal development although it may contribute only to approximately 20% of fetal size (10). In addition, maternal serum concentrations of placental GH (PGH) and insulin-like growth factor-I (IGF-I) strongly correlate throughout gestation, suggesting that PGH influences fetal growth through IGF-I (11). Chellakooty et al, evaluated the association of maternal serum levels of PGH and IGF-I with fetal growth in a prospective longitudinal study of 89 normal pregnant women (12).PGH levels were detectable in all samples from 5 wk gestation, increasing throughout pregnancy to approximately 37 wk when peak levels of 22 ng/ml (range, 4.6-69.2 ng/ml) were reached. Subsequently, PGH levels decreased until birth. The change in PGH during 24.5-37.5 wk gestation was positively associated with fetal growth rate, birth weight and IGF-I levels (12).
Growth Hormone Receptor (GHR)
GHR expression begins in fetal life. Hill et al demonstrated immunoreactive GHR in 14-16 weeks fetuses (8); later it was shown that in 15-20 week human fetuses" growth plate chondrocytes expressed GHR (9). Human fetal GHR expression appears to be regulated by the V3 promoter whereas in postnatal life GHR expression is regulated by hepatic V1 and V4 promoters and concomitant in vitro studies have demonstrated that these GHR are functional (10, 13). The cDNA encoding the GHR was cloned in 1987 (6). In humans the GHR is composed of 620 amino acid (AA) containing an extracellular (246 AA), a transmembrane (24 AA) and cytoplasmic (350 AA) domains (7). In addition to the membrane-bound receptor, a soluble GH binding protein, identical to the extracellular domain of the full-length receptors has been isolated in several species including humans (14). The availability of GHR cDNA and the identification of key intracellular proteins, tyrosine kinases, phosphatases and other transducers have allowed major advances in the understanding of the molecular aspects and the signaling pathways used by this receptor (15).
Regulation of GH secretion Several factors including hormones, neurotransmitters and metabolic modulators are known to regulate GH in man. There is extensive literature on the role of each of these factors that have been reviewed elsewhere (16) and in chapter 5c of EndoText (link). The best characterized include systemic factors and locally produced in the hypothalamus which are the primary regulating factors, GH releasing hormone (GHRH) and somatostatin (SRIF) (16). Initially it was thought that GH was only under the regulation of these two hypothalamic factors; however a recently identified small peptide hormone, ghrelin, also plays a role in GH secretion. The main regulatory factors are discussed in some detail below.
GHRH GHRH is the most important regulator of GH under physiologic conditions (17). GHRH stimulates GH synthesis by increasing both GH gene transcription and GH release (18-20). GHRH binds to its specific receptor, the GHRH receptor (GHRHR) which was cloned from a pituitary cDNA library (21). The GHRHR is a member of the seven transmembrane domain G-protein receptor superfamily (22). The critical role of GHRHR in human growth has been now fully documented with 1) the identification of functional receptors in fetal pituitary gland (23) and 2) the demonstration of molecular defects in the extracellular domain that are associated with profound GH deficiency and severe growth retardation (24, 25).
Somatostatin Krulich et al postulated the existence of somatostatin in 1968 using hypothalamic extracts that were able to inhibit the GH secretion (26) but it was finally isolated by Brazeau et al in 1973 (27) and subsequently the gene sequence was characterized in 1984 (28). Somatostatin binds to a family of specific receptors and inhibits adenyl cyclase via Gi, with net Ca influx reduction (16). Five somatostatin receptors which are regulated in a tissue specific manner have been cloned (29, 30). All subtypes are expressed in pituitary tumors and in normal fetal pituitary tissue (30, 31). It is worth noting that the primary function of somatostatin is to inhibit GH release but not its synthesis.
GH secretagogues (GHSs) Met-enkephalins are able to stimulate GH secretion in a specific manner (32). This observation led in the 1990s to the design of synthetic analogues that were not opiodergic but which were active on GH release (32). These growth hormone releasing peptides, such as GHRP-6 and GHRP-2, are quite potent stimulators of GH release compared to natural met-enkephalins precursors (33-35).
Ghrelin Ghrelin is an acylated peptide (MW of 3.3-kDa) and was first identified and characterized from the rat stomach (36). It is the endogenous ligand for the GH secretagogue receptor. Ghrelin is involved in a novel system for regulating GH release in a dose dependent manner in humans. Takaya et al demonstrated that the GH response to 0.2-"g/kg ghrelin was similar to 1.0 "g/kg GHRH and had a similar effect compared to GHRP-2, one of the most potent GH secretagogues (GHSs), indicating that per mol ghrelin is more potent for GH release (37, 38). Ghrelin levels in healthy lean children are similar to healthy lean adults but are reduced in obese children (38). Ghrelin levels are independent of gender and pubertal status but are negatively associated with obesity and insulin levels (38).
Thyroid hormones GH spontaneous nocturnal secretion is low in hypothyroidism and hyperthyroidism (39, 40). The rate and the amount of GH released are reduced in adolescent with untreated thyrotoxicosis compared with normal controls GH determined by spontaneous GH secretory profile (41). It has been proposed that the reduced GH release stimulated by GHRH in hyperthyroidism may be explained in part by an increase in hypothalamic somatostatin tone with a concomitant decrease in GHRH (41).
Hypoglycemia GHRH stimulation and insulin-induced hypoglycemia exert additive effects on GH release (42), which is consistent with a proposed somatostatin withdrawal during hypoglycemia. The clinically inhibitory effect of glucose on GH release may be due to a discharge of hypothalamic somatostatin (16, 42, 43)
Glucocorticoids The effect of glucocorticoids on growth is well established and many studies have examined the GH secretory profiles in prepubertal and pubertal children undergoing long-term glucocorticoid treatment or exposed to excess cortisol secretion (16, 44, 45). In these children GH responses assessed by GH spontaneous secretion and various pharmacological stimuli are decreased (16). The blunted GH response to stimulation tests in conditions of chronic exposure is well documented; in contrast the acute administration of glucocorticoids to normal subjects induces a transient increase in plasma GH levels reflecting a dual mode of action on GH secretion (45).
Sex steroid Androgens without the actions of GH are insufficient in the human to drive the fully normal tempo of puberty, as observed in hypopituitary boys who were replaced with testosterone but not with GH showing a long pubertal growth period (46). Studies in healthy pubertal boys have demonstrated the close relationship between rising serum androgen concentrations and increased GH peak amplitude (47). Serum GH concentration rises throughout puberty in both sexes and in healthy girls the increase in GH concentrations is proportional to the increase in serum oestradiol concentration (48).
The IGF system
The IGF system is composed of two IGF peptides, two specific receptors, a family of binding proteins and a glycoprotein named the acid-labile subunit (Figure 1). The insulin-like growth factors (IGF-I and IGF-II) are single chain polypeptides with structural similarity to proinsulin. In 1957 Salmon and Daughaday identified the "sulphation factor", later renamed "somatomedin" in the 1970s and referred as IGFs nowadays (49, 50). IGFs are essential regulators of normal fetal and postnatal growth (51-55). IGF-I and IGF-II, as many other growth factors, exert physiologic effects virtually in every organ and tissue during fetal and postnatal life (51-55). The development of IGF knockout models and the identification of four patients with IGF-I gene defects have provided clear evidence on the important role that circulating and locally produced IGF-I play in human and animal fetal and postnatal growth and development (55-60).
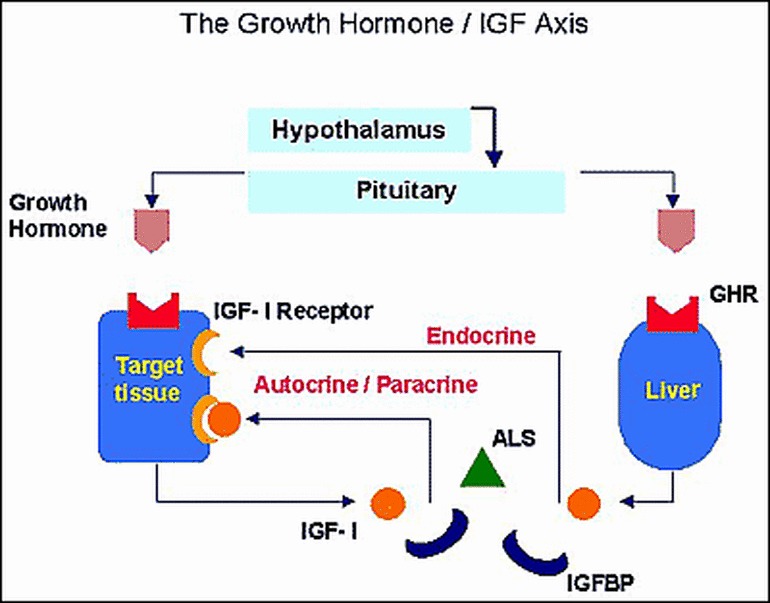
Figure 1Schematic representation of the GH-IGF axis.
IGF-I is a basic peptide with 70 amino acids and a molecular weight of 7.6-kDa (61). The human IGF-I peptide is the product of a single gene (~ 90-kb), that is located on the long arm of chromosome 12. It contains six exons and two promoters (61). The IGF-I transcribed by exon 1 is ubiquitously expressed, while the IGF-I transcribed by exon 2 is found exclusively in the liver, increasing with the onset of GH dependent linear growth (61). The expression of the IGF-I gene is determined by the activity of its promoters and by transcription factors that stimulate or inhibit their activity. The most potent regulator of IGF-I expression in postnatal life is GH (61). On the other hand IGF-I mediates growth hormone negative feedback (62). Nutritional status and supply of dietary energy and protein are also regulators of IGF-I and possibly the main regulators of IGF-I expression in fetal life (63, 64). IGF-II is a neutral peptide with 67 amino acids and molecular weight of 7.4-kDa (61) and is the product of a single gene (~ 30-kb) located on the short arm of chromosome 11. It contains nine exons (61, 65) with four different promoters (61). The mature IGF-II peptide is encoded by exons 7, 8 and 9. IGF-II is an imprinted gene, with only the paternal allele being expressed while the maternal allele is inactive. IGF-II gene is linked to H-19 gene, a tumor suppressor gene. Overall it is considered that the regulation of IGF-II expression is GH-independent. The IGF-1 Receptor (IGF-1R), located on chromosome 15, is a hetero-tetrameric glycoprotein of two a and β"subunits, product of single gene with 22 exons (65). The a-subunit corresponds to the extracellular cysteine-rich domain necessary for ligand recognition and binding with a transmembrane domain, and an intracellular domain with tyrosine kinase activity. Signaling activation of the IGF-1R is regulated through binding of the IGF ligands. IGF-I binds to the IGF-1R with high affinity, with dissociation constants in the range of 0.2-1.0 nM. The affinity of IGF-II for the IGF-1R is several folds lower, and the affinity of insulin for the IGF-1R is 100-fold lower (65-66). IGF-binding proteins (IGFBP) and acid-labile subunit (ALS): The IGFs are found in association with specific IGFBPs in blood and extracellular fluids. Six IGFBPs have been characterized IGFBP-1 through to IGFBP-6. All six IGFBPs have at least 50% homology among themselves and 80% homology among different species and are product of different genes with different chromosomal location (65). Most of the homology is found in the amino-and carboxy-terminal regions with a distinct mid-region among the six IGFBPs (65). The main functions of this family of proteins are 1) To extend IGFs half-life in the circulation 2) To transport the IGFs to the target cells and to modulate the biological actions of the IGFs (65). Most of the IGFs in circulation are found in a 150-kDa or ternary complex consisting of IGF-I or IGF-II, IGFBP-3 and acid-labile subunit (ALS) (67, 68). The human ALS gene is located on chromosome 16 and encodes a mature protein of 578 amino acids (69). GH is the main regulator of the ternary complex (70-72). In humans, ALS is undetectable in fetal serum at 27 weeks of gestation, but IGFs and IGFBPs can be detected from the first trimester of gestation. Serum IGFs and IGFBPs are present from early fetal life as determined from cord blood and are related to fetal size (70, 71). ALS is present at the end of fetal life, and increases five-fold from birth to puberty with little change during adulthood (73). A number of patients with short stature have been identified carrying homozygous mutations of the ALS gene, with no detectable serum ALS levels, very low IGF-I and IGFBP-3 levels and absence of 150-kDa ternary complex (74-76) The "somatomedin/IGF hypothesis" The original "somatomedin/IGF hypothesis" postulated that somatic growth was controlled by pituitary GH and mediated by circulating IGF-I expressed exclusively by the liver (49). The discovery that most tissues produce IGF-I in the late 1980"s supporting a role of autocrine/paracrine IGF-I led investigators to modify the original hypothesis to what is known today as the "dual effector" theory (77). Recent experiments using gene deletions and transgenic technologies have revealed new information that again has led investigators to revisit both hypotheses (78-85). These experimental studies have shown that the liver is the principal source of IGF-I in the circulation but is not required for postnatal body growth. This finding indicates that autocrine/paracrine IGF-I but not liver-derived IGF-I (endocrine IGF-I) is the major determinant of postnatal body growth (78-80). However, lack of liver-derived IGF-I results in disproportional organ growth. An early study showed that GH increases liver size in proportion to body weight. Thus, increased liver size in the LI-IGF-I -/- mice (inactivation of IGF-I gene in the liver) is a direct result of the increased GH levels. Decrease kidney size in LI-IGF-I -/- mice is also a result of the decreased serum IGF-I levels (80). In the last decade many more models of IGF-I conditional deletion alone or in combination with other mutants (with ALS knockout, GHR KO etc), have been produced to address the question of the contribution of endocrine vs. paracrine/autocrine IGF-I actions as well as the direct contributions of GH vs. IGF-I. Briefly, these models have led to the following conclusions 1. Tissue IGF-I plays an important role in prenatal and early neonatal growth 2. Over-expression of IGF-II cannot rescue the growth phenotype in IGF-I null mice 3. Endocrine or circulating IGF-I contributes approximately 30% of the adult body size but increased concentration of circulating IGF-I levels can compensate to some extent postnatal linear growth (78-83).
THE GROWTH PLATE
The most important target organ for linear growth is the epiphyseal growth plate, a layer of cartilage found in growing long bones between the epiphysis and the metaphysis. Longitudinal bone growth occurs at the growth plate by endochondral ossification, in which cartilage is formed and then remodeled into bone tissue (84-88). The cellular distribution is organized according to the stage of cell maturation and differentiation, under a tightly controlled programme, and consists of the following cell layers: germinative, proliferative, hypertrophic and degenerative (85). The germinative cell layer or resting zone consists of the stem cells that enter the proliferative cell layer organized in column following the longitudinal axis of the bone. The proliferative zone is crucial in endochondral bone formation, when the chondrocyte divides; the resulting two cells line up along the bone axis and as this process continues all columns are organized along this axis. Although it is still unclear which factor or factors determine this important spatial orientation. Recent studies using a microdissection technique into individual zones has shown that in the rat proximal tibia growth plate IGF-I mRNA expression is minimal compared to perichondrium, metaphyseal bone, muscle and liver whereas IGF-II mRNA was expressed at higher levels compared to bone and liver (85). IGF-II mRNA expression was significantly higher in the proliferative and resting zones compared to the hypertrophic zone. IGF-I and GH receptors are expressed throughout the growth plate. IGF-I and IGF-II mRNA expression is developmentally regulated; IGF-I mRNA levels increased and IGF-II mRNA levels decreased with increasing with age. These data suggest that in rodents IGF-I is not produced by the chondrocytes in the growth plate but rather by surrounding bone and perichondrium (85). Once cell division ceases these mature cells form part of the hypertrophic cell layer (86) and the final layer, the calcifying zone is where cartilaginous matrix is transformed into bone matrix.
ORGANISATION OF GROWTH
GH secretory patterns from birth until puberty
Given responsive somatotropes and adequately releasable GH pools within the anterior pituitary gland, analysis of the discrete secretory pulses of GH provides information about the frequency and amplitude of serum hormone concentration peaks. It gives insight into the neuroendocrine mechanisms that control both ultradian and circadian rhythms of pituitary hormone release (16). There is individual variability in GH concentration pulses in blood as discrete peaks but using new methodologies for GH analysis it has been possible to estimate the underlying hormone secretion rates from serial GH measurements and calculate the half- life of endogenous hormone released into the circulation (89-91). These new analytical methodologies together with the establishment of ultra-sensitive GH assays made it possible to demonstrate gender differences in the regularity of GH secretory activity in human (89), with greater irregularity in females compared to males (90).
GH dynamics: from birth to puberty
Within the first hours of postnatal life in the human markedly amplified GH secretory bursts are observed throughout day and night (92). Premature infants also exhibit marked GH secretion and reduced plasma IGF-I levels. Such amplified GH response may be due to relatively low IGF-I levels. Extensive literature exists on the pattern of GH secretion in puberty but only in the last decade studies have concentrated in the prepubertal period. In the decade before puberty 24h pulsatile GH secretion rates are stable from day to day and are estimated as 200-600ug/day (93-94). The onset of clinical and biochemical manifestations of puberty in boys and girls is associated with a marked increase in GH pulse amplitude and mass without changes in pulse frequency or changes in GH half-life (46-48; 93, 94). Studies in which either estrogen or testosterone treatment have been selectively used have shown that both cause an increase in GH released per secretory bursts. Thus, this is likely to represent the underlying neuroendocrine mechanism causing the increased GH secretion observed in puberty (46-48). This increased physiological GH secretion is associated with a significant increase in IGF-I and IGFBP-3 serum concentrations possibly all driven by sex steroid hormones. It has been postulated that this unusual event of concomitant rise in GH and IGF-I could be due to changes in central hypothalamus-pituitary and in auto-feedback sensitivity.
Growth phases
Human growth is a rather regular process characterized by a pattern of changing growth rate or height velocity from infancy to adulthood (Figure 2, 95). In the short-term the height velocity of a healthy individual fluctuates i.e. seasonal or longer periodicity has been shown (96, 97). It appears that some phases of growth are more important in their effect on final height. For example a child who is born short has a greater chance of being a short adult. The timing and duration of puberty are also crucial factors in determining final height.
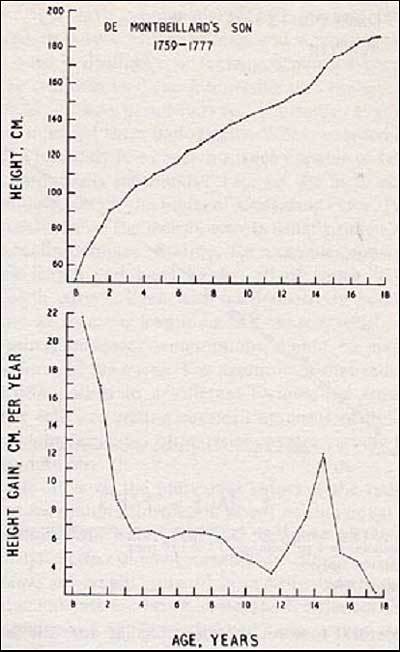
Figure 2
Growth in height of the Montebeillard"s son from birth to 18 years first described by Scanmon in 1927. Top panel: height attained at each age. Bottom panel: Growth velocity. Reproduced with permission from Tanner JM (95).
The infancy-childhood-puberty growth model divides the human growth process into three phases (95). Thus, the infancy phase begins in the middle of gestation and tails off at around 4 years of age, possibly representing the postnatal continuation of fetal growth. The childhood phase starts during the second half of the first postnatal year slowly decelerating and leading into the final phase at puberty. The puberty phase represents additional growth beyond the childhood phase with significant acceleration until the age of peak height velocity followed by a deceleration until linear growth finally ceases (Figure 3).
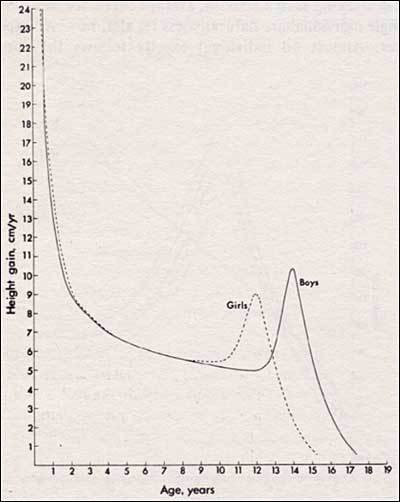
Figure 3
Growth velocity, indicating the adolescent height spurt for boys and girls. Reproduced with permission from Tanner JM (95).
Canalization and catch-up growth
The dynamic process underlying in the growth phases are under the control of complex regulatory mechanisms. The tendency to maintain a tight path in the growth channel and a prerequisite of catch-up growth was called "canalization" by Waddington while referring to the growth pattern of small animals and later adapted by Tanner in the context of human linear growth (95). In 1963, Prader et al described the phenomenon of catch-up growth observed in children after chronic illness or in those who suffered from starvation (98-100, 87). For reviews, the reader is referred to references and the article by Wit and Boersma (87) Catch-up growth is the phenomenon by which rapid growth occur after a period of illness or malnutrition allowing a child to return to the growth curve he/she had before the illness. Complete catch-up growth leads to a normal final height but incomplete catch-up growth will result in a compromised final height (87) The mechanisms underlying this important biological event still remain unclear. Tanner proposed the existence of a "time tally" a central regulatory mechanism that senses the organism"s height via a "humoral factor" (101) and which could be involved in the catch-up phenomenon. Mosier referred to it as "the age-appropriate set point for body size" (102). Although the mechanisms regulating catch-up growth remains unclear recent animal studies suggest a cross talk between central and peripheral mechanisms (87, 101) Therefore, some extrapolation from these studies appear reasonable.
OVERVIEW
Elegant studies in the last two decades established that the key hormonal axis, the GH-IGF axis, in which GH and IGF-I are produced from early fetal life play a significant role in human linear growth. More recent in vivo and in vitro studies have demonstrated that this axis is composed of hormones with highly specific binding proteins, which modulate their interactions with specific receptors. The development of the IGF system in the first trimester of gestation appears to be GH independent. However, in postnatal life GH is required for IGF-I synthesis and for the normal functional development of growth plate. It has been shown that endocrine, paracrine and autocrine mode of action both of GH and IGFs play important roles in human growth and development. The "revised somatomedin hypothesis" as described by Le Roith (51) provides the basis for a better understanding of the contribution of systemically secreted and locally acting peptides. However, there are many challenges and interactions at play, with short- and long-term consequences for human growth. Therefore, it is important to appreciate that in addition to the biological and physiological role of our genetic background, of interaction of several hormones and growth factors, other factors such as nutrition and socioeconomic status will have a significant effect in determining the attainment of a normal final height. This phenomenon, known as secular change, has been clearly shown in different populations in the world with a significant difference between current growth pattern and that observed in the early 20th century (103-104).
References
- 1.
- Matsuzaki F, Irie M, Shizume M. (1971) Growth hormone in human fetal gland and cord blood. J Clin Endocrinol Metab 33:908-911.
- 2.
- Parks JS. (1989) Molecular biology of growth hormone. Acta Paedatr Scand. Suppl 349:127-135
- 3.
- Baumann G. (1991) Growth hormone heterogeneity: genes, isohormones, variants and binding proteins. Endocrine Rev 12:424-449
- 4.
- Kaplan SL, Grumbach MM, Shepard TH. (1972) The ontogenesis of human fetal hormones. I. Growth hormone and insulin. J Clin Invest 51:3080-3093
- 5.
- Honnebier WJ, Swaab DE. (1973) The influence of anencephaly upon intrauterine growth of the fetus and the placenta, and upon gestational length. J Obstet Gyneco Br Common 80:577-588
- 6.
- Leung DW, Spencer SA, Cachianes G, Hammondss RG, Collins C, Henzel WJ, Barnard R, Waters MJ, Wood WI. (1987) Growth hormone receptor and serum binding protein: purification, cloning and expression. Nature 330:537.
- 7.
- Godowski PJ, Leung DW, Meacham LR, Galgani JP, Hellmiss R, Keret R, Rotwein PS, Parks JS, Laron Z, Wood WI. (1989) Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc Natl Acad Sci USA 86:8083-8087.
- 8.
- Hill DJ, Freemark M, Strain AJ et al. (1988) Placental lactogen and growth hormone receptors in human fetal tissues: relationship to fetal plasma hPL concentrations and fetal growth. J Clin Endocrinol Metab 66: 1283-1290.
- 9.
- Werther GA, Haynes K, Waters MJ. (1993) GH receptors are expressed on human fetal mesenchymal tissues-identification of mRNA and GH binding protein. J Clin Endocrinol Metab 76:1638-1646
- 10.
- Waters MJ, Kaye PL. (2002) The role of growth hormone in fetal development. GH and IGF Res 12:137-146
- 11.
- Caufriez A, Frankenne F, Hennen G, Copinschi G. (1993) Regulation of maternal IGF-I by placental GH in normal and abnormal human pregnancies. Am J Physiol. 265:E572-E577.
- 12.
- Chellakooty M, Vangsgaard K, Larsen T, Scheike T, Falck-Larsen J, Legarth J, Andersson AM, Main KM, Skakkebaek NE, Juul A. (2004) A longitudinal study of intrauterine growth and the placental growth hormone (GH)-insulin-like growth factor I axis in maternal circulation: association between placental GH and fetal growth. J Clin Endocrinol Metab. 89:384-91
- 13.
- Simard M, Manthos H, Giadid A, Lefebre Y, Goodyer CG. (1996) Ontogeny of GH receptors in human tissues: an immunohistochemical study. J Clin Endocrinol Metab 81:3097-3102
- 14.
- Baumann G, Stolar MW, Amburn K, Barsano CP, Devries BC (1986) A specific growth hormone-binding protein in human plasma: initial characterization. J Clin endocrinol Metab 62:134-141
- 15.
- Gofflin V, Kelly PA (1996) Prolactin and growth hormone receptors Clin Endocrinol 45:247-255
- 16.
- Giustina A, Veldhuis JD. (1998) Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocrine Rev 19:717-797
- 17.
- Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB. (1982) Growth hormone-releasing factor from a human pancreatic tumor that caused acromegaly. Science 218:585-587
- 18.
- Mayo KE, Vale W, Rivier J (1983) Expression, cloning and sequencing of a cDNA encoding human growth hormone-releasing factor. Nature 306:86-88
- 19.
- Barinaga M, Bilezikjian LM, Vale WW, Rosenfeld MG, Evans RM (1985) Independent effects of growth hormone releasing factor on growth hormone release and gene transcription. Nature 314:279-281
- 20.
- Grossman A, Savage MO, Lytras N (1984) response to analogues of growth hormone releasing hormone in normal subjects and in growth hormone deficient children and young adults. Clin Endocrinol (Oxf) 21:321-331
- 21.
- Gaylinn D, Harrison JK, Zysk JR, Lyons Jr CE, Lynch KR, Thorner MO (1993) Molecular cloning and expression of a human anterior pituitary receptor for growth hormone-releasing hormone. Mol Endocrinol 7:77-84
- 22.
- Mayo KE, Miller TL, De Almeida V, Zheng J, Godfrey PA (1996) The growth hormone-releasing hormone receptor: signal transduction, gene expression and physiological function in growth regulation. Ann NY Acad Sci 805:184-203
- 23.
- Shimon I, Yan X, Melmed S. (1998) Human fetal pituitary expresses functional growth hormone-releasing peptide receptors. J Clin Endocrinol Metab 58:212-214
- 24.
- Wajnrajch MP, Gertner JM, Harbison MD, Chua Jr SC, Leibel RL (1996) Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet 12:88-90
- 25.
- Aguiar-Oliveira MH, Gill MS, Barreto de A E, et al. (1999) Effect of severe growth hormone deficiency due to a mutation in the GH-releasing hormone receptor on insulin-like growth factors (IGFs), IGF-binding proteins and ternary complex formation throughout life. J Clin Endocrinol Metab 84:4118-4126.
- 26.
- Krulich L, Dhariwal AP, McCann SM (1968) Stimulatory and inhibitory effects of purified hypothalamic extracts on growth hormone release from rat pituitary in vitro. Endocrinology 83:783-790
- 27.
- Brazeau P, Vale W, Burgus R, Ling N, Butcher M, Rivier, Guillemin R. (1973) Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 179:77-79.
- 28.
- Shen LP, Rutter WJ (1984) Sequence of the human somatostatin I gene. Science 224:168-171
- 29.
- Yamada Y, Stoffel M, Espinosa III R, Xiang KS, Seino M, Seino S, Le Beau MM, Bell GI (1993) Human somatostatin receptor genes: localization to human chromosomes 14, 17 and 22 and identification of simple tandem repeat polymorphisms. Genomics 15:449-452
- 30.
- Miller GM, Alexander JM, Bikkal HA, Katznelson L, Zerva NT, Klibanski A. (1995) Somatostatin receptor subtype gene expression in pituitary adenomas. J Clin Endocrinol Metab 80:1386-1392
- 31.
- Roudenok B, Kuhnel W. (2001) Distribution of vasoactive intestinal polypeptide-, calcitonin gene-related peptide-, somatostatin- and neurofilament immunoractivities in sympathetic ganglia of human fetuses and premature neonates. Ann Anatomy 183:213-216.
- 32.
- Smith RG, Cheng K, Schoen WR, Pong S-S et al (1993) A non-peptidyl growth hormone secretagogue. Science 260:1640-1643
- 33.
- Korbonits M, Grossman AB (1995) Growth hormone-releasing peptide and its analogues. Novel stimuli to growth hormone release. Trends Endocrinol Metab 6:43-49
- 34.
- Casanueva FF, Dieguez C. (1999) Growth hormone secretagogues: physiological role and clinical utility. TEM 10:30-38.
- 35.
- Kojima M et al (1999) Ghrelin is a growth hormone-releasing acylated peptide from stomach. Nature 402:656-660
- 36.
- Takaya K, Ariyasu H, Kanamoto N, Iwakura H, Yoshimoto A, Harada M et al. (2000) Ghrelin strongly stimulates growth hormone release in humans. J Clin Endocrinol Metab 85:4908-4911
- 37.
- Hataya Y, Akamizu T, Takaya K, Kanamoto N et al. (2001) A low dose of ghrelin stimulates growth hormone (GH) release synergistically with GH-releasing hormone in humans. J Clin Endocrinol Metab 86:4552-
- 38.
- Bellone S, Rapa A, Vivenza D, Castellino N et al. (2002) Circulating ghrelin levels as a function of gender, pubertal status and adiposity in childhood. J Clin Endocrinol Investigation 25:RC13-15.
- 39.
- Katz HP, Youlton R, Kaplan SL, Grumbach MM (1969) Growth and growth hormone release in children with primary hypothyroidism and thyrotoxicosis. J Clin Endocrinol Metab 29:346-348
- 40.
- Chernausek SD, Turner R (1989) Attenuation of spontaneous, nocturnal growth hormone secretion n children with hypothyroidism and its correlation with plasma insulin-like growth factor-I concentration. J Pediatr 114:968-972
- 41.
- Iranmanesh A, Lizarralde G, Johnson ML, Veldhuis JD (1991) Nature of the altered growth hormone secretion in hyperthyroidism. J Clin Endocrinol Metab 72:108-115
- 42.
- Shibasaki T, Hotta M, Masuda A, Imaki T, Obara N, Demura H. (1985) Plasma responses to GHRH and insulin-induced hypoglycemia in man. J Clin Endocrinol Metab 60:1265-1267
- 43.
- Masuda A, Shibasaki T, Nakahara M, Imaki T et al (1985) The effect of glucose on growth hormone-releasing hormone-mediated GH secretion in men. J Clin Endocrinol Metab 60:523-526
- 44.
- Dieguez C, Mallo F, Senaris R,Pineda J, Martul P, Leal-Cerro A, Pombo M, Casanueva FF. (1996) Role of glucocorticoids in the neuroregulation of growth hormone secretion. J Pediatr Endocrinol Metab 3:255-260.
- 45.
- Magiakow M, Mastorakos G, Gomez M, Rese S, Chrousos G. (1994) Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing's disease before and after surgical cure. J Clin Endocrinol Metab 78:131-137.
- 46.
- Kerrigan JR, Rogol AD. (1992) The impact of gonadal steroid hormone action on growth hormone secretion during childhood and adolescence. Endocr Rev 13:281-298
- 47.
- Vandeer Werff ten Bosch JJ, Bot A (1990) Growth of males with idiopathic hypopituitarism without growth hormone treatment. Clin Endocrinol (Oxf) 32:707-717
- 48.
- Wennink JM, Delemarre-van der Waal HA, Schoemaker R, Blauw G, van den Brakern C, Shoemaker J. (1991) Growth hormone secretion patterns in relation to LH and estradiol administration throughout normal female puberty. Acta Endocrinol (Copenh) 124:129-135
- 49.
- Salmon W D, Daughaday WH (1957) A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med 49:825
- 50.
- Daughaday WH, Hall K, Raben MS et al (1972) Somatomedin: proposed designation for sulphation factor. Nature 235:107
- 51.
- LeRoith D Bondy C, Yakar S, Liu-JL, Butler A. (2001) The Somatomedin hypothesis:2001 Endocrine Rev 22:53-74
- 52.
- D'Ercole AJ, Hill DJ, Strain AJ, Underwood LE. (1986) Tissue and plasma somatomedin-C/insulin-like growth factor I concentrations in the human fetus during the first half of gestation. Pediatr Res. 20:253-255.
- 53.
- Han VK, Lund PK, Lee DC, D'Ercole AJ. (1988) Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetus: identification, characterization, and tissue distribution. J Clin Endocrinol Metab. 66:422-429.
- 54.
- Camacho-Hübner C, Clemmons DR, D'Ercole AJ (1991) Regulation of insulin-like growth factor (IGF) binding proteins in transgenic mice with altered expression of growth hormone and IGF-I. Endocrinology 129:1201-1206.
- 55.
- Camacho-Hübner C, Savage MO. (2001) Insulin-like growth factor-I deficiency. Horm Res 55 (Suppl) 1:17-20.
- 56.
- Woods KA, Camacho-Hübner C, Savage MO, Clark AJL. (1996) Intrauterine growth retardation and post natal growth failure associated with deletion of the IGF-I gene. N Engl J Med. 335:1363-1367.
- 57.
- Bonapace G, Concolino D, Formicola S, Strisciuglio P 2003 A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. J Med Genet 40:913-917
- 58.
- Walenkamp MJ, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, van Thiel SW, Sluimers CA, Bax JJ, de Laat JA, Breuning MB, Romijn JA, Wit JM 2005 Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. J Clin Endocrinol Metab 90:2855-2864
- 59.
- Netchine, I., Azzi, S., Houang, M., Seurin, D., Perin, L., Ricort, J.-M., Daubas, C., Legay, C., Mester, J., Herich, R., Godeau, F., Le Bouc, Y. 2009 Partial primary deficiency of insulin-like growth factor (IGF)-i activity associated with IGF-I mutation demonstrates its critical role in growth and brain development. J. Clin. Endocrinol. Metab. 94: 3913-3921
- 60.
- Baker J, Liu J-P, Robertson EJ, Efstriadis A. (1993) Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75:73-82.
- 61.
- Daughaday WH, Rotwein P (1989) Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum and tissue concentrations. Endocr Rev. 10:68-91
- 62.
- Berelowitz M, Szabo M, Frohman LA, Firestone S, Chu L (1981) Somatomedin-C mediates growth hormone negative feedback by effects on both the hypothalamus and the pituitary. Science 212:1279-1281
- 63.
- Chard T. (1989) Hormonal control of growth in the human fetus. J Endocrinol. 123:3-9
- 64.
- Gluckman PD. (1989) Fetal growth: and endocrine perspective. Acta Paediatr Scand. 349:21-25.
- 65.
- Jones JI, Clemmons DR. (1995) Insulin-like growth factors, and their binding proteins. Endocr Rev. 16:3-34.
- 66.
- Werner H, Adamo M, Roberts CT Jr, LeRoith, D. (1994) Molecular and cellular aspects of insulin-like growth factor action. Vitam. Horm. 48, 1-58.
- 67.
- Furlanetto RW. (1980) The somadomedin C binding protein: evidence for a heterologous subunit structure. J Clin Endocrinol Metab 51:12-19.
- 68.
- Baxter RC. (1990) Circulating levels and molecular distribution of the acid-labile (alpha) subunit of the high molecular weight insulin-like growth factor binding protein complex. J Clin Endocrinol Metab 10:1347-1353.
- 69.
- Leong SR Baxter RC, Camerato T, Dai J, Wood WI. (1992) Structure and functional expression of the acid-labile subunit of the insulin-like growth factor binding protein complex. Mol Endocrinol 6:870-876.
- 70.
- Lassarre C, Hardouin S, Daffos F, Forestier F, Frankennne F, Binoux M. (1991) Serum insulin-like growth factors and insulin-like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res. 29: 219-225.
- 71.
- Ashton IK, Zapf J, Einschenck I et al. (1985) Insulin-like growth factors (IGF) I and II in human fetal plasma and relationship to gestational age and fetal size during midpregnancy. Acta Endocrinol 110:558-563
- 72.
- Barrios V,Pozo J, Muñoz MT, Bruno M, Argente J. (2000) Normative data for total and free acid-labile subunit of the human insulin-like growth factor-binding protein complex in pre- and full-term newborns and healthy boys and girls throughout postnatal development. Horm Res 53:148-153.
- 73.
- Boisclair YR, Rhoads RP, Ueki I, Wang J, Ooi GT (2001) Regulation of the acid-labile subunit of the 150-kDa IGF-binding protein complex and its role in the circulating IGF system. J Anim Sci 79:E41-E47
- 74.
- Domené HM, Bengolea SV, Martinez AS, et al (2004) Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. N Engl J Med. 350:570-7.
- 75.
- Hwa V, Haeusler G, Pratt KL, Little B et al (2006) Total absence of functional acid labile subunit, resulting in severe insulin-like growth factor deficiency and moderate growth failure. J Clin Endocrinol Metab 91:1826-1831.
- 76.
- Domené HM, Hwa V, Argente J, Wit JM, Camacho-Hübner C, Jasper HG, Pozo J, van Duyvenvoorde HA, Yakar S, Fofanova-Gambetti OV, Rosenfeld RG; International ALS Collaborative Group. (2009) Human acid-labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm Res. 72:129-41.
- 77.
- Green H, Morikawa M, Nixon T (1985) A dual effector theory of growth hormone action. Differentiation 29:195-198
- 78.
- Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, LeRoith D. (1998) Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol Endocrinol 12:1452-1462.
- 79.
- Sjögren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO, Ohlsson C. (1999) Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but it is not requires for postnatal growth in mice Proc Natl Acad Sci 96:7088-7092
- 80.
- Yakar S, Liu J, Stannaard B, Butler A, Accili D, Sauer B, Le Roith D. (1999) Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 96:7324-7329.
- 81.
- Moerth C, Schneider MR, Renner-Mueller I, Blutke A, Elmlinger MW, Erben RG, Camacho-Hübner C, Hoeflich A, Wolf E 2007 Postnatally elevated levels of insulin-like growth factor (IGF)-II fail to rescue the dwarfism of IGF-I-deficient mice except kidney weight. Endocrinology 148:441-451
- 82.
- Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A (2008) The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci USA 105:19378-19383
- 83.
- Wu Y, Sun H, Yakar S, LeRoith D (2009) Elevated levels of insulin-like growth factor (IGF)-I in serum rescue the severe growth retardation of IGF-I null mice. Endocrinology. 150(9):4395-403
- 84.
- Kaplan SA, Cohen P. (2007) The somatomedin hypothesis 2007: 50 years later. J Clin Endocrinol Metab. 92(12):4529-35.
- 85.
- Parker EA , Hegde A , Buckley M , Barnes KM , Baron J , Nilsson O . (2007) Spatial and temporal regulation of GH-IGF-related gene expression in growth plate cartilage. J Endocrinol 194(1):31-40
- 86.
- Ohlsson C, Isgaard J, Tornell J, Nilsson A, Isaksson OGP, Lindahl RO (1993) Endocrine regulation of longitudinal bone growth. Acta Paediatr [Suppl] 391:33-40
- 87.
- Boersma B, Wit JM. (1997) Catch-up growth Endocr Rev 18: 646-661
- 88.
- Abad V, Meyers JL, Weise M, Gafni R, Barnes KM, Nilsson L, Bacher JD, Baron J (2002) The role of the resting zone in growth plate chondrogenesis. Endocrinology 143:1851-1857
- 89.
- Hartman ML, Faria AC, Vance ML, Johnson ML, Thorner MO, Veldhuis JD (1991) Temporal structure of in vivo growth hormone secretory events in man. Am J Physiol 260:E-101-E110
- 90.
- Pincus SM, Gevers E, Robinson ICAF, Van den Bergh G, RoelfsemaF, Hartman ML, Veldhuis JD (1996) Females secrete growth hormone with more process irregularity than males in both human and rat. Am J Physiol E107-E115
- 91.
- Hartman MI, Iranmanesh A, Thorner MO, Veldhuis JD. (1993) Evaluation of pulsatile patterns of growth hormone release in man. Am J Hum Biol 5:603-614
- 92.
- Ogilvy-Stuart AL, Hands Sj, Adcock CJ et al. (1998) Insulin, insulin-like growth factor I (IGF-I), IGF-binding protein-1, growth hormone and feeding in the newborn. J Clin Endocrinol Metab 83:3550-3557
- 93.
- Hindmarsh PC, Matthews DR, Brain C, Pringle PJ, Brook CG. (1990) The application of deconvolution analysis to elucidate the pulsatile nature of growth hormone secretion using a variable half-life of growth hormone. Clin Endocrinol (Oxf) 32:739-747
- 94.
- Albertsson-Wikland K, Rosberg S, Libre E, Lundberg LO, Growth T (1989) Growth hormone secretory rates in children as estimated by deconvolution analysis of 24-hr plasma concentration profiles. Am J Physiol 257:E809-E814
- 95.
- Tanner JM (1962) Growth at adolescence. 2nd Ed, Blackwell Scientific Publications, Oxford.
- 96.
- Martha Jr PM, Goorman KM, Blizzard RM, Garcia-Rubi E, Rogol AD, Veldhuis JD. (1992) Endogenous growth hormone secretion and clearance rates in normal boys as determined by deconvolution analysis: relationship to age, pubertal status an body mass. J Clin Endocrinol Metab 74:336-344
- 97.
- Mauras N, Rogol AD, Veldhuis JD. (1989) Specific, time dependent actions of low-dose estradiol administration on the episodic release of GH, FSH and LH in prepubertal girls with Turner's syndrome. J Clin Endocrinol Metab 69:1053-1058
- 98.
- Lampl M, Veldhuis JD, Johnson ML (1992) Saltation and stasis: a model of human growth. Science 258:801-803
- 99.
- Marshall WA (1971) Evaluation of growth rate in height over periods of less than one year. Arch Dis Child 46:414-420
- 100.
- Prader A, Tanner JM, von Harnack GA 1963 catch-up growth following illness or starvation - an example of developmental canalization in ma J Pediatr 62: 646-658
- 101.
- Tanner JM (1981) Catch-up growth in man. Br Med Bull 37:233-238
- 102.
- Mosier HD. (1971) Failure of compensatory (catch-up) growth in the rat. Ped Res 5;59-63.
- 103.
- Hauspie RC, Vercaauterren M, Susanne C. (1996) Secular changes in growth. Horm Res 45:8-17
- 104.
- Fredriks AM, Van Buuren S, Burgmeijer RJF, Meulmeester JF et al. (2000) Continuing secular growth change in the Netherlands 1955-1997. Pediatr Res 47:316-323.
- Body composition and physical fitness are major determinants of the growth hormone-insulin-like growth factor axis aberrations in adult Turner's syndrome, with important modulations by treatment with 17 beta-estradiol.[J Clin Endocrinol Metab. 1997]Body composition and physical fitness are major determinants of the growth hormone-insulin-like growth factor axis aberrations in adult Turner's syndrome, with important modulations by treatment with 17 beta-estradiol.Gravholt CH, Naeraa RW, Fisker S, Christiansen JS. J Clin Endocrinol Metab. 1997 Aug; 82(8):2570-7.
- Differential impact of simple childhood obesity on the components of the growth hormone-insulin-like growth factor (IGF)-IGF binding proteins axis.[J Pediatr Endocrinol Metab. 2004]Differential impact of simple childhood obesity on the components of the growth hormone-insulin-like growth factor (IGF)-IGF binding proteins axis.Ballerini MG, Ropelato MG, Domené HM, Pennisi P, Heinrich JJ, Jasper HG. J Pediatr Endocrinol Metab. 2004 May; 17(5):749-57.
- Review [The growth hormone axis and insulin-like growth factors].[Med Pregl. 2005]Review [The growth hormone axis and insulin-like growth factors].Radosavljević T, Todorović V, Vucević D, Sikić B. Med Pregl. 2005 Nov-Dec; 58(11-12):558-62.
- Mechanisms of thyroid hormone action on the insulin-like growth factor system: all thyroid hormone effects are not growth hormone mediated.[Endocrinology. 1993]Mechanisms of thyroid hormone action on the insulin-like growth factor system: all thyroid hormone effects are not growth hormone mediated.Näntö-Salonen K, Muller HL, Hoffman AR, Vu TH, Rosenfeld RG. Endocrinology. 1993 Feb; 132(2):781-8.
- Review Normal Physiology of Growth Hormone in Adults.[Endotext. 2000]Review Normal Physiology of Growth Hormone in Adults.Olarescu NC, Gunawardane K, Hansen TK, Møller N, Jørgensen JOL. Endotext. 2000
- Normal Physiology of Growth Hormone and Insulin-Like Growth Factors in Childhood...Normal Physiology of Growth Hormone and Insulin-Like Growth Factors in Childhood - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...