Study design
The study had two stages. Stage 1 was a cross-sectional study to establish the prevalence of a range of vision problems among people with dementia. Stage 2 was a qualitative study that used focus groups and interviews to explore and describe issues around detection and management of vision problems among people with dementia from the perspectives of affected individuals, family carers, professional care workers and optometrists.
Stage 1 was an observational cross-sectional study. PrOVIDe endeavoured to follow the STROBE statement for reporting of observational studies wherever possible. The STROBE recommendations have been developed to improve the quality of reporting of observational studies.89 An annotated version of the STROBE checklist, giving details of PrOVIDe’s compliance with each item, can be found in Appendix 1.
Throughout the report, the term ‘potential participant’ is used to describe individuals who were approached by a member of the research team for involvement in the study. The term ‘participant’ is used to describe individuals who were formally recruited into the study. The term ‘family carer’ is used to describe a family member or friend who cares, unpaid, for their relative or friend. The term ‘care worker’ is used to describe someone who is employed to support individuals with everyday tasks, in their own homes or in residential care settings. The term ‘formal consultee’ has been used to describe a personal consultee who was required to give approval on behalf of individuals who lacked mental capacity to provide informed consent to participate. The term ‘informal consultee’ refers to a family member or personal carer whose opinion was sought regarding an individual’s participation in the study; this was applicable for individuals who had the capacity to provide informed consent to participate. An informal consultee role was sought for all potential participants and participants to provide additional assurance that participation in the study was supported by an independent source concerned with the interests of the participant.
Setting
Participants were recruited from 20 NHS sites in six regions of England. Sites were selected to ensure the recruitment of participants living in rural, urban and city locations and to encourage the participation of people from black and minority ethnic groups. It was originally envisaged that sites in four regions would be included: East Anglia, the North East, North Thames and Thames Valley. Two more regions, Yorkshire and the Humber and the North West, were added later when recruitment rates were lower than predicted in the original regions and concerns were raised that the recruitment target might not be achieved within the time scale. The regions referred to were those defined by the NIHR Dementias and Neurodegenerative Diseases Research Network (DeNDRoN), which subdivided England into nine regions.
Regions and sites were selected in consultation with DeNDRoN. Local network co-ordinators identified local principal investigators willing to support the study and then advised the research team on the numbers of participants that they predicted could be recruited in the time frame, and site-specific recruitment targets were agreed.
Approvals
The study received NHS ethics approval in September 2012 in advance of the scheduled start date. NHS research and development approval was sought from the NHS sites including primary care trusts where participants would be recruited from care homes. The first sites granted approval in November 2012, with the remaining sites gradually joining the study over the next 3 months. Recruitment began in November 2012.
The Association of Directors of Adult Social Services (ADASS) was consulted on whether or not social care research ethics approval was also required for recruitment through care homes. ADASS concluded that NHS ethics approval was sufficient but advised the study team to write to the directors of adult social care in all (n = 47) participating local authorities to inform them of the study and offer additional information if required. One authority declined to support the study.
Sampling
Stage 1
The study sample was people with dementia (any type) aged 60–89 years. To ensure that the spectrum of care was represented, the sample included people living in their own homes and people living in residential care (such as care homes and hospital inpatient wards). An additional inclusion criterion was that, in the case of individuals lacking mental capacity to provide informed consent to participate, a formal consultee was required who could give approval on the individual’s behalf. The opinion of a family member or professional carer in the role of informal consultee was sought in respect of all participants as to whether or not participation would be appropriate. The exclusion criteria were as follows:
Individuals who had been in hospital in the preceding 2 weeks following an acute illness, or who had major delirium or a major infection.
Individuals unable to understand English, as consent procedures and the eye examination were conducted in English.
Individuals unable to comply with the requirements of the eye examination; although it was expected that some participants would not be able to comply with all elements of the examination, individuals were excluded if they were unable to co-operate with the simplest procedures.
Individuals participating in a clinical drugs trial, because the eye examination involved the administration of tropicamide eye drops (Mydriacyl, Alcon Laboratories Ltd, Surrey, UK) and all of the potential drug interactions could not be determined. This exclusion criterion was added on completion of the pilot study.
Sample size
As described in Chapter 1, the prevalence of conditions causing VI has been estimated to be as high as 50%.44 For the stage 1 cross-sectional study, a sample size of 385 was required to allow detection of an estimated prevalence of 50% with 5% precision and 95% confidence. If the prevalence of a condition was greater than or less than 50%, the study would have required a sample size smaller than 385. It was anticipated that some participants would be unable to perform all of the tests undertaken in a standard optometric examination, but there were no published data on the percentages of those with dementia who would be unable to perform individual elements of the examination. In the absence of published data, a decision was made to assume a worst-case scenario that up to 50% of the subjects might be unable to complete a particular test. This increased the maximum sample for stage 1 to 770 (385 × 2).
This led to a decision to divide the sample into two groups: group 1 was people living in their own homes and group 2 was people living in care. Although the group sizes were not defined to specifically support ‘between-group’ comparisons, the use of equal numbers in the two groups would provide scope for some comparative analysis between groups 1 and 2. With the determined sample size, there would be at least 80% power to detect a difference between proportions of 50% and 42% (or less) in two equal-sized groups. The initial sampling strategy for both groups was to stratify by age and sex to match as closely as possible the best estimates available at the time for the general dementia population. In both group 1 and group 2 the PrOVIDe target sample was one-third male and two-thirds female; 30% of males were aged between 60 and 74 years and 70% of males were aged between 75 and 89 years; and 15% of females were aged between 60 and 74 years and 85% of females were aged between 75 and 89 years.
Stage 2
The population under study in stage 2 was extended beyond people with dementia to include family carers, care workers working in care settings and optometrists. This process of triangulation – posing similar questions to different sources – was employed to increase the validity of findings. Stage 1 participants who had mental capacity to consent themselves into the study and who were able to participate in an interview were considered for inclusion in stage 2. Purposive sampling was applied to identify participants across the age range, of both sexes, and to include individuals who might be particularly informative because of a history of eye disease.
Family carers were also identified through their involvement in stage 1 in that they were the named next of kin or the informal consultee of stage 1 participants. The method of data collection with family carers was focus groups, and the named carers of stage 1 participants living in an area where focus groups were planned were invited to participate. Similarly, the care workers approached for stage 2 worked in care homes that had assisted with recruitment of stage 1 participants.
Optometrists were sampled using the membership of the College of Optometrists (CoO). The College had 10,086 members representing in excess of 70% of the members of the profession registered to practise. The method of data collection used for optometrists was focus groups. All College members living or working in an area where focus groups were planned were invited to participate.
Recruitment of participants
In stage 1, the initial approach to potential participants was made by a member of the direct care team, a member of DeNDRoN, local co-ordinating research staff or a member of the CoO research team. The aim of this initial contact was to inform the potential participant and carers of the study and its remit. This was achieved by direct face-to-face contact (e.g. in a clinic), by telephone or by letter. Potential participants and carers were provided with written information on the study’s purpose, methods and risks, and what was required of participants.
Subsequently, potential participants and personal and professional carers were contacted by either a DeNDRoN staff member or a member of the research team to determine if the potential participant and family member/carer were willing to be involved in the study. They were also given the opportunity to ask any questions or raise any concerns they might have had at that stage. Potential participants were given a minimum of 1 week to consider their possible participation in the study. If the potential participant and carer felt confident and comfortable to participate in the study, consent forms were completed.
Ethically, and for validity of the findings, it was important and appropriate to include participants who lacked mental capacity to consent within the tenets of the Mental Capacity Act (2005).90 In recognition of the complexities surrounding informed consent and capacity in people with dementia, all research workers involved in the study received formal NIHR Clinical Research Network training in good clinical practice, informed consent, the ethics of consent and the Mental Capacity Act.90 In the case of individuals unable to consent for themselves, a formal consultee (family member/dependant/friend) was asked for their opinion. The formal consultee was asked to take into account any advance decisions or previously expressed wishes and feelings, and consider the potential participant’s best interests. In group 1, 67.9% of participants were able to consent themselves into the study and 32.1% required a consultee. In group 2, 27.6% were able to consent themselves into the study and 72.4% required a consultee. The consultee was provided with an information sheet, containing the same information as that for a participant able to consent for themselves, and was asked to sign a consultee declaration form. As an additional measure to safeguard participants’ interests, for participants with the capacity to consent an informal consultee was asked to give their opinion on the potential participant’s suitability to take part in the study.
In stage 2, potential participants were contacted by a member of the research team. During stage 1, the examining optometrist assessed participants for their ability to cope with an interview, and those who were considered to be able to do so were asked if they were willing to be contacted about stage 2. Individuals who were subsequently selected through purposive sampling were initially telephoned by a member of the research team to check their willingness to consider participation in stage 2. This was followed by a letter of invitation and further written information about what the interview would entail. A second telephone call was made at least 1 week later to answer any questions and, if the individual agreed, to set a date and time for the interview to take place. The appointment was confirmed with a follow-up letter. Formal consent was obtained on the day of the interview prior to commencing data collection.
Potential focus group participants (family carers and optometrists) were sent a letter of invitation and an information sheet about the focus groups. The sheet included details of how to contact the research team with any queries. Those wishing to attend the focus groups indicated this by returning a contact form (by fax, e-mail or prepaid reply envelope). On receipt of this form, an acknowledgement was sent to the participant, together with further details about the focus group venue. Non-responders were not contacted again.
Early pilot work suggested probable difficulties in recruiting care home care workers to attend focus groups. Only one focus group was arranged, with the assistance of a care home group manager. Additional data from care workers were collected through interviews. With assistance from stage 1 recruiters, purposive sampling was used to identify and target care homes that might be willing to take part. The initial invitation to participate in stage 2 was made by a letter accompanied by a participant information sheet. Formal consent to take part in focus groups or interviews was taken on the day of the data collection procedure, prior to any data collection taking place.
Data collection
Stage 1
Stage 1 comprised three elements:
a sMMSE
functional and behavioural assessment questionnaires completed by a carer
an eye examination, that is, a full optometric examination in line with requirements of the GOS sight test and the CoO guidance document C4.
91
Standardised Mini-Mental State Examination
The MMSE is a tool used by clinicians to help them diagnose and assess dementia. It was originally developed by Marshall Folstein92 in 1975. The MMSE is available in several versions. A standardised version (sMMSE) of the test was used for the PrOVIDe study. The sMMSE consists of a series of 12 questions or tests designed to assess orientation, memory, attention and calculation, recall and language. Each element is scored and a maximum score of 30 is possible. The study recruiter carried out sMMSE on the day that the participant was consented into the study, once consent had been obtained.
Functional and behavioural assessment questionnaires
Two questionnaires were used to provide background information on the participant’s functional and behavioural state. This allowed comparison between cases with and without VI, as well as providing background information on individuals for whom it was not possible to perform certain elements of the eye examination.
The two questionnaires employed were established tools: the Bristol Activities of Daily Living Scale (BADLS)93 and the Cambridge Behavioural Inventory – Revised (CBI-R).94 The BADLS was selected as it is a well-established, validated, carer-rated instrument. It was selected as a means of gathering additional information about the extent to which participants’ dementia impacted their day-to-day life. It was considered to offer scope for some evaluation of the interactions between dementia, vision and daily living activities. The BADLS assesses the performance of 20 activities of living, using four statements describing different levels of performance and a fifth ‘not applicable’ option. The respondent is asked to select the statement that most closely describes the individual’s performance over the preceding 2 weeks.
The CBI-R was selected to provide scope to conduct analyses to explore the possible relationships between vision, vision problems and the levels of cognitive impairment encountered within the sample population. The CBI-R describes behavioural changes using 45 items arranged into 10 sections. It is designed to be completed by a carer who states the frequency of a behaviour using a five-point scale ranging from ‘never’ to ‘constantly’. If a question does not apply, a ‘not applicable’ response should be used.
The eye examinations
Each optometrist aimed to perform a full optometric examination in line with the requirements of the GOS sight test and professional guidance.91 Participants were offered a choice between having their eye examination in an optometric practice (normal community setting) and having it in their own home (domiciliary examination). A number of community optometrists volunteered to assist with the study by performing practice-based eye examinations. However, the number was small for the wide geographical spread of the recruitment area, which meant that most study participants would have been required to travel some distance to their appointment if they wished to have an examination in an optometric practice. All study participants chose to have a domiciliary eye examination.
Examinations were carried out by an optometrist from The Outside Clinic (TOC), a company specialising in providing domiciliary eye care nationwide. During recruitment of optometrists, a number of TOC optometrists expressed interest. TOC was willing to support its optometrists interested in working on PrOVIDe; it extended this support to allow the study to use TOC software to gather participants’ data from the eye examinations and its experienced logistics team to co-ordinate the arrangement of appointments.
Further factors that contributed to the research team’s decision to involve TOC included the rigorous training its staff undergo, the clearly evidenced and rigorous audit and clinical governance mechanisms used by the company, its bespoke tablet-based electronic patient data-management system, and its familiarity with working in Clinical Commissioning Groups across England. The strong clinical governance framework in place at TOC provided assurance that the research data collected were reliable, valid, comprehensive and consistent. The record-keeping system for clinical records was electronic, facilitating data extraction and analysis for the study.
The Outside Clinic’s optometrists are trained and experienced in dealing with people with illness or disability, including people with dementia, and all had the necessary and specialised equipment for conducting eye examinations in the home. Following several meetings and a review of TOC’s processes, systems and clinical governance framework, it was determined that working with TOC and its optometrists would offer significant advantages to PrOVIDe.
Fourteen optometrists were recruited to perform the eye examinations and data collection for the study. Three optometrists were required in each of the initial four regions to cover the wide geographical areas in which participants lived. Two more were recruited later to cover the recruitment of participants from the two additional regions. The optometrists received additional training from the PrOVIDe project manager on study-specific requirements. Additional tests that the optometrists were required to perform on study participants included using the logMAR chart for recording vision and VA and the use of a specific cataract grading scale when grading cataracts.
Participants and family members/care workers received advance notice of the optometrist’s visit. They were contacted 3–5 days before the appointment to confirm the suitability of the appointment date. If the date was not suitable, TOC sent another appointment date by post. On the day of the examination, the examining optometrist telephoned the named point of contact approximately 1 hour before the appointment to give an accurate time of arrival.
On arrival at the participant’s place of residence, the optometrist introduced him- or herself to the participant and family member or carer, explaining who they were and the reason for their visit. The optometrist set up his or her equipment in the room allocated for the eye examination (in care homes) or in the most appropriate available room (in the participant’s own home). Where there was a choice of rooms, the ability to achieve dim illumination was a major factor in the final selection.
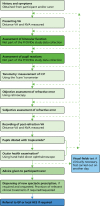
Prior to the eye examination commencing, participants were asked if there were any tasks (vision related) that they found particularly difficult. Where a participant was unable to respond appropriately, advice was taken from carers and/or care home staff as to the cognitive and other abilities of the patient. The tests constituting the eye examination were normally conducted in the order described in , but the over-riding principle was that the optometrist should adopt a flexible approach throughout, and the order of tests was adapted as required to suit the individual patient. The examinations were performed according to the cognitive and physical abilities of the participant, with the emphasis on objective assessments and omitting tests (both objective and subjective) as required by the needs of individual participants. This approach was consistent with the current CoO professional guidance on examining patients with dementia,91 although not all examination data were required for the PrOVIDe study.
Elements of the eye examination. a, The ocular health examination and pupil dilatation were able to be carried out at any of the points indicated by the dashed arrows: their place in the order of examination elements was not rigidly fixed.
Tests carried out in a typical PrOVIDe eye examination
History and symptoms
These were obtained from the participant and/or the carer. For care home residents, the optometrist requested access to the participant’s care record or asked a member of staff to provide the relevant information from the record. Where necessary, input was sought from the carer or care home staff regarding the participant’s history and symptoms. Information was gathered regarding the participant’s current visual status and spectacle wear, their general health, previous ocular history and family general health.
Vision and visual acuity
Distance vision
Distance vision and VA were recorded using either the Thomson Test Xpert 3Di Test chart (Thomson Software Solutions, Welham Green, UK) or TOC’s own computerised test chart. Both test charts allow the presentation of letters using either logMAR or Snellen progression of letter sizes. The optometrist would normally use a 3-m testing distance, and if shorter testing distances were required both test charts would adjust the letter size to compensate for the distance at which the chart was used. No mirror was used during VA testing in this study. LogMAR VA was assessed using letters from the Roman alphabet, with five letters on each line of letters presented. However, the optometrist would attempt to obtain vision and VA measurements using symbols if standard letters did not prove successful. Vision and VA were measured on a letter-by-letter basis.
The procedure followed by the research optometrists when deciding when to terminate the acuity measurement is described in the following example. The participant in this illustrative example correctly identified three of the five letters on the smallest line of letters that they could read (i.e. all of the letters on the next smallest line of letters were read incorrectly or could not be read at all). The optometrist would then present a different line of five letters of the same size and check how many letters the participant could read on this second line. If this number was again three letters out of five, the acuity measurement would terminate at that point for that eye. However, if the participant read a different number of letters on this second line of letters of the same size, for example four letters, the optometrist would present a third line of five letters of the same size. If three letters were read on this third line, the acuity measurement would be terminated, and three letters on this line would contribute to this participant’s VA. If four letters were read on this third line, the acuity measurement would be terminated and four letters on this line would contribute to this participant’s VA. In summary, the optometrist would always check the ability of the participant to correctly identify letters on the lowest line of letters that they could read by presenting a second line of letters of the same size, and would present a third line (or more) of letters of that size if required and if co-operation allowed. The aim of this approach was to seek confirmation of the number of letters the participant correctly identified on the lowest line of letters they could read. However, with the PrOVIDe participant base it was unlikely that these methods would be successful with every participant. Co-operation levels would vary between participants, and could vary in a single participant during the eye examination. Therefore, the optometrist was instructed to take a pragmatic approach to the recording of vision and VA, using their professional judgement to decide when a test should be terminated if co-operation was no longer adequate to justify further assessment.
Whenever participant co-operation permitted, the optometrist measured the distance vision (if no previous spectacles were available) or distance VA with the participants’ existing spectacles and with the prescription determined during the PrOVIDe examination. The optometrists recorded vision and VA for the right and left eyes separately. This allowed the prevalence of VI to be calculated based on best monocular VA.
Near vision
NVA was measured by means of a conventional eye examination approach using a standard Faculty of Ophthalmologists near-vision chart. The near-point scale used in these charts does not have a consistent progression between sizes of print, and in this respect it is similar to Snellen charts used for distance VA measurement. For example, the difference in size between N6 print and N5 print on the near vision chart is 1.2×, while the difference between N24 and N18 is 1.33×.
Near visual acuity was recorded as the smallest print size a participant could read at their preferred reading distance. Angular near vision reading acuity, which would require measurement of the smallest print size read at a fixed working distance with a fixed addition, was not recorded in this study. Rather than specify a particular number of words or lines of print that had to be read in order to give credit for that print size, the optometrists regarded the participant as being able to read a line of print if that line of print could be read fluently. All participants were encouraged to read the smallest size of print possible. The optometrist always aimed to provide optimum NVA at the preferred near working distance. Letters were available on the back of the near vision chart for those participants unable to read words.
Whenever participant co-operation permitted, the optometrist also measured NVA with the participants’ existing spectacles for near vision. The optometrists recorded NVA for the right and left eyes separately. This allowed the prevalence of presenting near-vision loss to be calculated based on best monocular VA.
Binocular function
This was assessed using the cover test and by testing ocular motility. The cover test is a dissociation test in which each eye is covered in turn while the patient fixates on a specified target at a given distance. The practitioner observes the eye movements and diagnoses the anomaly, if any. The ocular motility test is used to assess the extraocular muscles and their associated neural pathways. These tests were included in the eye examination but were not part of the PrOVIDe study.
Pupil reactions
The integrity of the pupillary pathways is assessed by shining a torch into each of the participant’s eyes in turn. Testing pupil reactions was included in the eye examination but was not part of the PrOVIDe study.
Tonometry
This is performed to measure the IOPs in the eyes. Tonometry is an objective measurement during which the only requirement from the participant is to keep their eyes open and maintain reasonably steady fixation. This test is used as part of a battery of tests (the others being visual field examination and optic nerve head examination) to detect glaucoma. The Icare tonometer was used (Icare Finland Oy, Vantaa, Finland). The Icare is a contact technique (i.e. involves contact between the tonometer probe and the cornea) that can be performed without the use of a topical anaesthetic. A comparison between the Icare tonometer and the Goldmann applanation tonometer (Haag Streit AG, Bern, Switzerland), the current reference standard method of measuring IOP, concluded that there is a good agreement between the two methods of IOP measurement. In addition, the Icare tonometer is easy to use and records consistent readings rapidly, with minimal training required.95 The TOC optometrists working on the study were all familiar with the Icare and used this device routinely prior to their participation in the project.
The objective and subjective determination of refractive error
This allows the strength of the spectacle prescription to be determined. Retinoscopy is an objective technique used by optometrists to determine the refractive error of the eye by observing the movement of light reflected from the fundus. It was attempted in all the study examinations to determine the prescription objectively. Wherever possible, the retinoscopy result was modified subjectively using a conventional monocular subjective refraction with the patient reading letters or symbols presented on the logMAR test chart.
On completion of the determination of each participant’s refractive error (their spectacle prescription), the distance VAs for the right and left eyes were recorded (whenever participant co-operation permitted) and, in most cases, a binocular VA (VA with both eyes together) was also recorded. The participant’s working distance for doing near tasks was recorded and informed the determination of the near addition, which is the extra element of power required by older people to provide comfortable, clear vision for near visual tasks. This initial estimate of near addition was refined, where possible, and NVAs were recorded.
Ocular health examination
At this stage in the test the participant’s pupils were dilated using tropicamide eye drops to facilitate the examination of the eye’s structures behind the iris, notably the lens, the retina and other back-of-the-eye structures. Both 0.5% and 1% Tropicamide Minims® (Bausch + Lomb, Surrey, UK) (single-dose containers) were available to the optometrist, who chose the concentration based on their clinical assessment of the participant. An examination of the anterior eye (eyelids, cornea, conjunctiva and iris) would take place prior to examination of the posterior surfaces of the eye. Prior to pupillary dilatation, the likelihood of the pupil dilatation provoking an acute angle-closure glaucoma (ACG) attack (see Glossary) was assessed using the pen light test for anterior chamber depth estimation.96 The risk of provoking an acute ACG attack with tropicamide is very low, with zero cases of ACG reported in almost 4000 dilatations.97 However, participants judged at risk of an ACG attack were not dilated and were examined through undilated pupils.
Following the instillation of tropicamide eye drops, the participant’s pupils were normally sufficiently dilated after approximately 20 minutes. Tropicamide is a drug that can reduce accommodation (the ability to alter the focus of the eyes) but PrOVIDe participants were aged > 60 years and would therefore have no accommodation. Once the pupils were fully dilated, the examination of the fundus followed, using a direct hand-held ophthalmoscope. As a further check on the possibility of a dilatation-induced acute ACG attack, post-dilatation IOPs were measured using the Icare tonometer. A significant increase in IOP could be an indicator of a pending ACG attack, and if this was suspected the optometrist remained and monitored the participant for a further hour.
Optometrists use a variety of methods to grade the severity of AMD, diabetic retinopathy and cataract. For this study, AMD was classified into dry and wet (neovascular) AMD and then graded as mild, moderate or severe. Diabetic retinopathy was graded as background diabetic retinopathy; mild, moderate or severe non-proliferative retinopathy; proliferative retinopathy; and diabetic maculopathy. When calculating the prevalence of AMD, diabetes and glaucoma in this study, a participant was considered as being positive for the condition irrespective of the severity recorded by the optometrist.
Optometrists regularly detect and monitor patients with different types and grades of cataract. The three most common types of age-related cataract (nuclear, posterior subcapsular or cupuliform, and cortical) are often graded by community optometrists as mild, moderate or severe. In addition to this grading, a sketch of the lens changes observed may form part of the clinical record. Some community optometrists use more sophisticated slit-lamp based grading systems for cataract, such as The Lens Opacities Classification System (LOCS) III.98 Systems such as LOCS III, also often used in research studies, require the use of a slit-lamp biomicroscope. The slit lamp normally used in community optometric practices is a bulky piece of equipment, unsuitable for domiciliary use, and was not available for PrOVIDe.
PrOVIDe optometrists used the cataract grading section of the Optometric Grading Scale () as the main method for grading cataract for PrOVIDe participants. The research team contacted the designer, Mr RM Pearson, for approval to use the Optometric Grading Scale99 when grading cataracts in study participants. Free-text descriptions and diagrams were also used if necessary.
Optometric Grading Scale for grading age-related cataract. © Richard M Pearson. Reproduced with permission.
Visual fields
This was by far the most demanding of the tests attempted in the study eye examination. In the first half of the recruitment and data collection period, optometrists were advised to request this test for all participants for whom the test was clinically necessary or if the optometrist felt that the participant would be able to understand and complete the test. This requested test was carried out by a member of TOC’s field staff team on a different day from the actual eye examination. The visual field tests were conducted using either the Humphrey® Frequency Doubling Technology perimeter (Carl Zeiss Meditec AG, Jena, Germany) or the OCULUS Easyfield® perimeter (OCULUS Optikgeräte GmbH, Wetzlar, Germany). In the second half of the data collection period, optometrists were advised to return to their normal practice of requesting the test only when it was clinically necessary, owing to practical and logistical difficulties encountered when arranging the fields appointments and the additional burden on participants of having a second visit.
Conclusion
The examination concluded with verbal and written advice to the participant and/or carer based on the optometrist’s findings. This included advice on any referral required, on the use of the participant’s existing or any new spectacles or other treatments advised, and on the recommended date of the next eye examination. Information explaining the effects of the tropicamide drops, including the symptoms and signs of an ACG attack and advice on the action to take in the unlikely event of this occurring, were printed on the form containing the spectacle prescription.
Where participant co-operation was poor, the optometrist moved the ocular health examination forwards in the eye examination. This allowed the optometrist to focus on ophthalmoscopy through dilated pupils whenever possible, which is the key test for detection of eye disease and does not require high levels of participant co-operation.
Optometrists asked carers for their views on whether or not the extent to which a participant was able to co-operate with the examination reflected his or her usual state. These views were noted on the record card.
Participants, their relatives and carers were informed in writing of how to raise any concerns they might have regarding the study or any aspect of the eye examination. Concerns were reported initially to the PrOVIDe research team, who liaised with TOC and recruiters as required. All concerns or complaints were documented and addressed.
Pilot study
Recruitment of participants for the pilot study commenced at the end of November 2012 and the main study began on 15 February 2013. The pilot largely involved group 1 participants, as there were some outstanding approvals relating to group 2 participants. Data from the pilot are not presented separately in this report. Feedback from recruiters during the pilot study led to minor changes being made to procedures relating to arranging and scheduling eye examinations, but no changes were made to data collection.
A recruiter query arose during the pilot study about whether or not to include subjects who were already taking part in other studies involving clinical drug trials of pharmaceutical products. This was discussed at the Steering Group meeting held at the end of the pilot study; the group agreed a modification to the exclusion criteria to exclude those participating in clinical trials, because the possible interactions between trial drugs, particularly if a new product, and the tropicamide eye drops were unknown.
Main study
As there were no changes to data collection after the pilot study, the eye examination data collected from the 21 participants who took part in the pilot were used in the main study; therefore, the eye examination data collection period began in December 2012 and finished in April 2014.
Stage 2
Qualitative data collection techniques were employed in stage 2 through a combination of focus groups and interviews. Focus groups are particularly useful when the aim of data collection is to bring together individuals with a shared knowledge to explore common or contrasting opinions and experiences.100 For this reason, focus groups were employed with two populations: family carers of people with dementia, and optometrists. With any population a minimum of three focus groups is recommended to reduce selection bias,101 and in this study there was a total of 10 focus groups from these two populations: five with family carers and five with optometrists.
Each focus group lasted for between 1 and 1.5 hours. The key question areas were the frequency and relevance of sight tests for people with dementia, reported experiences of eye care and barriers to eye care. The focus group question schedules are in Appendix 2.
Focus groups were also planned with professional carers. The recommended number of participants is a minimum of four and a maximum of 12, but pilot work with care homes indicated that there would be difficulties in recruiting a sufficient number of participants. Indeed, it was possible to arrange only one focus group, and so interviews were used to elicit the views of additional care home representatives. Each interview was conducted face to face by the same interviewer using a semistructured interview schedule, which can be found in Appendix 3.
Interviews were also used to collect the views of people with dementia. Data saturation can usually be achieved with around 30 interviews.102 In this study, 36 interviews were conducted with an equal number of male and female participants, covering the full sample age spectrum of 60–89 years. Interviews took place in participants’ homes and participants were invited to have a carer present if they wished. Nearly all interviewees (n = 30) exercised this option, which proved to be helpful, as carers were able to answer recall questions which participants found difficult. Carers often also offered their own contributions to the interview discussion, thereby providing additional data. The length of interviews varied but usually ranged between 15 and 30 minutes; the variation was largely dependent on the individual’s ability to respond as a result of their cognitive state and the extent of the carer’s contribution to the discussion. Focus groups and interviews were audio-recorded and then transcribed in preparation for data analysis.
Interviews with people with dementia were conducted over a 12-month period between May 2013 and April 2014. Two focus groups were arranged in November 2013 to facilitate the piloting of the sampling and recruitment procedures and the focus group schedule. The remaining eight focus groups with family carers and optometrists took place between February and May 2014. Data collection with care workers was completed in June 2014.
Stage 1 data analysis plan
Study outcomes
Distance vision
The calculation and analysis of the prevalence of the different types of VI in our sample addressed several of the PrOVIDe study objectives, namely primary objectives 1 and 2, and secondary objectives 1, 2 and 3 (see Chapter 1, Aims and objectives). In PrOVIDe, the three types of VI investigated were as follows:
Presenting VI, based on best monocular VA recorded with the participant wearing their current distance spectacles or unaided if no spectacles were worn for distance vision.
Post-refraction (or best corrected) VI, based on best VA recorded with the participant wearing the prescription determined by the PrOVIDe optometrist.
Uncorrected or undercorrected VI, which includes those participants who had presenting VI but who were not visually impaired wearing the prescription determined by the PrOVIDe optometrist.
As discussed in Chapter 1 (see Visual impairment), a variety of definitions of VI have been used in prevalence studies. All of these definitions use criteria for VI based on levels of VA as recorded on Snellen progression VA charts. The two most commonly used cut-off points used to define VI in terms of VA are (1) VA < 6/12 Snellen and (2) VA < 6/18 Snellen. The logMAR equivalent of 6/12 Snellen is 0.30, so a VA of < 6/12 is equivalent to 0.32 or worse logMAR acuity, and this was one of the cut-off points taken in the PrOVIDe study to define VI. There is no full logMAR line equivalent to 6/18 Snellen; reading all of the letters of a 0.5 logMAR line is equivalent to 6/19 Snellen, and reading all the letters on the 0.4 logMAR line is equivalent to 6/15 Snellen. The logMAR equivalent of 6/18 Snellen is 0.4771, so in mathematical terms a logMAR acuity of 0.48 is worse than 6/18 Snellen. Therefore, 0.48 or worse logMAR acuity was the second cut-off point used in the PrOVIDe study to define VI.
For distance vision, our data analyses included descriptive statistics of VI prevalence in the whole sample and for groups 1 and 2 separately, with tests for differences in proportions between groups. Multivariable ORs were calculated for each type of VI by age, sex and group, and for uncorrected/undercorrected VI by sMMSE score. Prevalence data were presented using descriptive statistics for our four target eye conditions (AMD, cataract, diabetic retinopathy and glaucoma) for the whole sample and for groups 1 and 2 separately, with tests for differences in proportions between groups. Multivariable ORs were calculated for each eye condition by age, sex and group. The clinically determined causes of post-refraction VI were presented using descriptive statistics. The proportion of carers providing support to the participant with dementia during the eye examination was tabulated for the whole sample and for groups 1 and 2 separately, with tests for differences in proportions between groups. Improvements in VA between pre and post refraction were presented for the sample as a whole and for groups 1 and 2 separately, with tests for the statistical significance of any improvements between groups.
Near vision
The two cut-off points in terms of NVA used to define impaired near vision were (1) best monocular NVA of worse than N8 and (2) best monocular NVA of worse than N10. Our analyses included descriptive statistics of the prevalence of near vision loss in the whole sample and for groups 1 and 2 separately, with tests for differences in proportions. Multivariable ORs were calculated for each type of near vision loss by age, sex and group, and for uncorrected/undercorrected VI by sMMSE score. Improvements in NVA between pre and post refraction were presented for the sample as a whole and for groups 1 and 2 separately, with tests for the statistical significance of any improvements between groups.
Ability of participants to complete individual elements of the eye examination
This was secondary objective 2 of the study (see Chapter 1, Aims and objectives). Our data analyses included descriptive statistics of the proportions of the whole sample able to complete each key test in the eye examination, and proportions for groups 1 and 2 separately, with tests for differences in proportions between groups. Multivariable ORs were calculated for each key test with age, sex and group as covariates to assess their independent associations with the completion of each examination component. Any association between participants’ level of cognition, as assessed by their sMMSE score, and their ability to complete key elements of the eye examination was summarised using descriptive statistics and multivariable ORs calculated for each test, with age, sex and group as covariates.
Patient management on completion of the eye examination
Descriptive statistics were presented for the proportions of the sample referred to participants’ GP or to the HES.
Analysis of the effects of visual impairment on function (Bristol Activities of Daily Living Scale) and behaviour (Cambridge Behavioural Inventory – Revised)
The results from the visually impaired and non-visually impaired participants were compared for both instruments and ORs were derived, with age, sex and group as covariates. This was secondary objective 3 of the study (see Chapter 1, Aims and objectives).
Statistical methods
In stage 1, for descriptive statistics, the mean and SD were used to describe data that were close to normally distributed, and the median and interquartile range (IQR) were used to describe data that were strongly non-normally distributed. All relevant data reported in the text of Chapter 3 are in the following style ‘count/percentage (95% CI)’ and will appear as ‘N/N, PP% (PP to PP)’. When data are stated as proportions in tables, the 95% CI is given. Pearson chi-squared tests were performed to test the difference between group proportions, using Yates’ continuity correction when appropriate (two-group comparisons), and through Monte Carlo simulation (empirical distribution) when the number of cases in a given group was low. When the results of chi-squared tests are reported, the degrees of freedom have been recorded where appropriate. Logistic regression was used to calculate adjusted ORs when attempting to control for covariates and when investigating independent effects on binary outcomes. For our exploratory analyses of VI and behavioural and functional ability, ordinal logistic regression was used in some of the item responses to the BADLS and CBI-R instruments. We have used ‘binary logistic regression’ and ‘logistic regression’ synonymously, as is conventional, with an extra qualification for ordinal logistic regression for the analyses indicated. Initial analyses as described above were performed on complete cases: that is, only on participants in whom all relevant data were present for a particular tabulation, estimate, model or test.
For all tests, p < 0.05 was considered significant, with the Bonferroni correction applied when required. All data were analysed using SPSS version 21 (IBM Corporation, Armonk, NY, USA) and R (an open-source programming package from the R Foundation for Statistical Computing, Vienna, Austria).
Missing data plan
For all proportion estimates, and for all regression analyses, examining the independent associations of age, sex, sMMSE scores (where appropriate) and residential status (living in residential care or own home) with study outcomes (i.e. VI status, near vision loss, presence of eye conditions and the effects of VI on behaviour and function), the primary analysis was a complete-case analysis (i.e. it included only participants in each analysis in whom the relevant variables were fully observed).
Given that different study outcomes would be missing in different participants and for varying reasons (e.g. some participants might not have been able to complete the recording of presenting or post-refraction VA at distance and/or near vision, while for others dilatation of the pupil might not have been possible), this may lead to some inconsistency in our separate analysis of each study outcome. Complete-case analysis also leads to less efficient estimates, because it does not use the partial information available on participants. Perhaps most crucially, it may lead to bias in unadjusted estimates if the data are not missing completely at random. For example, our VI rate estimates from complete cases would be unbiased only if those participants in whom we did/could not measure VA were expected to have the same rate of VI as those with observed VA. That assumption seems implausible. Missing data can even lead to bias in regression modelling on complete cases if the causes of missing data are not fully explained by the covariates in each model (missing at random assumption).103 This assumption is unlikely to be realistic given that our regression models adjust only for age, sex, sMMSE (where appropriate) and residential status.
To improve the plausibility of our prevalence estimates and our regression analysis assumptions, a multiple imputation procedure employing chained equations104,105 was performed as a sensitivity analysis. Simply stated, under multiple imputation, observed values of variables are used to predict missing values in other variables. For our data, we used age, sex, location (living in residential care or own home), region, site and examining optometrist as predictor variables, all of which were fully observed in the sample. Other variables with some missing data included in the imputation model were time since last eye examination; presenting and post-refraction distance and NVA; sMMSE score; presenting and post-examination AMD; glaucoma and diabetes; post-examination diabetic retinopathy; presenting cataract and post-examination cataract severity (left and right eye); BADLS total score; and CBI-R total score. Thus, for each variable with missing data, the observed values of all other variables in this set would be used to predict the missing values. A total of 30 multiply imputed data sets were generated and the estimates in each imputed data set were pooled to obtain a result, the standard error of which accounted for the random variation across data sets owing to the uncertainty from missing data. All proportion estimates and their standard errors were calculated on the logistic scale within each imputation and combined similarly.
Additionally, for cognitive performance, we operated under the assumption that missing sMMSE scores were indicative of lower cognitive performance and were informatively missing.103 Thus, even if two participants had similar characteristics but one had missing and the other had non-missing sMMSE scores, we would expect there to be a difference in their cognitive performance. To address this, we performed a pattern mixture model with a conditional expectation difference of 10 units between observed and missing sMMSE scores.
Extrapolating prevalence to the UK dementia population
Prevalence estimates for VI and its causes in the wider UK population of those aged 60–89 years with dementia were calculated by comparing the estimated distributions of characteristics in this target population with the PrOVIDe sample characteristics and calibrating estimates of rates of VI and of ocular conditions (AMD, cataract, diabetic retinopathy and glaucoma) accordingly. The two-stage clustered sampling of six regions and then 20 sites used in PrOVIDe was accounted for in this prevalence estimation, as was the stratified sampling by age (60–74 years, 75–89 years), sex and location in each site (see Chapter 3, Demographics and characteristics of the sample, Recruitment). The use of clustered sampling would result in larger standard errors and wider CIs of estimates from the so-called design effect, whereas stratification acts to reduce sampling variability and, in effect, reduce the standard error and the width of CIs.106
Post-stratification population calibration weights were derived using the estimate counts of the population aged between 60 and 89 years with dementia in various strata. Two reliable joint distributions (cross-tabulations) of the population were obtained2 and used for calibration: (1) age group and sex; and (2) age group and residency (residential care or own home). Age groups were in 5-year bands – 60–64 years, 65–69 years, 70–74 years, 75–79 years, 80–84 years and 85–89 years – and residency was dichotomised as ‘living in the community’ (own home) or ‘living in care’ (residential care). A raking procedure107 was performed to derive weights to calibrate on these two population distributions (age–sex and age–residency). Multiply imputed data were used (see Missing data plan) to account for non-completion of components of the eye examination or other sources of missing data in outcomes within the sample. Thus, a single weight factor could be calculated for all outcomes, with the imputation addressing representativeness in the sample. All proportions and their standard errors were calculated on the logistic scale within each imputation108 and combined using Rubin’s rules.109
All analyses were performed using the survey package in R.110
Comparison of prevalence with other UK studies of visual impairment in older people
Using a similar approach to the prevalence estimation for the wider UK dementia population, estimated rates of VI in PrOVIDe were also reweighted for greater comparability with two nationally representative studies of VI in the elderly.56,57 This was achieved by applying post-stratification weights to the PrOVIDe data. Rather than being derived using estimated distributions of characteristics from the UK population aged 60–89 years with dementia, these weights were instead derived from the distributions of participant characteristics in the NDNS and MRC studies.
The NDNS reported the cross-tabulations of age–sex and age–residency, where age groups were in bands of 65–74 years, 75–84 years and ≥ 85 years, and residency was ‘community’ or ‘institution’ (the latter has been interpreted as equivalent to living in residential care). The MRC study reported cross-tabulations of age–sex, where age groups were in bands of 75–79 years, 80–84 years, 85–89 years and ≥ 90 years, and participants lived in the community only. NDNS and MRC participants in age categories that fell outside the age range of PrOVIDe were excluded for these comparative analyses (≥ 85 years for NDNS and ≥ 90 for MRC), as were participants living in residential care for the MRC comparison. In turn, participants not in the age ranges of NDNS or MRC were excluded in each respective comparison. Thus, the age range used when comparing NDNS and PrOVIDe was 65–84 years and the age range used when comparing MRC and PrOVIDe was 75–89 years.
An alternative approach would have been to simply examine the rate of VI in each age–sex stratum available; however, these strata become increasingly small, and estimates within them become increasingly imprecise, with additional issues of multiple testing. Both of these issues would limit the ability to conclude whether or not any differences were credible. The rationale, therefore, was to compare as large an overlapping sample as possible for each of these two comparator studies in order to maximise the power to detect any meaningful differences, while using weighting in the PrOVIDe estimates to control for differences in sampling proportions of age and sex (and residency for NDNS).
From a statistical perspective, both comparator studies had differences in estimating and reporting VI prevalences. The MRC reported that non-response was higher in females than in males, and in older age groups than in younger age groups, but did not adjust for this in estimates. The NDNS used a weighting factor to adjust and report overall prevalence to reflect the UK population, but this was not accompanied by a CI to assess its precision. The NDNS did not report any CIs for any of their estimated prevalences, whereas the MRC reported CIs with an adjustment for the clustered sampling of the study. For the comparison of PrOVIDe estimates with the NDNS in the comparable/overlapping age group of 65–84 years, it was only possible to use VI rates and CIs calculated crudely from reported counts in age strata and there was no information on the weights in the subset of age strata. From reported MRC estimates the mean VI rate in those aged 75–89 years was calculated and combined with the cluster-adjusted standard errors across the three relevant age strata to obtain approximate cluster-adjusted CIs on the rate of presenting VI for distance vision.
Stage 2 data processing and qualitative analysis
In stage 2, audio recordings of the interviews and focus group discussions were transcribed and then analysed using framework analysis.111 The process consists of five stages:
Familiarisation with the data – key ideas and recurrent themes are listed.
Identifying a thematic framework – data are sifted and sorted into key issues, concepts and themes.
Indexing – systematic application of the framework to the data while judging the significance and meaning of the data.
Charting – lifting the data from the original source into charts.
Mapping and interpretation of the chart contents – perceptions, accounts and experiences are compared and contrasted, patterns and connections are sought and explanations for differences are considered.
Reliability and validity were achieved through independent analysis of the data by several members of the research team. There was public and participant involvement (PPI) in data analysis: transcripts, data interpretation and reporting were reviewed by the PPI member of the Project Steering Group.