

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.

Once the Gag, Gag-Pro-Pol, and Env polyproteins are synthesized, they come together (along with two copies of viral RNA and tRNA primers) at a common site on the cell membrane to assemble viral particles. It is the Gag protein, in groups of approximately 2000 interacting molecules (Stromberg et al. 1974), that alone provides the driving force for the release of uniform particles from the plasma membrane, and how it does so will be discussed in this section.
Gag proteins must function on several levels. One set of functions serves to direct the budding process. A second set of functions is needed to control the size of the virion, and in their absence, particles of great heterogeneity are released. A third set of Gag functions encompasses all those that are needed to mediate the packaging of the other components of the virus (Gag-Pro-Pol, Env, RNA, etc.). A fourth set of Gag functions is not needed until the fully assembled and mature virion infects the next host cell.
Organization and Sequence Similarities of Gag Proteins
One of the goals in investigating Gag proteins is to assign these functional domains to amino acid sequences on the protein. Despite the fact that all Gag proteins (with the exception of the spumaviruses) share functional domains and cleavage products, comparative analyses among different genera of retroviruses has limited utility because of the absence of amino acid sequence identity over most of the proteins.
Sequence Relationships among Gag Proteins
The most conserved region of Gag, the major homology region (MHR), is found in the second half of CA (see Fig. 5). This small sequence contains 20 residues, about half of which are conserved (Patarca and Haseltine 1985; Wills and Craven 1991), although this motif is absent in spumaviruses (Maurer et al. 1988; Renne et al. 1992). The MHR is discernible in the retrotransposons that are most similar to retroviruses by other criteria, such as Ty3, but not in all retrotransposons (Clare and Farabaugh 1985; Warmington et al. 1985; Hansen et al. 1988; Orlinsky et al. 1996; see also Chapter 8). The function of this conserved, and therefore important, sequence is unknown. Conflicting answers have been obtained in examining a role in particle release. In the cases of ASLV (Craven et al. 1995) and HIV-1 (Srinivasakumar et al. 1995), the entire MHR can be deleted without affecting the release of particles, although partial deletion of the MHR is deleterious to particle release (Reicin et al. 1995). In the case of ASLV, the cores of the mutant particles are unstable in nonionic detergent, suggesting that they may not have achieved the proper CA-CA interactions, whereas in the case of HIV-1, they have an aberrantly light density. Similarly, certain amino acid substitutions in the MHR severely reduce infectivity without affecting particle release; however, other substitutions within the MHR were found that have dramatic effects on particle release (Strambio-de-Castillia and Hunter 1992; Mammano et al. 1994; Craven et al. 1995). These latter mutations may indicate a subtle role for the MHC in budding or they may exert their effect indirectly on the folding and function of assembly (budding) domains located elsewhere in Gag.
A second similarity among Gag sequences is the one or two copies of a cysteine array (Cys-X2-Cys-X4-His-X4-Cys) found within NC (see Fig. 5 and Chapter 2. Each array coordinates a zinc atom (Bess et al. 1992; Summers et al. 1992), and the arrays are involved in packaging the RNA genome, but they are not required for assembly of the viral core (see, e.g., Aldovini and Young 1990; Gorelick et al. 1990, 1996; Rein et al. 1994a). Of all retroviruses, only members of the spumaviral genus lack these motifs. As in the case of the MHR, the retrovirus-like retrotransposons such as Ty3 have the characteristic cysteine arrays, whereas other retrotransposons, such as Ty1 and Ty2 in yeast, do not (see Chapter 8. It is not known what sequence elements functionally replace the cysteine arrays when they are absent. Recently, a glycine-arginine-rich motif near the carboxyl terminus of the spumaviral Gag protein has been shown to have nucleic-acid-binding properties (Yu et al. 1996b).
Additional Gag Cleavage Products
The dissimilarity among Gag proteins is underscored by the variety of additional cleavage products (other than MA, CA, and NC) derived from various positions in Gag proteins from different genera (see Chapter 2). The greatest variation in this regard is found in the region between MA and CA, where the number and sizes of extra cleavage products seem to be completely different from one viral genus to the next (see Fig. 5). In this region of HIV-1 Gag (and most other lentiviral Gag proteins), there are no extra sequences, and thus MA is linked directly to CA. Additional sequences are often found at the carboxyl terminus of Gag as well (M-PMV and HIV-1 Gag), and in the case of ASLV Gag, this is the position of the viral protease. Additional small peptides are likely to be discovered as the carboxyl termini of more retroviral cleavage products are determined. Moreover, new PR-mediated cleavage products may be generated after the virus enters the host cell, as has been suggested for the NC protein of EIAV (Roberts and Oroszlan 1989; Roberts et al. 1991).
Budding Functions of Gag Proteins
Because of the limited sequence similarity among Gag proteins, it has not been possible to predict the location of functions important for particle assembly. Studies of the mature Gag cleavage products, which are certain to acquire new shapes and properties upon release from the precursor (as appears to be the case for the amino terminus of CA; Gitti et al. 1996), have revealed only limited insights about how budding takes place. The alternative has been to genetically map for each virus domains that are important for assembly. In this context, it has been useful to define discrete events that must occur during budding as a way of identifying unifying features in studies of different retroviruses. One scheme that has been used identifies three discrete types of interactions. These have been named the M (membrane binding), I (interaction), and L (late) domains to reflect their roles in assembly (Fig. 10). The M domain specifically directs the Gag protein to the plasma membrane; the I domain represents regions of Gag-Gag interaction; and the L domain provides functions needed very late in budding, at the cell-virus separation step. The order of some of the assembly domains within the sequence of Gag proteins appears not to be absolutely conserved between viruses from different genera.

Interactions among Gag Proteins
The striking change in morphology that occurs as immature virions acquire their mature appearance implies that the interactions among Gag proteins are dynamic. That Gag protein-protein interactions change after proteolytic cleavage is suggested by crosslinking experiments with MLV (Pepinsky et al. 1980; Pepinsky 1983). MA-MA contacts and NC-NC contacts appeared to be similar in mature and immature particles, but CA-CA contacts appeared to be different. Thus, some interactions may be established prior to proteolytic processing and then maintained, whereas others may be gained or lost. It is the interactions among uncleaved Gag proteins that are essential for budding and that are the focus here. These issues have been reviewed recently (Craven and Parent 1996).
Sites of Initial Interactions
Newly synthesized Gag molecules must find other molecules with which to interact so that the formation of a particle can begin. Little is known about when and where in the cell these interactions first begin. There is a high local concentration of nascent Gag molecules associated with the ribosomes translating Gag mRNA, but genetic complementation experiments have clearly demonstrated that Gag proteins encoded by different mRNAs (and thus made on different polysomes) can be copackaged with high efficiency (see, e.g., J.W. Wills et al. 1991; Park and Morrow 1992; Smith et al. 1993; Yuan et al. 1993; Srinivasakumar et al. 1995). This does not rule out the possibility that some interactions begin cotranslationally, but it seems likely that most occur later.
In the case of type-B/D retroviruses (e.g., MMTV and M-PMV), which assemble particles in cytosolic compartments, Gag interactions must take place prior to transport to the membrane (see Figs. 3 and 4). Assembly seems to take place deep in the cell at perinuclear locations (Rhee and Hunter 1987), but how Gag proteins arrive at these positions is unclear. It may be that the membrane-targeting information of the type-B/D Gag proteins is initially masked, and the proteins passively accumulate to concentrations high enough to drive self-assembly near the sites of synthesis. Alternatively, there may be a specific transport mechanism to direct these Gag proteins to their cytoplasmic sites of assembly. In either case, completion of the ICAP must bring about a change in Gag that functionally and perhaps physically exposes the membrane targeting information.
Where do the Gag proteins of viruses with the type-C pattern of morphogenesis first interact? Electron microscopy suggests that this occurs after the molecules have been transported to the plasma membrane, since electrondense aggregates of viral proteins are observed only at this location for these viruses. Experiments using myristate-minus derivatives of MLV or spleen necrosis virus (SNV) Gag proteins seem to support this interpretation. These mutant proteins fail to bind to membranes and are unable to be rescued into viral particles when coexpressed with assembly-competent Gag proteins, as would be expected if membrane binding were a prerequisite for incorporation (Schultz and Rein 1989; Weaver and Panganiban 1990). In contrast, membrane-binding mutants of ASLV and HIV-1 Gag are readily rescued in this type of complementation experiment (J.W. Wills et al. 1991; Yuan et al. 1993; Morikawa et al. 1996; Parent et al. 1996), which suggests that the Gag proteins of at least some type-C viruses may begin to interact prior to their arrival at the membrane. It is possible that these Gag molecules form regular (but electron-lucent) structures that become visible only after condensing against the hydrophobic surface of the plasma membrane. The idea that membrane interactions are not required for the assembly of some type-C Gag proteins is further supported by the finding that soluble ASLV and HIV-1 Gag proteins can be assembled into virus-like structures in vitro (see below).
The I Domain
A major region of Gag-Gag interaction (the I domain) maps to the NC sequences of Gag proteins (Fig. 10). Evidence for I domains is derived from several lines of experimentation. First, deletions that remove the NC regions of various Gag proteins typically reduce budding (see, e.g., Gheysen et al. 1989; Weldon et al. 1990; Dupraz and Spahr 1992; Jowett et al. 1992; Weldon and Wills 1993; Carrière et al. 1995; Craven et al. 1995), and such mutants cannot be rescued into particles by coexpressing assembly-competent molecules (Weldon and Wills 1993). Although a few NC deletion mutants have been found that appear to produce particles at levels similar to those of wild type (see, e.g., Royer et al. 1991; Bennett et al. 1993; Gonzalez et al. 1993; Weldon and Wills 1993), these invariably have lower than normal density in isopycnic sucrose gradients, which indicates that they have fewer Gag molecules packed together along the internal surface of the virion membrane (i.e., a reduced ratio of Gag to lipid). Such low-density particles are important because they reveal that interactions outside of NC can be sufficient to drive some sort of budding process; nevertheless, the normal tight packing of Gag proteins within a particle requires the NC sequence.
Additional evidence for the existence of I domains within the NC sequence is derived from studies of Gag chimeras. For example, when small portions of the NC sequences of ASLV, HIV-1, or MLV are attached to the ends of ASLV mutants that lack NC and produce low-density particles, functional chimeras are obtained that are capable of producing dense particles with high efficiency (Bennett et al. 1993; Weldon and Wills 1993; R.P. Bennett and J.W. Wills, unpubl.). This approach has revealed the presence of two copies of the I domain in the Gag proteins of ASLV and HIV-1, but only one within MLV. Evidence for the positions of I domains has also been obtained through the use of the yeast two-hybrid system (Luban et al. 1992; Franke et al. 1994), and this approach has also revealed that unrelated Gag proteins are capable of interacting.
Although I domains of several retroviruses map to a common region of Gag (NC), it has been difficult to discern a sequence motif that is common to all. The Cys-His boxes are not part of the I domain since these can be removed without affecting particle density (see, e.g., Gorelick et al. 1988; Rein et al. 1994a; Ottmann et al. 1995); moreover, spumaviruses have readily identified I domains (R.P. Bennett et al., unpubl.) even though they do not have Cys-His boxes. The one factor that these I domains seem to have in common is a high concentration of basic residues, and the addition of a short basic peptide containing a repeat of the sequence R-K-K has been found to be capable of restoring dense particle production when substituted for the I domains of ASLV (R.P. Bennett and J.W. Wills, unpubl.).
How do these I domains mediate interactions among Gag molecules? The clustering of basic charges suggests that the interactions are mediated by RNA binding, a well-studied function of the NC protein (see below Viral RNA Packaging, Viral Proteins Involved in Packaging Genomic RNA). In this model, RNA would provide a scaffold upon which Gag proteins could tightly condense and pack together. In cases where viral genomic RNA is not available in the cell, this function could be served by host-cell RNA, which is found in particles under such conditions (see, e.g., Gallis et al. 1979; Levin and Seidman 1979; Gorelick et al. 1988; Méric and Goff 1989; Linial and Miller 1990).
Other interactions in addition to NC appear to have a role in Gag assembly. For both MLV (Lobel and Goff 1984; Schwartzberg et al. 1984; Goff and Lobel 1987; Hansen et al. 1990) and HIV-1 (Jowett et al. 1992; Hong and Boulanger 1993; von Poblotzki et al 1993; Wang et al. 1993; Chazal et al. 1994; Reicin et al. 1995), certain deletions or insertions within CA (and specifically the carboxy-terminal region of the HIV-1 CA) are deleterious to particle assembly. This is in contrast to ASLV where CA can be deleted without affecting particle release (Weldon and Wills 1993; J.W. Wills et al. 1994).
In Vitro Particle Assembly
Recent in vitro assembly experiments further support the notion that RNA is required for particle assembly. These experiments have revealed that the CA-NC portion of both the ASLV and HIV-1 Gag proteins (purified from E. coli expression systems) can rapidly aggregate to form long hollow tubes, but only in the presence of RNA (Campbell and Vogt 1995). There is not a requirement for specific viral RNA, and the longer the RNA, the longer the tubes. Treatment of the structures with ribonuclease results in disassembly, as does separation of CA from NC using the viral protease. Tubes have also been produced using just the CA sequence of HIV-1 (Ehrlich et al. 1992), but the inclusion of the NC sequence appears to enable efficient assembly under conditions that do not work for CA alone. If the sequences normally upstream of CA-NC are included in the ASLV Gag protein in this reconstituted assembly system (i.e., p10), spherical particles are generated that closely resemble real immature particles. As with the cylinders, RNA remains essential, consistent with the notion of an RNA scaffold. Thus, there are interactions among Gag proteins that provide polymerizing activity (e.g., I domains) and others that constrain that activity to form spheres (Campbell and Vogt 1997).
In vitro assembly of particles has also been obtained using the Gag protein of M-PMV (Klikova et al. 1995; Sakalian et al. 1996). In this case, purified proteins were not used, rather the particles were found to assemble in a cell-free protein synthesis system. Because of the large amount of RNA present in the translation reaction, it is not clear whether it is essential for M-PMV assembly in vitro. Interestingly, when HIV-1 Gag proteins are made in the cell-free translation system, they do not appear to assemble into particles as readily as those of M-PMV (Sakalian et al. 1996), although assembled particles have been detected (Spearman and Ratner 1996). Since the concentrations of proteins after in vitro translation are low, this difference may reflect a higher association constant for M-PMV Gag proteins as compared with HIV-1 Gag proteins.
Membrane Binding and Targeting of Gag Proteins
One of the biggest mysteries of Gag proteins is how they are targeted to the proper membrane for budding. Infectious retroviruses do not bud from all of the available membrane surfaces within an infected cell, but primarily from the plasma membrane, which constitutes a small proportion of the total membrane surface in most cells. In polarized cells, the sites of budding are further restricted to the basolateral membrane (see below Incorporation of Env into the Viral Particle, Active Packaging). Consistent with this, the lipid composition of retrovirions has been found to be distinct from that of the plasma membrane as a whole (Quigley et al. 1971; Aloia et al. 1993).
Although interactions between Env and Gag may influence the selection of assembly sites on the plasma membrane (see below Incorporation of Env into the Viral Particle, Active Packaging), other factors seem to be more important because of the absence of budding from internal membranes, even when Gag proteins are expressed without Env. This principle is further illustrated by the IAPs, which lack Env proteins but nevertheless exhibit striking membrane specificity in budding exclusively at the ER (see Fig. 3). These observations hint at the existence of a reliable system for controlling the targeting of Gag proteins within the cytoplasm of the cell; however, the mechanisms are not understood.
Gag molecules would most likely follow the same “tracks” used by those cellular proteins that are targeted to the cytoplasmic faces of specific membrane surfaces after synthesis on free ribosomes. Two examples of such proteins are NADH cytochrome b 5 reductase, which is involved in lipid biosynthesis on the ER, and pp60c-src, which is involved in signaling events on the plasma membrane. Targeting is thought to involve unique features of the membrane-binding domain located at the amino terminus of each of these proteins, but the basis of specificity is not understood (Ozols et al. 1984; Silverman and Resh 1992; Resh 1993, 1994; Buser et al. 1994; Sigal et al. 1994).
The M Domain
The membrane-binding domains of Gag proteins are also located at the amino termini and are contained entirely within the MA sequence (Fig. 10). Deletions within M abolish membrane binding and budding. As mentioned above (see Principles of Particle Assembly, Interactions among Gag Proteins), M mutants of ASLV and HIV-1 (but not MLV or SNV) retain the ability to interact with other Gag proteins and thereby can be rescued into particles, suggesting that membrane association is not a prerequisite for Gag multimerization for all viruses with the type-C pattern of morphogenesis.
Although M domains are poorly understood, they can usually be moved from one Gag protein to another without affecting the efficiency of budding. For example, various protein fragments containing the M domain of HIV-1 have been used to replace the M domains of ASLV (Bennett et al. 1993; Parent et al. 1996), MLV (Deminie and Emerman 1993, 1994), and visna virus (Dorfman et al. 1994a). Moreover, M domains can be replaced with heterologous membrane-binding domains from myristylated cellular proteins (e.g., pp60c-src) which are targeted to the cytoplasmic faces of membranes (see, e.g., J.W. Wills et al. 1991; Lee and Linial 1994). Binding constants for M domains have not been measured, although the ability of Gag proteins to aggregate in large numbers should allow significant cooperativity. In this regard, it has been found that Gag proteins lacking their interaction domains bind more poorly to membranes in vivo (J.W. Wills et al. 1991) and in vitro (Platt and Haffar 1994). Weak membrane interactions may be essential for allowing mature MA proteins to perform functions during the entry steps of an infection (e.g., nuclear targeting of the integration complex).
The best understood M domain is that of HIV-1, which maps to the first 32 residues of Gag and includes the terminal myristate. This segment can direct heterologous proteins such as pp60v-src and DHFR to the membrane and can also replace the M domain of ASLV Gag (Zhou et al. 1994; Parent et al. 1996). Myristate probably inserts into the lipid bilayer when Gag binds to the membrane, but this alone is insufficient for a stable membrane interaction (Kim et al. 1991; Peitzsch and McLaughlin 1993). Further binding energy is provided by a cluster of basic amino acids, which lies within a pleated sheet structure (Massiah et al. 1994; Matthews et al. 1994; Rao et al. 1995; Hill et al. 1996; Chapter 2. In a manner similar to that of Src and other myristylated peptides (Kim et al. 1991; Mosior and McLaughlin 1992; Silverman and Resh 1992; Resh 1993; Buser et al. 1994; Sigal et al. 1994), the positive charges of the arginines and lysines in the HIV-1 M domain are thought to participate in electrostatic interactions with the negative charges associated with acidic phospholipids on the inner leaflet of the plasma membrane and, together with myristate (Kim et al. 1991; Peitzsch and McLaughlin 1993), stabilize the association of the Gag protein with the membrane (Yuan et al. 1993; Zhou et al. 1994). It has been proposed that membrane binding associated with MA is regulated in the context of the Gag structure, being exposed in Gag to allow membrane targeting but hidden after processing to MA to permit membrane dissociation (Zhou and Resh 1996).
Certain alterations of the HIV-1 MA sequence have been found that result in Gag-mediated budding at the ER membrane (Fäcke et al. 1993; Freed et al. 1994; Gallina et al. 1994). This principle is reminiscent of IAPs, except that low levels of budding can be seen to continue at the plasma membrane. Because the surface area of the ER is large, it is possible that these MA mutants have lost the signals necessary for specific membrane binding and exhibit nonspecific budding preferentially to the biggest target.
The only other M domain that has been clearly mapped is that of ASLV Gag. In contrast to HIV-1, this membrane-binding domain is much larger and resides in the first 85 residues of Gag (Fig. 10). It is also capable of directing heterologous proteins to the plasma membrane (Nelle and Wills 1996; Verderame et al. 1996). How the M domain of ASLV works is a mystery because it is not myristylated and does not possess an obvious cluster of positively charged amino acids. It does contain 11 basic residues scattered along its length, and it is possible that protein folding brings them together to form a cluster of positive charges in three-dimensional space. Alternatively, a novel mechanism for membrane binding could be at play.
Transport of Gag Proteins
The actual mechanism by which Gag proteins are transported to the plasma membrane is unknown. Simple diffusion does not seem likely since M-PMV mutants have been found that are blocked for particle release while retaining the ability to assemble cores at nonrandom locations in the cytoplasm, far from the plasma membrane (Rhee and Hunter 1987). By what mechanism are Gag proteins transported? One suggestion is that newly synthesized Gag proteins travel to the plasma membrane by interacting with elements of the cytoskeletal system (Edbauer and Naso 1983, 1984). Although this has been difficult to prove, it remains an attractive model in view of the finding that certain cytoskeletal proteins contain WW domains—motifs that have been implicated in retroviral budding (see below). Another suggestion for transport is that Gag molecules bind to the outer surface of the Golgi apparatus and then travel to the plasma membrane on transport vesicles (Hansen et al. 1990); however, this model appears not to apply to the type-B/D Gag proteins, whose assembled cores interact only with the plasma membrane. In addition, the Gag proteins of ASLV and HIV-1 cannot be transported in this manner because they direct budding with full efficiency in high concentrations of brefeldin A, a drug that disrupts the secretory pathway (Pal et al. 1991; Krishna et al. 1996). It is also possible that interactions with chaperone proteins are required to facilitate folding and transport of Gag proteins to the membrane (see, e.g., Rothman 1989; Becker and Craig 1994; Hartl et al. 1994).
Two general models can be imagined to explain the membrane specificity of Gag proteins. In one, there is a specific receptor for each Gag protein that is located on the specific membrane surfaces to which the Gag proteins are targeted. Older models of assembly which suggested that the viral glycoproteins serve this purpose cannot be correct since Env glycoproteins are dispensable for particle formation. Moreover, the idea that basic charges in the amino terminus of Gag interact with acidic phospholipids on the inner leaflet of the plasma membrane (Zhou et al. 1994) is also not sufficient to explain the specificity of targeting, because these negatively charged lipids are also found on the cytoplasmic faces of internal membranes, such as the ER (for review, see Op den Kamp 1979; Bishop and Bell 1988). In the other model for specific membrane targeting, the transport mechanism itself is involved in sorting proteins to the proper surface; i.e., a specific cytoplasmic protein (“receptor”) might recognize the membrane-binding domain of a Gag protein, bind to it, and mediate transport to the appropriate membrane location in the cell. If host-specified Gag receptors do exist, they are likely to be conserved since Gag proteins appear to be capable of directing particle formation in cells derived from many species, including insects.
Emergence and Release of the Bud
After Gag proteins arrive at the cell membrane, whether individually or within a preassembled particle, they somehow cause buds to emerge and separate from the cell surface. How this occurs is not understood. One hypothesis is that the spherical structure made by interacting Gag proteins has a high enough affinity for the membrane that it can coat itself with lipid, much as warm caramel completely surrounds and coats an apple when a spoonful is placed on top. In this model, high affinity, progressive lipid-particle interactions bring about the separation of a membrane-enclosed particle from the surface of a cell. An alternative explanation is based on the observation that some emerging spheres sit on top of narrow stalks just prior to being released, rather than directly abutting the cell surface (see Figs. 4 and 11). The contents of these stalks and the molecular forces that maintain their rod shape are unknown.
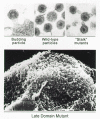
Stalk Mutants
The evidence for a stalk intermediate in budding comes from certain ASLV capsid mutants that appear to be released from the cell with a protruding rod (Craven et al. 1995). The attached structures (Fig. 11) are thought to correspond to the stalk not only because of their size and morphology, but also because there is never more than one per particle. Some investigators have found that wild-type particles have a single, stick-like structure, which may be a vestige of the stalk (Fig. 11). These thin structures are occasionally seen in the mutant and the thick stalks are occasionally seen in the wild type. This is consistent with the mutants accumulating at an intermediate step along the normal pathway. Despite the physical abnormalities of these stalk mutants, they are released at the normal rate and hence are not defective for the pinching-off step (Craven et al. 1995).
The L Domain
Other Gag mutants have been found that fail to produce particles, even though the Gag proteins accumulate underneath the membrane. Electron microscopy has revealed in the case of HIV-1 that the mutant proteins form buds that remain tethered to the cell surface (Fig. 11) (Göttlinger et al. 1991). The region of Gag that appears to be critical for virus-cell separation has been designated the L domain because of its apparent role at a late step in budding. L domains are very small and can be found at different positions in different retroviruses (Fig. 10). In HIV-1, the L domain maps to the p6 sequence, and the critical amino acids include P-T-A-P (Göttlinger et al. 1991; Huang et al. 1995). In contrast, the L domain of ASLV Gag is located closer to the amino terminus, within the p2b sequence (J.W. Wills et al. 1994), and its critical residues are P-P-P-P-Y (Xiang et al. 1996). The L domain of the EIAV Gag protein is located near the carboxyl terminus (in the p9 sequence), and it resembles neither P-T-A-P nor P-P-P-P-Y (Parent et al. 1995). The L domains of HIV-1 and EIAV can restore budding competence to L mutants of ASLV when placed at the carboxyl terminus, and thus the L domains are interchangeable between unrelated retroviruses (Parent et al. 1995). Moreover, the L domains of ASLV and HIV-1 can function even when moved to the ends of Gag opposite their normal location; hence, L domains are positionally independent (Parent et al. 1995; Xiang et al. 1996).
Not all investigators have observed defects in particle release with HIV-1 p6 deletion mutants, and the reason for the discrepancy is not clear (see, e.g., Hoshikawa et al. 1991; Royer et al. 1991; Jowett et al. 1992; Paxton et al. 1993). Particles made in the absence of p6 have a dramatically higher sedimentation rate (i.e., larger size), which suggests that they are released by a different mechanism (L. Garnier and J.W. Wills, unpubl.). Evidence also suggests that the late function in p6 is only needed when the viral PR is active (Huang et al. 1995). In contrast, the L domain of ASLV is needed whether or not the PR is active (J.W. Wills et al. 1994; Parent et al. 1995; Xiang et al. 1996). In certain “leaky” ASLV L mutants, inactivation of PR has the effect of prolonging the half-life of the nascent buds on the membrane (Bowles et al. 1994; J.W. Wills et al. 1994). Because the steps in budding mediated by the L domain take place at about the same time that the viral PR turns on, dissection of these two events has been difficult. In the case of HIV-1, the situation is further complicated by the observation that PR activity is required for maximal efficiency of particle release (Kaplan et al. 1994a).
The Vpu protein of HIV-1 can also enhance the release of viral particles (Cohen et al. 1988; Strebel et al. 1988, 1989; Terwilliger et al. 1989b; Klimkait et al. 1990; Göttlinger et al. 1993; Maldarelli et al. 1993; Jabbar 1995). Although there seems to be a cell-type dependence for this effect (Geraghty et al. 1994; Sakai et al. 1995), in those situations where Vpu mutants do exhibit defects, the concentration of tethered particles observed at the cell surface is elevated, suggesting that the block is late in budding. How Vpu works is not clear. It is an integral membrane protein but is not packaged into the virion. Moreover, it is not HIV-1-specific but can enhance the release of retroviruses from different genera (Göttlinger et al. 1993). Vpu is phosphorylated, but this modification is not required for the enhancement of particle release (Schubert and Strebel 1994; Friborg et al. 1995). Vpu also has the ability to interact with CD4 at the ER membrane to induce its degradation (see above Synthesis and Organization of Env Glycoproteins, Premature Receptor Interactions); however, this function is contained in a region of the protein different from the one involved in particle release at the plasma membrane (Schubert et al. 1996). Vpu is unique to HIV-1; however, a Vpu-like activity has been reported to be associated with the cytoplasmic domain of the HIV-2 TM protein (Bour et al. 1996).
Potential Role of Host Proteins
The means by which L domains mediate the pinching-off step is not known, but a recent clue from studies of the Yes protein tyrosine kinase suggests that interactions with specific host proteins may be involved. Yes is located on the cytoplasmic face of the plasma membrane. In mediating signal transduction, it interacts with Yap (Yes-associated protein) via an SH3 domain. Yap in turn contains an amino acid motif called WW, characterized by two conserved, widely spaced tryptophan residues (Bork and Sudol 1994). This motif has been found to interact with the sequence PPPY (Chen and Sudol 1995) and thus would be predicted to recognize Gag proteins with this sequence in their L domain. Indeed, it has been shown directly that peptides carrying the ASLV L domain, but not peptides carrying the HIV-1 L domain, bind to Yap in vitro (Garnier et al. 1996). Thus, it is possible that a host protein with a WW motif plays a part in the late stages of ASLV budding. By analogy, other host proteins would recognize the different L motifs of HIV-1 and EIAV. No systems have yet been developed to address how such host proteins could function in retroviral assembly.
Cyclophilin A is incorporated into virions of HIV-1 but not other primate lentiviruses, including HIV-1 of type O (Luban et al. 1993; Franke et al. 1994; Thali et al. 1994; see also Chapter 2. Although cyclophilin A is incorporated into virions during viral assembly, it appears to have a role during the early steps of viral replication, after viral binding but before DNA synthesis (Steinkasserer et al. 1995; Braaten et al. 1996). The nuclear magnetic resonance (NMR) structure of the amino-terminal two thirds of the HIV-1 CA has recently been determined, and the region of CA that binds cyclophilin A was found in two distinct conformations (Gitti et al. 1996 and references therein). Binding of cyclophilin A fixes Pro-90 of CA in a transconformation which is unusual for proteins bound to cyclophilin A (Gamble et al. 1996; Zhao et al. 1997). The requirement to bind cyclophilin A is hardly absolute, since mutations of HIV-1 to cyclophilin independence are readily obtained (see Aberham et al. 1996).
Several lines of evidence point to a role for the cytoskeleton in viral assembly and budding. For both MLV (Edbauer and Naso 1983, 1984) and HIV-1 (Rey et al. 1996), significant amounts of Gag remain associated with the cytoskeleton during fractionation of cells. Disruption or rearrangement of the cytoskeleton affects particle budding (Pearce-Pratt et al. 1994; Sasaki et al. 1995). These observations are reinforced by the ability of HIV-1 Gag to interact with actin directly (Rey et al. 1996) and by the demonstration of several cytoskeletal proteins, including actin, in highly purified viral particles (Ott et al. 1996).
Determinants of Particle Size
The functions of Gag needed for budding are insufficient for controlling the size of the viral particle. For example, large deletions within the ASLV Gag protein have been found that have no effect on rates of budding or density, but the particles that are released are very heterogeneous and much larger than the wild type as measured in rate-velocity sucrose gradients (Weldon and Wills 1993; J.W. Wills et al. 1994). Systematic analysis of this Gag protein using a large number of deletion mutants indicates that the most important determinant of particle size maps to the CA sequence, along with the spacer peptides that are subsequently cleaved from its carboxyl terminus (N.K. Krishna and J.W. Wills, unpubl.). Alterations outside of this region have no effect on particle size. These findings suggest that interactions among CA sequences begin during particle assembly, even though they are not absolutely needed for particle release (J.W. Wills et al. 1994; Srinivasakumar et al. 1995).
A role for the CA sequence in influencing particle size is further supported by the release of filaments and other anomalously shaped virions by M-PMV mutants that have substitutions at the less strongly conserved positions in MHR (Strambio-de-Castillia and Hunter 1992). Furthermore, several mutants of HIV-1 CA have been found that also appear to make abnormally large particles (see, e.g., Göttlinger et al. 1989; Dorfman et al. 1994b; Kräusslich et al. 1995).
Packaging of Gag-Pro-Pol during Assembly
The Gag portion of the Gag-Pro-Pol protein is thought to provide the targeting signals that direct these fusion proteins to the site of assembly. This idea is supported by the finding that foreign (nonviral) proteins also can be directed into particles when fused to the carboxyl terminus of Gag (see, e.g., Jones et al. 1990; Weldon et al. 1990; Wang et al. 1994). However, the Gag-Pro-Pol proteins of all retroviruses tested (HIV-1, MLV, and ASLV) are incapable of directing particle formation by themselves; rather, packaging requires the coexpression of Gag molecules (see, e.g., Felsenstein and Goff 1988; Craven et al. 1991; Park and Morrow 1991, 1992; Stewart and Vogt 1991; Smith et al. 1993; Srinivasakumar et al. 1995).
It is not clear why Gag-Pro-Pol proteins are incapable of directing budding. One hypothesis is that a retroviral particle simply does not have the room to accommodate the extra amino acids encoded by pro and pol if these are present on every Gag molecule (i.e., steric hindrance). Another possibility is that Gag-Pro-Pol proteins fold in a manner that prevents one or more of their Gag-associated assembly domains from functioning but without hiding the region required for interaction with Gag. Moreover, it could be that Gag-Pro-Pol proteins actually interact with Gag in a manner distinct from the interactions employed when Gag proteins interact with themselves. For example, the MHR region of HIV-1 Gag can be deleted without greatly affecting the release of particles; however, the same MHR deletions prevent packaging of the Gag-Pro-Pol molecule (Srinivasakumar et al. 1995). Similarly, the MHR region and the adjacent carboxy-terminal region of CA must be present to allow incorporation of a chimeric Gag-Pro-Pol precursor (Huang and Martin 1997).
Although the Gag sequence is of central importance for the packaging of the Gag-Pro-Pol protein, there are examples of pol mutations in HIV-1 (e.g., IN mutants) that adversely affect particle release (Shin et al. 1994; Engelman et al. 1995; Englund et al. 1995). This phenotype has been difficult to explain because Gag-Pro-Pol is made at levels 10–20-fold less than Gag and is not needed for budding. Particles that are released by these pol mutants generally have immature morphology (Shin et al. 1994; Engelman et al. 1995), suggesting that reduced amounts of PR (and hence, Gag-Pro-Pol) have been packaged. More recently, it has been found that PR mutations and PR inhibitors can restore budding to pol mutants that have defects in particle release (Bukovsky and Göttlinger 1996), suggesting that alterations in Pol can adversely affect the regulation of PR. Perhaps premature activation of PR as a result of downstream pol mutations alters the internal structure of the nascent particles, thereby reducing the number of particles released.
- Principles of Particle Assembly - RetrovirusesPrinciples of Particle Assembly - Retroviruses
Your browsing activity is empty.
Activity recording is turned off.
See more...