
 and
and The Authors.
This open access article is licenced under Creative Commons Attribution 4.0 International (CC BY 4.0). https://creativecommons.org/licenses/by-nc/4.0/

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Scott JF, Gerstenblith MR, editors. Noncutaneous Melanoma [Internet]. Brisbane (AU): Codon Publications; 2018 Mar. doi: 10.15586/codon.noncutaneousmelanoma.2018.ch1
Melanoma is a life-threatening malignancy that may involve different organs such as the skin and the eye. The primary intraocular form is called uveal melanoma. Its etiology, mutation profile, and clinical behavior are distinct from that of cutaneous melanoma. In most cases, the malignancy originates from the choroid. Usually at the time of detection, no metastatic disease is found. Therapy is therefore focused on the primary tumor and depends on different factors such as the size and location of the malignancy and other individual factors like patient age and visual acuity of the fellow eye. Since vision loss and even loss of the eye may be the consequences of therapy, diagnosis needs a strong base. The primary tumor is effectively controlled in most cases. Metastatic disease, however, will develop in about half of the patients many years thereafter. Many steps regarding evolution, dissemination, and metastatic disease are still unknown. Several prognostic factors are used to evaluate the risk for metastatic disease. Survival of patients with metastasis is less than 1 year. As of now, there is neither a therapy that bears sufficient evidence for a prophylactic effect nor a therapy that can reduce the mortality rate. Recent understanding of the biology, the initiating mutations in the G-alpha subunits GNAQ and GNA11, the alterations of chromosomes 3 and 8, the mutation of the tumor suppressor gene BAP1 and the splicing factor SF3B1, as well as the role of the tumor-immune privilege may aid in the development of efficacious adjuvant therapies.
Uveal melanoma is a rare cancer but the most frequent noncutaneous melanoma and primary malignancy of the eye in adults. Worldwide, it is estimated that there are 7095 new cases of uveal melanoma annually with a mean age-adjusted incidence of 4.3 per million (1, 2). The mean age at presentation is 60 years with a range of 6–100 years. The malignancy affects male and females at a similar rate, with a slight predominance among males (3). Caucasians are affected in 98% of the cases, Hispanics in about 1%, and in Asians, Africans, and Native Americans, the incidence rate is less than 1%. Uveal melanoma is usually diagnosed in the sixth decade of life, with a median age of 55 in most series (4). The incidence rate has been shown to progressively increase with age, peaking at 70–75 years and then reaching a plateau (2, 5).
In 90% of the cases, the choroid is affected. Rarely does the primary malignancy originate from the ciliary body (6%) or the iris (4%) (6).
The etiology of uveal melanoma is still unclear. Concomitant with the fact that Caucasians are the ethnic group mostly affected, light skin and eye color have been found to be predisposing factors for the development of the malignancy (5, 7). This is relevant when chronic sunlight exposure is added. In Europe, a north–south decreasing gradient of melanoma incidence among European population does support the protective role of pigmentation (5). However, molecular data seem to exclude a typical UV-associated mutational spectrum for uveal melanoma, as it is known for cutaneous melanoma. Therefore, if light exposure plays a role in uveal melanoma carcinogenesis, it seems to act in a different way than in cutaneous melanoma (8–15). Preexisting uveal nevi may be the base for the development of the malignancy. It has been estimated that 5–10% of the Caucasian population have nevi in their eyes and that 1 in 8845 nevi may transform into uveal melanoma (16).
Uveal melanoma arises from pluripotent neural crest cells that migrate out and populate different anatomical locations (e.g., epidermis, dermis, and uveal tract). The melanocytes that reside in the uveal tract appear to have a distinct developmental lineage and cytogenetic profile compared to their epidermal skin melanocyte counterparts.
Uveal melanoma tumors show alterations in chromosomes 1, 3, 6, and 8. By far the most salient chromosomal aberration associated with metastatic uveal melanoma is the loss of chromosome 3. The presence of monosomy 3 in a primary tumor strongly correlates with the risk of metastatic disease. The gain of 8q, which is also associated with a reduced survival, occurs frequently in combination with monosomy 3 and is considered to be a later event induced by the loss of chromosome 3. Other chromosomal alterations such as the loss of 6q or 1p also augment the metastatic risk, while the gain of 6p occurs almost in a mutually exclusive manner with monosomy 3 and is associated with a better prognosis (17–23).
The development of the malignancy has been associated with oncogenic mutations that influence cell cycle and programmed cell death. With the exception of chromosome 3, the identification of specific genes that correlate functionally with these chromosomal aberrations has been elusive.
Uveal melanomas show an enhanced expression of the important cell-cycle regulatory protein cyclin D (CCND) involving the RAF/MEK/ERK pathways. These pathways are important for melanocyte homeostasis. Activation of these pathways leads to the phosphorylation and inactivation of the retinoblastoma tumor suppressor gene (24). Another molecular event associated with dysfunction of the retinoblastoma protein is the inactivation of the INK4A gene, which encodes the cyclin-dependent kinase inhibitor 2A. The destabilization of the retinoblastoma protein by these mechanisms allows affected cells to reenter the cell cycle (25).
An oncogene mutation affecting the RAF/MEK/ERK pathway is a mutation of the genes GNAQ and GNA11 in codon 29. In addition, GNAQ is also involved in endothelin signaling which is essential for melanocyte survival early in development (26). Activation of GNAQ mimics growth factor signaling in the RAF/MEK/ ERK pathway, leading to the transcriptional activation of CCND1 that was found to be overexpressed in uveal melanoma. GNAQ mutation was noted in 45–49% of uveal melanoma biopsy samples, whereas GNA11 mutations have been noted in 31.9% of uveal melanoma samples (27).
A recent finding in primary uveal melanomas are nontruncating (missense, in-frame deletions, and termination read-through) and truncating (nonsense, splice, and insertion/deletion) mutations in the nuclear ubiquitin carboxyl-terminal hydrolase BAP1 (28). This apparently results in the loss of BAP1 protein expression. Previous studies have shown BAP1 to have tumor suppressor activity. BAP1 has been revealed to regulate cell proliferation by deubiquitinating hcf-1, a cell-cycle regulator (29). It is part of the polycomb group repressive deubiquitinase complex involved in the removal of monoubiquitin from histone H2A, and ultimately stem cell pluripotency and organismal development (30, 31).
Since the BAP1 gene is localized on chr3p21.1, these genetic alterations appear to have functional consequences primarily in the tumors with monosomy 3. Within tumors that showed monosomy 3, BAP1 mutations were present in 81% of cases. In addition, BAP1 mutations were highly correlated with class 2 tumor status (a gene-expression profiling test for high metastatic risk), chromosome 3 loss in primary tumors, and ultimately the emergence of metastatic disease in patients (28). Therefore, the heterozygosity of BAP1 may be a major factor controlling metastatic disease. The exact mechanism(s) by which loss of BAP1 mediates primary uveal melanoma metastasis is currently being investigated. However, a recent study indicates that the loss of BAP1 results in the accumulation of mono-ubiquitinated histone H2A and a more de-differentiated cellular phenotype (32). The molecular events in uveal melanoma that have been associated with the inhibition of programmed cell death (apoptosis) include the inactivation of the p53 pathway (33), activation of the prosurvival PI3K-AKT pathway (34), and defects in the Bcl-2 pathway (35).
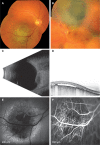
Uveal melanoma may be characterized by its location, pigmentation, shape, and size. In most of the cases (>90%), the choroid is involved. These tumors are pigmented in approximately half of the cases, nonpigmented in 15%, and show a mixed (pigmented and nonpigmented) appearance in about one-third of the cases (6). In 75% of the cases, the choroidal tumor is dome shaped. When it breaks through the Bruch’s membrane and continues to grow into the subretinal space, it acquires a mushroom-like appearance (20%). In 5% of the cases, it shows a more diffuse growth, remains flat, and can be misinterpreted as a choroidal nevus (36). The mean basal dimension is 11.1 mm and the mean thickness is 5.5 mm (6). The tumor is often associated with subretinal fluid, nondetectable at fundoscopy, and can extend to a large exudative retinal detachment obscuring the causative malignancy. In some cases, hemorrhages may develop at the tumor site and rarely also involve the vitreous space when the retina is eroded (Figure 1).
The ciliary body melanoma is less common (6%). Depending on its size, it may remain long unrecognized and asymptomatic. However, lens tilting and cataract development may occur, leading to vision impairment and clinical diagnosis. It is often associated with dilated episcleral “sentinel” vessels, but only rarely with extrascleral extension. Depending on its intraocular extension, it may be classified as iridociliary melanoma (involving the anterior chamber) or as ciliochoroidal melanoma. The growth is usually dome shaped, less common is the circumferential ring pattern.
The rare iris melanoma (4%) shows variability in pigmentation, size, and shape. In 80% of cases, the tumor is located in the inferior portion of the iris. Rare variants show a diffuse growth with an infiltration of the trabecular meshwork, the primary trabecular meshwork ring melanoma, and the tapioca melanoma with a gelatinous, nodular structure of the iris.
A detailed classification of uveal melanoma is provided by the American Joint Committee on Cancer (AJCC) (37). The tumor size is evaluated and defined in the T category (1–4), the lymph node involvement in the N category (NX, N0, N1), and the presence of distant metastases in the M category (MX, M0, M1a, M1b, M1cI). Furthermore, it distinguishes anterior (iris) uveal melanoma from posterior (ciliary body and choroid) uveal melanoma for prognostication. An iris tumor is classified as T1-4, depending on whether it is confined to the iris or has expanded to the ciliary body, choroid and/or sclera, or is already extended extraocularly. Some stages are divided into smaller groups that help describe the tumor in even more detail. The AJCC classification for posterior uveal melanoma involves grading according to the size category based on a combination of basal diameter and thickness, labeled as T1-4, and subclassification (a–e), judged by the absence of ciliary body involvement and extraocular extension (EOE) and the presence of the following: ciliary body involvement, EOE or thickness less than 5 mm, and ciliary body involvement and EOE.
In contrast to the basic principles of oncology, histological or cytologic evaluation is not routinely used in the diagnosis of intraocular neoplastic lesions. The diagnosis of uveal melanoma is based primarily on clinical examination by biomicroscopy and indirect ophthalmoscopy, and experienced clinicians can diagnose a uveal melanoma based on clinical examination. However, it is imperative that additional diagnostic testing is done. Ancillary testing will include ultrasonography, color fundus photography, fundus fluorescein angiography, indocyanine green angiography, optical coherence tomography, fundus autofluorescence, and ultrasound biomicroscopy.
Ultrasound sonography is the most often used auxiliary method. Ultrasonography helps to measure size and detect growth, which is highly valuable in the follow-up. The A-mode examination of the tumor typically shows a medium to low internal reflectivity. In B mode, the tumor appears as an acoustically hollow dome-shaped or mushroom-shaped choroidal mass. A choroidal excavation and orbital shadowing may be observed, especially in large tumors. This helps enforcing clinical diagnosis and allows discrimination from hemangioma that typically shows high reflectivity. The presence of EOE can be recognized by areas of hyporeflectivity compared to normal orbital tissue. However, this should be verified by additional computer tomography (CT) and magnetic resonance imaging (MRI) (38–41).
Ultrasound biomicroscopy is useful for the evaluation of tumors that originate from the ciliary body and to detect extension and differentiate it from a cyst. This technique allows the visualization and evaluation of hyporeflective plaques on the tumor surface, internal reflectivity, tumor-specific vasculature, and, if present, EOE (42, 43).
Fundus fluorescein angiography and indocyanine green angiography may help visualize uveal melanomas that feature intrinsic tumor circulation as well as choroidal circulation (Figure 1). The observation of this double circulation pattern or leakage from tumoral vasculature is occasionally necessary in order to confirm the diagnosis. Fundus fluorescein angiography is also used in the detection and follow-up of complications such as radiation retinopathy and radiation maculopathy after radiotherapy (40).
Spectral domain optical coherence tomography allows for the detailed evaluation of changes in the retina and retinal pigment epithelium overlying lesions in choroidal melanoma. It helps to detect subretinal fluid, which is considered to be one of the high-risk features predicting transformation into melanoma (44, 45). With enhanced depth imaging (EDI), it is now possible to examine deeper tissues like the choroid and sclera and to measure the thickness of flat tumors (46, 47).
On fundus autofluorescence imaging, pigmented tumors exhibit moderate hypoautofluorescence, whereas nonpigmented (amelanotic) tumors show moderate hyperautofluorescence. However, in both tumor types, the orange pigment can be distinguished from drusen using this method (48, 49).
CT and MRI have an important role in the evaluation of EOE. On CT it appears as a hyperdense mass with mild/moderate contrast and distinct margins. On MRI, the tumor characteristically returns a hyperintense signal on T1-weighted images and hypointense signal on T2-weighted images. However, this can also be observed in the subacute phase of a circumscribed hemorrhage and choroidal hemangioma. These imaging methods are not strictly necessary in the diagnosis stage but are a requirement in the planning stage of proton beam therapy or stereotactic radiotherapy (SRT) (39).
In some cases, a tumor biopsy is useful and provides material not only for a final diagnosis but also for cytogenetic analysis that can provide a prognostic value. A biopsy can be performed in different ways. Anterior segment tumors can be evaluated by aqueous humor sampling, and incisional or excisional biopsy. Fine-needle biopsy (transscleral, transvitreal, or transcameral), vitrectomy biopsy (Figure 2), and incisional or excisional biopsy (endoresection or transscleral resection) can be done in order to evaluate posterior segment intraocular tumors (50).
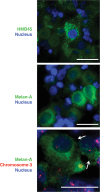
Immunohistology of a tumor biopsy disclosing HMB-45- (upper) and Melan-A- (middle) positive cells and Immuno-FISH for chromosome 3 (lower). Arrows indicate Melan-A-positive cells with monosomy 3. Scale = 25 μm.
Studies on tumor doubling time of choroidal melanoma indicate that micrometastases occur several years before diagnosis (51, 52). An early detection, for example, in cases of suspicious lesions would therefore be useful to avoid a crescendo malignancy and the continuous dissemination of malignant cells into the systemic circulation. However, usually, small lesions are only observed until growth has been demonstrated. A reason for this strategy is that even a fine-needle biopsy is quite invasive for a small and complex organ such as the eye. Therefore, an evolving and less invasive new approach in diagnostics aims at the detection of circulating melanoma cells. This approach is referred to as “liquid biopsy” (see Chapter 3).
The therapy of uveal melanoma depends on the tumor size, location, associated features, status of the other eye, patient’s systemic status, and the patient’s desire. Management choices include transpupillary thermotherapy (TTT), plaque radiotherapy, charged particle irradiation, stereotactic radiotherapy (SRT), local resection, enucleation, or orbital exenteration.
TTT is a treatment method that utilizes a modified diode laser delivery system to induce hyperthermia in tumor by delivering light in the infrared range. Tumor is heated to a temperature of 60–65°C (53). When TTT was introduced in the treatment of choroidal melanoma, short-term follow-up data impressively showed that in appropriate cases, tumor regression may be achieved in more than 90% of the patients (54). Studies with a longer follow-up, however, have dampened the initial enthusiasm, showing that conventional photocoagulation and TTT may not be as different as previously thought. Singh et al. found an average recurrence rate of 17% (8–56%) after primary TTT in small melanoma patients and reported that 7% of these recurrences involved extrascleral extension (55). The therapy shall be limited to flat tumors (2.5 mm). With the high risk of tumor recurrence (56, 57), patients should be selected carefully and monitored closely when treated with TTT. TTT has been currently adapted in combination with plaque radiotherapy (58) or is applied as secondary treatment to local tumor recurrence after radiotherapy or local resection (59). Shields et al. combined plaque radiotherapy and TTT for choroidal melanoma in 270 consecutive patients and found that plaque radiotherapy combined with TTT provides excellent local tumor control, with only 3% recurrence at 5-year follow-up (60). Complications of this therapeutical approach include macular traction, vascular occlusion, and hemorrhage (61).
Radiotherapy is currently the most common treatment for uveal melanoma, especially in posterior uveal melanoma. In clinical application, radiotherapy can be administered in the form of radioactive plaque, external beam radiotherapy, or SRT with a linear accelerator.
Plaque brachytherapy is commonly performed with the radioisotopes ruthenium-106 (beta-source), iodine-125 (gamma source), or a combination of both. Other less used gamma-particle-emitting isotopes are Cobalt-60 and Palladium-103. The tumoricidal dose that needs to be delivered at the apex of the tumor is around 100 Gy. Brachytherapy is the most often used treatment modality, but it depends on tumor thickness and location. Ru-106 plaques have been found effective for small and medium tumors (basal diameter up to 16 mm and thickness up to 6 mm) when applied alone or up to 8 mm in thickness when used in combination with TTT (62).
The medium tumor arm of the Collaborative Ocular Melanoma Study (COMS) included tumors 2.5–10 mm thick with a basal diameter less than 16 mm and compared patients treated by I-125 plaque brachytherapy versus enucleation. There was no significant difference between the two groups in 10-year mortality. Melanoma-related mortality rates at 5, 10, and 12 years were 10, 18, and 21%, respectively, in the brachytherapy group, versus 11, 17, and 17% in the enucleation group (63).
Studies using Ru-106 plaques have shown that this isotope carries an increased risk of local recurrence with tumors having a thickness over 5 mm (64). The therapy-related complications include cataract, radiation retinopathy and opticopathy, maculopathy, neovascular glaucoma, and an exudative tumor response (65, 66).
Charged particle irradiation is indicated when tumor size and location does not allow to proceed with brachytherapy. This modality can be used to treat tumors up to 14 mm thick with a basal diameter up to 28 mm. Desjardins et al. reported 5- and 10-year metastasis rates of 18.5 and 26.6%, respectively. Local recurrence was observed in 4% of the patients at 5 years and 10% at 10 years, with most occurring in the first 3 years after treatment (67).
Though - very large tumors may be treated with this technique (68, 69), tumor necrosis following irradiation may either lead to surgical resection or eventual secondary enucleation. This therapy appears attractive for small tumors at the posterior pole involving the macula and/or the optic nerve. However, vision loss will occur in 68% of patients at 5 years after treatment (70). Radiation-induced complications are similar to brachytherapy but include also loss of eyelashes in 12%, retinal detachment in 8.5%, glaucoma in 23.4%, dry eye in 6%, cataract requiring surgery in 15%, optic neuropathy in 18%, and maculopathy in 37% of the patients after a follow-up period of 8 years (67).
An alternative to proton beam therapy is the stereotactic irradiation with a photon beam. Although proton beam therapy is theoretically not proven superior in terms of sparing healthy tissue from the effects of radiation, stereotactic radiosurgery is more advantageous, as it does not require preoperative surgical marking and is more cost-effective (71, 72).
In SRT, the radiation is delivered either as a single dose or fractionated SRT in smaller equal doses. The devices used in stereotactic photon beam irradiation are the Gamma Knife, linear accelerator, and the Cyber Knife. An advantage of the stereotactic approach is that the tumor borders are determined by MRI and CT and no surgical procedure is required to determine the tumor’s location (73).
Gamma Knife has been used to treat uveal melanomas with successful results (74, 75). However, it is not a preferred treatment modality due to high reported rates of radiation retinopathy and neovascular glaucoma (8.6–64%) (76). The linear accelerator is used to treat uveal melanoma by stereotactic hypofractionated radiotherapy. The advantages of this approach are less radiation exposure to the healthy tissues adjacent to the tumor and avoidance of long-term effects. Noninvasive fixation systems designed for use with linear accelerators have increased patient comfort and compliance with treatment (73).
Using SRT, Zehetmayer et al. achieved a local tumor control in 98% of cases and tumor height reduction in 97%. The mean relative tumor volume reductions were 44, 60, and 72% after 12, 24, and 36 months, respectively. Seven patients developed metastases (11%). Secondary enucleation was performed in eight eyes (13%). Morbidity was significant in tumors exceeding 8 mm in initial height. With tumors larger than 8 mm and a dose of 10 Gy/fraction arose a high risk for radiation-induced inflammation (77).
A common complication of all types of radiotherapy is radiation retinopathy and opticopathy. The underlying pathomechanism is a chronic, progressive vasculopathy of the capillaries resulting from radiotherapy-induced damage to the vascular endothelium (78).
This damage causes capillary dilation, increased vascular permeability, thrombosis, and retinal exudate and hemorrhage, eventually leading to full thickness retinal atrophy and capillary nonperfusion. The first sign may be a decrease in visual acuity due to subclinical macular edema. Ischemic retinopathy can often progress to proliferative retinopathy and vitreous hemorrhage. Guyer et al. reported the incidence of radiation maculopathy after proton beam radiotherapy as 90% (79). Radiation-induced optic neuropathy typically causes sudden, painless, unilateral vision loss starting as early as 3 months or up to 8 years after radiation exposure (80, 81).
Primary enucleation is generally indicated for large melanomas that occupy most of the intraocular space or for tumors that have invaded the optic nerve. In terms of survival, many studies have demonstrated no significant difference in mortality between eye-conserving therapies and enucleation. Comparison of the COMS medium uveal melanoma patients treated with plaque brachytherapy and those that underwent enucleation revealed no significant difference in long-term survival (63, 82).
Therefore, in recent years, eye-conserving treatments have gained favor over enucleation. Local resection is an alternative treatment choice for choroidal melanoma patients, which spares the eye. Choroidectomy is currently only performed by a small number of surgeons due to the technical challenges involved. Tumors can be surgically removed via a transretinal (endoresection) or transscleral (exoresection) route. Major complications such as retinal detachment and vitreous hemorrhage have been reported with both techniques (83).
Secondary enucleation is indicated in some cases of recurrence or otherwise nonmanageable complications associated with other therapies. In most cases after enucleation, an orbital implant is inserted into the socket. No implants shall be used when extrascleral extension is detected and no orbital exenteration is indicated.
Despite the availability of different treatment modalities and usually good local tumor control, patients with uveal melanoma are at risk for metastatic disease, and survival rates have not changed in 40 years. Metastatic dissemination occurs hematogenously and typically involves the liver in about 90% of cases. Metastasis, however, can also appear in lung (24%), bone (16%), and other organs (84, 85). Death usually occurs mostly within 12 months after detection of metastatic disease (86). Patients with liver metastases survive for an average of 4–6 months, with a 1-year survival rate of 10–15%. Reported survival time for patients with other metastases is 19–28 months (87, 88).
Treatment by systemic or local chemotherapy and/or partial hepatectomy rarely prolongs life (89). In order to have the chance of an impact on the poor survival rate, early detection of metastatic disease is necessary. Therefore, systemic monitoring that includes primarily the liver function and imaging using ultrasonography or MRI twice yearly is advised.
Based on the facts that patients may develop metastatic disease even after enucleation of the primary tumor-bearing eye and with regard to studies demonstrating circulating melanoma cells (see Chapter 3 on “liquid biopsy”) in patients without clinically evident metastases, it is to be assumed that many patients already have undetectable micrometastases at the time of detection and treatment of the primary tumor.
The risk for metastatic disease depends on clinical factors. It increases with the tumor size, location at the ciliary body, and EOE with involvement of the lymphatic pathway. A comparative analysis of uveal melanoma has indicated that the 5-year survival rates after enucleation were 84% for small, 68% for medium-sized, and 47% for large tumors (90).
The prognosis is also dependent on the histological type of the tumor, with a higher risk of metastasis when the tumor shows an epithelioid cell type, microvascular networks, mitotic activity, and lymphocytic infiltration. Cytogenetic analysis definitively helps in prognostication. Monosomy 3 has a significant risk for metastatic disease (19). Abnormalities associated with chromosomes 1, 6, 8, and 11 add to the increased risk. Gene-expression profiling identifies two classes of melanoma, in which disomy-3-positive class 1 (low grade) showed 95% survival and monosomy-3-positive class 2 (high grade) showed only 31% survival at 8 years (91–93).
Estimation of the survival probability is important in many ways. Good prognosis may be helpful for the psychological support of the patient. With high risk for metastases, a more intense monitoring and eventually recruitment of patients in ongoing studies for systemic adjuvant therapy shall be suggested. However, in the absence of a proven beneficiary treatment, opinions differ about informing patients on their prognosis.
Despite advances in the diagnosis and treatment of uveal melanoma, the general mortality remains high due to metastatic disease that is still resistant to treatment. Adjuvant therapies that target micrometastases instead of macrometastases may be therefore a more successful approach. Because of possible side effects and part of the patients being at low risk for metastatic disease, it is important to select the high-risk patients that may benefit from therapy and therefore tolerate this negative facet. Systemic treatment options include chemotherapy, immunotherapy, hormone therapy, biologic therapy, and targeted therapy. Nonrandomized studies conducted so far have not reported promising results (94–98).
The MAPK pathway, activated by GNAQ mutations, has been considered a potential therapeutical target. The MEK-inhibitor selumetinib administered to uveal melanoma patients with GNAQ mutation extended progression-free survival. However, one limitation of MAPK inhibitors is that the drug is effective for an average of 6–10 months, and it is believed that this leads to more aggressive recurrences (99, 100).
Studies on preventing metastasis and extending survival in high-risk uveal melanoma patients are currently in progress for ipilimumab, dacarbazine, recombinant interferon alpha-2b I, c-Ros oncogene inhibitor crizotinib, sunitinib and valproic acid, and arylsulfonamides (101–105).
Dendritic cells and vaccine therapies are a different approach as they aim to elicit therapeutically relevant immune responses in patients. This approach has been extensively investigated throughout the past decade for different types of cancers, including uveal melanoma. However, as of now, their value has not been conclusively proven (106).
Although this chapter gives an overview of uveal melanoma, the pathogenesis and therapy of this intriguing malignancy is far away from being fully understood. Various controversial hypotheses challenge pathologists and clinicians. These affected the way the patients were treated in the past and even today various centers have different algorithms on diagnostics, monitoring, and therapy. What has not changed is the lethal outcome when the malignancy exhibits features of a high metastatic risk. However, a “crescendo malignancy” is assumed and supported by intra-tumoral genetic heterogeneity, which suggests an ongoing evolutionary process (93, 107). Understanding the cells and the mechanisms leading to micrometastases is of paramount importance to find an appropriate and effective therapy to avoid melanoma-related deaths. Until this is accomplished, screening for suspicious lesions, intensified diagnostic steps, and early treatment may save lives.
Conflict of interest: The authors declare no potential conflict of interest with respect to research, authorship, and/or publication of this article.
Copyright and permission statement: To the best of our knowledge, the materials included in this chapter do not violate copyright laws. All original sources have been appropriately acknowledged and/or referenced. Where relevant, appropriate permissions have been obtained from the original copyright holder(s).
This open access article is licenced under Creative Commons Attribution 4.0 International (CC BY 4.0). https://creativecommons.org/licenses/by-nc/4.0/
Your browsing activity is empty.
Activity recording is turned off.
See more...