New business models for the development of products to treat rare and neglected diseases have developed within a legal and regulatory framework that has been shaped largely by the 1983 Orphan Drug Act. Dr. Coté provided an overview of the Food and Drug Administration’s (FDA) orphan drug program.
The year 2008 marks the twenty-fifth anniversary of the passage of the Orphan Drug Act and the establishment of the FDA’s Office of Orphan Products Development (OOPD). In the decades before 1983, those with rare diseases suffered what Coté termed “pharmacologic neglect.” It was impossible for pharma to earn a reasonable return on its research investment in therapies for such conditions given the small number of patients who would benefit. Academic laboratories would occasionally discover promising new therapies, but without the capital required to conduct the clinical trials necessary for FDA approval and without the interest of a pharmaceutical company in bringing these compounds to market, these potential new products remained undeveloped. While the science of drug discovery progressed rapidly during this period, yielding powerful insights into human biology and the pathologic processes of diseases, rarely were these insights applied to research on rare diseases.
The irony is that rare diseases provide a critically important window into disease processes that can be of benefit across the full range of medical research. For example, much has been learned about hemoglobin from people with hemoglobinopathies, such as sickle cell anemia and thalassemia. Understanding of the urea cycle was gleaned from the experiences of patients with urea cycle disorders. Patients with diseases such as phenylketonuria (PKU) provided knowledge about amino acid metabolism. These are but a few of the hundreds of known examples. Absent patients with rare diseases, many basic aspects of medical science would be less well understood. Despite these benefits, however, opportunities for research on rare diseases and the ability of patients to receive licensed therapies for these illnesses have historically been limited.
In this context emerged the National Organization for Rare Disorders (NORD), a powerful political movement founded by grassroots organizer Abbey Meyers. She knew that rare diseases were individually infrequent but collectively common. Meyers and several others who shared her vision drafted legislation that would change the way drugs are developed. The most important feature was an allowance for 7 years of market exclusivity, during which a company could recoup some of the expense of drug development. Additionally, tax credits and exemptions from fees made it possible to build a sound business model on investments in products for rare diseases. Clarifying terminology, Coté explained that “rare” is defined by regulation as diseases that affect fewer than 200,000 people in the United States; “neglected” is the term used by the tropical medicine community. While tropical diseases have significant impact in the developing world, all tropical diseases are rare diseases as defined in the U.S. Orphan Drug Act.
Biotechnology as an industry was propelled into a major expansion by the Orphan Drug Act. Thousands of scientific, commercial, and humanitarian opportunities were made possible by the act that could otherwise not have existed. Historically, pharma has been less than fully responsive to these opportunities, but this situation is changing as the country’s larger scientific and fiscal drug enterprise recognizes the value of investing in orphan drug development.
The crafters of the Orphan Drug Act also intended to jumpstart the science behind rare diseases. The establishment of the National Institutes of Health’s Office of Rare Diseases is a prime example of this. Congress also established the Orphan Products Grant Program at FDA, administered by OOPD, which Coté believes “has become the single most tangibly productive grants program in the entire U.S. government.” The program is currently funded at only $14 million per year and has been declining in buying power over the past 15 years; nonetheless, it has yielded 41 FDA-approved therapies.
During the 25-year history of OOPD, the program has been successful, granting more than 1,850 orphan drug designations, 326 of which have received full FDA marketing approval (see Figure 3-1). And as noted above, 41 of these drugs came out of the OOPD grants program. FDA estimates that collectively, these therapies have benefited about 12 million Americans with rare diseases, many of them children. Despite the program’s accomplishments it has only resulted in therapies for less than 10 percent of such diseases—approximately 6,000 diseases are designated as rare. If one looks at orphan drug designations by organ system, the largest category is oncology drugs, but virtually every organ system has been impacted by an orphan drug designation (see Figure 3-2). As noted in the introduction to the workshop, FDA approvals overall have been decreasing. However, the proportion of FDA approvals for orphan drugs has been increasing, and now amounts to roughly one-third of all FDA approvals.

FIGURE 3-1
The number of products that received orphan designation and the number of new drugs approved from 1983 to 2005. Note that the numbers on the charts do not match the numbers in the text because the charts show data only through 2005, while Dr. Coté (more...)
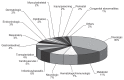
FIGURE 3-2
Diseases treated by orphan drugs, 2000 to 2006. SOURCE: Coté, 2008.
STRATEGIES
Coté emphasized that the objective of the workshop, and of his office, is to accelerate the development of therapies for rare and neglected diseases. To this end, he offered ten strategies for consideration.
Don’t fix what isn’t broken. The Orphan Drug Act is working well through the core activities of OOPD—making orphan designations, awarding grants, providing advocacy, and shepherding products through the FDA approval process. OOPD connects sponsors with the relevant review divisions, which can advise on the design of clinical trials. It also protects the spirit of the Orphan Drug Act, considering all designations carefully to prevent specious products.
Nurture and expand FDA’s relationships with industry. There are a host of reasons why pharma needs to be more involved in the development of orphan products. While the vast majority of the 1,850 orphan compounds came from academia and biotechnology companies, pharma has greater resources to develop these compounds and bring them to market. Orphan drugs can be part of a viable business model, as evidenced by the 326 orphan drug approvals, and can yield the occasional blockbuster (e.g., Gleevec®, Botox®, and synthetic erythropoetin [EPO], all of which started as orphan products). And for an industry currently struggling with its public image, there can be a positive payoff from addressing the unmet medical needs of people with rare diseases. In addition, the orphan drug process is an intermediate step in the growth of personalized medicine. Mastering the development of orphan drugs positions a company to develop products for smaller and smaller populations—the essence of personalized medicine. Moreover, individual pharmaceutical companies have vast libraries of compounds that have not yet been developed, primarily for commercial reasons.
Work together, and make the other party’s job easier. OOPD wants new orphan drugs to reach those who need them, and sponsors want to have new products in their portfolio. OOPD needs the help of all applicants to advance the process as effectively as possible. With designation applications, for example, brevity is key. Although applicants must demonstrate that a drug has potential efficacy for a rare disease or condition that affects fewer than 200,000 people in the United States, Coté suggested this could be done succinctly in two or three pages. In addition, FDA needs to make the process more transparent to applicants by, for example, issuing guidance and performing more outreach. With regard to involving patients and patient advocates, although OOPD does work with patient advocacy groups, there are 6,000 rare diseases, while OOPD has a staff of 25. Therefore, the office relies on interactions with umbrella organizations, such as NORD and the Genetic Alliance.
Understand the value of the grants program. As noted earlier, 41 drug approvals came out of OOPD’s grants program, a clear demonstration of its significant success. Yet the total budget for the program has remained essentially flat when inflation and the increasing costs of conducting clinical trials are taken into account, and the program is currently funded at only $14 million. During the open discussion following Coté’s presentation, one participant who is not currently a federal employee urged workshop participants to discuss with their members of Congress how increasing the grants funding for OOPD could yield a substantial payoff for human health.
Focus on tropical infectious diseases. The last 50 years has seen very limited development of anti-infective drugs for the developing world. However, with the passage of the Food and Drug Administration Amendments Act (FDAAA) of 2007 and the establishment of the priority review voucher, this situation could finally change. The priority review voucher uses current market forces to create new development incentives. Prior to FDAAA, once FDA had approved a new orphan drug to treat a tropical disease, the sponsor found itself in the difficult position of having a lifesaving new drug that it could not sell since the people who needed it could not afford it. Under FDAAA, the sponsor of a New Drug Application (NDA)/Biological License Application (BLA) for a drug to treat a tropical disease receives a priority review voucher that is redeemed on a subsequent NDA/BLA and ensures review and action by the agency within 6 months of submission of that application. The critical aspect of the priority review voucher is that it is transferable and can be sold to another sponsor, which can then apply it to any other drug. Getting a product to market more rapidly can be highly valuable to a large pharmaceutical company with a potential blockbuster in the pipeline (some say a blockbuster can earn $5–10 billion per year), and selling the voucher generates income for the sponsor of the orphan drug. In addition to the new priority review voucher system, FDA is working with the World Health Organization (WHO), OneWorld Health, the Sabin Institute, and the Gates Foundation on the possibility of establishing an “orphanage” of early-stage drug candidates for tropical diseases. This entity would be housed within an existing nongovernmental organization and created fairly rapidly, perhaps through a workshop involving no more than 12 professionals who would generate 40–50 orphan designation applications over the course of 1 week. Drug candidates entered into the orphanage would already be eligible for the priority review voucher and have both FDA and European Medicines Agency (EMEA) orphan status, making this a valuable resource for pharmaceutical and biotechnology companies wishing to acquire orphan drug candidates for the purposes of obtaining their own priority review vouchers.
Know thyself. While the numbers of orphan designations and approvals are known, there are other important statistics that OOPD has not tracked. Moving forward, OOPD plans to determine the proportion of designated orphans for which Investigational New Drug (IND) applications have been submitted; the number of NDAs or BLAs that have been filed; the phase of development of each product and the locations of ongoing clinical trials; and the projected timeline for progression between development Phases I, II, and III. Lastly, OOPD plans to explore whether it can identify predictors for successful development and approval.
Know the disposition of all designees. Over 1,850 products have received orphan designation. Although 326 orphan products have been approved, OOPD is interested in determining the status of the remaining 1,525 orphans that have not yet received approval. While some are probably in development, others have been abandoned for various scientific or business reasons, and it is likely that some of these designees still hold promise. OOPD plans to develop criteria that could be used to screen the regulatory submissions for the remaining designated orphans to determine which hold promise.
Explore the possibility of orphan policy in drug review. There is no special policy for the review of drugs to treat rare diseases; prior to approval, all drugs are required to have shown “substantial evidence of effectiveness” (21 CFR § 314.50). And while the FDA review divisions have, according to Coté, sense, sensibility, and sensitivity with regard to the issues surrounding orphan drugs, the level of understanding is not homogeneous. FDA would like to consider what if any policies might be needed to better facilitate the review of orphan drugs and is beginning discussions to that end internally and with Institute of Medicine (IOM) leadership.
Mentor the review divisions in the fundamental science underlying small clinical trials. In 2001, the IOM released a report on conducting and interpreting small clinical trials (IOM, 2001). Expanding on this study, OOPD would like to establish a curriculum to enhance the knowledge of reviewers regarding the fundamental science underlying small clinical trials. As highlighted in the IOM report, there are new methodologies, each with its own strengths and weaknesses, with which reviewers should be familiar. OOPD hopes to create a cadre of go-to reviewers who would have expertise in small clinical trials and interpretation of the data that orphan drug sponsors would submit, and would employ these new methodologies.
Act globally. OOPD has worked to establish relationships with regulatory agencies around the world, particularly EMEA. Globally, there are patients with the same diseases and researchers working to understand the same underlying science. EMEA passed its own orphan drug act in 1999, and while there are some important differences, the EMEA and U.S. acts have many similarities. EMEA and FDA now share a joint application form for orphan product designation that can be submitted to either or both EMEA and FDA. The processes are still independent, but the agencies hold monthly teleconferences and are reviewing many of the same applications. There is also an exchange program whereby people from the European orphan drug office and OOPD meet to learn how each operates. Coté noted that OOPD has not yet reached out significantly to Japan and other countries, but that such interactions are planned for the future.
Footnotes
- 1
This chapter is based on the presentation of Timothy Coté, M.D., M.P.H., Director, FDA Office of Orphan Products Development.
Publication Details
Copyright
Publisher
National Academies Press (US), Washington (DC)
NLM Citation
Institute of Medicine (US) Forum on Drug Discovery, Development, and Translation. Breakthrough Business Models: Drug Development for Rare and Neglected Diseases and Individualized Therapies: Workshop Summary. Washington (DC): National Academies Press (US); 2009. 3, The Food and Drug Administration’s Orphan Drug Program.

