

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking. Lyon (FR): International Agency for Research on Cancer; 2004. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 83.)

4.1. Absorption, distribution, metabolism and excretion
4.1.1. Humans
The Working Group attempted to provide extensive coverage of the published literature since 1985, in some cases referring to recent reviews.
(a) Introduction
Most carcinogens are enzymatically transformed to a series of metabolites as the exposed organism attempts to convert them to forms that are more readily excreted. The initial steps are usually carried out by cytochrome P450 (P450) enzymes that oxygenate the substrate (Guengerich, 1997). Other enzymes such as lipoxygenases, cyclooxygenases, myeloperoxidase and monoamine oxidases may also be involved, but less commonly. If the oxygenated intermediates formed in these initial reactions are electrophilic, they may react with DNA or other macromolecules to form covalent binding products known as adducts. This process is called metabolic activation. Alternatively, these metabolites may undergo further transformations catalysed by glutathione S-transferases, uridine-5′-diphosphate (UDP)-glucuronosyltransferases, epoxide hydrolase (EH), N-acetyltransferases (NATs) (Kadlubar & Beland, 1985), sulfotransferases and other enzymes (Armstrong, 1997; Burchell et al., 1997; Duffel, 1997). Such reactions frequently, but not always, result in detoxification.
Figure 4.1 presents an overview of the metabolism of the six tobacco smoke carcinogens for which the formation of DNA adducts has been demonstrated in human tissues, namely, benzo[a]pyrene (IARC, 1983a, 1987), 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone (NNK) (IARC, 1985a; Hecht et al., 1994), N-nitrosodimethylamine (NDMA) (IARC, 1978a; Shuker & Bartsch, 1994), N′-nitrosonornicotine (NNN) (IARC, 1985b; Hecht et al., 1994), ethylene oxide (IARC, 1994a), and 4-aminobiphenyl (4-ABP) (IARC, 1972; Kadlubar, 1994). The major metabolic activation pathway of benzo[a]pyrene is conversion to a 7,8-diol-9,10-epoxide, which is highly carcinogenic and reacts with DNA to form adducts with the exocyclic N2 of guanine (Cooper et al., 1983). In competition with this process are detoxification pathways leading to phenols, diols and their conjugates as well as other metabolites. The major metabolic activation pathways of NNK and its main metabolite, 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanol (NNAL), involve hydroxylation of the carbons adjacent to the N-nitroso group (α-hydroxylation) which leads, via diazonium ions, to the formation of two types of DNA adduct: methyl adducts such as 7-methylguanine and O6-methylguanine (IARC, 1985a), and pyridyloxobutyl adducts (Hecht, 1998). Glucuronidation of NNAL and pyridine-N-oxidation of NNK and NNAL are detoxification pathways. The metabolic activation of NDMA occurs by α-hydroxylation leading, via methyl diazonium ions, to the formation of 7-methylguanine and O6-methylguanine. Denitrosation, producing nitrite and methylamine, is considered to be a detoxification pathway (Preussmann & Stewart, 1984). Aldehydes are also formed in the metabolism of NNK and NDMA. Their role in carcinogenesis is unclear. α-Hydroxylation of NNN can lead to the formation of pyridyloxobutyl adducts whereas detoxification occurs by β-hydroxylation, pyridine-N-oxidation and denitrosation/oxidation to produce norcotinine (Hecht, 1998). Ethylene oxide reacts directly with DNA to form 7-(2-hydroxyethyl)guanine and other adducts. There are competing detoxification pathways involving glutathione conjugation (IARC, 1994b). 4-ABP is metabolically activated by N-hydroxylation. Conjugation of the resulting hydroxylamine with acetate or other groups such as sulfate ultimately produces nitrenium ions that react with DNA to produce adducts mainly at C-8 of guanine. Acetylation of 4-ABP can be a detoxification pathway if it is not followed by N-hydroxylation. Ring hydroxylation and conjugation of the phenols result in detoxification (Kadlubar & Beland, 1985).
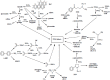
Figure 4.1
Metabolism of six tobacco smoke carcinogens which produce DNA adducts that have been identified in the lungs of smokers
The balance between metabolic activation and detoxification varies between individuals exposed to these genotoxic components of tobacco smoke and is likely to affect cancer risk because DNA adducts are absolutely central to the carcinogenic process induced by these agents (Hecht, 1999; Tang et al., 2001). DNA adducts, if unrepaired, can cause miscoding during replication resulting in permanent mutation.
Cells have DNA repair systems that can remove adducts and restore the DNA to its normal structure (Memisoglu & Samson, 2000; Pegg, 2000; Hanawalt, 2001; Norbury & Hickson, 2001). There are interindividual differences in their capacity for DNA repair that can affect cancer risk (Wei et al., 2000). Moreover, DNA repair systems are not completely efficient or error-free, and some adducts escape repair and persist in DNA. These persistent DNA adducts can cause miscoding. For example, when the O6-position of guanine in DNA is methylated following metabolic activation of NNK, the resulting DNA adduct, O6-methylguanine, is misread by DNA polymerases as adenine, and thymine is inserted during replication (Loechler et al., 1984). The consequence is the permanent conversion of a G:C base pair to an A:T base pair. This mutation and others can activate oncogenes such as KRAS or inactivate tumour-suppressor genes such as TP53. There are considerable data to indicate that mutations in KRAS and TP53 result directly from the reaction of these genes with metabolically activated carcinogens (Hecht, 1999). Many genetic abnormalities occur during the process of lung cancer induction. These include loss of heterozygosity, microsatellite alterations, mutations in RAS oncogenes, MYC amplification, BCL-2 expression, mutations in the TP53, RB, CDKN2A and FHIT tumour-suppressor genes, expression of telomerase activity and others (Figure 4.2) (Wistuba et al., 1997; Sekido et al., 1998). Although the temporal sequence of mutations is somewhat unclear, we do know that carcinogens can, through the process just described, cause irreversible damage to critical genes involved in the control of cellular growth. Smokers are subjected to a chronic barrage of metabolically activated carcinogens that cause these multiple changes (Figure 4.2). This constant assault on genes is entirely consistent with genetic derangements that lead to six proposed hallmarks of cancer:
- self-sufficiency in growth signals;
- insensitivity to anti-growth signals;
- evasion of apoptosis;
- tissue invasion and metastasis;
- sustained angiogenesis; and
- limitless replicative potential (Hanahan & Weinberg, 2000).

Figure 4.2
Scheme linking cigarette smoke carcinogens with genetic changes in lung and lung cancer development
Urinary carcinogen metabolites, carcinogen-protein adducts, and carcinogen-DNA adducts have been used as biomarkers to assess the uptake, metabolic activation and detoxification of tobacco carcinogens in humans. These are discussed in the following sections.
(b) Effects of tobacco smoke on human enzyme activities and metabolism
(i) Enzyme induction
In-vivo studies
The induction by tobacco smoke of several phase I and phase II enzymes in human tissues, including P4501A1, P4501A2, P4502E1 and some isoforms of UDP-glucuronosyltransferase has been widely reported (Anttila et al., 1991; Guengerich et al., 1991; Bock et al., 1994). Similarly, the pharmacological interactions between tobacco smoking and drugs, many of them as a consequence of enzyme induction, have been reviewed (Zevin & Benowitz, 1999). The consequences of this induction are evident in the altered rate of metabolism of many drugs and carcinogens (Zevin & Benowitz, 1999). For example, smoking decreases the activity of H2-receptor antagonists in reducing nocturnal gastric secretion (Boyd et al., 1983). The metabolic enzymes are also induced by poly-cyclic aromatic hydrocarbons (PAHs), which may therefore be the components of tobacco smoke that are largely responsible for its enzyme-inducing activity. Cigarette smoke contains a variety of ligands that bind to the Ah receptor, which is known to mediate induction of P4501A1 and P4501A2 (Zevin & Benowitz, 1999).
Pulmonary P4501A1 activity (reflected by aryl hydrocarbon hydroxylase (AHH) activity) is elevated in smokers, and a highly significant correlation between enzyme activity and levels of PAH–DNA adducts has been reported (Geneste et al., 1991; Alexandrov et al., 1992). Among smokers, women with lung cancer had higher levels of CYP1A1 mRNA and bulky DNA adducts than men with lung cancer (Mollerup et al., 1999).
Smoking results in induction of P450 activity in the human placenta. Evidence for this comes from in-vitro experiments in which placental microsomes were incubated with benzo[a]pyrene in the presence of calf thymus DNA; higher levels of DNA adducts result from incubation of microsomes of smokers than of those from nonsmokers (Kim et al., 1992). AHH activity is higher in placental microsomes from women who smoke than from women who do not, but EH activity is not (Vaught et al., 1979). Placental P450 activity, as measured by in-vitro oxidation of 7-ethoxyresorufin, was found to be 10- to 30-fold higher for female smokers than for nonsmokers (Manchester & Jacoby, 1981). In another study, placental microsomes from 32 female smokers were twice as active in benzo[a]pyrene metabolism as microsomes prepared from the placentas of 25 nonsmokers, but the in-vitro formation of DNA adducts in the presence of calf thymus DNA was not significantly higher in smokers (Sanyal et al., 1994). In a recent study, maternal smoking was found to be significantly associated with levels of placental CYP1A1 mRNA (p < 0.01) but not with levels of PAH–DNA adducts measured by competitive enzyme-linked immunosorbent assay (ELISA) (Whyatt et al., 1998).
In humans, cigarette smoking significantly enhances CYP2E1 activity in alcoholic patients, as measured by increased metabolism of chlorzoxazone in vivo (Girre et al., 1994). CYP2E1 bio-activates some substrates in tobacco smoke and other pro-carcinogens and several hepatotoxins (Guengerich et al., 1991).
The levels of three enzymes involved in DNA repair in the peripheral blood cells of 20 smokers and 17 nonsmokers were compared. O6-Alkylguanine–DNA–alkyltransferase activity was the same in both groups, but the activities of methylpurine (MeP) and 2,6-diamino-4-hydroxy-5N-formamidopyrimidine–DNA glycosylase were lower in nonsmokers; the difference was statistically significant only for MeP (Hall et al., 1993). Exposure of human buccal cell cultures to organic extracts of tobacco (bidi) smoke condensate and betel leaf decreased O6-methylguanine–DNA methyltransferase activity (Liu et al., 1997).
In-vitro studies
Although there have been many in-vitro studies of the properties of tobacco carcinogens, their effects are not always the same as those of the complex mixture of tobacco smoke. For example, exposure of human U937 cells (human [histiocytic] lymphoma cell line) to tobacco smoke increased the expression of haeme oxygenase-1 (HO-1) and inhibited the activity of nuclear factor-κB (NF-κB) (Favatier & Polla, 2001). However, the tobacco-specific nitrosamine NNK activates NF-κB and induces cyclooxygenase (COX)-1 expression in U937 cells (Rioux & Castonguay, 2000).
(ii) Enzyme inhibition
In-vivo studies
Cigarette smoking has been demonstrated to reduce the level of monoamine oxidase B in the brain by about 40%, relative to the levels in nonsmokers or former smokers, although the effect is probably not caused by nicotine. This enzyme plays a key role in dopamine pharmacokinetics (Fowler et al., 1996a). Measurable reductions in monoamine oxidase B were not observed when nonsmokers smoked only one cigarette, implying that the reduction seen in smokers requires chronic exposure and that it may be a gradual response (Fowler et al., 1999). The activity of the enzyme was also significantly lower in the platelets of 23 heavy smokers (≥ 20 cigarettes/day) than in those of 41 nonsmokers (p < 0.001) (Yong & Perry, 1986). Smoking cessation resulted in an increase in the activity of platelet monoamine oxidase B, which started after a week of abstinence and was approximately back to normal values after 4 weeks. Low baseline activity of monoamine oxidase B is related to more intense withdrawal symptoms (Rose et al., 2001). Similarly, the activity of monoamine oxidase A was markedly reduced (average, 28%) in the brains of smokers compared with nonsmokers (Fowler et al., 1996b). The emerging view is that, whereas nicotine does not reduce monoamine (A and B) oxidase activity by itself, the reduction of this enzyme by other components of tobacco smoke may lead to the potentiation of nicotine's effect by slowing down the catabolism of certain neurotransmitters, i.e. norepinephrine, dopamine and serotonin (Berlin & Anthenelli, 2001).
The activity of EH in human lung was measured in samples obtained from patients undergoing open chest surgery for lung cancer and non-neoplastic pulmonary disease. In 10 ‘non-recent’ smokers (who had not smoked for > 1 month prior to surgery), levels of cytosolic EH activity, but not of microsomal EH activity, were significantly higher than in nine ‘recent’ smokers (who had smoked in the month prior to surgery). Cytosolic EH activity was positively correlated with the number of days since smoking cessation (p < 0.05) and was inversely correlated with the number of cigarettes smoked per day (p < 0.01) (Petruzzelli et al., 1992).
Emphysema is associated with decreased levels of α1-antitrypsin, a plasma protease that inhibits neutrophil elastase activity. Cigarette smoking is associated with a decrease in neutrophil elastase inhibitory capacity in the lower respiratory tract, increasing the risk for the development of emphysema (Ogushi et al., 1991). Elastase activity in human leukocytes can be suppressed in vitro by cigarette smoke extract (Ejiofor et al., 1981). Serum antioxidant activity is significantly lower in smokers than in nonsmokers and, although there is a compensatory increase in ceruloplasmin levels, this increase is insufficient to prevent the suppression of elastase inhibitory capacity of α1-antitrypsin by cigarette smoke extract (Galdston et al., 1984).
In-vitro studies
Numerous studies have reported enzyme inhibition in cultured cells treated with tobacco smoke, tobacco smoke condensate or extract, or known components of tobacco smoke.
A number of tobacco alkaloids, including N-n-octanoylnornicotine and N-(4-hydroxyundecanoyl) anabasine have been shown to inhibit aromatase activity in cultures of the human breast cancer cell lines, MDA-MB-231 and SK-BR-3, decreasing the estrogen biosynthesis (Kadohama et al., 1993). In cultured MA-10 Leydig tumour cells, tobacco alkaloids (nicotine, cotinine, anabasine) and aqueous extract of cigarette smoke inhibited progesterone synthesis and cell growth by a cytotoxic mechanism; this mechanism could reduce fertilization, implantation and early development of human embryos (Gocze & Freeman, 2000).
The activity of lecithin:cholesterol acyltransferase, which is believed to play a pivotal role in facilitating high-density lipoprotein-mediated removal of cholesterol from peripheral tissues such as arterial cells, is inhibited in human blood plasma by cigarette smoke extract and by a number of aldehydes present in tobacco smoke (i.e. acrolein (IARC, 1995a), hexanal, formaldehyde (IARC, 1995b), malonaldehyde (IARC, 1999a) and acetaldehyde (IARC, 1999b)). The inhibition of the enzyme activity is probably involved in the mechanism of smoking-induced atherosclerosis (Chen & Loo, 1995). There is evidence that the mechanism for this inhibition is based on the covalent modification of the two free cysteine residues (Cys-31 and Cys-184) of the enzyme (Bielicki et al., 1995). Further studies have shown that the activity of another enzyme implicated in protecting against atherosclerosis, plasma paraoxonase, is also inhibited through modification of its thiol groups by cigarette smoke extract (Nishio & Watanabe, 1997). However, exposure to gas-phase cigarette smoke from four cigarettes over an 8-h period did not inhibit paraoxonase in human plasma under conditions in which the activity of plasma lecithin:cholesterol acyltransferase was inhibited by > 80% and platelet-activating factor acetylhydrolase by 50% (Bielicki et al., 2001).
Thiol depletion by cigarette smoke may be induced by α,β-unsaturated aldehydes such as acrolein. The activation of respiratory burst by neutrophils stimulated by phorbol myristate acetate (PMA) was impaired by exposure to gas-phase cigarette smoke, either through depletion of cellular glutathione or by inhibition of NADPH oxidase activation. These effects also occurred when neutrophils were exposed to acrolein (Nguyen et al., 2001). Subsequent experiments demonstrated that acrolein inhibits neutrophil apoptosis and that enzymes relevant to this process were either induced by acrolein, e.g. extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinases (p38 MAPKs), or their activation was prevented, e.g. caspase-3 (Finkelstein et al., 2001). In human bronchial fibroblasts, reactive aldehydes such as acrolein have also been shown to be cytotoxic and to inhibit the DNA repair enzyme O6-methylguanine–DNA methyltransferase, possibly by reacting with the cysteine thiol group at the active site of the enzyme; no effect on uracil–DNA glycosylase was observed (Krokan et al., 1985).
The activity of aldehyde dehydrogenase (ALDH) in human blood cells in vitro was found to be inhibited by cigarette smoke condensate in a dose-dependent manner. This inhibition was associated with the non-volatile fraction of the condensate. The lower levels of ALDH activity observed in alcoholics could be due, in part, to smoking, as alcoholics are frequently also heavy smokers (Helander et al., 1991).
Platelet-activating factor (PAF) (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine) is a potent pro-inflammatory agent whose degradation is regulated by PAF acetyl hydrolase (PAF-AH), a plasma enzyme. The activity of this enzyme is inhibited in a dose-dependent manner by cigarette smoke extract, which may explain the increase in plasma PAF concentration noted in smokers and may be relevant to the development of smoking-induced cardiovascular and pulmonary diseases (Miyaura et al., 1992).
(c) Biomarkers of tobacco smoke carcinogens
(i) Urinary compounds
Urinary carcinogens and their metabolites are practical and useful biomarkers of the uptake of tobacco smoke constituents. They can provide important information about exposure to tobacco smoke carcinogens, carcinogen doses and mechanisms of carcinogenesis. The use of urinary compounds as biomarkers for investigating the links between tobacco and cancer has been reviewed (Hecht, 2002b). The structures of the compounds discussed below are illustrated in Figure 4.3.
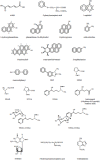
Figure 4.3
Structures of compounds discussed in Section 4.1.1.(c)
trans,trans-Muconic acid (trans, trans-2,4-hexadienedioic acid), S-phenylmercapturic acid and other benzene metabolites
S-Phenylmercapturic acid (S-PMA) and trans, trans-muconic acid (tt-MA) are believed to be the most sensitive biomarkers for exposure to low levels of benzene (IARC, 1982, 1987; Stommel et al., 1989; van Sittert et al., 1993; Boogaard & van Sittert, 1995, 1996; Qu et al., 2000). A recent review has summarized the literature on this subject (Scherer et al., 1998). One pathway of benzene metabolism proceeds via ring oxidation and ring cleavage to trans, trans-muconaldehyde and finally to tt-MA. This metabolite has been widely used as a biomarker of benzene uptake. Significantly elevated levels of tt-MA were found in the urine of smokers in 11 of 13 studies (Lee, B.L. et al., 1993; Melikian et al., 1993; Lauwerys et al., 1994; Melikian et al., 1994; Ong et al., 1994; Rauscher et al., 1994; Ghittori et al., 1995; Ruppert et al., 1995; Boogaard & van Sittert, 1996; Buratti et al., 1996; Ghittori et al., 1996; Kivistö et al., 1997; Ruppert et al., 1997). The levels of tt-MA were 1.4–4.8 times higher in smokers than in nonsmokers and the additional amount of tt-MA excreted by smokers ranged from 0.022 to 0.20 mg/g creatinine (Scherer et al., 1998). However, sorbic acid (trans, trans-2,4-hexadienoic acid), a widely used food preservative that can be transformed metabolically to tt-MA, can also contribute to background levels of tt-MA, thereby decreasing its specificity as a benzene biomarker (Scherer et al., 1998; Pezzagno et al., 1999).
S-PMA is formed by normal degradation of the glutathione conjugate of benzene epoxide. Levels of S-PMA were reported to be significantly higher in smokers (1.71 µmol/mol creatinine) than in nonsmokers (0.94 µmol/mol creatinine) whereas levels of tt-MA in smokers and nonsmokers were not significantly different (Boogaard & van Sittert, 1996).
Phenol (IARC, 1999c), hydroquinone (IARC, 1999d), catechol (IARC, 1999e) and 1,2,4-trihydroxybenzene are also urinary metabolites of benzene. Mixed results have been obtained in studies of the relationship of the levels of these metabolites in urine to occupational exposure to benzene because background levels are high (Inoue et al., 1988, 1989; Ong et al., 1995, 1996; Qu et al., 2000). There was no difference in levels of urinary catechol between smokers and nonsmokers; diet being a major source of these metabolites, the contribution of smoking is comparatively small (Carmella et al., 1982).
1- and 2-Naphthol
1-Naphthol and 2-naphthol (1- and 2-hydroxynaphthalenes) are metabolites of naphthalene (IARC, 2002) that are excreted in urine as glucuronide and sulfate conjugates. The levels of these metabolites are higher in smokers than in nonsmokers (Hansen et al., 1994; Heikkilä et al., 1995; Jansen et al., 1995; Andreoli et al., 1999; Kim et al., 1999; Yang, M. et al., 1999; Nan et al., 2001). For example, Nan et al. (2001) found levels of 3.94 ± 1.89 µmol (geometric mean ± geometric standard deviation)/mol creatinine 2-naphthol in smokers as opposed to 1.55 ± 2.19 µmol/mol creatinine in nonsmokers (p < 0.01). There is some indication that urinary naphthols may be particularly appropriate as biomarkers of inhalation exposure to PAHs, possibly because of the high volatility of naphthalene (Jansen et al., 1995; Yang, M. et al., 1999; Nan et al., 2001). It has been proposed that urinary 2-naphthol is a better biomarker of inhalation exposure than urinary 1-naphthol (Nan et al., 2001), because it correlated more closely with urinary cotinine level than did 1-naphthol (Yang, M. et al., 1999). Levels of urinary 2-naphthol can be affected by genetic polymorphisms in carcinogen metabolizing enzymes such as CYP2E1 and GSTM1 (Yang, M. et al., 1999; Nan et al., 2001).
Polycyclic aromatic hydrocarbons
Hydroxyphenanthrenes and phenanthrene dihydrodiols
Phenanthrene (IARC, 1983b, 1987) is the simplest of the PAHs with a bay region and is a reasonable model for studies of the metabolism of carcinogenic molecules of PAHs with bay regions. Hydroxyphenanthrenes and phenanthrene dihydrodiols have been quantified in human urine. Heudorf and Angerer (2001), using high-performance liquid chromatography (HPLC) with fluorescence detection, reported highly significant differences in concentrations of 2-, 3- and 4-hydroxyphenanthrene between smokers and nonsmokers and dose–response relationships to cigarettes smoked per day, but such relationships were not found with 1-hydroxyphenanthrene. For example, the amounts of 3-hydroxyphenanthrene were 473 ± 302 ng (mean ± SD)/g creatinine in 100 smokers and 305 ± 209 ng/g creatinine in 288 nonsmokers (p = 0.001). Jacob et al. (1999) measured phenanthrene metabolites by gas chromatography–mass spectrometry (GC–MS). They found no significant differences in urinary concentrations of hydroxyphenanthrenes or phenanthrene dihydrodiols between 20 smokers and 10 nonsmokers. [The Working Group noted that the study size was small.] It should be noted that there are important sources of phenanthrene exposure other than smoking (Grimmer et al., 1993; Angerer et al., 1997; Grimmer et al., 1997).
Jacob et al. (1999) found a lower ratio of phenanthrene-1,2-dihydrodiol to phenanthrene-3,4-dihydrodiol in the urine of smokers than in nonsmokers suggesting that smoking induces formation of phenanthrene-3,4-dihydrodiol via induction of P4501A2. Similar results were obtained by Heudorf and Angerer (2001) who found that the ratio of 1- plus 2-hydroxyphenanthrene to 3- plus 4-hydroxyphenanthrene decreased with increased number of cigarettes smoked per day. Both studies also reported a decreased ratio of phenanthrene metabolites to 1-hydroxypyrene with increased smoking, reflecting greater intake of pyrene than phenanthrene in smokers.
1-Hydroxypyrene
Pyrene (IARC, 1983c, 1987) is a non-carcinogenic component of all environmental mixtures of PAHs. The major urinary metabolite of pyrene is 1-hydroxypyrene glucuronide (Sithisarankul et al., 1997). Jongeneelen and colleagues pioneered the development of a method for measurement of 1-hydroxypyrene in urine (Jongeneelen et al., 1985). It has been measured in hundreds of studies of occupational and environmental exposure to PAHs. Several reviews of the data on the effects of smoking have been published (Jongeneelen, 1994; Van Rooij et al., 1994; Levin, 1995; Heudorf & Angerer, 2001; Jongeneelen, 2001). Most studies have measured significantly higher levels of 1-hydroxypyrene in smokers than in nonsmokers. Some representative data from recent investigations of urinary 1-hydroxypyrene are summarized in Table 4.1; these data are from non-occupationally exposed individuals. The levels of 1-hydroxypyrene in the urine of nonsmokers vary considerably and are likely to be influenced by environmental pollution and diet. In most studies, the concentrations of 1-hydroxypyrene in the urine of smokers are about twice as high as those in the urine of nonsmokers, although greater differences have been reported. These concentrations may be influenced by genetic polymorphisms in carcinogen metabolizing enzymes (Alexandrie et al., 2000; Nerurkar et al., 2000; Nan et al., 2001; van Delft et al., 2001).
Table 4.1
1-Hydroxypyrene in the urine of smokers and nonsmokers: selected recent studies.
Benzo[a]pyrene metabolites
The concentrations of benzo[a]pyrene in cigarette smoke are quite low. In laboratory animals its metabolites are excreted mainly in the faeces. Therefore, benzo[a]pyrene metabolites are difficult to quantify in the urine of smokers.
3-Hydroxybenzo[a]pyrene is a major metabolite of benzo[a]pyrene in vitro and is excreted in urine as its glucuronide. Several methods for quantitation of 3-hydroxybenzo[a]pyrene in human urine have been described (Grimmer et al., 1997; Gündel & Angerer, 2000; Simon et al., 2000). In occupationally exposed workers, the levels reported are quite low, ranging from about 1 to 14 (median value) ng/L urine. Limited data are available on the levels of 3-hydroxybenzo[a]pyrene in smokers. One small study reported 0.1–0.8 ng/L urine in three smokers as opposed to < 0.1–0.2 ng/L in three nonsmokers (Simon et al., 2000). In workers in China occupationally exposed to coal smoke, elevated levels of 3-hydroxybenzo[a]pyrene and 9-hydroxybenzo[a]pyrene have been reported in the urine (Mumford et al., 1995).
r-7,t-8,9,c-10-Tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene (trans-anti-BaP-tetraol) is a hydrolysis product of r-7,t-8-dihydroxy-t-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene, the major established ultimate carcinogenic metabolite of benzo[a]pyrene. This metabolite has been quantified in the urine of psoriasis patients treated with a coal-tar ointment and in coke-oven workers and smokers. Concentrations of trans-antiBaP-tetraol in the urine of smokers were lower than in the other two groups, ranging from not detected to 0.2 fmol/µmol creatinine. It was detected in urine samples of nine out of 21 smokers (Simpson et al., 2000).
Benzo[a]pyrene metabolites can be converted to benzo[a]pyrene by treatment with hydrogen iodide. This reaction has been employed as the basis for a technique to determine urinary metabolites of benzo[a]pyrene and several other PAHs (Becher & Bjørseth, 1983; Becher et al., 1984; Venier et al., 1985; Haugen et al., 1986; Buckley et al., 1995). In individuals who were not exposed occupationally, the concentrations of benzo[a]pyrene measured by this method ranged from 4 to 19 ng/mmol creatinine in nonsmokers (n = 5) and 18 to 102 ng/mmol creatinine in smokers (n = 4) (Becher et al., 1984). The disadvantages of this method include different conversion rates for various metabolites and low analytical recoveries (Buckley et al., 1995).
The presence of an unstable benzo[a]pyrene–DNA adduct, 7-(benzo[a]pyren-6-yl)adenine, was reported in the urine of three out of seven smokers and measured in one as 0.6 fmol/mg creatinine (the concentrations were not quantifiable in the other two smokers) (Casale et al., 2001).
Aromatic amines and heterocyclic aromatic amines
Aromatic amines, but not their metabolites, have been quantified in human urine. In one study, smokers excreted 6.3 ± 3.7 µg/24 h 2-toluidine (ortho-toluidine, see IARC, 2000), whereas the concentrations in the urine of nonsmokers were 4.1 ± 3.2 µg/24 h, a non-significant difference (El Bayoumy et al., 1986). Another investigation reported higher concentrations of ortho-toluidine in the urine of smokers than in that of nonsmokers (0.6 and 0.4 ng/L urine, respectively) (Riffelmann et al., 1995). There appear to be important sources of human uptake of ortho-toluidine other than cigarette smoke. Smokers excreted amounts of 4-ABP (78.6 ± 85.2 ng/24 h) similar to those measured in nonsmokers (68.1 ± 91.5 ng/24 h) and the amounts of 2-aminonaphthalene (2-naphthylamine) (IARC, 1974, 1987) excreted by smokers (84.5 ± 102.7 ng/24 h) and nonsmokers (120.8 ± 279.2 ng/24 h) were also similar (Grimmer et al., 2000).
DNA adducts have been detected by 32P-postlabelling in urinary bladder biopsies and in exfoliated urothelial cells isolated from the urine of smokers and nonsmokers. At least four adducts may have been related to smoking, one of which was qualitatively similar to the N-(deoxyguanosin-8-yl)-4-aminobiphenyl adduct (Talaska et al., 1991a,b).
Of the heterocyclic aromatic amines found in cigarette smoke, analyses of urinary metabolites have been carried out on only one, 2-amino-1-methyl-6-phenylimidazo-[4,5-b]pyridine (PhIP) (IARC, 1993a) (reviewed in Kulp et al., 2000). Most studies have focused on the effects of diet, but one found no effect of smoking on levels of PhIP in urine, after adjustment for ethnicity (Kidd et al., 1999).
N-Nitrosamines
N-Nitrosoproline (IARC, 1978b, 1987) and other nitrosamino acids
Ohshima and Bartsch (1981) demonstrated that N-nitrosoproline (NPRO) could be formed endogenously in humans after the ingestion of proline and nitrate. This finding led to the development of a test for endogenous nitrosation by measurement of NPRO in the urine of people who had ingested proline and nitrate, or proline alone with or without ascorbic acid, an inhibitor of nitrosation (Bartsch et al., 1989). The test is safe because NPRO is not metabolized and has not been shown to be carcinogenic. The NPRO test has been applied in several studies designed to compare endogenous nitrosation between smokers and nonsmokers. The results indicate that endogenous formation of NPRO occurs in smokers, and that thiocyanate catalysis may be important (Bartsch et al., 1989; Tsuda & Kurashima, 1991; Tricker, 1997). However, mixed results have been obtained in population-based studies (Tricker, 1997).
Several other nitrosamino acids are present in human urine. The major ones are Nnitrososarcosine (NSAR) (IARC, 1978c), N-nitrosothiazolidine 4-carboxylic acid (N-nitrosothioproline) (NTCA) and trans- and cis- isomers of N-nitroso-2-methylthiazolidine 4-carboxylic acid (NMTCA) (Bartsch et al., 1989; Tsuda & Kurashima, 1991). NTCA and NMTCA are formed by the reaction of formaldehyde or acetaldehyde with cysteine, followed by nitrosation. Some studies have demonstrated increased concentrations of urinary NTCA and NMTCA in smokers (Tsuda & Kurashima, 1991). Total nitrosamino acids correlated with urinary nicotine plus cotinine in smokers (Malaveille et al., 1989), but mixed results have been obtained in other studies (Tricker, 1997). Collectively, the available data support the concept that higher concentrations of nitrosamines can be formed endogenously in smokers than in nonsmokers under some conditions.
4-(N-nitrosomethylamino)-4-(3-pyridyl)butyric acid (iso-NNAC) was suggested as a potential monitor of the endogenous nitrosation of nicotine (Djordjevic et al., 1991). iso-NNAC was found in the urine of four of 20 cigarette smokers (at levels of 44, 65, 74 and 163 ng/day). However, no evidence for its formation after the oral administration of nicotine or cotinine to abstinent smokers could be found (Tricker et al., 1993).
Volatile nitrosamines
Low-molecular-weight nitrosamines such as NDMA and N-nitrosopyrrolidine (NPYR) (see IARC, 1978d, 1987) are extensively metabolized, but small amounts of the unchanged compounds have been quantified in urine (Tricker, 1997). One investigation found that smokers excreted higher levels of NDMA than nonsmokers (Conney et al., 1986), but two other studies reported no effect of smoking on the amounts of volatile nitrosamines in urine (Mostafa et al., 1994; van Maanen et al., 1996).
4-(N-Nitrosomethylamino)-1-(3-pyridyl)-1-butanol, its glucuronides and other metabolites of 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone (see Figure 4.1)
4-(N-Nitrosomethyl(amino)-1-(3-pyridyl)-1-butanol (NNAL), like NNK, is a pulmonary carcinogen with particularly strong activity in the rat (Hecht, 1998). Glucuronidation of NNAL at the pyridine nitrogen gives NNAL-N-Gluc while conjugation at the carbinol oxygen yields NNAL-O-Gluc (Carmella et al., 2002). NNAL-N-Gluc and NNAL-O-Gluc both exist as a mixture of two diastereomers and each diastereomer is a mixture of S- and R-rotamers (Upadhyaya et al., 2001). The NNAL glucuronides are collectively referred to as NNAL-Gluc.
NNAL and NNAL-Gluc can readily be determined in urine by gas chromatography–thermal energy analysis (GC–TEA) with nitrosamine-selective detection (Carmella et al., 1993, 1995; Hecht et al., 1999a). The presence of these metabolites in human urine has also been established by mass spectrometry methods, but these are less convenient and sensitive than GC–TEA (Carmella et al., 1993; Parsons et al., 1998; Carmella et al., 1999; Hecht et al., 2001). The amounts typically excreted are about 1 nmol NNAL/24 h and 2.2 nmol NNAL-Gluc/24 h (Hecht et al., 1999a); unchanged NNK is not detected. The investigations of NNAL and NNAL-Gluc in the urine of smokers are summarized in Table 4.2. In all studies to date, these biomarkers have been found to be absolutely specific to tobacco exposure and have not been detected in the urine of nonsmokers unless they had been exposed to secondhand tobacco smoke. Because NNAL is not present in cigarette smoke (see Section 1), the NNAL and NNAL-Gluc in urine originate from the metabolism of NNK. Most investigations to date have demonstrated a correlation between NNAL plus NNAL-Gluc and cotinine. The ratio of NNAL-Gluc:NNAL varies at least 10-fold in smokers and, because NNAL-Gluc is a detoxification product of NNK whereas NNAL is carcinogenic, this ratio could be a potential indicator of cancer risk (Carmella et al., 1995; Richie et al., 1997). In human urine, (R)-NNAL-O-Gluc is the predominant diastereomer of NNAL-O-Gluc (68%) whereas (R)-NNAL is only slightly in excess (54%) over (S)-NNAL (Carmella et al., 1999). (R)-NNAL is the more tumorigenic enantiomer of NNAL in A/J mouse lung in which it is as tumorigenic as NNK (Upadhyaya et al., 1999). NNAL and NNAL-Gluc are released only slowly from the human body after smoking cessation (Hecht et al., 1999a) and this finding has been linked to the particularly strong retention of (S)-NNAL (Upadhyaya et al., 1999) [possibly at a receptor site].
Table 4.2
4-(N-Nitrosomethylamino)-1-(3-pyridyl)-1-butanol (NNAL) and its glucuronides (NNAL-Gluc) in urine of smokers: biomarkers of 4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone (NNK) uptake.
In rodents, NNK and NNAL undergo metabolic oxidation at the pyridine nitrogen giving NNK-N-oxide and NNAL-N-oxide, respectively. Both metabolites are less tumorigenic in rodents than NNK and NNAL (Hecht, 1998; Upadhyaya et al., 1999). In humans, analysis of the urine of smokers for NNAL-N-oxide found that it was present at lower concentrations than NNAL; NNK-N-oxide was not detected in the urine of smokers. Thus, pyridine-N-oxidation is a relatively minor detoxification pathway of NNK and NNAL in humans (Carmella et al., 1997).
4-Hydroxy-4-(3-pyridyl)butanoic acid (hydroxy acid) and 4-oxo-4-(3-pyridyl)butanoic acid (keto acid) are metabolites of NNK resulting from the α-hydroxylation metabolic activation pathway. These metabolites were investigated as potential biomarkers of NNK metabolic activation in humans, but they are also formed from nicotine, which is 1400–13 000 times more abundant in cigarette smoke than is NNK. Since hydroxy acid is chiral, it was thought possible that one enantiomer would be formed preferentially from NNK, whereas the other would be produced from nicotine. Studies in rats demonstrated that this was plausible (Trushin & Hecht, 1999). However, hydroxy acid is a more abundant nicotine metabolite in humans than in rats and consequently even the minor enantiomer, as formed from nicotine, was far greater in concentration than that which would be produced from NNK. Because of the abundant metabolism of nicotine to hydroxy acid and keto acid (see Section 4.1.1.e), these metabolites cannot be used as biomarkers of tobacco-specific nitrosamine metabolism in humans (Hecht et al., 1999b; Trushin & Hecht, 1999).
Products of oxidative DNA damage
Cigarette smoke contains free radicals and induces oxidative damage (Pryor, 1997; Arora et al., 2001). The gas phase of freshly generated cigarette smoke contains large amounts of nitric oxide (NO) and other unstable oxidants. The particulate phase is postulated to contain long-lived radicals which are an equilibrium mixture of semiquinones, quinones and hydroquinones that causes redox cycling (Pryor, 1997; Hecht, 1999). The presence of such free radicals and oxidants can lead to oxidative DNA damage resulting in the formation of products such as 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG)1, [8-hydroxy-2′-deoxyguanosine (8-OHdG)], thymine glycol, thymidine glycol and 5hydroxymethyluracil. Repair of these modified DNA constituents ultimately leads to their being excreted in urine. 8-OHdG has been quantified frequently in the urine of smokers and nonsmokers and the results have been reviewed. Cigarette smoking usually results in modest increases in the levels of 8-OHdG in urine; 16–50% higher than those in nonsmokers, although negative results have also been reported (Loft & Poulsen, 1998; Priemé et al., 1998; Renner et al., 2000; Besarati Nia et al., 2001). Four weeks of smoking cessation caused a decrease in the excretion of 8-OHdG by 21% (Priemé et al., 1998). A longitudinal study showed that intra-individual variation in the levels of urinary 8-OHdG was greater than the increase due to smoking, suggesting that there may be a complex pattern of factors determining the levels of this biomarker in urine (Kasai et al., 2001; Pilger et al., 2001). No effect of smoking on urinary concentrations of 5-hydroxymethyluracil was observed (Pourcelot et al., 1999).
Thioethers and mercapturic acids
Conjugation of electrophiles with glutathione ultimately results in the excretion of mercapturic acids (N-acetylcysteine conjugates) in the urine (van Doorn et al., 1979, 1981). A method for determination of total thioethers in urine has been applied in numerous studies comparing smokers and nonsmokers reviewed by van Doorn et al. (1981), IARC (1986) and Scherer et al. (1996). Cigarette smokers excrete significantly higher levels of thioethers in urine than nonsmokers. There is considerable interindividual variation when diet, a major source of sulfur-containing compounds, is not controlled (Aringer & Lidums, 1988; Scherer et al., 1996). The assay of total thioethers in urine provides no information about the structure of the electrophiles which are ultimately detected in urine as conjugates.
More specific methods have been applied to investigate the presence of mercapturic acids in human urine (Scherer et al., 2001a). 3-Hydroxypropylmercapturic acid, a likely detoxification product of acrolein, has been detected in the urine of smokers (Mascher et al., 2001).
Alkyladenines and alkylguanines
Reaction of alkylating agents with DNA results in the formation of alkyladenines and alkylguanines among other products (Singer & Grunberger, 1983). Alkylation at the 3-position of deoxyadenosine or at the 7-position of deoxyguanosine weakens the glycosidic bond, which readily breaks either spontaneously or is cleaved by glycosylases, ultimately resulting in the excretion of 3-alkyladenines and 7-alkylguanines in urine. 3-Alkyladenines have been more extensively investigated as biomarkers of exposure to alkylating agents than 7-alkylguanines because background levels in urine are expected to be lower. However, substantial amounts of 3-methyladenine occur in the diet (Prévost et al., 1993; Fay et al., 1997). Nevertheless, two controlled studies have demonstrated increased excretion of 3-methyladenine in the urine of smokers compared with that of nonsmokers (Kopplin et al., 1995; Prévost & Shuker, 1996). Background levels of 3-ethyladenine are lower than those of 3-methyladenine (Prévost et al., 1993). Two studies demonstrated convincing increases in urinary concentrations of 3-ethyladenine in smokers, indicating the presence in cigarette smoke of an unidentified ethylating agent (Kopplin et al., 1995; Prévost & Shuker, 1996). There was no effect of smoking on the concentration of 3-(2-hydroxyethyl)adenine in urine (Prévost & Shuker, 1996). One population-based study found greater amounts of both 3-methyladenine and 7-methylguanine in the urine of smokers than of nonsmokers (Stillwell et al., 1991) while a second found no difference between 3-methyladenine concentrations in smokers and nonsmokers (Shuker et al., 1991).
Metals
Large studies in Germany and the USA demonstrated that urinary cadmium increased with age and smoking (Hoffmann et al., 2000; Paschal et al., 2000). The study in the USA involved 22 162 participants in the Third National Health and Nutrition Examination survey (NHANES III 1988–94), and urine cadmium, expressed either as uncorrected (µg/L) or creatinine corrected (µg/g creatinine), increased with age and smoking (Paschal et al., 2000). The German study, involving 4021 adults, found that active cigarette smoking was the predominant factor affecting cadmium concentrations in the blood and urine of adults. Environmental and occupational exposure to cadmium played only a minor role (Hoffmann et al., 2000). These results are consistent with those of other studies of cadmium uptake in smokers (IARC, 1993b). The limited data available do not indicate consistent significant differences in levels of urinary nickel (IARC, 1990a), chromium (IARC, 1990b), or lead (IARC, 1980, 1987) between smokers and nonsmokers (Morimoto et al., 1977; Schaller & Zober, 1982; Minoia et al., 1988; Jin et al., 1997; Huang et al., 2000).
Conclusion
Representative levels (nmol/24 h) of eight urinary biomarkers discussed here are summarized in Table 4.3. The amounts of these compounds in urine are generally proportional to the levels of their parent compounds in cigarette smoke, with the possible exception of 8-OHdG and 3-ethyladenine, the precursors for which are not known. It should be noted that there are a number of established tobacco smoke carcinogens for which no urinary biomarkers have been validated. Examples include formaldehyde, acetaldehyde, 1,3-butadiene (IARC, 1999f), ethylene oxide and vinyl chloride (IARC, 1979, 1987; Hoffmann et al., 2001).
Table 4.3
Representative concentrations of biomarkers in urine of smokers.
Of the urinary biomarkers discussed here, the following are consistently higher in smokers than in nonsmokers: tt-MA, S-PMA, 1-naphthol, 2-naphthol, 1-hydroxypyrene, NNAL and NNAL-Gluc, 8-OHdG, 3-ethyladenine and cadmium. For all of these biomarkers, analytical methods with the requisite sensitivity and specificity are available, but only NNAL and NNAL-Gluc have high specificity as biomarkers of exposure to tobacco smoking because sources other than tobacco smoke, including environmental, dietary and occupational exposure, can contribute to the urinary concentration of the other possible biomarkers. Because NNAL and NNAL-Gluc are metabolites of the tobacco-specific nitrosamine NNK, they are not found in the urine of nonsmokers unless they have been exposed to environmental tobacco smoke (see the monograph on involuntary smoking, this volume). It is possible that other sources, such as nicotine replacement products, could, under certain conditions, contribute to the concentrations of urinary NNAL and NNAL-Gluc but this has not been demonstrated to date (Hecht et al., 2000). Levels of NNAL plus NNAL-Gluc of less than 1 pmol/mL of urine have seldom been observed in smokers whereas the highest levels in nonsmokers exposed to environmental tobacco smoke seldom exceed 0.4 pmol/mL (Hecht et al., 2001; Hecht, 2002b).
(ii) Protein adducts in human tissues (blood)
Although proteins are not the target molecules for mutagenic events, protein modifications can be useful biomarkers because of the greater abundance of proteins than of DNA and the fact that protein modifications are not subject to enzymatic repair. The rate of turnover of the proteins and/or of the cells that harbour them implies that carcinogen–protein adducts can provide evidence of exposure on a time-scale of up to several months. In theory, haemoglobin, serum albumin, histones and collagen are suitable proteins to study for this purpose (Skipper et al., 1994), but in practice only haemoglobin and albumin have been extensively studied as biomarkers of human exposure to environmental carcinogens.
Protein adducts are generally detected in one of three ways:
- -
mass spectrometric detection of the carcinogen moiety after its release from protein by mild acid or base hydrolysis, or release and detection of the modified N-terminal valine of haemoglobin;
- -
immunochemical analysis using antibodies raised against protein adducts; and
- -
HPLC with fluorescence detection of the released carcinogen (Phillips & Farmer, 1995).
The use of protein adducts as biomarkers of human exposure to carcinogens has been extensively reviewed elsewhere (Strickland et al., 1993; Skipper et al., 1994; Wild & Pisani, 1998; Poirier et al., 2000).
The entire literature on smoking-related protein and DNA adducts in human tissues has been produced since the previous IARC evaluation of tobacco smoking (IARC, 1986). These studies are summarized below.
Adducts formed by aromatic amines
In an early study of 22 smokers and 24 nonsmokers, 4-ABP–haemoglobin adducts1 were measured by GC–MS. The adduct levels of smokers were significantly higher than those of nonsmokers and there was a significant correlation with average number of packs smoked per day. It is noteworthy that the range of values for smokers (75–256 pg/g haemoglobin) did not overlap with that for nonsmokers (7–51 pg/g) (Perera et al., 1987). The absence of overlap was also reported in another study involving 19 smokers (mean, 154 pg/g) and 26 nonsmokers (mean 28 pg/g); the difference between the two groups was highly significant (p < 0.001). The finding of detectable levels of adducts in all the nonsmokers is consistent with the existence of environmental sources of 4-ABP other than tobacco smoking (Bryant et al., 1987).
In a group of male volunteers from a case–control study of bladder cancer in Turin, Italy (Vineis et al., 1984), haemoglobin adducts of 15 different aromatic amines were measured in nonsmokers and in smokers of blond- or black-tobacco cigarettes. The smokers of blond tobacco (n = 40) and black tobacco (n = 18), and three subjects who smoked both types, had significantly higher levels of 4-ABP adducts than 25 nonsmokers. Furthermore, adduct levels were significantly higher (40–50%) for smokers of black tobacco (mean, 288 pg/g) than for those of blond tobacco (mean, 176 pg/g). Adduct levels were found to be correlated with amount smoked per day for all smokers (p = 0.0015) and this correlation was also significant for smoking blond tobacco only (p = 0.0074). Of other aromatic amines, the levels of 3-ABP adducts were also significantly elevated in smokers (p < 0.0001) and associated with numbers of cigarettes smoked by the blond tobacco users (p = 0.02). The levels of adducts derived from a further five aromatic amines, 2-naphthylamine, ortho-toluidine, para-toluidine, 2-ethylaniline and 2,4-dimethylaniline, were significantly higher in the smokers, while those of meta-toluidine, 2,5-, 2,6-, 2,3-, 3,5- and 3,4-dimethylaniline and 3- and 4-ethylaniline were not (Bryant et al., 1988). In a subsequent analysis, the authors sought explanations for variability observed between individuals. There appeared to be a distinct difference between the levels measured for the binuclear compounds and those obtained with the mononuclear compounds. Correlations between the levels of the three binuclear amines (2-naphthylamine, 4-ABP and 3-ABP) were significant (p < 0.05) and 49/54 of the correlations between different mononuclear amines were significant, but only 2/33 correlations between a binuclear and a mononuclear amine were significant. These results suggest the existence of two distinct pathways of metabolic activation of aromatic amines and explain a part, but not all, of the interindividual variation in adduct levels observed (Ronco et al., 1990).
As part of a validation exercise in a study of 4- and 3-ABP–haemoglobin adducts in nonsmokers exposed to secondhand smoke (see the monograph on involuntary smoking, this volume), seven smokers who quit were monitored on the day they stopped smoking and again at least 2 months later. The mean levels of 4-ABP adducts fell by 75% from 130.4 pg/g at baseline to 33.3 pg/g and the mean level of 3-ABP fell by at least 80% (from 16.0 pg/g to a baseline of 1.7 pg/g) (in four cases levels of 3-ABP fell below the limit of sensitivity of the assay) (Maclure et al., 1989). A further study of smokers enrolled at a cessation clinic found that the 34 subjects starting the programme had a mean level of 4-ABP–haemoglobin of 120 ± 7 (SE) pg/g which declined to 82 ± 6 pg/g after 3 weeks. In the 15 smokers who remained abstinent after 2 months, the level was 34 ± 5 pg/g. The rate of decline was slightly more rapid than would be expected from the assumption that erythrocytes have a lifespan of 120 days. There was little correlation between plasma cotinine levels and haemoglobin adducts (Maclure et al., 1990). Similar findings were reported from another study of smoking cessation. The mean level of haemoglobin adducts at baseline was four-fold higher than the mean value 8 months later. Depending on the model used, the half-life of the adducts was estimated to be 7–12 weeks (Mooney et al., 1995).
In a study of 50 nonsmokers, 31 smokers of blond tobacco, 16 smokers of black tobacco and three pipe smokers, the relationship between smoking blond or black tobacco and 4-ABP–haemoglobin adduct levels was confirmed (p = 0.0001 in both tests). A linear relationship between adduct levels and the numbers of cigarettes smoked in the preceding 24 h was also observed. Furthermore, when the subjects were divided into slow and rapid acetylators (by measuring their urinary excretion of caffeine after administration of a test dose), it was found that the ratio of adduct levels in slow acetylators to that in rapid acetylators was 1.6 for nonsmokers, 1.3 for blond-tobacco smokers and 1.5 for black-tobacco smokers. This approximates to the estimated relative risk of the slow acetylator phenotype for bladder cancer of 1.3 (95% CI, 1.0–1.7) (Evans, 1986; Vineis et al., 1990). A further study of this group, as well as confirming these findings, also found that urinary mutagenicity was associated with the number of cigarettes smoked, but not with the acetylator phenotype (Bartsch et al., 1990). Subsequently, 4-ABP–haemoglobin adduct levels in 21 nonsmokers, 11 blond-tobacco smokers and seven black-tobacco smokers were found to correlate with the total amount of DNA adducts in exfoliated urothelial cells (p = 0.03) (Talaska et al., 1991b).
In a case–control study of lung cancer with 53 cases (23 smokers) and 56 controls (18 smokers) (33 with non-cancer pulmonary disease and 23 with non-pulmonary cancer), levels of 4-ABP–haemoglobin adducts were higher in smokers than in nonsmokers and reflected recent exposure to tobacco smoke. There was no association between adduct levels and lung cancer (Weston et al., 1991). [The control group may have biased the results.] However, in a case–control study of bladder cancer, the mean adduct level in the cases (n = 13, all smokers) was significantly higher than in the controls (n = 13, all smokers) (103 ± 47 [SD] and 65 ± 44 pg/g haemoglobin, respectively; p = 0.04) (Del Santo et al., 1991).
The levels of 4-ABP-haemoglobin adducts were also significantly higher in pregnant women who smoked (n = 15) than in those who did not (n = 40) (183 ± 108 and 22 ± 8 pg/g haemoglobin; p < 0.001), respectively, in a study in which levels of the adduct in fetal blood were also measured (see the monograph on involuntary smoking, this volume) (Coghlin et al., 1991). Similarly, a study of 74 nonsmokers and an equal number of smokers measured paired maternal–fetal blood samples. The levels of 4-ABP-haemoglobin adducts were significantly higher in the smokers, and the ratios of adduct levels in maternal and fetal cord blood were found to be similar to those reported in the previous study. There was also a correlation between adduct levels in maternal blood and number of cigarettes smoked per day (Myers et al., 1996).
In another study comparing 27 pregnant women who smoked with 78 who did not, significantly higher levels of haemoglobin adducts of 3-ABP (p < 0.001), 4-ABP (p < 0.001), ortho-toluidine (p < 0.001), para-toluidine (p < 0.001) and 2,4-dimethylaniline (p < 0.05) were found in smokers than in nonsmokers (Branner et al., 1998).
A study of smokers and nonsmokers, in which an analysis of the haemoglobin adducts of several aromatic amines was made, revealed significantly higher levels in smokers of those adducts formed by 4-ABP (p < 0.001), 3-ABP (p < 0.001) and 2,4-dimethylaniline (p < 0.05), but not of those formed by aniline, ortho-toluidine, meta-toluidine, para-toluidine, 2-ethylaniline or ortho-anisidine (IARC, 1999g). For many of these comparisons, the number of subjects was small (4–22 smokers, 4–16 nonsmokers) (Falter et al., 1994).
Comparisons of 3- and 4-ABP-haemoglobin adducts in three different racial groups (white, black and Asian) led, as expected, to the detection of higher levels of both adducts in 61 smokers than in 72 nonsmokers (these levels were highest in whites, intermediate in blacks and lowest in Asians). There was also a correlation with the numbers of cigarettes smoked per day (p < 0.0005). Subjects from all three ethnic groups with the slow acetylator phenotype had higher adduct levels than those in rapid acetylators (2.5-fold higher for 3-ABP, p < 0.0005; 1.2-fold higher for 4-ABP, p = 0.19) (Yu et al., 1994).
Differences in the levels of 3- and 4-ABP–haemoglobin adducts between men and women have also been reported. When plotted against number of cigarettes smoked per day, the slopes of the linear regression lines of adduct levels were significantly steeper for women than for men in a cohort of 1514 patients with bladder cancer and 1514 matched controls (p < 0.001 and 0.006 for 3-ABP and 4-ABP, respectively) (Castelao et al., 2001).
In a group of 55 smokers and four nonsmokers, the levels of 4-ABP–haemoglobin adducts were related to the number of cigarettes smoked, although saturation of adduct formation was apparent at > 30 cigarettes/day. GSTM1 and NAT2 polymorphisms did not affect the levels of haemoglobin adducts (Dallinga et al., 1998). The same authors investigated other genetic polymorphisms in 67 smokers and found no overall effects of polymorphisms in NAT1, NAT2, GSTM1 or GSTT1 on 4-ABP–haemoglobin adduct levels, except that in smokers of < 25 cigarettes/day, NAT2 slow acetylators had significantly higher adduct levels than fast acetylators (p = 0.03) (Godschalk et al., 2001).
Another study also reported that NAT2 slow acetylators had significantly higher levels of 4-ABP-haemoglobin adducts than rapid acetylators, but there was no association between the NAT1*10 genotype and the levels of 3- or 4-ABP–haemoglobin adducts, after adjustment for NAT2 phenotype. As in previous studies, smokers had significantly higher levels of 3- and 4-ABP–haemoglobin adducts than nonsmokers and the levels increased with increasing numbers of cigarettes smoked per day (p < 0.0001) (82 smokers, 321 nonsmokers) (Probst-Hensch et al., 2000).
Adducts formed by polycyclic aromatic hydrocarbons
In a study involving 87 mothers and their 87 children in which PAH–albumin adducts in the plasma were investigated as a biomarker of the children's exposure to secondhand smoke from smoking by their mothers (see the monograph on involuntary smoking, this volume), it was also noted that adduct levels were significantly higher in the mothers who smoked than in those who did not (Crawford et al., 1994).
A GC–MS method was used to measure benzo[a]pyrene-diol epoxide (BPDE)–globin adducts in 10 smokers and 10 nonsmokers. Subjects were also monitored for formation of globin adducts of chrysene diol epoxide. In both cases the procedure involved measurement of the respective tetrols of PAHs released from globin by acid hydrolysis. Levels of BPDE adducts in smokers were 2.7-fold higher than those in nonsmokers (p < 0.01); although the levels of the chrysene diol epoxide adducts were 1.25-fold higher in smokers, this difference was not statistically significant (p = 0.06) (Melikian et al., 1997).
BPDE adducts with both haemoglobin and serum albumin were determined by GC–MS of tetrols released by acid hydrolysis in a study of 44 men with incident lung cancer. Individuals who were positive for haemoglobin adducts (n = 6) all had detectable albumin adducts, but not vice versa (24 subjects were positive for albumin adducts). Those who were carriers of a CYP1A1 variant allele were more frequently positive for albumin adducts (p = 0.03) and those with two ‘slow’ mEH alleles had a lower frequency of these adducts (Pastorelli et al., 1998).
In another study, 27 smokers were found to have significantly higher levels of BPDE–albumin adducts (p < 0.05) and non-significantly higher levels of BPDE–haemoglobin adducts than 42 nonsmokers (Scherer et al., 2000) (see also the monograph on involuntary smoking, this volume).
Adducts formed by tobacco-specific nitrosamines (TSNA)
Treatment of NNK– or NNN–haemoglobin adducts with mild bases releases 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB), which is derivatized to its pentafluorobenzoate, which is detectable by GC–MS after purification. The mean value in 40 smokers was 79.6 ± 189 fmol HPB/g haemoglobin (undetectable in 11 individuals; limit of detection, 5 fmol/g), whereas that in 21 nonsmokers was 29.3 ± 25.9 fmol/g (undetectable in four individuals) [statistical significance not stated]. The HPB levels in 22 snuff dippers were 517 ± 538 fmol/g, i.e. significantly higher than those in smokers (p < 0.0001). No relationship was apparent between HPB levels in smokers and levels of plasma cotinine or number of cigarettes smoked (Carmella et al., 1990).
In a comparison of 20 smokers and 15 nonsmokers, the HPB levels derived from haemoglobin were significantly higher in smokers (69.2 ± 43.9 versus 34.4 ± 16.0 fmol/g haemoglobin; p < 0.005) (Falter et al., 1994).
In another study of 18 smokers and former smokers and 52 never-smokers, the levels of HPB–haemoglobin adducts were significantly higher in the smokers (26 ± 12 [SD] fmol HPB/g haemoglobin) than in never-smokers (19 ± 8 fmol/g) (p = 0.02). No differences were found between the sexes or between current smokers and former smokers [duration of smoking cessation not stated] (Atawodi et al., 1998).
Significantly higher levels of haemoglobin adducts of TSNA (p < 0.001) were found in pregnant women who smoked than in pregnant women who did not smoke (Branner et al., 1998).
Adducts formed by other compounds
Ethylene (IARC, 1994b) is a major gaseous constituent of tobacco smoke that converts, via ethylene oxide, the N-terminal valine of haemoglobin to N-(2-hydroxyethyl)valine (HOEtVal). The level of this adduct correlates linearly with the alkylating activity occurring in DNA: 10 pmol HOEtVal/g globin corresponds to 0.33 pmol adduct/g DNA (Farmer et al., 1987; Bono et al., 1999). In 11 smokers who smoked more than 20 cigarettes/day, the levels of HOEtVal in haemoglobin were in the range 217–690 pmol/g haemoglobin (mean ± SD, 389 ± 138), whereas in 14 nonsmokers the range was 27–106 pmol/g (58 ± 25). It is noteworthy that there was no overlap in values between the two groups (Törnqvist et al., 1986). In a controlled experiment, two volunteers who were regular smokers (of 29 and 18 cigarettes/day, respectively) abstained from smoking for 7 days and then resumed. From the measurements of levels of HOEtVal before stopping, 7 days after stopping and 7 days after resuming, the investigators calculated that in the smoker of 29 cigarettes/day, each cigarette smoked increased the level of HOEtVal by 0.12 pmol/g globin. For the smoker of 18 cigarettes/day, the increase was 0.08 pmol/g (Granath et al., 1994).
In a study of 26 smokers and 24 nonsmokers, the background levels of HOEtVal in the nonsmokers averaged 49.9 pmol/g haemoglobin (range, 22–106). In the smokers, the levels were significantly higher by an estimated 71 pmol/10 cigarettes/day (Bailey et al., 1988).
As part of a maternal–fetal comparison of HOEtVal in haemoglobin (see the monograph on involuntary smoking, this volume), samples from 10 pregnant women who did not smoke and 13 pregnant women who smoked 15 or more cigarettes/day were analysed. In nonsmokers, 63 ± 20 (mean ± SD) pmol/g haemoglobin were measured, whereas in the smokers, the levels were 361 ± 107 pmol/g (p < 0.01) (Tavares et al., 1994).
In a study of 146 adults (82 men and 64 women), 44 smokers were reported to have significantly higher levels of HOEtVal in haemoglobin than did 74 nonsmokers and 29 self-reported passive smokers (mean ± SD: 59.5 ± 53, 17 ± 21 and 17 ± 23 pmol/g haemoglobin, respectively). Linear regression analysis of adduct levels and the number of cigarettes smoked per day showed a highly significant correlation coefficient (r = 0.63, p < 0.0001). The adduct levels were also significantly higher in men than in women (mean value 2.2-fold higher, p = 0.00001) (Bono et al., 1999).
Ethylene oxide is a substrate for GSTT1. To determine whether polymorphisms in GSTT1 modulate HOEtVal formation, blood samples from 10 women (one smoker) and 17 men (five smokers) were analysed. The median level of the adduct was 5.6-fold higher in smokers than in nonsmokers, but no correlation with daily cigarette consumption was found. GSTM1 and GSTT1 genotypes did not influence the adduct levels in the smokers, although the GSTT1 genotype did influence the adduct levels in nonsmokers (a two-fold higher median HOEtVal value was detected in GSTT1-null individuals than in GSTT1-positive individuals) (Müller et al., 1998).
The effect of GSTT1 was also investigated in a study involving 14 nonsmokers, 16 smokers of one pack of cigarettes a day and 13 smokers of two packs a day. HOEtVal levels increased with increasing number of cigarettes smoked, and the differences between the three groups were statistically significant. In addition, levels of N-(2-cyanoethyl)valine (CEVal), which is formed from acrylonitrile, were also significantly correlated with increasing smoking in these subjects. HOEtVal and CEVal levels were found to be significantly correlated in smokers (p = 0.003). There was no effect of GSTM1 or GSTT1 genotypes on the levels of CEVal in any of the groups or of GSTM1 on HOEtVal levels; however, GSTT1-null individuals had higher levels of HOEtVal compared with GSTT1-positive individuals (slope of regression line 50% higher) (Fennell et al., 2000).
A study of the effects of exposure to acrylamide and acrylonitrile in laboratory workers (smokers and nonsmokers) included the measurement of ethylene oxide–haemoglobin adducts. The mean levels of adducts from acrylamide, acrylonitrile and ethylene oxide in 10 smokers were 116, 106 and 126 pmol/g, respectively; these levels were significantly higher than those in eight nonsmokers (31, < 2 and 17 pmol/g, respectively). In smokers, the levels of acrylamide, acrylonitrile and ethylene oxide adducts correlated with the number of cigarettes smoked per day (Bergmark, 1997).
Two other studies have reported on acrylonitrile–haemoglobin adducts in smokers. In the first, four smokers had 75–106 pmol CEVal/g haemoglobin (mean, 88) whereas in four nonsmokers the levels were all below the limit of detection of 20 pmol/g (Osterman-Golkar et al., 1994). In the second study, 13 mothers who smoked had adduct levels in the range 92.5–373 pmol/g haemoglobin (mean ± SD: 217 ± 85.1), but the levels in 10 nonsmoking mothers were below the limit of detection (1 pmol/g). In the smokers, there was a linear correlation with number of cigarettes smoked per day (p = 0.02) and also with the levels of the adducts in their newborn babies (see the monograph on involuntary smoking, this volume) (Tavares et al., 1996).
A study was conducted in 43 Chinese workers exposed to benzene and 44 unexposed controls. The haemoglobin and albumin adducts of two metabolites, benzene oxide and 1,4-benzoquinone, were measured in these subjects. Tobacco smoking was found to have an additive effect on 1,4-benzoquinone–albumin formation (p = 0.034), but not on benzene oxide–albumin formation (p = 0.23) (Yeowell-O'Connell et al., 2001).
Human haemoglobin contains relatively high background levels of N-methylvaline, which could limit its sensitivity as a biomarker of exposure to environmental carcinogens. However, in a study of 11 pairs of monozygotic twins discordant for tobacco smoking, both HOEtVal and N-methylvaline were found to be higher in the smokers than in the nonsmokers. In the smokers the levels of HOEtVal and N-methylvaline were 143 ± 24 (mean ± SE) and 268 ± 13 pmol/g haemoglobin, respectively, and in the nonsmokers 15.6 ± 1.9 and 225 ± 11 pmol/g, respectively. Thus, the levels of N-methylvaline adduct were significantly different between smokers and nonsmokers (p = 0.006) and there was a highly significant correlation with the number of cigarettes smoked per day (p < 0.001) (Törnqvist et al., 1992).
Similarly, levels of N-methylvaline were found to be higher in a group of 32 smokers than in a group of 37 nonsmokers (mean ± SD: 1546 ± 432 versus 1175 ± 176 pmol/g haemoglobin; p < 0.001); for the smokers there was also a linear correlation between adduct levels and the number of cigarettes smoked per day (p < 0.001). An increment of 42 pmol/g haemoglobin per cigarette per day was calculated. A significant difference between smokers and nonsmokers in levels of HOEtVal was also observed in this study (Bader et al., 1995).
Because cigarette smoke contains reactive species that can cause oxidative and nitrative damage in cellular macromolecules, samples of plasma protein from 52 lung cancer patients (24 smokers, 28 nonsmokers) and 43 control subjects (18 smokers, 25 never-smokers) were analysed for nitrotyrosine and carbonyl groups as markers of nitration and oxidation, respectively. The quantities of nitrated proteins were significantly higher in the patients than in the controls (p = 0.003), but were not related to smoking status. In contrast, the amounts of oxidized proteins were higher in smokers (p < 0.001), but were not related to disease status (Pignatelli et al., 2001).
(iii) DNA adducts and other types of DNA damage in human tissues
The induction of DNA damage, frequently in the form of chemically stable adduct formation, is an early and essential step in the sequence of events by which genotoxic carcinogens initiate the carcinogenic process. The detection of DNA adducts in human tissues is therefore a useful and appropriate means to assess human exposure to such agents and several different procedures have been used for this.
- 32P-Postlabelling analysis, in which DNA is digested and the resultant carcinogen-modified nucleotides are radiolabelled enzymatically with [32P]ortho-phosphate, provides a sensitive method that requires no prior knowledge of the structures of the adducts formed. A common feature of the postlabelled DNA obtained from cells or tissues exposed to complex mixtures of carcinogens, such as tobacco smoke, is the appearance of a diagonal radioactive zone (DRZ) seen when the material is resolved by two-dimensional thin-layer chromatography. Although the material in the DRZ has not been fully characterized, its properties are compatible with those of a complex mixture of aromatic and/or hydrophobic DNA adducts.
- Another method used to detect DNA adducts in human tissues employs antibodies raised against DNA or nucleotides modified by various carcinogens. Immunohistochemistry allows locating of adducts within a tissue specimen and measurement of the intensity of fluorescent staining permits a semi-quantitative estimate of adduct levels.
- Fluorescence detection of adducts in DNA, or of products released from DNA by hydrolysis, has been used for carcinogens that have strongly fluorescent properties, for example PAHs.
- Mass spectrometry has been used for the chemical-specific detection of certain adducts in DNA samples.
- Electrochemical detection has been used for some smaller DNA lesions, for example 8-OHdG, formed in DNA by oxidative processes.
The uses, strengths and limitations of the various techniques have been extensively described (Beach & Gupta, 1992; Strickland et al., 1993; Weston, 1993; Phillips, 1997; Kriek et al., 1998; Wild & Pisani, 1998; Poirier et al., 2000). Earlier studies of tobacco-related DNA adducts in human tissues have also been reviewed (Phillips, 1996).
Respiratory tract
Lung and bronchus
Many studies have been published comparing the levels and characteristics of DNA adducts in the lung and bronchus of smokers and nonsmokers. These studies are summarized in Table 4.4.
Table 4.4
Studies of smoking-related DNA adducts in human lung and bronchus. Non-tumorous tissue was analysed except where indicated.
In most of these studies, significantly elevated levels of DNA adducts were detected in the peripheral lung, bronchial epithelium or in cells obtained by bronchial lavage of the smokers. This is the case for total bulky DNA adducts (as detected by the 32P-postlabelling method) and for DNA adducts detected by more chemical-specific methods including HPLC/fluorescence and GC–MS. However, the levels of 4-ABP-DNA adducts in lung did not correlate with smoking status (Culp et al., 1997). In some studies in which smoking status was not known, but was inferred from plasma cotinine levels (indicating smoking habit at the time of death of the subjects), there was a poor correlation with the detection of only one specific type of adduct, such as 7-methyldeoxyriboguanosine (7-MedG) (Kato et al., 1995; Blömeke et al., 1996) or those formed by benzo[a]pyrene (Weston & Bowman, 1991; Kato et al., 1995).
Some studies have found a linear correlation between adduct levels and daily or lifetime consumption of cigarettes (Phillips et al., 1988, 1990a; Dunn et al., 1991; Asami et al., 1997) but, in other studies, no such relationship was found (Godschalk et al., 1998; Schoket et al., 1998). In one study in a group of male smokers with lung cancer, a linear regression analysis showed that adduct levels were inversely related to the number of years of smoking (Ryberg et al., 1994). Mean levels of adducts in former smokers (usually with an interval of at least 1 year since smoking cessation) were generally found to be intermediate between the levels found in smokers and in life-long nonsmokers. From these comparisons, the half-life of adducts in the lung is estimated to be 1.7 years in bronchial tissue from former smokers (Schoket et al., 1998). This is somewhat longer than would be estimated from a consideration of the biochemical processes of DNA repair and the rate of cell turnover, and may be a consequence of the slow clearance of carcinogen-containing tar and particulate deposits from the lung, resulting in continued metabolic activation of tobacco carcinogens after smoking cessation.
In a study in Norway, adduct levels were higher in the lungs of women smokers than in those of men, a difference that was greater after adjustment for intensity of smoking (Ryberg et al., 1994; Mollerup et al., 1999). However, in a study in Hungary, adduct levels in male and female smokers with lung cancer were found to be the same (Schoket et al., 1998).
Larynx
32P-Postlabelling analysis of DNA from larynx mucosa revealed the presence of hydrophobic adducts (DRZ) in 21 smokers but not in four nonsmokers. The ability of both the nuclease P1 digestion and the butanol extraction procedures to detect these adducts suggested that they were formed primarily by PAHs rather than aromatic amines. Adduct levels correlated with levels of P4501A1, 2C and 3A4, but not with levels of P4502E1 and 2A6 (Degawa et al., 1994).
Another study compared adduct levels in laryngeal tumours with those in non-tumour tissue from 43 patients, 38 of whom were smokers. Adduct levels, as determined by 32P-postlabelling, were higher in tumour tissue than in normal tissue, and the few non- or former smokers had lower levels than the smokers (differences were not significant) (Szyfter et al., 1994). In a subsequent study, 32P-postlabelling analysis was used to measure levels of 7-alkylguanine in DNA from laryngeal biopsies. In larynx tumour tissues, the levels reported in heavy smokers (> 40 cigarettes/day) were 2.5-fold higher than those in moderate smokers (∼20 cigarettes/day) and 5.5-fold higher than in nonsmokers and former smokers. There was a significant correlation between 7-alkylguanine levels and aromatic/hydrophobic adduct levels in these tissue samples (Szyfter et al., 1996).
An immunohistochemical study of laryngeal biopsies from 38 patients, using antibodies against 4-ABP–DNA adducts, included analyses of tumours (9), polyps (28) and surrounding tissue (1). The investigators noted a significantly higher staining intensity of polyps and surrounding tissue from smokers than of that from nonsmokers, but the differences between tumour tissue from smokers and nonsmokers were not significant [limited number of samples]. The study showed that 4-ABP–DNA adducts, related to smoking exposure, are formed in the larynx (Flamini et al., 1998).
An analysis of laryngeal tumour biopsies from 33 patients was carried out by 32P-postlabelling. DNA from tumour tissue, non-tumour tissue and the interarytenoid area was analysed separately. Large interindividual differences in adduct levels were reported, with highest levels in the interarytenoid area. Adduct levels were reported to be correlated with age (the highest levels in all tissues were noted in the 50–60-year and 60–70-year age groups), sex (28 of the patients were men, five were women; adduct levels were higher in the men), cigarette smoking (all patients were smokers, 19 smoked 20 cigarettes/day, 14 smoked 30–40) and stage of tumour progression (p values were not reported) (Banaszewski et al., 2000).
Sputum
DNA isolated from sputum, induced by inhalation of nebulized saline solution, was compared in a group of 20 smokers and 24 nonsmokers using the nuclease P1 digestion method of 32P-postlabelling analysis. All smokers, but only one nonsmoker, showed a DRZ in their adduct patterns, and adduct levels were significantly higher in smokers than in nonsmokers (3.1 ± 1.4 versus 0.6 ± 0.8/108 nucleotides; p = 0.0007) (Besarati Nia et al., 2000a). The same authors also found that the adduct levels in the induced sputum of nine smokers (monitored on three occasions) were significantly higher than those in nine nonsmokers (monitored once) (Besarati Nia et al., 2000b). When immunohistochemical analysis of induced sputum was carried out, the cells of 20 smokers were found to have significantly greater intensity of staining when using antibodies against 4-ABP–DNA than the cells of 24 nonsmokers (p = 0.001), but not when BPDE–DNA antibodies were used (p = 0.07) (Besarati Nia et al., 2000c).
Oral and nasal cavities
In two early 32P-postlabelling studies of DNA from oral mucosal cells, varying amounts of a variety of aromatic and/or hydrophobic adducts were seen in smokers and nonsmokers, but none of them were specific to exposure to tobacco smoke (Dunn & Stich, 1986; Chacko & Gupta, 1988).
DNA from clinically normal oral tissue (mucosa) from patients undergoing surgery for intraoral squamous-cell carcinoma was analysed by 32P-postlabelling using both nuclease P1 digestion and butanol extraction enhancement methods. Both the range and levels of adducts measured were greater with the latter method, and the mean level of adducts in the smokers (n = 19 + 1 pipe smoker) was statistically significantly higher than that in the four former smokers and nine nonsmokers combined (Jones et al., 1993). A comparison between DNA obtained from oral biopsies and from buccal mucosa from the same individuals showed that there was a good correlation between adduct levels from the two sources, with a significantly higher level of adducts in DNA from 20 smokers than that from 10 nonsmokers (Stone et al., 1995).
In a small study of nasal mucosal DNA from nine smokers, two former smokers and 10 nonsmokers, 32P-postlabelling analysis detected the DRZ in eight of the smokers, one of the former smokers and one of the nonsmokers. The mean adduct level in smokers was significantly higher than that in the nonsmokers (p < 0.01; 4.8 versus 1.4/108 nucleotides using the nuclease P1 digestion) and a linear relationship between adduct levels and daily cigarette consumption was observed among the smokers (Peluso et al., 1997). In another study, the levels of DNA adducts detected by 32P-postlabelling analysis in nasal cells from six smokers were also significantly higher than the levels in cells of 14 nonsmokers (mean value, 17.0 versus 6.8/108 nucleotides) (Zhao et al., 1997).
Using a 32P-postlabelling method to detect cyclic adducts such as 1,N2-propanodeoxyguanosine, possible products of the reaction of tobacco smoke constituents such as acrolein (A) and crotonaldehyde (C) with DNA, significantly higher levels of each of three such adducts (AdG, CdG1 and CdG2) were detected in the DNA of gingival tissue of 11 smokers than in that of 12 nonsmokers; the total levels of the adducts were 4.4-fold higher in smokers than in nonsmokers (Nath et al., 1998).
Using the alkaline comet (single-cell gel electrophoresis) assay to detect DNA strand breaks in exfoliated buccal cells, significantly higher DNA migration was found to occur in the cells of 11 smokers than in those of nine nonsmokers (Rojas et al., 1996).
A study of exfoliated oral cells from healthy volunteers used an immunohistochemical technique to stain cells with antibodies raised against BPDE–DNA adducts. The intensity of staining was significantly greater in cells from 33 smokers than in those from 64 nonsmokers, and increased intensity of staining was observed with increasing number of cigarettes smoked per day by the smokers (Romano et al., 1999a). Similar results were obtained in another study that used this technique. Both buccal cavity and mouth floor cells were investigated; staining intensity in both cell types was found to be significantly higher in 26 smokers than in 22 nonsmokers (Besarati Nia et al., 2000d). When antibodies to 4-ABP–DNA adducts were used, staining intensity was also found to be significantly higher in the exfoliated oral cells of 12 smokers than in those of 12 nonsmokers (Romano et al., 1997).
Immunohistochemical staining of human oral mucosal cells, using antibodies against BPDE–DNA adducts, revealed significantly higher intensities of staining in 16 smokers than in 16 matched nonsmokers (Zhang et al., 1995). A second study, using antibodies against BPDE–DNA adducts or against 4-ABP-modified DNA in exfoliated oral cells, showed significantly higher levels of DNA damage in 20 smokers than in 20 matched nonsmokers for both DNA adducts (Hsu et al., 1997). Subsequently, an analogous analysis using antibodies to malonaldehyde–DNA adducts has demonstrated higher levels of this adduct, derived from lipid peroxidation, in the oral mucosal cells of 25 smokers than in 25 matched nonsmokers (Zhang et al., 2002).
Urogenital tissues
Bladder
Investigations of the presence of smoking-related DNA adducts in bladder cells have made use of autopsy and biopsy specimens and of exfoliated epithelial cells excreted in urine.
When the levels of DNA adducts were determined in bladder biopsy samples using 32P-postlabelling with both butanol extraction and nuclease P1 enhancement procedures, the mean levels of several specific adducts were higher in 13 smokers than in 20 former smokers and nine never-smokers; some adducts were present at similar levels in all groups and thus were not smoking-related. One of the smoking-related adducts was chromatographically identical to the major adduct formed in DNA by 4-ABP (Talaska et al., 1991a).
Exfoliated urothelial cells recovered from the urine of 39 of 73 individuals yielded sufficient DNA for 32P-postlabelling analysis (21 from nonsmokers, 18 from current smokers). The DNA was found to contain several DNA adducts, of which at least four were present in the DNA of smokers at levels 2–20 times higher than those in the DNA of nonsmokers. One of these corresponded chromatographically to N-(deoxyguanosin-8yl)-4-ABP (Talaska et al., 1991b). In a further study of the same subjects, two DNA adducts detected in exfoliated uroepithelial cells were found to be specifically associated with smoking and with the levels of 4-ABP–haemoglobin adducts (Vineis et al., 1996).
In a study of bladder biopsies of 20 bladder cancer patients and of exfoliated bladder cells of 36 healthy individuals, a dose–response relationship between smoking levels and adduct levels was found for both groups [statistics not given and the sizes of the groups studied were small]. The levels of adducts were not higher in the cases than in the controls (Talaska et al., 1994).
Bladder tissue samples taken at autopsy from 56 individuals were analysed by 32P-postlabelling of DNA. A positive but weak association (p = 0.09) between adduct levels and tobacco smoking was found, and there was a correlation between the levels of adducts in lung and bladder (p < 0.01, Spearman rank correlation test) (Routledge et al., 1992) (see also Table 4.4).
A comparison of the adducts detected by 32P-postlabelling in bladder biopsies from 30 smokers with those from 24 nonsmokers revealed that, overall, adduct levels were not significantly different between the two groups; 3–5-fold higher levels of adducts were detected by the butanol extraction procedure than by nuclease P1 digestion, implying a preponderance of aromatic amine-like adducts over those formed by PAHs. In a more detailed analysis, however, the level of one minor DNA adduct was found to be twice as high in samples analysed from 17 smokers than in those from eight nonsmokers (p < 0.005, one-tailed) (Phillips & Hewer, 1993).
Biopsy samples from human bladder were subjected to immunohistochemical analysis with antibodies to 4-ABP–DNA adducts to determine the relationship between smoking history and intensity of staining. The adduct levels were significantly higher in 24 current smokers than in 22 nonsmokers (p < 0.0001). There was also a linear relationship between mean levels of adduct staining and number of cigarettes smoked per day (Curigliano et al., 1996). In another study using the same technique, higher levels of 4-ABP–DNA adducts were also observed in bladder specimens from 30 smokers than in those from 41 former smokers and 24 nonsmokers, but the statistical significance of this difference was borderline (p = 0.07) (Romano et al., 1999b).
Immunohistochemical staining of human exfoliated urothelial cells from urine, using antibodies to BPDE–DNA adducts or to 4-ABP–DNA adducts, revealed significantly higher (p < 0.0005) intensities of staining in 20 smokers than in 20 matched nonsmokers in both cases (Hsu et al., 1997).
The presence of 3-alkyladenines in human urine was determined in two smokers who underwent a period of voluntary abstinence from smoking. In one subject, the excretion of 3-ethyladenine was significantly lower on nonsmoking days (p < 0.01). Smoking-dependent differences in levels of 3-methyladenine were apparent only when volunteers consumed a diet low in 3-alkyladenines during the study period. Thus, although both adducts are produced by alkylating agents in tobacco smoke, natural dietary levels of 3methyladenine make this an insensitive biomarker of smoking (Prévost & Shuker, 1996).
In a study of depurinated adducts of benzo[a]pyrene in urine, detectable levels of BaP-6-N7 Ade1 were found in the urine of three of seven smokers (not quantifiable but detectable up to 0.1–0.6 fmol/mg creatinine), but not in the urine from any of 13 nonsmokers (Casale et al., 2001).
The comet assay was used to analyse urinary bladder cells for DNA damage. A significant increase in comet tail moments for 18 smokers and former smokers was found as compared with 12 nonsmokers (p < 0.03) (Gontijo et al., 2001).
In a study of oxidative DNA damage measured by 8-OHdG excretion in urine, 30 smokers were reported to have levels of the nucleoside that were 50% higher (corrected for body weight) than 53 nonsmokers (Loft et al., 1992).
Taken together, the studies analysing DNA adducts in bladder tissue or exfoliated urothelial cells demonstrate that smoking-related formation of DNA adducts is detectable, but that some of the adducts present derive from other sources of environmental carcinogens.
Cervix
A pilot study of DNA isolated from cervical scrapes of 22 women and analysed by the butanol extraction enrichment procedure of 32P-postlabelling showed the presence of the DRZ and higher levels of adducts in three of nine smokers than in 13 nonsmokers (Phillips et al., 1990b). In a subsequent study, sufficient DNA was isolated from 33 of 38 cervical smear samples and subjected to 32P-postlabelling analysis. Adduct levels were significantly higher in the DNA from 18 smokers than in that from 15 nonsmokers (Simons et al., 1995).
Using DNA isolated from cervical biopsies, 32P-postlabelling analysis with butanol extraction was carried out on samples from 39 women (11 smokers, seven former smokers and 21 never-smokers). Adduct levels were significantly higher in the smokers and former smokers than in the nonsmokers (p = 0.048). Exclusion of those women whose urinary cotinine levels did not confirm their self-reported nonsmoking status (n = 7) increased the differences in levels of adducts between smokers and nonsmokers (p = 0.03) (Simons et al., 1993).
A study of cervical biopsy samples from 35 women (19 smokers, five former smokers and 11 nonsmokers) found that DNA adduct levels, determined by 32P-postlabelling analysis with the butanol extraction procedure, were significantly higher in the smokers than in the nonsmokers (p = 0.002), and an intermediate mean value was obtained for the former smokers (Phillips & Ni She, 1993). This study population was subsequently increased to 22 smokers, four former smokers and 14 nonsmokers and analysed by both butanol extraction and nuclease P1 digestion procedures of 32P-postlabelling. Adduct levels were significantly higher in smokers when the former procedure was used (p = 0.005), but not with the latter (Phillips & Ni She, 1994). The lack of a difference between smokers and nonsmokers using nuclease P1 digestion was also reported in another small study in women not using oral contraceptives (15 smokers, eight nonsmokers) (King et al., 1994). However, in another larger study, 32P-postlabelling analysis using nuclease P1 digestion found significantly higher levels (p = 0.07) in DNA from histologically normal cervical biopsy tissue from 48 current smokers than in that from 48 non-current smokers (non-current smokers included nonsmokers, former smokers and passive smokers) (Ali et al., 1994).
Immunohistochemical analysis of human cervical cells, using antibodies against BPDE–DNA adducts, demonstrated significantly higher staining intensity (p = 0.04) in smears from 16 smokers than in smears from 16 nonsmokers (Mancini et al., 1999).
GC–MS analysis of BPDE–DNA adducts in cervical epithelial cells found that the level of adducts was approximately twofold higher in seven smokers than in seven nonsmokers and this difference was statistically significant (p = 0.02) (Melikian et al., 1999).
Other tissues
Breast
In a pilot study, a total of 31 samples of human breast tissue were analysed by 32P-postlabelling using nuclease P1 digestion to determine the levels of aromatic/hydrophobic DNA adducts. The breast tissues included samples from tumours and from tissues adjacent to tumours from 15 women with breast cancer and four samples from women who had had reduction mammoplasties. The characteristic DRZ was detected in tumorous and non-tumorous tissues from five of 15 cancer patients, all of whom were current smokers. None of the eight former smokers and nonsmokers displayed this pattern (p < 0.01) (Perera et al., 1995).
Another 32P-postlabelling study using nuclease P1 digestion of aromatic DNA adducts was conducted on normal (non-tumour) breast tissue from 87 breast cancer patients undergoing mastectomy and from 29 women who did not have cancer (controls) undergoing reduction mammoplasty. The characteristic DRZ was detected in 29 of 87 normal tissues from breast cancer patients (17/17 smokers, 5/8 former smokers, 4/52 nonsmokers, 3/10 with unknown smoking status), and in two of 10 control tissues. In addition, a benzo[a]pyrene-like DNA adduct was observed in 36 normal adjacent breast tissue samples (27 of them from nonsmokers) from cancer patients and its presence was not significantly associated with smoking (Li et al., 1996).
Higher levels (p = 0.0001) of DNA adducts putatively derived from malonaldehyde and detected by 32P-postlabelling using nuclease P1 digestion were determined in tissue adjacent to tumours from 51 breast cancer patients than in tissue from 28 controls (undergoing reduction mammoplasty). The levels of these adducts were also associated with the previously detected benzo[a]pyrene-like DNA adduct (see above), but were significantly lower in smokers than in former smokers and in nonsmokers (Wang et al., 1996).
Immunohistochemical staining intensity of breast tumour tissue using antibodies against BPDE-DNA adducts was not significantly different between 35 smokers, 72 former smokers who had smoked one cigarette or more per day for 6 months or longer and 75 nonsmokers. However, among smokers and former smokers, there was a trend towards higher intensities of staining with greater levels of exposure or earlier age at starting smoking (Santella et al., 2000). Positive staining using antibodies against BPDE–DNA adducts was also obtained with tissue samples from 48 patients with breast tumours and tissue samples from 30 patients with benign disease (with higher adduct levels being found in tissues from patients with benign breast disease than in cancer tissues). This study was carried out on inhabitants of Upper Silesia, a region of Poland that has been subject to high levels of environmental pollution. However, in neither group of patients were staining intensities higher in smokers than in nonsmokers (Motykiewicz et al., 2001).
In a study of PAH–DNA adducts in breast tumour tissue from 119 cancer patients and from 108 patients with benign breast disease, immunohistochemical analysis indicated a significant association between adduct levels and breast cancer, but there was no relationship between smoking status, stage or tumour size and adduct levels (Rundle et al., 2000).
Pancreas
32P-Postlabelling analysis of 20 pancreatic tumours and 13 samples of normal tissue adjacent to the tumour, using nuclease P1 digestion, led to the detection of significantly higher levels of DNA adducts in the normal tissues surrounding the tumour than in either the tumour tissue or in normal pancreatic tissue from five patients with non-pancreatic cancer and 19 previously healthy organ donors who served as controls. Two novel clusters of adducts were observed: 11/13 in adjacent normal tissues from pancreatic cancer patients, 12/20 in tumour tissues and 2/24 in normal pancreatic tissues from controls. The presence of these adducts was positively correlated with smoking status (Wang et al., 1998).
Another study that used fluorescence spectral analysis for the presence of BPDE–DNA adducts in pancreatic samples revealed no detectable levels in any of 11 samples (six from smokers) (Alexandrov et al., 1996).
Colon
Fluorescence spectral analysis of samples of colon mucosa for the presence of BPDE–DNA adducts resulted in their detection in three of four samples from smokers and one of three samples from nonsmokers. Adduct levels were estimated to be in the range 0.2–1.0 adducts/108 nucleotides (Alexandrov et al., 1996).
Stomach
Tumour DNA from 26 patients (18 smokers) with gastric cancer was analysed for DNA adducts using 32P-postlabelling with butanol extraction. In men only, the numbers of DNA adducts were found to be significantly higher in the samples from 14 smokers than in that from four nonsmokers (Dyke et al., 1992). [The Working Group noted that the number of nonsmokers was small.]
Placenta and fetal tissue
In the first study of placental DNA for the presence of DNA adducts, 32P-postlabelling analysis and ELISA with antibodies against BPDE–DNA adducts were used. The use of the ELISA found a small but non-significant increase in adducts in placental tissue from smokers when compared with the numbers found in nonsmokers. Using 32P-postlabelling analysis, one particular DNA adduct was found to be present in the DNA from 16 of 17 smokers, but in only three of 14 nonsmokers (Everson et al., 1986). In a subsequent study using 32P-postlabelling, up to seven different adducts were detected in 53 specimens of human placental tissue, three of these adducts were found ‘almost exclusively’ in smokers (Everson et al., 1988).
The combined use of immunological, fluorescence spectroscopic and mass spectrometric methods has led to the detection and characterization of BPDE–DNA adducts in human placental DNA and also provided evidence for the presence of adducts formed by other PAHs (Weston et al., 1989b; Manchester et al., 1990). Synchronous fluorescence spectroscopy for the detection of BPDE–DNA adducts gave positive results in 10 of 28 human placental samples, but there was no correlation with smoking status (Manchester et al., 1988). In a subsequent study, five of seven placentas from smokers and three of nine from nonsmokers gave positive results when analysed for these adducts (Manchester et al., 1992).
Placental DNA from five smokers was found to contain adduct levels, detected as a DRZ by 32P-postlabelling, 1.8-fold higher than that in placental DNA from five nonsmokers (4.3 ± 1.7 versus 2.3 ± 0.4 adducts/108 nucleotides) (Gallagher et al., 1993a) [statistical significance not stated].
Using 32P-postlabelling with nuclease P1 digestion, DNA adducts were detected in DNA from placenta and umbilical cord regardless of whether or not the mothers were smokers. Adduct levels were significantly higher in maternal than in fetal tissue (and were higher in placenta than in umbilical cord) and, combining data for all tissues, total levels of DNA adducts were significantly higher in eight smokers than in 11 nonsmokers. Although individual tissues showed a trend towards increased levels of adducts in smokers, the differences were not statistically significant (Hansen et al., 1992, 1993). DNA adducts derived from PAHs were detected by a competitive ELISA in six of 14 placentas and in five of 12 matched fetal lung samples from spontaneous abortions. None of the samples were from women who reported smoking during pregnancy; thus smoking was not a likely source of the adducts (Hatch et al., 1990). However, in another study using ELISA with antibodies against BPDE–DNA adducts, a linear correlation was found between levels of adducts and levels of urinary cotinine for both placental DNA and umbilical cord DNA, the former tissue having the higher adduct levels. Overall, adducts were detected in 13 of 15 placental samples and 12 of 15 samples of umbilical cord blood from smokers and three of 10 placental samples and one of 10 samples of umbilical cord from nonsmokers (Arnould et al., 1997).
When placental DNA was analysed both for bulky DNA adducts by 32P-postlabelling with nuclease P1 digestion and for 8-OHdG by electrochemical detection, neither method showed a difference between 11 smokers, 10 nonsmokers and nine nonsmokers who were exposed to secondhand smoke (Daube et al., 1997).
Thus, although some studies indicate the presence of smoking-related DNA adducts in human placenta, overall the association between smoking status and adduct levels, determined by a variety of different methods, is weak. The results appear to indicate that there are significant sources of environmental carcinogens other than tobacco smoke that result in DNA adducts being formed in this tissue.
Sperm
In a study in which sperm DNA from 12 heavy smokers (> 20 cigarettes/day), 12 moderate smokers (1–19 cigarettes/day) and 12 nonsmokers was subjected to 32P-postlabelling analysis, no discernible differences were observed between the patterns or levels of DNA adducts between the three groups of subjects (Gallagher et al., 1993b).
However, immunohistochemical analysis of sperm, with antibodies against BPDE–DNA adducts showed significantly higher intensity of staining in the sperm of 11 smokers than in the sperm of 12 nonsmokers (Zenzes et al., 1999a). Furthermore, in-vitro fertilization experiments using sperm from smokers and oocytes from nonsmoking partners suggested that there was transmission of benzo[a]pyrene-modified DNA to the embryos (Zenzes et al., 1999b). In related studies, granulosa-lutein cells from women undergoing in-vitro fertilization were analysed. Immunostaining for BPDE–DNA adducts was confined to the nucleus and was significantly greater in cells from 14 smokers than in those from seven passive smokers and 11 nonsmokers (Zenzes et al., 1998) (see also the monograph on involuntary smoking, this volume).
The sperm DNA of 28 smokers contained levels of 8-OHdG about 1.5 times higher than that of 32 age-matched nonsmokers, a difference that was statistically significant (6.19 ± 1.71 versus 3.93 ± 1.33 8-OHdG/105 dG, p < 0.001) (Shen et al., 1997). The same authors reported a correlation between sperm defects and levels of 8-OHdG in sperm DNA, indicating that this lesion is useful for assessing sperm quality and male fertility (Shen et al., 1999).
In another study, the sperm DNA of smokers (n = 35) was found to be more sensitive to acid-induced denaturation (p < 0.02) and to possess higher levels of DNA strand breaks (p < 0.05) than that of nonsmokers (n = 35) (Potts et al., 1999).
Cardiovascular tissues
In a pilot study investigating the presence of DNA adducts by 32P-postlabelling in a number of human tissues at autopsy, the DRZ characteristic of smoking-related DNA damage was observed in heart tissue from two smokers, but not in that of a nonsmoker who chewed tobacco. Moreover, of all the tissues examined, which included lung and bronchus, the levels of adducts were highest in heart tissue (Randerath et al., 1989). A study comparing 32P-postlabelling, HPLC-fluorescence detection of BPDE–DNA adduct hydrolysis products and synchronous fluorescence spectroscopy (SFS) detection of the same products demonstrated that all three methods were capable of detecting DNA adducts in atherosclerotic lesions in smooth muscle of human abdominal aorta. However, because of the limited number of samples investigated (four from smokers, three from former smokers), no conclusion could be drawn regarding the origins of the adducts detected (Izzotti et al., 1995a). Another study measured DNA adducts by 32P-postlabelling with nuclease P1 digestion in the thoracic aorta of 133 victims of sudden or accidental death. Those with significant atherosclerotic changes and for whom atherosclerosis was classified as the main cause of death were designated as 76 cases, those with few atherosclerotic changes as 57 controls. Smoking status was determined by measuring cotinine levels in plasma. Levels of bulky aromatic DNA adducts were significantly higher in the cases than in the controls, but when smokers and nonsmokers were compared, significant differences were observed in the controls, but not in the cases (Binková et al., 2001).
Immunohistochemical staining of endothelial and smooth muscle cells of blood vessels using antibodies against BPDE–DNA adducts resulted in higher intensity of the endothelial cells. However, the correlation with smoking habits among the 33 subjects studied (samples from nine of 11 smokers showed positive staining, compared with 12 of 22 nonsmokers) was not statistically significant (Zhang et al., 1998).
In a study of DNA from the right atrial appendage of 41 patients undergoing open heart surgery, the levels of aromatic DNA adducts detected by 32P-postlabelling with nuclease P1 digestion were significantly higher in 15 smokers than in 15 former smokers (p < 0.01) and in 11 nonsmokers (p < 0.001). A significant linear relationship between adduct levels and daily cigarette smoking was observed in the smokers (Van Schooten et al., 1998).
Blood cells
Although nucleated peripheral blood cells are not generally considered to be target cells for tobacco-induced tumorigenesis, the relative ease with which they can be obtained from human subjects, when compared with many target organs, has led to a large number of studies exploring the differences between cells from smokers and nonsmokers. The studies in which smoking-related DNA adducts have been determined in blood cells are summarized in Table 4.5.
Table 4.5
Studies of smoking-related DNA adducts in human blood cells.
The picture that emerges from these many studies is an inconsistent one. Measurements made in the longer-lived cells (lymphocytes and monocytes) are more likely to reveal significant differences between smokers and nonsmokers than measurements made in whole white blood cell preparations (see Table 4.5). However, a study of adduct levels in smokers following cessation of the habit did show a decline over time (Mooney et al., 1995). It is clear from studies of populations occupationally exposed to agents such as PAHs that there are many sources of exposure that result in DNA adducts in blood cells and whether or not the influence of tobacco smoking is discernible probably depends on the extent of the contribution of other occupational or environmental sources in particular study populations. There have also been reports of the influence of nutritional factors on levels of DNA adducts in blood cells in some studies (Grinberg-Funes et al., 1994; Mooney et al., 1997; Jacobson et al., 2000), but not in others (Wang et al., 1997). The modulating effects of genetic polymorphisms in xenobiotic metabolizing genes and DNA repair genes have been implicated in several studies. In general, these observations have been made on rather small numbers of subjects and the magnitude of the effects noted was not large. Confirmation of the findings from larger studies is required.
A few reports have suggested that the levels of adducts in the blood of smokers correlate with the levels of adducts in the lung (Tang et al., 1995; Wiencke et al., 1995, 1999). However, there are also conflicting studies in which no association was found between levels of blood BPDE–DNA adducts and levels of adducts in lung (Van Schooten et al., 1992) or in cells obtained by bronchio-alveolar lavage (Van Schooten et al., 1997).
A number of studies in lymphocytes have reported an increase in oxidative damage in the DNA of smokers (Kiyosawa et al., 1990; Asami et al., 1996). The magnitude of this increase, although significant, is generally small (less than twofold) and its biological significance is unclear. In contrast, there have been two reports in which smokers had lower levels of this lesion in DNA from their leukocytes or lymphocytes (van Zeeland et al., 1999; Besarati Nia et al., 2001).
Several studies that have compared smokers with cancer with smokers who are free of the disease have reported an approximately twofold higher level of lymphocyte adducts in the cases. This has been observed in two case–control studies of lung cancer (Perera et al., 1989; Vulimiri et al., 2000) and in one prospective study (Tang et al., 2001), but not in another case–control study (Hou et al., 1999). The effect was also observed in a case–control study of bladder cancer (Peluso et al., 2000). These findings suggest that DNA adducts can be considered as biomarkers of risk in some populations, although the magnitude of the changes in levels is small when seen against a background of wide inter-individual variation, making the assessment of risk on an individual basis problematic.
Another approach to monitoring human exposure to carcinogens has been to detect the antigenic response to BPDE-DNA adducts in blood cells. Serum antibodies to these adducts have been detected in a number of groups of occupationally exposed workers, but in a study of smokers only four of 50 heavy smokers initially tested positive for the antibodies. Paradoxically, the frequency of positivity increased during a cessation programme in which 28 subjects quit smoking and 22 reduced cigarette consumption by 75% (Pulera et al., 1997). Nevertheless, in a larger population study of 1345 individuals tested for the presence of detectable BPDE–DNA antibodies in serum, there was a positive association with smoking in 17.8% of nonsmokers, 21.5% of former smokers and 25.7% of smokers. There was also a synergism between smoking and family history of lung cancer in determining the prevalence of antibodies (p = 0.02) (Petruzzelli et al., 1998).
(iv) Blood compounds
Tobacco use is associated with increased levels of several carcinogens in blood. These compounds may prove useful as biomarkers of exposure to tobacco smoke when blood samples are collected in epidemiological studies.
Tobacco is a major source of exposure to cadmium in the general population and blood cadmium concentrations correlate positively with current tobacco smoking habits (WHO, 1992). Cadmium levels increase with the number of cigarettes smoked per day (Hoffmann et al., 2000). Current smokers have, on average, levels of cadmium that are four to five times higher than those of never-smokers (Ellingsen et al., 1997; Hoffmann et al., 2000). Cigarette smoking or both sheesha (tobacco–fruit mixture cooked) and cigarette smoking lead to significantly higher blood cadmium levels than those observed in nonsmokers or in smokers of only sheesha (Al-Saleh et al., 2000). Although smoking has less effect on lead levels than on those of cadmium, slightly higher levels of lead in blood have generally been reported for smokers than for nonsmokers or former smokers (Brockhaus et al., 1983; Al-Saleh, 1995; Ellingsen et al., 1997).
Many studies have shown that the levels of certain volatile organic compounds in blood were significantly higher in smokers than in nonsmokers (Ashley et al., 1996). The mean levels of benzene and styrene in blood were significantly higher among smokers than nonsmokers. The concentrations of benzene in blood were significantly associated with the number of cigarettes smoked per day (Ashley et al., 1995). The difference in the levels of benzene in blood persisted when comparisons between smokers and nonsmokers in the general public and in occupationally exposed groups were made (Brugnone et al., 1999).
The levels of NNAL (a metabolite of NNK) has been measured in the blood (unhydrolysed plasma) of smokers (Hecht et al., 1999a). NNAL and its detoxification product, NNAL-glucuronide, can be detected in the blood plasma for up to 2 days after quitting tobacco use. Levels of NNAL and NNAL-glucuronide in blood are 18.8% and [4.1%] of those in urine, respectively (Hecht et al., 2002).
(v) Breath compounds
Exhaled carbon monoxide
The measurement of exhaled carbon monoxide (CO) has often been used as a tool for validation of self-reported cigarette consumption and smoking histories and to distinguish smokers from nonsmokers (Robertson et al., 1987; Muranaka et al., 1988; Morabia et al., 2001).
The concentration of CO in exhaled air was significantly correlated with the self-reported number of cigarettes smoked per day. The average concentration of expired CO was 4.9 ppm (v/v) (n = 20) in nonsmokers and 18.5 ppm in smokers (n = 20; p < 0.001). The concentration of expired CO in male smokers (n = 12) was 22.7 ppm and in female smokers was 12.0 ppm (n = 8; p < 0.05) (Muranaka et al., 1988).
Various measures of exhaled CO as a marker of exposure to cigarette smoke were compared between 400 current regular male and female smokers. Cigarette consumption was categorized as moderate (< 15 cigarettes/day for at least 5 days per week for the last 2 years) or heavy (> 15 cigarettes/day for at least 2 years). Individuals with genetic variants of the CYP2A6 gene (CYP2A6*2 and CYP2A6*A genes) were not included in the analysis (n = 25). No difference was found in the number of cigarettes per day or levels of exhaled CO between the sexes among Caucasian smokers. Women smoked 19.3 cigarettes per day and exhaled 19.6 ppm (v/v) CO, whereas men smoked 19.8 cigarettes/day and exhaled 20.6 ppm CO. The ratio of nicotine:CO was determined using plasma nicotine levels. Women had significantly lower levels of nicotine in the blood than men (17.3 versus 20.9 ng/mL; p < 0.01). Although no significant difference was found in levels of CO in breath, women tended to smoke cigarettes with a lower nicotine content and their nicotine:CO ratios were lower than those measured in male smokers. Among other ethnic groups, there were no statistically significant gender differences in the number of cigarettes smoked per day, but men had higher concentrations of CO in exhaled air and higher concentrations of blood nicotine and cotinine (p < 0.01) than women (Zeman et al., 2002).
The CO level measured in exhaled breath condensate was significantly higher in smokers (12.5 ppm) than in nonsmokers (3.4 ppm) and increased significantly to 19.6 ppm 30 min after exposure to tobacco smoke (Balint et al., 2001).
At the time of delivery, pregnant women who smoked exhaled 8.42 ppm (v/v) CO corrected for inhaled air as opposed to 1.33 ppm exhaled by pregnant women who did not smoke (p < 0.0001) (Seidman et al., 1999). The concentration of exhaled CO in mothers who smoked was strongly correlated with the number of cigarettes smoked per day (p < 0.000001). Within hours of delivery, the newborns of women smokers exhaled 10 ppm CO, whereas 1.74 ppm CO was exhaled by newborns of nonsmoking mothers (p < 0.0001). Maternal exhaled CO correlated strongly (p < 0.000001) with neonatal exhaled CO.
No differences in concentrations of exhaled CO were found between smokers of either three mentholated or three non-mentholated cigarettes with different nicotine content (the three cigarettes were smoked 45 min apart, in a single session) (Pickworth et al., 2002). According to Clark et al. (1996), African American smokers had significantly higher levels of cotinine and CO per cigarette smoked and per millimetre of tobacco rod smoked than white American smokers (p < 0.001). After adjustment for race, number of cigarettes smoked per day and mean length of each cigarette smoked, the presence of menthol was associated with higher concentrations of cotinine (p = 0.03) and CO (p = 0.02).
A preliminary evaluation of a novel smoking system that employs electrical heating of tobacco (600 °C) instead of combustion (900 °C) (Accord®) was carried out by enrolling 10 smokers of ‘light’ cigarettes (≥ 10 cigarettes/day). The novel system limited users to eight puffs per cigarette instead of their usual 10–11 puffs. The levels of expired CO were lower in subjects using the novel system than when normal cigarettes were smoked (Buchhalter & Eissenberg, 2000).
Eclipse® is a novel nicotine delivery device that primarily heats, rather than burns, tobacco. In a cross-over study, the number of cigarettes smoked per day decreased from 19 at baseline to 2.1 after two weeks of using Eclipse® (p < 0.001), but exhaled CO concentrations increased from 21 ppm to 33 ppm (p < 0.001). During use of Eclipse®, the concentration of nicotine in blood remained fairly stable although it increased slightly from 16.8 ng/mL to 18.0 ng/mL (Fagerström et al., 2000).
Smoking fewer cigarettes may reduce exposure to toxins even if the smoker adopts a more intensive smoking behaviour to compensate for the reduced number of cigarettes. If consumption is reduced from an average of 37 cigarettes/day to an average of five cigarettes/day, the intake of tobacco toxins per cigarette increased roughly threefold and daily exposure to tar and CO declined by only 50% (Benowitz et al., 1986). The reduction of cigarette consumption from 40 cigarettes/day to 20 cigarettes/day was not followed by any consistent reduction in the levels of biomarkers of exposure to tobacco carcinogens (Hurt et al., 2000).
Exhaled nitric oxide
Nitric oxide (NO) is formed from L-arginine by the enzyme NO synthase (NOS). NO is a highly reactive molecule that can be oxidized or complexed with other biomolecules depending on the microenvironment. The stable oxidation products of NO metabolism are nitrite and nitrate. Cigarette smoking is associated with an increased risk for respiratory tract infection, chronic airway disease and cardiovascular diseases, all of which may be modulated by endogenous NO (which acts as an endogenous vasodilator). Cigarette smoking reduces the levels of NO exhaled by healthy subjects, but the mechanism for this is unclear (Schilling et al., 1994; Yates et al., 2001). In the study by Yates et al. (2001), the amount exhaled by active smokers was significantly less than the amounts of NO exhaled by subjects who were exposed to sham and passive smoking for the same length of time (49 ppb (v/v) after 15 min of active smoking, a 30.3% decrease from the baseline level of 71 ppb versus 115 ppb in subjects exposed to sham smoking and 102 ppb in subjects exposed to passive smoking). Schilling et al. (1994) reported the same trend, with concentrations of exhaled NO being lower in smokers than in nonsmokers exposed to secondhand smoke (mean, 16 ppb in women who smoked versus 21 ppb in women who did not smoke and 15 ppb in men who smoked versus 19 ppb in men who did not smoke). However, the absolute values were much lower than those reported by Yates et al. (2001). In a single-breath analysis, smokers exhaled about 20 ppb NO, healthy controls, 40 ppb and asthmatic patients, 60 ppb (Persson et al., 1994). Kharitonov et al. (1995) reported that smokers exhaled 42 ppb NO, whereas the concentration exhaled by nonsmokers was 88 ppb (p < 0.01). There was a significant relationship between the amount of exhaled NO and cigarette consumption (r = – 0.77, p < 0.001).
Measurement of exhaled NO may yield information about the mechanisms underlying cigarette-induced lung damage. Balint et al. (2001) found that there was no difference between smokers and nonsmokers in the levels of nitrite, nitrite + nitrate, Snitrosothiols and nitrotyrosine in the exhaled breath condensate at the baseline visit. Thirty minutes after smoking two cigarettes, the levels of nitrite + nitrate were significantly increased (from 20.2 to 29.8 µM, p < 0.05) and returned to the baseline within 90 min. There was no significant change in the levels of exhaled NO, nitrite, S-nitrosothiols or nitrotyrosine 30 and 90 min after smoking two cigarettes. There was no correlation between levels of exhaled NO and NO metabolites in breath condensate before and after smoking.
Benzene
Breath analysis under controlled conditions has been used to measure the exposure to benzene associated with active smoking (Jo & Pack, 2000). The mean benzene concentrations in exhaled breath measured 1 min after smoking 5.0 cm of a cigarette (to a remaining butt length of 3.3 cm) ranged from 58.1 to 81.3 µg/m3, depending on the commercial cigarette brand, whereas benzene concentrations measured prior to smoking ranged from 15.9 to 19.2 µg/m3. These post-exposure concentrations of benzene in breath were much higher than the mean concentrations of benzene in breath reported in some previous studies (e.g. 16 µg/m3 among smokers versus 2.5 µg/m3 among nonsmokers; p < 0.001) (Wallace & Pellizzari, 1986) in which the exposure conditions and post-sampling times were not controlled. The concentration of benzene in breath increases with the number of cigarettes smoked: from 9.4 µg/m3 (no cigarettes) to 47 µg/m3 (> 50 cigarettes/day). The increase in concentrations of benzene in breath after active smoking is due to benzene being absorbed through the lung while an individual is smoking. From direct measurements of benzene in mainstream smoke, it was calculated that a typical smoker (30 cigarettes/day) inhales 2 mg benzene daily, whereas a nonsmoker inhales 0.2 mg/day (Wallace et al., 1987).
Volatile organic compounds
Volatile organic compounds (VOC) other than benzene are often measured in expired breath to determine the exposure to cigarette smoke. Among 20 VOC analysed by Wallace and Pellizzari (1986), the levels of meta + para-xylene, ethylbenzene, ortho-xylene, styrene and octane were found to be statistically significantly higher (p < 0.05) in smokers (n = 198) than in nonsmokers (n = 322). Significant increases in the concentration in breath with number of cigarettes smoked were noted for styrene, ethylbenzene and meta + para-xylene (Wallace et al., 1987).
Gordon (1990) analysed VOC in the exhaled breath of 26 smokers and 43 nonsmokers to identify possible biochemical markers of exposure to cigarette smoke. Among 230 GC–MS peaks, 2,5-dimethylfuran was found to have sufficient discriminatory power to allow almost complete distinction to be made between smokers and nonsmokers. Several other compounds could also be used to distinguish between the two groups with a high level of accuracy. However, the half-lives of these compounds must be determined before they can be exploited as practical indicators of exposure to smoking.
Ethane, often used routinely as a marker of lipid peroxidation, has the potential to be a non-invasive marker of free-radical activity. Ethane levels in exhaled air were found to be higher in active smokers than in former smokers and nonsmokers (2.9 pmol/min/kg versus 1.55 and 1.11 pmol/min/kg, respectively), reflecting the presence of ethane in cigarette smoke (Habib et al., 1995).
The levels of isoprene in exhaled breath increased on average by 70% after smoking one cigarette. The concentration of acetone increased by 22%, whereas that of ethanol decreased by 28% after smoking one cigarette (Senthilmohan et al., 2001).
(vi) Other
Polycyclic aromatic hydrocarbons
Several studies have quantified PAHs in lung tissue (Tomingas et al., 1976; Seto et al., 1993; Tokiwa et al., 1993; Lodovici et al., 1998). In a recent investigation, the quantities of 11 PAHs were measured in 70 lung tissue samples from 37 smokers and 33 nonsmokers (defined by serum cotinine concentration). The sum of PAH concentrations was higher in smokers and there was a dose–response relationship for smoking characterized by increasing serum cotinine (Goldman et al., 2001).
Benzo[a]pyrene
Benzo[a]pyrene and several of its metabolites were detected in the cervical mucus of both smokers and nonsmokers (Melikian et al., 1999).
N-Nitrosamines
The tobacco-specific nitrosamine, NNK, was detected in 16 samples of cervical mucus from 15 women who were smokers at concentrations of 11.9–115 ng/g (mucus) (two samples were collected from one smoker at different times) and in nine of 10 samples from nonsmokers at concentrations of 4.1–30.8 ng/g (mucus), but the concentrations of NNK in specimens from cigarette smokers were significantly higher than in those obtained from nonsmokers (Prokopczyk et al., 1997). Further studies demonstrated that human cervical tissue can metabolize NNK by both α-hydroxylation and carbonyl reduction (Prokopczyk et al., 2001).
NNK was detected in 15 of 18 samples of pancreatic juice from smokers at concentrations of 1.4–604 ng/mL and in six of nine samples from nonsmokers (range of concentrations, 1.13–97 ng/mL), and the levels were significantly higher in smokers than in nonsmokers. NNAL was present in 11 of 17 samples from smokers and in 3/9 from nonsmokers. NNN was found in two of 17 samples from smokers (68 and 242 ng/mL) (Prokopczyk et al., 2002).
Particulate matter (tar)
A problem in the analysis of exposure to tobacco smoke is that there is no direct marker for tar uptake (exposure). The biomarkers most frequently used for measuring the uptake of tobacco smoke (nicotine and cotinine) reflect the exposure to particulate-phase constituents.
Russel et al. (1986) and Woodward and Tunstall-Pedoe (1992) used an indirect measure, based on the assumption that the intake of different smoke components relative to one another is proportional to their concentration in the smoke, to evaluate the exposure of smokers in large population studies. Two tar indices were calculated:
- tar index (CO) = eCO × tar yield/CO yield; and
- tar index(cot) = serum cotinine × tar yield/nicotine yield
The atherogenic potential of mainstream smoke is associated with the particulate and vapour-phase components and not with CO. The thrombogenic potential is also associated with the particulate matter and vapour phase (Smith & Fisher, 2001).
Nicotine and its metabolites as markers of exposure to tobacco smoke
Nicotine is the principal alkaloid present in tobacco. Nicotine is also the main addictive constituent of tobacco products. It also has other pharmacological activities and toxic effects (Domino, 1999). Manufactured cigarettes contain 1–7% nicotine by weight (6–12 mg nicotine/cigarette) and, of this, 15–25% enters the mainstream smoke while 75% is emitted into the air as sidestream smoke (Benowitz, 1999a). Nicotine is also found in exhaled smoke (Curvall & Enzell, 1986; IARC, 1986; California Environmental Protection Agency, 1997) (see Section 1).
An overview of the pathways of nicotine metabolism is presented in Figure 4.4. Nicotine is hydroxylated at the 5′-position by cytochromes P450 to yield an unstable intermediate, 5′-hydroxynicotine (5; see Figure 4.4), which exists in equilibrium with Δ1′(5′) iminium ion (6). 5′-Hydroxynicotine is oxidized by aldehyde oxidase to cotinine (8). Cotinine, in turn, is metabolized further to cotinine–glucuronide (cotinine–Gluc, 9), trans-3′hydroxycotinine (12) and trans-3′-hydroxycotinine–glucuronide (trans-3′-hydroxycotinine–Gluc, 13) (Gorrod & Schepers, 1999). 2′-Hydroxylation of nicotine, mediated by cytochromes P450, gives 2′-hydroxynicotine (4), which spontaneously yields Δ1′(2′) iminium ion (3) and 4-(methylamino)-1-(3-pyridyl)-1-butanone (aminoketone, 7), also known as pseudooxynicotine. Aminoketone (7) is then converted to 4-oxo-4-(3-pyridyl)butyric acid (keto acid, 11) and 4-hydroxy-4-(3-pyridyl)butyric acid (hydroxy acid, 14) (Hecht et al., 2000).
Figure 4.4
Pathways of mammalian nicotine metabolism initiated by 5′-hydroxylation and 2′-hydroxylation
The major urinary metabolite of nicotine in smokers is trans-3′-hydroxycotinine (12) (about 40% of total urinary nicotine and metabolites). This is followed by cotinine (8) and cotinine–Gluc (9) (12–15% of each), trans-3′-hydroxycotinine–Gluc (13) (10%), hydroxy acid (14) (8%) and keto acid (11) (2%). Unchanged nicotine (8% of total urinary nicotine plus metabolites) and nicotine–Gluc (2) (2–3%) also occur (Byrd et al., 1992; Benowitz et al., 1994; Hecht et al., 1999b).
Since the mid-1980s, studies that have measured exposure to tobacco smoke and its biological effects have predominantly used nicotine and its metabolite cotinine determined in blood, urine or saliva as biological markers of exposure and uptake of tobacco smoke (IARC, 1986; National Research Council, 1986; US Department of Health and Human Services, 1986; Environmental Protection Agency, 1993; California Environmental Protection Agency, 1997). The main advantage of using nicotine and, consequently that of cotinine, as a marker compound over other markers is their specificity for tobacco and tobacco smoke. The measurements of all the metabolites in Figure 4.4 provides a more complete assessment of nicotine uptake.
The half-life of nicotine in the body is short, approximately 2 h in nonsmokers (IARC, 1986; Benowitz, 1996; Benowitz & Jacob, 1997; Benowitz, 1999b). Nicotine concentration can therefore reflect only acute exposure. This limits its use as a biomarker (IARC, 1986; US Environmental Protection Agency, 1993; Benowitz, 1996). The concentrations of nicotine in blood or plasma are higher in smokers than in nonsmokers, but vary considerably, mainly depending on the number of cigarettes smoked recently. Other variables, such as the tar content, yield of nicotine, average number of cigarettes smoked per day or type of filter, have been found to be poor predictors of nicotine concentrations in the blood (IARC, 1986).
The concentrations of cotinine can be measured in blood, urine or saliva samples. Because of its specificity and the availability of sensitive methods for reliable measurements, cotinine, regardless of the body fluid, is currently the most widely used biomarker for measuring tobacco smoke uptake (reviewed in IARC, 1986; National Research Council, 1986; Jarvis et al., 1987; Muranaka et al., 1988; Etzel, 1990; US Environmental Protection Agency, 1993; Benowitz, 1996, 1999b). It has a sensitivity of 96–97% and a specificity of 99–100% (Jarvis et al., 1987). The half-life of cotinine in smokers is longer than that of nicotine, on average about 16–20 h, with a range of approximately 10–40 h (US Environmental Protection Agency, 1993; Benowitz & Jacob, 1994; Benowitz, 1996; Pérez-Stable et al., 1998; Benowitz, 1999a,b). Cotinine concentrations are a good measure of average daily exposure to tobacco smoke and reflect the exposure to tobacco smoke over the past 2–3 days (IARC, 1986; National Research Council, 1986; US Environmental Protection Agency, 1993). Cotinine concentration, however, is not suitable for measuring more long-term exposure, although it may serve as a surrogate measure for that purpose.
The main determinant of cotinine levels in smokers and nonsmokers, like that of nicotine, is exposure to tobacco smoke (reviewed in IARC, 1986; National Research Council, 1986; US Environmental Protection Agency, 1993; Benowitz, 1996, 1999a) and also exposure from use of products designed for nicotine replacement therapy (Leyden et al., 1999). However, some variability due to individual differences in uptake, metabolism, distribution and elimination occurs (Benowitz et al., 1982; Benowitz, 1996; Benowitz & Jacob, 1997; Benowitz et al., 1999, 2002). The amount of nicotine converted to cotinine in humans varies, and this variability is attributable to dietary, environmental and genetic factors (Benowitz et al., 1996; Benowitz & Jacob, 1997; Benowitz, 1999a; Hecht et al., 1999b; Sellers et al., 2000). Differences in the metabolism of nicotine and cotinine have been reported to occur between various racial and ethnic groups (Wagenknecht et al., 1993; Caraballo et al., 1998; Pérez-Stable et al., 1998; Benowitz et al., 1999; Kwon et al., 2001). These differences have been associated with slower metabolism and lower uptake of nicotine in some populations (Benowitz et al., 1999, 2002), whereas in others uptake may be higher (Caraballo et al., 1998).
Many studies have demonstrated that cotinine levels are clearly higher in smokers than in nonsmokers, leading to increased levels of cotinine being detected in the urine, serum and/or saliva of smokers. Generally, urinary cotinine concentrations are more than one hundred times greater in smokers than those in nonsmokers (e.g. IARC, 1986; National Research Council, 1986; US Department of Health and Human Services, 1986; Jarvis et al., 1987; Etzel, 1990; Tunstall-Pedoe et al., 1991; Environmental Protection Agency, 1993; Pirkle et al., 1996; Jarvis et al., 2001). The large studies have been consistent in showing a positive correlation between cotinine levels and number of cigarettes smoked per day regardless of the body fluid in which cotinine was measured; however, the magnitude of the correlation varies to some extent (IARC, 1986; National Research Council, 1986; US Department of Health and Human Services, 1986; US Environmental Protection Agency, 1993; Law et al., 1997a; Etter et al., 2000).
Carboxyhaemoglobin and thiocyanate
The uptake of tobacco smoke constituents from the gaseous and particulate phases of mainstream smoke, inhaled by smokers, and of secondhand smoke, breathed in by nonsmokers, was investigated by Scherer et al. (1990). Tobacco smoke uptake was quantified by measuring COHb in erythrocytes and measuring nicotine and cotinine in plasma and urine, and the data obtained were correlated with urinary excretion of thioethers (biomarkers of electrophilic mainstream smoke carbonyls such as acrolein) and with mutagenic activity. An increase in all biochemical parameters was observed in smokers who inhaled the whole smoke of 24 cigarettes over a period of 8 h, whereas, after smoking the gas phase of mainstream smoke under the same conditions, only an increase of COHb and, to a minor degree, in urinary thioethers was found.
Serum thiocyanate has often been used for the validation of smoking histories as well as for the assessment of passive exposure to cigarette smoke. There was a significant increase (p < 0.01) in serum thiocyanate with increased smoking. The concentrations of serum thiocyanate in plasma were positively associated with moderate and heavy smoking, but could not distinguish between nonsmokers, light smokers and passive smokers. Fourteen smokers enrolled in this study had serum thiocyanate concentrations higher than 70 µM. The complementary assay of exhaled CO was helpful in confirming nonsmoking status. Using a combination of serum thiocyanate and exhaled CO levels, nonsmoking status was confirmed in 98% of the cases (Robertson et al., 1987).
Genetic factors affecting the metabolism of nicotine to cotinine
Genetic factors are among the major determinants of the variation in the metabolism of nicotine to cotinine. Poor metabolism of nicotine with lack of cotinine formation has been seen and shown to have a genetic basis (Benowitz et al., 1995). Many P450 enzymes are involved in the oxidative metabolism of nicotine; they include P4502A6, P4502B6, P4502D6, P4502E1, P4501A2, P4503A4 and P4501A1 (Yamazaki et al., 1999), but, in contrast to the findings reported from earlier studies, P4502D6 probably plays only a minor role (Benowitz et al., 1996; Nakajima et al., 1996; Yamazaki et al., 1999). According to present data, the most important and rate-limiting gene in 5′-hydroxylation [c-oxydation] of nicotine to cotinine is P4502A6 (Nakajima et al., 1996; Messina et al., 1997; Yamazaki et al., 1999). In addition, there are data indicating that P4502A6 is involved in 2′-hydroxylation of nicotine potentially leading to endogenous formation of NNK. This was reported to occur in human liver microsomes incubated with nicotine at a rate of about 6% of that of cotinine formation (Hecht et al., 2000).
P4502A6 enzyme activity can be lost as a result of a genetic polymorphism that can give rise to homozygosity for the inactive alleles of CYP2A6. In addition, individuals who are carriers of alleles with an inactivating mutation in a heterozygous condition and/or carry defective alleles of decreased enzyme activity are deficient in metabolizing nicotine to cotinine. Subjects with low P4502A6 enzyme activity have significantly lower levels of cotinine, as seen in smokers and in those administered nicotine via another route (Fernandez-Salguero et al., 1995; Oscarson et al., 1998; Kitagawa et al., 1999; Nunoya et al., 1999; Oscarson et al., 1999; Nakajima et al., 2000; Kwon et al., 2001; Nakajima et al., 2001; Yang et al., 2001; Xu et al., 2002). The prevalence of the homozygous genotypes with two inactive alleles varies in different populations. They are very rare among Caucasians and Japanese populations, but have been reported to be somewhat more common in the Chinese population (Yokoi & Kamataki, 1998; Kitagawa et al., 1999; Oscarson et al., 1999). More than 95% of members of Caucasian populations who show the CYP2D6 poor-metabolizer phenotype can be diagnosed by gene analysis, whereas about 20–30% of poor metabolizers can be diagnosed by the known mutant alleles in the Japanese population. Subsequently to this finding, a new mutation, CYP2D6 J9 was found in the Japanese population (Yokoi & Kamataki, 1998). In addition, a gene duplication genotype associated with increased plasma cotinine has recently been reported to occur at a low frequency in Caucasians (Rao et al., 2000). It has been proposed that CYP2A6 polymorphism may be a major determinant of an individual's smoking behaviour and may lead to less desire to smoke (Pianezza et al., 1998; Sellers et al., 2000).
In addition to the individual and racial variation that occurs in the oxidative metabolism of nicotine, variation in the glucuronide conjugation of nicotine and cotinine also exists (Benowitz et al., 1999). Both nicotine and cotinine are metabolized by N-glucuronidation catalysed by uridine-5′-diphosphate-glucuronosyltransferase (UGT) enzymes (see Figure 4.4; Byrd et al., 1992; Benowitz et al., 1994). Although the specific UGTs involved in these metabolic steps are so far not known, it has been observed that N-glucuronidation of nicotine and cotinine is polymorphic, with evidence of slow and fast N-glucuronidation (Benowitz et al., 1999). Environmental factors may influence N-glucuronidation, but with current data indicating that many of the genes encoding UGTs are polymorphic (Burchell et al., 2000; Mackenzie et al., 2000), genetic polymorphism may also be involved. Distribution of slow and fast glucuronidation has been found to vary according to ethnicity; the extent of N-glucuronidation is generally less in blacks than in whites (Benowitz et al., 1999).
4.1.2. Experimental systems
(a) Effects of tobacco smoke on enzyme activities
In-vivo studies
Most investigations of the effects of tobacco smoke on enzyme activities in animals have measured changes in levels of phase I, phase II and antioxidant enzymes in the lung and liver of mice and rats (Table 4.6). Some studies have also measured its effects on enzyme activities in other organs such as the kidney, stomach, heart and brain.
Table 4.6
Effects of tobacco smoke on enzyme activities.
Enzyme effects
The induction of several phase I enzymes, including P4501A1 (aryl hydrocarbon hydroxylase), P4501A2, P4502B, P4502C, P4502D and P4502E1 by tobacco smoke in mouse lung and liver has been reported (Table 4.6). An increase of at least twofold in the activity of most of these enzymes is induced within a few days after initial exposure of the mice to tobacco smoke, and the enzyme levels usually remain elevated throughout exposure. It is probable that the induction of these enzymes can be attributed to the presence of many carcinogens and toxins in tobacco smoke. P4502E1 appears to be induced to a greater extent (5–13-fold) than other phase I enzymes both in the lungs and liver of mice (Villard et al., 1994, 1998a,b). Similarly, P4502E1 is strongly induced by tobacco smoke in mouse kidney (Seree et al., 1996). To investigate the role of nicotine in the induction of CYP2E1, rats were treated once daily with saline or nicotine bitartrate (0.1, 0.3 and 1.0 mg/kg bw, subcutaneously) for 7 days. After nicotine administration, immunostaining for CYP2E1 was increased in the centrilobular region of the liver. Western-blot analyses revealed that hepatic levels of CYP2E1 were increased 1.3–1.7-fold by nicotine. In-vitro analysis of the metabolism of chlorzoxazone (i.e. 6-hydroxylation) showed that Vmax values were higher than those measured in saline-treated rats when hepatic microsomes from nicotine-treated rats were used (2.35 ± 0.04 versus 1.32 ± 0.55 nmol/mg/min, p < 0.005), with no change in affinity. The magnitude of the enhancement of metabolism by microsomes from nicotine-treated animals is consistent with the observed increase in CYP2E1 protein as measured by immunoblot analysis. The data suggest that nicotine may increase CYP2E1-induced toxicity (Howard et al., 2001). Although not detectable in mouse lung, P4503A is induced by tobacco smoke in mouse liver (Villard et al., 1994). Some investigations have reported conflicting results on the extent of phase I enzyme induction by tobacco smoke. For example, in one study, P4501A1 was induced in the liver of NMRI mice exposed to tobacco smoke (Villard et al., 1994) whereas, in another study in the same laboratory, P4501A1 activity was either reduced or not detected in the liver of NMRI mice (Villard et al., 1998b). These opposing results might be explained by the length of the exposure period: P4501A1 was induced in mice exposed to tobacco smoke for 4 or 8 days, and was reduced in mice exposed for 30 days. In one report on the effects of tobacco smoke on phase II enzyme activity in the tissues of mice exposed to tobacco smoke, UGT was induced in the lung and liver, but not in the kidney (Villard et al., 1998a).
The induction of phase I and phase II enzymes in rats has been thoroughly investigated (Table 4.6). An early study demonstrated a three- to fourfold induction of Ahh in lung microsomes of Sprague-Dawley rats exposed daily to either mainstream or side-stream whole smoke for 16 weeks (Gairola, 1987). The magnitude of Ahh induction in animals exposed to mainstream and sidestream smoke was similar, although the dose of smoke particulates received from rats exposed to sidestream smoke was significantly less than the amount inhaled by the corresponding group exposed to mainstream smoke. In another study, Ahh was induced in both the lung and liver of Sprague-Dawley rats given intraperitoneal injections of condensates of mainstream and sidestream smoke. The induction of the enzyme in the lung exceeded that in the liver, and the sidestream-smoke condensate was a more effective inducer than the mainstream-smoke condensate. The activity of lung and liver NDMA demethylase was unaffected by treatment with either condensate (Pasquini et al., 1987). Ethoxyresorufin O-deethylase (associated with P4501A1/2) activity was induced to a similar extent in both the lung and liver of Wistar rats after a single, short exposure to cigarette smoke (Godden et al., 1987). In another study, Sprague-Dawley rats were exposed to mainstream smoke produced by a commercial filter-tipped cigarette for 8 consecutive days, amounting to a cumulative exposure to 75 cigarettes. The most pronounced changes in enzyme expression consisted of a 2.6-fold induction of Ahh in the lung and an eightfold induction of ethoxyresorufin O-deethylase in the liver (Bagnasco et al., 1992). In addition to Ahh and ethoxyresorufin O-deethylase, other phase I enzymes that were induced by tobacco smoke in either the lung or liver of rats included aniline 4-hydroxylase, para-nitroanisole O-demethylase, and aminopyrine Ndemethylase (Eke et al., 1996). With the exception of aniline 4-hydroxylase, the inducibility of these enzymes in the lung and liver was similar in rats ranging in age from 20 days to 360 days. Aniline 4-hydroxylase was induced in the lung and liver of 20-day-old rats, but not in 360-day-old rats (Eke et al., 1997). A more recent study in Fischer 344 rats reported on the inducibility by tobacco smoke of P4501A1, P4501A2 and P4502B1/2 in the nasal mucosa, lung and liver. Rats were exposed to mainstream cigarette smoke or to filtered air for 2 or 8 weeks. The inducibility of the three enzymes varied significantly: P4501A1 levels were increased in all three tissues; P4501A2 was increased slightly in nasal mucosa and liver and decreased in lung; and P4502B1/2 was increased in liver and decreased in nasal mucosa and lung (Wardlaw et al., 1998). These and other data suggest that the regulation of xenobiotic-metabolizing enzymes varies from one rodent tissue to another.
In a study of the effects of tobacco smoke on the activity of phase II enzymes in rat tissues, liver glutathione S-transferase (GST) activity with regard to ethacrynic acid was increased by tobacco smoke whereas that with regard to 1,2-epoxy-3-(para-nitrophenoxy)-propane was decreased (Eke et al., 1996). Hepatic GST activities against 1-chloro-2,4-dinitrobenzene or 1,2-dichloro-4-nitrobenzene were unaltered. In the lung, however, the activity of GST against all substrates was decreased by tobacco smoke. These results indicate that the regulation of hepatic and pulmonary GSTs is differentially influenced by tobacco smoke. Cigarette smoke has the potential to depress severely the detoxification capacity of the lung.
The effects of tobacco smoke on the activities of the antioxidant enzymes catalase, superoxide dismutase, glutathione peroxidase and glutathione reductase in rat tissues have been investigated (Table 4.6). In one study in which Sprague-Dawley rats were exposed to mainstream cigarette smoke for 65 weeks, oxidative stress was increased whereas the activities of catalase and glutathione peroxidase in the lung were not significantly altered by exposure to smoke (Wurzel et al., 1995). In another study, the activities of glutathione reductase and catalase were reported to be decreased in the kidney of rats exposed to tobacco smoke for 3 months, whereas the activity of glutathione peroxidase and lipid peroxide levels were increased. The excretion of glutathione and lipid peroxides in urine was also increased. The authors concluded that the reduced activity of glutathione reductase and the increased activity of glutathione peroxidase may perturb the ratio of reduced glutathione:oxidized glutathione, which in turn could lead to the increased levels of lipid peroxide in the kidney and urine seen in chronic exposure to tobacco smoke (Anand et al., 1996). In a more recent study, the activities of catalase, superoxide dismutase and glutathione peroxidase were increased in the lung, liver and kidney of albino rats exposed to tobacco smoke for 2 × 15 min per day for 30 days. The activity of GST was also increased in the lung, liver and kidney. Interestingly, the activities of the antioxidant enzymes were unchanged in the brain and heart of the animals, but GST was increased in the brain. The authors concluded that tobacco smoke induces lipid peroxidation in the lung, liver and kidney, and the levels of the antioxidant enzymes are enhanced to protect these tissues against the deleterious effects of oxygen-derived free radicals (Baskaran et al., 1999).
Nitric oxide synthase (Nos) catalyses the production of NO which, through its conversion to peroxynitrite, can cause damage to cellular macromolecules. The effects of tobacco smoke on Nos gene expression and protein production have been examined in the lung of rats exposed to tobacco smoke either once only or daily and killed after 1, 2, 7 or 28 days of exposure. Nos1, Nos2 (inducible Nos (iNos)) and Nos3 mRNAs in whole lung were quantified using reverse transcription polymerase chain reaction, and Nos protein levels were determined by Western blotting. Neither Nos1 gene expression nor protein levels were altered by exposure to tobacco smoke. Levels of Nos2 expression were more than doubled in animals exposed to smoke on day 1 and had decreased to control values by 28 days, whereas protein levels did not change. Nos3 expression was increased by approximately 35% after 2 days of exposure and remained at this level to 28 days, whereas protein levels were increased by approximately 60% at day 7 and remained elevated to 28 days. In-situ hybridization showed that Nos2 was diffusely expressed in the lung parenchyma, airways and vessels, and that Nos3 was strongly expressed in vascular endothelium. The authors concluded that tobacco smoke could directly and rapidly affect Nos expression, and thus potentially affect the function of the pulmonary vasculature (Wright et al., 1999). In another study, the expression of Nos2, NF-κB, the mitogen-activated protein kinases Mek1 (mitogen-activated extracellular kinase 1) and Erk2 (extracellular signal-regulated kinase 2), phosphotyrosine protein and c-Fos was increased in the terminal bronchioles of rats exposed to gas-phase tobacco smoke in association with an increase in lipid peroxidation. In contrast, the levels of protein kinase C, Mekk1, Jnk, p38, c-Jun and c-Myc in the terminal bronchioles were unchanged. The authors concluded that exposure to tobacco smoke results in oxidative stress (NOx and ROS) that leads to the induction of Nos2 (iNos) and c-Fos together with the induction of transduction signalling proteins, protein tyrosine phosphorylation and Mek1/Erk2 which, in turn, may promote lung pathogenesis (Chang et al., 2001).
The effects of tobacco smoke on enzyme activities have also been investigated to a limited extent in guinea-pigs. In contrast to the results obtained in mice and rats, daily exposure of Hartley strain guinea-pigs to either mainstream or sidestream tobacco smoke for 16 weeks did not result in an increase in Ahh activity in the lung (Gairola, 1987). In another study, however, tobacco smoke-induced emphysema in the lungs of guinea-pigs was associated with an increase in the activity of a proteolitic enzyme–collagenase activity in the alveolar walls and interstitium (Selman et al., 1996).
(b) Biomarkers of tobacco smoke carcinogens
(i) Urinary compounds
Urinary compounds are useful markers of the uptake of tobacco smoke constituents (Hecht, 2002b). Most, if not all, of the compounds detected in the urine of human smokers have also been found in the urine of animals exposed to mainstream tobacco smoke. Since 1985, however, there have been relatively few reports of the identification and quantitation of urinary compounds in animals exposed to tobacco smoke (Table 4.7). Rats exposed to four dilutions of cigarette smoke over a period of 4 weeks had a lower output of hydroxyproline (relative to creatinine) for all dilutions of smoke and showed a negative dose–response relationship (Read & Thornton, 1985). cis-3′-Hydroxycotinine was detected as a nicotine metabolite in the urine of human smokers as well as in the urine of rats and hamsters dosed with nicotine. This was the first report of the identification of cis-3′-hydroxycotinine as a urinary metabolite of nicotine (Voncken et al., 1990). The compound 4-(methylnitrosamino)-4-(3-pyridyl)-butyric acid (iso-NNAC) was identified in the urine of rats exposed to tobacco smoke and in the urine of human smokers (Pachinger et al., 1993).
Table 4.7
Biomarkers affected by exposure to mainstream tobacco smoke.
(ii) DNA adducts in animal tissues
Mouse
There have been several reports of the detection of DNA adducts in tissues of mice exposed to tobacco smoke (Table 4.7). Synchronous fluorescence spectrophotometry was used to investigate the formation of the BPDE–DNA adducts in the lung and liver of genetically responsive C57BL/6 and non-responsive DBA/2 mice exposed to cigarette smoke for 3–16 days. Interestingly, BPDE–DNA adducts were not detected in the lung and liver of either mouse strain, although Ahh activity, an indicator of benzo[a]pyrene metabolism, was clearly induced in the lungs of C57BL/6 mice. Thus, there appeared to be no clear correlation between Ahh activity and the formation of BPDE–DNA adducts (Bjelogrlic et al., 1989).
The effect of tobacco smoke on the formation of promutagenic O6-methyldesoxyguanosine (O6-MeG) adducts from the TSNA NNK, in the lungs and liver of A/J mice has been investigated. Mice were exposed to smoke generated from Kentucky 1R4F reference cigarettes at 0, 0.4, 0.6 or 0.8 mg wet total particulate matter per litre of air for 2 h, and a single intraperitoneal injection of NNK (0, 3.75 or 7.5 µmol/mouse) was administered midway through the exposure. Tobacco smoke alone did not yield detectable levels of O6-MeG but NNK did form this adduct. The number of O6-MeG adducts following intraperitoneal injection of NNK during cigarette smoke exposure was significantly (p < 0.05) reduced in both lung and liver. The same effect was seen in mice co-exposed to NNK and cotinine. The authors hypothesized that the reduction of O6-MeG in liver and lung results from competitive inhibition by cotinine of the cytochrome P450 enzyme system for NNK activation (Brown et al., 1999). The effect of exposure to tobacco smoke on the formation of DNA adducts in the lungs and heart of B6C3F1 mice was determined using the 32P-postlabelling assay. Mice were exposed for 1 h per day, 5 days per week for a period of 4 weeks to mainstream smoke at concentrations of 0, 0.16, 0.32 and 0.64 mg total particulate matter per litre of air. There was an exposure-dependent increase in the numbers of DNA adducts in lung and heart at all three concentrations; increases found in the mid- and high-exposure groups were significant (p < 0.05) (Brown et al., 1997).
Mainstream-smoke condensate and sidestream-smoke condensate were applied topically to mouse skin, and DNA adducts were quantified in skin, lung, kidney, liver and bladder tissues by 32P-postlabelling. Mainstream-smoke condensate produced higher levels of DNA adducts in mouse heart and bladder than sidestream-smoke condensate (but the difference was not statistically significant). However, sidestream-smoke condensate produced higher adduct levels in mouse skin, lung and kidney than mainstream smoke (Carmichael et al., 1993).
Rat
The effect of mainstream tobacco smoke on the formation of DNA adducts in various tissues of rats has been investigated extensively by use of 32P-postlabelling and SFS (Table 4.7). The formation of DNA adducts in the nasal cavity, lung and liver of Sprague-Dawley rats exposed daily to fresh smoke from a University of Kentucky reference cigarette (2R1) for up to 40 weeks was examined by 32P-postlabelling (Gupta et al., 1989). The amount of total particulate matter inhaled with the smoke was 5–5.5 mg per animal per day. The average concentration of COHb in blood was 5.5%. Mainstream smoke induced at least four new DNA adducts in the nasal mucosa of rats and the amount of these adducts increased with length of exposure. In the lung, smoke induced an accumulation of one DNA adduct, which upon cessation of exposure for 19 weeks was reduced by about 75%. Smoke-related adducts were not detected in the liver. Selective chromatography and butanol extractability suggested that the DNA adducts in all tissues examined were aromatic and/or lipophilic. In a more recent study, Gupta et al. (1999) reported increases in the levels of DNA adducts in various tissues of Sprague-Dawley rats exposed to both mainstream and sidestream tobacco smoke, but not in liver. Mean total adduct levels in the various tissues ranked from heart > lung > trachea > larynx > bladder. Izzotti et al. (1995b) employed 32P-postlabelling analysis to evaluate the effect of N-acetyl-l-cysteine (NAC) on the formation of carcinogen–DNA adducts in tracheal epithelial cells of Sprague-Dawley rats exposed (whole-body) to mainstream tobacco smoke for either 40 or 100 consecutive days. DNA adducts were observed after 40 days of exposure and no further increase was noted after 100 days. NAC, given by gavage in the 40-day study and in the drinking-water in the 100-day study, reduced the formation of smoke-related carcinogen–DNA adducts in the tracheal epithelium to the levels seen in sham-exposed control rats. These results indicate the considerable efficacy of oral NAC, a chemopreventive agent, in inhibiting the formation of smoke-related carcinogen–DNA adducts (Izzotti et al., 1995b).
32P-Postlabelling was also used to investigate the effects of exposure mode, sex and time (adduct persistence) on the level of DNA adducts in tissues of Fischer 344 rats (Bond et al., 1989). Rats were exposed to tobacco smoke for 6 h per day, 5 days per week for 22 days by intermittent nose-only, continuous nose-only or continuous whole-body exposure. The animals were killed at either 18 h or 3 weeks after the 22-day exposure period and DNA adducts in lung tissues were quantified. Significant (p < 0.05) increases in the levels of DNA adducts were observed in both male and female rats exposed to tobacco smoke. No significant effects of exposure mode or sex on the induction of lung DNA adducts were observed. Significantly fewer clearly resolved DNA adducts were found in the lungs of rats killed 3 weeks after exposure, suggesting that smoke-induced adducts were repaired by DNA repair mechanisms. A single unidentified adduct accounted for about 20% of the total resolved lung DNA adducts and occurred at levels nine- to 14-fold higher than those in control animals (Bond et al., 1989). It is not certain whether this adduct is the same one as that described in the lung of Sprague-Dawley rats by Gupta et al. (1989) or in the skin of mice by Randerath et al. (1986). Izzotti et al. (1998) used 32P-postlabelling to investigate the effects of tobacco smoke and alcohol consumption on DNA adduct levels in the oesophagus, lung, liver and heart of BD6 rats. Groups of female rats were exposed to ethanol (5% in drinking-water for 8 consecutive months) and/or whole-body to mainstream tobacco smoke (1 h per day, 5 days per week for 8 months). As expected, ingestion of alcohol alone did not affect the levels of DNA adducts in any of the four organs studied. Exposure to tobacco smoke induced formation of DNA adducts in the lung and heart, but not in the oesophagus or liver. Combined exposure to alcohol and smoke, however, resulted in the significant formation of smoke-related DNA adducts in the oesophagus and in a further increase in the number of adducts in the heart. Therefore, a likely interpretation is that ethanol may solubilize water-insoluble smoke components in the upper aerodigestive tract, thereby determining a first-pass effect in the oesophagus (Izzotti et al., 1998). Another important mechanism may be the induction by ethanol of CYP2E1-dependent microsomal monooxygenases that catalyse the metabolism of a variety of xenobiotics (Tsutsumi et al., 1993; Gonzalez & Gelboin, 1994).
The formation of smoke-related DNA adducts and their chemoprevention were investigated in tissues of Sprague-Dawley rats by SFS which, as mentioned above, can detect BPDE–DNA adducts (Izzotti et al., 1992). Animals were exposed (whole-body) to mainstream cigarette smoke once daily for up to 40 consecutive days. No adduct was detected in liver DNA, whereas smoke-related DNA adducts were detectable in the lung from the day 8 of exposure and continued to increase until the study ended at day 40. The levels of adducts in heart DNA were even higher than those found in the lung. The daily administration of NAC by gavage significantly inhibited the occurrence of the same adducts in both heart and lung DNA, as measured by 32P-postlabelling of DNA adducts in tracheal epithelial cells (Izzotti et al., 1995b).
It is currently not clear why rats exposed to cigarette smoke form lung DNA adducts when previous studies have not demonstrated smoke-induced pulmonary carcinogenesis in rats. However, it is possible that the previous studies failed to demonstrate smoke-induced cancer because:
- -
a smaller dose of particulate matter was delivered per gram of lung tissue than in human heavy smokers;
- -
less than lifetime exposures were used; and
- -
the experimental groups used were too small in number (Bond et al., 1989).
Interspecies comparison
32P-Postlabelling has also been used to detect and quantify DNA adducts in the lung of guinea-pigs and mice exposed to mainstream and sidestream tobacco smoke. DNA adducts were identified in lung, but less DNA-adduct formation was seen in guinea-pigs than in either mice or rats at the same level of exposure to tobacco smoke (Gupta et al., 1999).
(iii) DNA damage in cultured human lung cells
In cultured human lung cells, bubbling cigarette smoke through phosphate-buffered saline was found to induce DNA single-strand breaks and formation of 8-OHdG in DNA. Evidence was presented that this was mediated, in part, by the formation of reactive oxygen species (Leanderson, 1993). It was shown that cigarette tar promotes neutrophilinduced DNA damage in cultured human lung cells and that this activity is further enhanced by iron and inhibited by catalase (Leanderson & Tagesson, 1994).
Treatment of human fetal lung cells with the TSNAs NNN and NNK caused single-strand breaks in DNA. Inhibition of this effect by oxygen radical scavengers suggested that the hydroxyl radical was an important intermediate in the process (Weitberg & Corvese, 1993).
DNA damage, as measured by the alkaline single-cell gel microelectrophoresis (Comet) assay, has also been shown to be induced in human embryo lung cells treated with water-soluble compounds from cigarette smoke (Wang, Q. et al., 2000).
Treatment of tracheobronchial epithelial cells with gas-phase cigarette smoke caused DNA strand breakage that was accompanied by increases in the levels of a number of DNA lesions, including 8-OHdG, xanthine and hypoxanthine. These latter lesions can arise from the deamination of guanine and adenine by a mechanism involving reactive nitrogen species. Thus, DNA damage induced by cigarette smoking may be mediated by both reactive oxygen species and reactive nitrogen species (Spencer et al., 1995).
(c) Other data
(i) Effects on particle clearance
Cigarette smoking induces a variety of carcinogenic and non-carcinogenic effects in humans and laboratory animals. An issue of concern is the extent to which smoking might influence pulmonary responses to other inhaled toxic materials. This influence can take the form of a direct alteration of the deposition or clearance of another inhaled agent. For example, it has been reported that cigarette smoking delays the pulmonary clearance of inhaled, insoluble particles in humans (Bohning et al., 1982) and in laboratory animals (Mauderly et al., 1989).
Finch et al. (1995) investigated the influence of cigarette smoke exposure of Fischer 344 rats on the pulmonary clearance of inhaled, relatively insoluble radioactive tracer particles. Following 13 weeks of whole-body exposure to air or mainstream tobacco smoke for 6 h per day, 5 days per week at concentrations of 0, 100 or 250 mg/m3 total particulate matter, rats were acutely exposed pernasally to 85Sr-labelled fused aluminosilicate (85Sr-FAP) tracer particles; exposure to air or smoke was then resumed. A decreased clearance of 85Sr-FAP from the lungs, which was smoke concentration-dependent, was observed. By 180 days after exposure to the tracer aerosol, about 14, 20 and 40% of the initial activity of the tracer was detected in the control, 100-mg/m3 and 250-mg/m3 groups, respectively. Exposure to mainstream smoke produced lung lesions that contained increased numbers of pigmented alveolar macrophages throughout the parenchyma, and focal collections of enlarged alveolar macrophages with concomitant alveolar hyperplasia. The severity of lesions increased with duration of exposure. These data confirm previous findings that exposure to cigarette smoke decreases the ability of the lung to clear inhaled materials. In a subsequent publication, Finch et al. (1998) reported that chronic exposure to cigarette smoke containing 100 or 250 mg/m3 total particulate matter increased the pulmonary retention and radiation dose of 239plutonium inhaled as 239PuO2 in two groups of Fischer 344 rats. Assuming a linear dose–response relationship between radiation dose and the incidence of lung neoplasms, the exposure to 239PuO2 was predicted to increase the incidence of lung tumours relative to that in controls by 20% or 80%, depending upon the concentration of total particulate matter in smoke.
(ii) Carboxyhaemoglobin
There have been several reports on the relationship between levels of exposure to carbon monoxide from tobacco smoke and levels of COHb adducts in the blood of animals. For example, Attolini et al. (1996) exposed male NMRI mice to tobacco smoke for 2, 4, 8 or 31 days. The levels of COHb in blood increased significantly after 4 or 8 days of exposure and decreased after 31 days to a level which remained higher than the level of the controls. Two hypotheses were proposed to explain the decrease in COHb levels at 31 days.
(1) The mice may develop a state of tolerance against compounds in tobacco smoke: the increase in respiratory rhythm leads to hyperventilation and to a concomitant decrease in the quantity of CO fixed to haemoglobin.
(2) Chronic exposure to tobacco smoke increases the thickness of the airway epithelium and alveolar septae, as well as mucus hypersecretion and ciliostasis, which increases the barrier to smoke constituents, reducing their accessibility to the blood circulation and therefore decreasing the level of CO fixed to haemoglobin.
Loennechen et al. (1999) exposed Sprague-Dawley rats to CO at 100 ppm for 1 week or to 100 ppm CO for 1 week followed by 200 ppm for 1 week. The formation of COHb was found to be dependent upon the amount of CO exposure; COHb was approximately 13% in the group exposed to the low level of CO and 23% in the group exposed to the higher level. Exposure to a high level of CO increased the expression of endothelin-1 mRNA by more than 50% in both the left and right ventricles of the heart. The authors concluded that chronic exposure to CO leading to COHb levels similar to those observed in smokers increases endothelin-1 gene expression and induces myocardial hypertrophy in the rat.
4.2. Toxic effects
4.2.1. Humans
(a) Nicotine addiction
Cigarette smoking is the single largest avoidable cause of premature death and disability. Many smokers express a desire to stop smoking, and many have made one or more unsuccessful attempts to quit, supporting the evidence that tobacco smoking is addictive. Research has been focused on nicotine because it is the most addictive constituent of tobacco products (see also Section 4.1.1(c)(vi)). Therefore, cigarette smoking should be understood as a manifestation of nicotine addiction. This topic has been extensively reviewed (Benowitz, 1988; US Department of Health and Human Services, 1988; Moxham, 2000).
Nicotine is an addictive drug and smoking of tobacco rapidly delivers a dose of nicotine to receptors in the brain. The effects on the central nervous system are more important than those on the peripheral nervous system (Le Houezec & Benowitz, 1991). With repeated experience, consolidation into physiological and psychological addiction is reinforced by pronounced withdrawal symptoms.
To achieve a psychoactive impact, nicotine must be delivered rapidly to the brain which is best achieved by inhalation of tobacco smoke. The speed of nicotine delivery is a fundamental difference between cigarettes and products aimed at nicotine replacement. The nicotine-replacement products deliver nicotine at lower, subaddictive rates and are only effective in reducing cravings and withdrawal symptoms from tobacco-delivered nicotine dependence.
It is far from clear that the benefits attributed to nicotine use, such as stress relief, improved mood and enhanced cognitive performance, are real. Many of the perceived benefits are actually attributable to the relief of nicotine withdrawal symptoms (Le Houezec & Benowitz, 1991).
The addictive properties of nicotine imply that analytical measurements of tar and nicotine yields from cigarettes do not reflect the true exposure to tar and nicotine experienced by smokers. Smokers adjust the way they smoke in order to self-administer a satisfactory dose of nicotine (Benowitz, 1995).
(b) Health effects other than cancer
Besides its carcinogenic effects, tobacco smoke has a number of other pathogenic properties. Causal associations have been established between active smoking and a number of specific diseases.
(i) Effects on the cardiovascular system
Cigarette smoking is a major independent risk factor for coronary heart disease and the most important risk factor for atherosclerotic peripheral vascular disease. There is a dose–response relationship between cigarette smoking and cardiovascular disease: the risk increases with the number of cigarettes smoked daily, the total number of years for which a person has smoked, the degree of inhalation and earlier age of initiation of the smoking habit. Cigarette smoking has been found to elevate significantly the risk for sudden death (US Department of Health and Human Services, 1983, 1989). Cigarette smoking accounts for about half of deaths from coronary disease in women during middle age. It has been shown that premenopausal women have lower rates of heart disease than postmenopausal women. This protection is presumed to be provided by the presence of circulating estrogens, but it is unknown whether estrogens have the same protective effect in women who smoke (Villablanca et al., 2000). A synergistic effect between smoking and the use of oral contraceptives has also been reported (US Department of Health and Human Services, 1983). In female smokers who take oral contraceptives, particularly those over the age of 35 years, there is a well-established increase in risk of myocardial infarction and cerebrovascular disease (Villablanca et al., 2000).
Cigarettes that nominally deliver less tar or nicotine have not consistently been shown to reduce the risk of cardiovascular disease (reviewed by Burns et al., 2001).
(ii) Effects on the cerebrovascular system
Cigarette smoking is a major cause of cerebrovascular disease (ischaemic stroke). It is estimated that as many as 25% of all strokes can be attributed to smoking. The relative risk of stroke is similar in male and female smokers and is maximal near middle age (Hankey, 1999). Although nicotine has strong and potentially harmful effects on cerebral and peripheral vascular tissues, it is not certain whether and how these effects are related to stroke (Hawkins et al., 2002).
(iii) Effects on the respiratory system
Cigarette smoking is the most important cause of cough, sputum production, chronic bronchitis and asthma (Hargreave & Leigh, 1999; Maestrelli et al., 2001; Ulrik & Lange, 2001). It increases the risk for dying from chronic bronchitis and pulmonary emphysema (US Department of Health and Human Services, 1984; Aubry et al., 2000; Seagrave, 2000; Fraig et al., 2002).
(iv) Gastrointestinal effects
Smoking increases the risk for peptic ulcer and mortality from this disease (Ma et al., 1998). It delays peptic ulcer healing and increases the risk of recurrence after healing (Ashley, 1997). Nicotine has been shown to potentiate aggressive gastric factors and to attenuate defensive ones; it also increases acid and pepsin secretions, gastric motility, duodenogastric reflux of bile salts, the risk of Helicobacter pylori infection, levels of free radicals and platelet-activating factor, endothelium generation and vasopressin secretion (Endoh & Leung, 1994). Although the mechanisms by which smoking or nicotine adversely affect the gastric mucosa have not been fully elucidated, the available evidence supports the hypothesis that nicotine is harmful to the gastric mucosa (Endoh & Leung, 1994). Smoking is also a risk factor for Crohn disease in both men and women, but the excess risk is higher among female smokers. There is growing evidence that smoking is inversely related to ulcerative colitis (Westman et al., 1995; Rubin & Hanauer, 2000) and some evidence that it is a risk factor for gallstones (Ashley, 1997).
(v) Neurological disorders
The relationship between smoking and some neurological diseases has been controversial. A number of epidemiological studies have found a significant, negative association between cigarette smoking and Parkinson or Alzheimer disease, the risk among nonsmokers being approximately twice that of smokers (Fratiglioni & Wang, 2000). However, whereas a community-based longitudinal study of elderly people found a higher risk of Alzheimer disease in smokers than in nonsmokers (Merchant et al., 1999), an analysis based on a comparison of persons with Alzheimer dementia with their unaffected siblings suggests that smoking does not decrease the risk for the disease (Debanne et al., 2000). Both retrospective and prospective epidemiological studies have demonstrated an inverse association between cigarette smoking and Parkinson disease, leading to theories that smoking and nicotine may be neuroprotective. Coffee and caffeine consumption have been reported to have a similar effect (Ross & Petrovitch, 2001; Hernan et al., 2002).
(vi) Other inverse associations of smoking with health effects
Some studies suggest that there may be inverse associations of smoking with uterine fibroids and endometriosis, and protective effects against hypertensive disorders and vomiting during pregnancy are likely. Inverse associations of smoking with venous thrombosis after myocardial infarction are probably not causal, but indications of positive effects with regard to recurrent aphthous ulcers and control of body weight may reflect a genuine benefit. A variety of mechanisms for the potentially beneficial effects of smoking have been proposed; of these three predominate:
- the anti-estrogenic effect of smoking;
- alterations in prostaglandin production; and
- stimulation of nicotinic cholinergic receptors in the central nervous system.
It should be noted that even established inverse associations cannot be used as a rationale to encourage cigarette smoking because overall effects on health and mortality are clearly negative (Baron, 1996).
4.2.2. Animals
(a) Nicotine addiction/dependence
(i) Studies with animal models
Various animal models have been described that mimic nicotine dependence and withdrawal syndromes. In one such model, dependence is induced in rats by continuous subcutaneous infusion of nicotine (3 or 9 mg/kg bw per day as nicotine hydrogen tartrate) over 7 days by the use of implanted osmotic minipumps. The nicotine is absorbed quickly and almost completely. Abstinence is initiated through termination of infusion or by injection of nicotine antagonists. The resulting abstinence syndrome involves a pattern of behaviour somewhat resembling opiate abstinence, with weight gain and reduced locomotor activity. The model has been replicated in a number of laboratories. It is sensitive to various abstinence-alleviating therapeutic approaches, such as nicotine replacement and the administration of nitric oxide synthase inhibitors and serotonergic compounds. A strong reduction of abstinence symptoms was seen with bupropion and acetyl-l-carnitine, both of which are used clinically as part of smoking cessation regimens (Malin, 2001).
Nicotine has also been given orally to experimental animals either in liquid diets or in drinking fluids, or by forced oral administration (rats show an aversion to the taste of nicotine). When nicotine is given orally, it is absorbed slowly by the gastrointestinal tract and the concentrations in blood remain considerably lower than those observed after subcutaneous infusion (Le Houezec et al., 1989).
In another experimental model, mice were given drinking-water containing gradually increasing concentrations (50–500 µg/mL) of nicotine for 7 weeks. After replacement of the nicotine solutions with tap-water, a significantly higher fluid intake was seen in the nicotine-treated mice than in control animals, but this effect disappeared within 1 week. Plasma nicotine concentrations in the treated mice were found to be similar to those reported in heavy smokers. The results of a pharmacological study with this mouse model suggested that the effects of nicotine in striatal dopamine metabolism are critical for its stimulating and reinforcing effects (Pietilä & Ahtee, 2000).
(ii) Studies with genetically modified mice
Numerous studies have shown that nicotine is likely to be responsible for the addictive properties of tobacco. In addition, nicotine has effects on locomotion, cognition, affect and sensitivity to pain. In recent studies with transgenic mice, molecular biology has been combined with pharmacology, electrophysiology and behavioural analysis to elucidate the specific role of nicotine in these phenomena. The physiological effects of nicotine are mediated by binding to and activation of nicotine acetylcholine receptors. These receptors are pentamers made up of subunits with distinct expression patterns in different neurons. More than 10 different neuronal receptor subunits have been identified, and for seven of these subunits, knock-out mice lacking one receptor subunit have been constructed. These mice are being used in studies to identify the receptor subtypes responsible for the different effects of nicotine. As an example, nicotine self-administration is abolished in mice lacking the β2 subunit of the receptor, which implies that this subunit is a component of the receptor that mediates nicotine reinforcement (Marubio & Changeux, 2000; Picciotto et al., 2000).
(b) Other effects in experimental animals
(i) Effects on the cardiovascular system
To determine whether chronic exposure to tobacco smoke for less than 2 months alters cardiovascular regulation, male Sprague-Dawley rats were exposed to tobacco smoke from low-nicotine cigarettes (1 mg/cigarette) for 4–6 weeks, and a second group served as sham controls receiving only puffs of room air. Reflex adjustments in mean arterial blood pressure after bilateral common carotid occlusion were compared between the two groups. In the anaesthetized control state, there was no significant difference between the cardiovascular parameters measured in the two groups. However, the increase in mean arterial blood pressure after carotid occlusion was significantly greater in the smoke-treated than in the control animals (p < 0.05). In addition, the time required to reach maximum arterial blood pressure after carotid occlusion was significantly less (p < 0.05) for the smoke-treated animals (8.5 ± 0.2 s) than in the controls (11.2 ± 0.3 s). The results show that chronic exposure to tobacco smoke in experimental animals for periods as short as 4–6 weeks alters the reflex regulation of the cardiovascular system (Bennett & Richardson, 1990).
To determine whether exposure to sidestream cigarette smoke promotes atherogenesis in a mouse model of human atherosclerosis, female ApoE-deficient mice, fed a western diet, were exposed to sidestream smoke in a whole-body exposure chamber for a total of 6 h/day, 5 days per week, for 7, 10 and 14 weeks. Animals exposed to filtered ambient air served as controls. Elevated concentrations of blood COHb and pulmonary CYP1A1 were indicative of effective exposure. There were no consistent changes in serum concentrations of cholesterol between control and exposed mice. Morphometric assessment of grossly discernible lesions covering the intimal area of the aorta showed remarkable increases in exposed mice, at all three durations of exposure studied. Increases in the area of the lesion were accompanied by higher levels of esterified and unesterified cholesterol in the aortic tissues of exposed mice. The results clearly demonstrate promotion of the development of atherosclerotic lesions by tobacco smoke in an atherosclerosis-susceptible mouse model (Gairola et al., 2001).
(ii) Effects on the cerebrovascular system
Initial investigations with a rat model of nicotine exposure in adolescents have demonstrated that the vulnerable developmental period for nicotine-induced brain cell damage extends into adolescence. The effect of nicotine on cholinergic systems in adolescent male and female rats was investigated with a nicotine-infusion protocol designed to produce nicotine plasma concentrations similar to those measured in human smokers or in users of transdermal nicotine patches. Choline acetyltransferase activity (ChAT), a static marker that closely reflects the density of cholinergic innervation, and binding of [3H]hemicholinium-3 (HC-3), which labels the presynaptic high-affinity choline transporter, were monitored in the midbrain (the region most closely involved in reward and addiction pathways), as well as in the cerebral cortex and hippocampus. During nicotine treatment and for 1 month after termination of treatment, ChAT activity was significantly reduced and HC-3 binding was significantly increased in the midbrain, but not in the other regions. The levels returned to normal immediately after cessation of nicotine exposure and subsequently showed a transient suppression of activity. Although the cerebral cortex showed little or no change in HC-3 binding during or after nicotine administration, activity was persistently reduced in the hippocampus. The regionally selective effects of nicotine treatment of adolescent rats on cholinergic systems support the concept that adolescence is a vulnerable developmental period for determining ultimate effects on behaviour (Trauth et al., 2000).
(iii) Effects on the respiratory system
To study the role of transforming growth factor-β1 (TGF-β1) in the pathogenesis of chronic bronchitis and emphysema, an animal model was used in which hamsters were exposed by chronic inhalation of cigarette smoke. The expression of TGF-β1 mRNA and protein in the pulmonary tissue was measured. In a parallel experiment, bronchial epithelium was stimulated with cigarette smoke extract in vitro and the expression of TGF-β1 was determined. After 3 months of exposure, the animals developed chronic bronchitis and emphysema. The increase in TGF-β1 immunoreactivity in the pulmonary tissue and in the cultured bronchial epithelial cells was significantly higher than in the controls (p = 0.001). The expression of TGF-β1 mRNA was also increased in the pulmonary tissue of exposed animals. The results indicate that exposure to cigarette smoke can induce over-expression of TGF-β1 in bronchial epithelia; this may be one of the mechanisms for smoking-induced chronic bronchitis and emphysema (Li et al., 2002).
To assess induction of emphysema in the rodent lung, B6C3F1 mice and Fischer 344 rats were exposed, whole-body, to cigarette smoke at a concentration of 250 mg/m3 total particulate matter for 6 h per day, 5 days per week, for either 7 or 13 months. Morphometry included measurements of parenchymal air-space enlargement and tissue loss. In addition, centriacinar intra-alveolar inflammatory cells were counted to assess species differences in the type of inflammatory response associated with the exposure. In mice, significant differences in many of the morphometric parameters indicating emphysema were noted between smoke-exposed and control animals. In rats exposed to cigarette smoke, only some of the parameters differed significantly from control values. Morphological evidence of tissue destruction in the mice included alveoli that were irregular in size and shape and alveoli with multiple foci of septal discontinuities and isolated septal fragments. There were more morphometric anomalies in the mice at 13 months than at 7 months, suggesting a progression of the disease. Inflammatory lesions in the lungs of mice contained significantly more neutrophils than these lesions in rats. These results suggest that B6C3F1 mice are more susceptible than Fischer 344 rats to the induction of emphysema by this exposure regimen and that the emphysema may be progressive in mice. Furthermore, the type of inflammatory response may be a determining factor for species differences in susceptibility to the induction of emphysema by exposure to cigarette smoke (March et al., 1999).
The hypothesis was tested that variations in α1-antitrypsin expression modulate the pattern of emphysema and functional consequences in mice exposed to cigarette smoke. The effects of cigarette smoke were investigated in C57BL/6J (C57) mice and in low-α1-antitrypsin, C57BL/6J pa+/pa+ (pallid) mice. After 4 months of exposure, a significant increase in the extent of emphysema was seen in pallid mice, but not in C57 mice. After 6 months, mechanical properties of lung, the extent and type of emphysema, and the cellular inflammatory response were measured. C57 mice and pallid mice had similar degrees of emphysema, whereas pallid mice, but not C57 mice, had developed a T-cell inflammation in the alveolar wall (p < 0.01). Although lung compliance was not changed in C57 mice after exposure to smoke, it increased significantly in pallid mice over the 6 months of exposure (p < 0.0082). In summary, exposure to cigarette smoke induced emphysema in C57 and pallid mice, but the emphysema, inflammatory infiltrate and the resulting physiological abnormalities were substantially different in the two strains, with the C57 and pallid mice exhibiting features similar to centrilobular and panlobular emphysema, respectively (Takubo et al., 2002).
To determine whether smoking affects the clearance of asbestos fibres, guinea-pigs were given amosite asbestos by intratracheal instillation. They were divided into groups that received (a) no further treatment, (b) were exposed to tobacco smoke after asbestos instillation, or (c) were exposed to smoke both before and after asbestos instillation. The numbers and sizes of the asbestos fibres were measured in respiratory tract tissue and in lavage samples at 1 week and 1 month after exposure. During this time, the asbestos burden in the first group decreased sixfold on average, whereas no significant decrease was seen in either of the smoke-exposed groups. The mean length of retained fibres increased in the first group (asbestos only), but decreased in both the smoke-exposed groups. This phenomenon was seen in tissue samples and lavage samples, although the fibres in the lavage fluid were consistently shorter than those in tissue. The authors concluded that, in this model, cigarette smoking impeded asbestos clearance, largely by increasing retention of short fibres. This increased pulmonary fibre burden may be important in the increased rate of parenchymal fibrosis and carcinoma of the lung seen in asbestos workers who smoke (McFadden et al., 1986).
(iv) Neurological disorders
In view of the suggested inverse relationship in humans between cigarette smoking and the risk for Parkinson and Alzheimer disease, which are both characterized by enhanced oxidative stress, the antioxidant potential of nicotine was investigated in rats. Initial chromatographic studies suggested that nicotine can affect the formation of the neurotoxin 6-hydroxydopamine resulting from the addition of dopamine to Fenton's reagent (i.e. Fe2+ and hydrogen peroxide). Under certain circumstances, nicotine can strongly affect the course of the Fenton reaction. In in-vivo studies, adult male rats treated with nicotine showed greater memory retention than controls in a water-maze task. However, neurochemical analysis of neocortex, hippocampus and neostriatum from these animals revealed that nicotine had no effect on the formation of reactive oxygen species or on lipid peroxidation in any brain region studied. In an in-vitro study with rat neocortical homogenates, there were no differences in lipid peroxidation between nicotine-treated rats and controls. The results of these studies suggest that the beneficial/protective effects of nicotine in both Parkinson disease and Alzheimer disease may result, at least partly, from antioxidant mechanisms (Linert et al., 1999).
The effects of nicotine on the central nervous system are mediated by the activation of neuronal heteromeric acetylcholine-gated ion channel receptors (also termed nicotinic acetylcholine receptors). The neuroprotective effects of nicotine were studied in two animal models of parkinsonism: diethyldithiocarbamate-induced enhancement of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice and methamphetamine-induced neurotoxicity in rats and mice. The neuroprotective effect of nicotine was very similar to that of the non-competitive N-methyl-d-aspartate receptor antagonist, MK-801. In parallel experiments, nicotine was shown to induce the basic fibroblast growth factor-2 (FGF-2) and the brain-derived neurotrophic factor in rat striatum. The effect on the induction of FGF-2 was prevented by the nicotinic acetylcholine receptor antagonist, mecamylamine, whereas MK-801 induced FGF-2 in the striatum. As trophic factors have been reported to be neuroprotective for dopaminergic cells, these data suggest that the increase in neurotrophic factors is a possible mechanism by which nicotine provides protection from experimental parkinsonism (Maggio et al., 1998).
(v) Effects on the immune system
Five groups of 2-month-old male Syrian golden hamsters were exposed to cigarette smoke for three 10-min periods per day, on 5 days per week, for the duration of their lives. Three of the groups were also chronically exposed to aerosols of chrysotile asbestos, cobalt oxide or nickel oxides. The fourth group was exposed to smoke and sham dust and was compared with the control group exposed to sham smoke and sham dust. The fifth group received 12 weekly injections of 0.25 mg of N-nitrosodiethylamine. Each cigarette smoke-exposed group was compared with the group exposed to sham smoke and the respective aerosol treatment. The cigarette smoke-exposed groups lived significantly (p < 0.01) longer than the sham-exposed groups and the untreated controls. Their mean body weights were significantly (p < 0.01) lower than those of the sham-exposed groups. The delayed onset of amyloidosis and the lower body weight in the smoke-exposed hamsters may have been responsible for their increased lifespans. The results suggest that cigarette smoke affects the immune system of the animals, resulting in retardation of amyloidosis, a frequent cause of death in hamsters (Wehner et al., 1976).
In rats, chronic inhalation of cigarette smoke preferentially inhibited the plaque-forming cell response of lung-associated lymph nodes (LALN) to sheep erythrocytes rather than anatomically distant lymph nodes. Inhibition of the antibody response in LALN of smoke-exposed animals was first detected after 21 weeks of smoke inhalation and was well established by the 27th week of exposure to smoke. After prolonged exposure (> 34 weeks) to cigarette smoke, similar changes in the plaque-forming cell response were also observed in other lymphoid tissues. Cigarette smoke affected the response of LALN cells to sheep erythrocytes, a T cell-dependent antigen, but did not alter the relative percentages of W3/13-positive (T cells) or Ig-positive (B cells) cells, or those of T-cell subsets as scored by their surface phenotypes, i.e. T helper (W3/25+) or T suppressor/cytotoxic (OX-8+) cells. The percentage of phagocytic cells and the accessory cell functions of macrophages remained comparable between sham-exposed and smoke-exposed animals. Exposure to cigarette smoke did not significantly alter the response of LALN cells to T-cell mitogens (concanavalin A and phytohaemagglutinin). However, response to trinitrophenyl Brucella abortus, a T-cell-independent antigen, was also significantly reduced. The results show that exposure to cigarette smoke results in a decreased antibody response in the rat, primarily affecting the B-cell function (Sopori et al., 1989).
Chronic exposure of mice and rats to cigarette smoke affects T-cell responsiveness which may account for the decreased T-cell proliferative and T-dependent antibody responses in humans and animals exposed to cigarette smoke. However, the mechanism by which cigarette smoke affects the T-cell function is not clearly understood. Chronic exposure of rats to nicotine has been shown to inhibit the antibody-forming cell response, to impair the antigen-mediated signalling in T cells and to induce T-cell anergy. To study cigarette smoke-induced immunosuppression and to compare it with the effects of chronic nicotine exposure, rats were exposed to diluted, mainstream cigarette smoke for up to 30 months or to nicotine (1 mg/kg bw per day) by osmotic minipumps for 4 weeks, and were evaluated for immunological function in vivo and in vitro. The T cells from rats subjected to long-term exposure to cigarette smoke showed decreased antigen-mediated proliferation and constitutive activation of protein tyrosine kinase and phospholipase C-γ1 activities. Moreover, spleen cells from smoke-exposed and nicotine-treated animals had depleted inositol-1,4,5-trisphosphate-sensitive Ca2+ stores and a decreased ability to raise intracellular Ca2+ concentrations in response to T-cell antigen receptor ligation. The results suggest that chronic smoking causes T-cell anergy by impairing the antigen receptor-mediated signal transduction pathways and depleting the inositol-1,4,5-trisphosphate-sensitive Ca2+ stores. Moreover, nicotine may be responsible for or contribute to the immunosuppressive properties of cigarette smoke (Kalra et al., 2000).
(vi) Toxic effects of kreteks
Kreteks are a type of small cigarette containing approximately 60% tobacco and 40% ground clove buds (Stratton et al., 2001). The typical chemical composition of mainstream smoke per kretek is total particulate matter, 52.3 mg; nicotine, 2.4 mg; and CO, 23.7 mg. In the several inhalation studies that have looked specifically at exposure of rats and hamsters to kreteks smoke, few signs of toxicity (focal alveolitis, bronchiolar epithelial hyperplasia, alveolar haemorrhage) were observed after 1 or 14 days' exposure (LaVoie et al., 1986; Clark, 1989, 1990), but the routes and methods of exposure used do not replicate human exposure (Guidotti, 1989).
4.3. Reproductive and hormonal effects
4.3.1. Humans
(a) Reproductive effects
Cigarette smoking has clearly been associated with a wide range of adverse effects on reproduction, some of which may have implications for cancer risk.
(i) Effects on female fertility
Women who smoke cigarettes have an increased risk for both primary and secondary infertility, with an odds ratio of 1.6 (95% CI, 1.3–1.9) (Augood et al., 1998 (meta-analysis of 12 studies); US Department of Health and Human Services, 2001). The resulting decrease in parity could have implications for risk for cancers of the breast, endometrium and ovary. There is also evidence that women who smoke cigarettes are more likely to be subfertile, and to take longer to get pregnant than women who do not smoke (Baird, 1992; Hughes & Brennan, 1996; Jensen et al., 1998, Hull et al., 2000; US Department of Health and Human Services, 2001).
(ii) Effects on pregnancy
The use of tobacco products by pregnant women is associated with placenta praevia, placental abruption, premature rupture of membranes, pre-term birth, intrauterine growth restriction and sudden infant death syndrome (Castles et al., 1999; Andres & Day, 2000; US Department of Health and Human Services, 2001). The association between smoking and pre-term delivery appears to be more pronounced for older women (US Department of Health and Human Services, 2001). There is also a relatively modest increase in the risk for spontaneous abortion and stillbirth among women who smoke during pregnancy (Windham et al., 1999). Smoking may be responsible for 15% of all pre-term births, 20–30% of all infants of low birth weight, and for a 1.5-fold increase in overall perinatal mortality (Andres & Day, 2000).
Women who smoke cigarettes during pregnancy have been observed to have a decreased risk for various pregnancy-related disorders, including hypertension of pregnancy and pre-eclampsia, even after control for relevant covariates (Castles et al., 1999; US Department of Health and Human Services, 2001).
Pre-eclampsia is caused by damage to the placenta and develops in the second half of pregnancy, usually in the last few weeks, or immediately after delivery. Initially, the main symptoms are raised blood pressure and the presence of protein in the urine of the mother, and a slower-than-normal growth of the unborn child. In rare cases, women can go on to develop fits, known in pregnancy as eclampsia. Cigarette smoking has been associated with a lower rate of pre-eclampsia among women who were pregnant for the first time, independent of other maternal factors (Newman et al., 2001). For pre-eclampsia, however, dose–response relationships have not regularly been found (US Department of Health and Human Services, 2001). The limited data available regarding the association between cigarette smoking and the risk of eclampsia are mixed (US Department of Health and Human Services, 2001).
(iii) Effects on menopause
Cigarette smoking has repeatedly been found to be associated with an earlier menopause. Current female smokers are about twice as likely to reach menopause at an earlier age as nonsmokers; the relative risk increases with the average number of cigarettes smoked. When the effect is expressed as a difference in the median (or mean) age at menopause, women who smoke reach menopause about 0.8 to 1.7 years earlier than never-smokers, again with a greater effect among heavier smokers. Women who have stopped smoking reach menopause at an age similar to that of never-smokers, or somewhat later (Midgette & Baron, 1990; US Department of Health and Human Services, 2001).
The association between smoking and earlier age-at-menopause has been very consistently found in studies from Asia (Kato et al., 1988), northern Europe (Andersen et al., 1982; Luoto et al., 1994; Torgerson et al., 1994), southern Europe (Parazzini et al., 1992; Meschia et al., 2000) and the USA (Jick et al., 1977). Studies using various designs have reported this association, including cross-sectional surveys of patient populations (Jick et al., 1977), population-based surveys (Andersen et al., 1982; McKinlay et al., 1985; Luoto et al., 1994; Gold et al., 2001), retrospective cohort studies (Kaufman et al., 1980) or prospective cohort studies (Willett et al., 1983; Bromberger et al., 1997; Kato et al., 1998). In a study from Nigeria, the effect of smoking on age-at-menopause could not be demonstrated because of the small numbers of female smokers in that country, where smoking by women is a social taboo (Okonofua et al., 1990).
The mechanisms underlying this association are not entirely clear. However, at least in animals, the PAHs contained in cigarette smoke are toxic to ovarian follicles (Mattison & Thorgeirsson, 1978) and the human ovary probably contains the microsomal monooxygenases required to convert toxic chemicals into reactive species that affect oocytes. Smoking has been implicated in the increased rate of follicular atresia in women (Matisson, 1982). The higher concentrations of follicle-stimulating hormone in premenopausal smokers than in premenopausal nonsmokers are consistent with ovarian toxicity (Velasco et al., 1990; Cooper et al., 1995; Cooper & Thorp, 1999; Cramer et al., 2002). The occurrence of a diminished ovarian reserve is significantly higher among women who smoke, as was shown in studies of infertile women undergoing assisted reproduction (Sharara et al., 1994; El-Nemr et al., 1998). Premenopausal women who smoke may have fewer ovarian follicles than nonsmokers (Westhoff et al., 2000), although the data are conflicting. One study reported that smoking is associated with smaller ovarian volume (Syrop et al., 1995), whereas another failed to demonstrate this association (Flaws et al., 2000).
(iv) Effects on male reproductive potential
Cigarette smoking has repeatedly been associated with modest reductions in sperm density, motility and morphology (Vine et al., 1994; Wong et al., 2000). It has also been associated with increases in the levels of estrone, estradiol, testosterone and free testosterone in serum (Vine, 1996; Trummer et al., 2002). This effect may be mediated in part by constituents of seminal plasma. In one study, exposure of spermatozoa from nonsmokers to seminal plasma from smokers was associated with decreased viability. On the other hand, when spermatozoa from smokers were incubated in seminal plasma from nonsmokers, an improvement in the quality of semen was seen (Zavos et al., 1998). Despite the relatively clear effects of smoking on sperm parameters and the possible delay in conception, smoking by the male partner has not been found to have a consistent impact on fertility (Bolumar et al., 1996; Curtis et al., 1997; Hull et al., 2000).
(b) Hormonal effects
(i) Sex hormones
Estrogens
Smoking is thought to exert an ‘antiestrogenic’ effect in women (Baron et al., 1990; US Department of Health and Human Services, 2001). This is based on the observation that smoking increases the risk for estrogen-deficiency disorders, and decreases the risk for many disorders associated with estrogen-excess. Generally, this effect seems more pronounced in postmenopausal women. Of the estrogen-deficiency disorders, smoking has been found to be associated with an increased risk for hip fracture (Law & Hackshaw, 1997; Law et al., 1997; US Department of Health and Human Services, 2001). Of the estrogen-excess disorders, smoking has been found to be inversely associated with endometrial cancer, uterine fibroids, endometriosis, vomiting during pregnancy and hypertensive disorders of pregnancy (Baron et al., 1990; US Department of Health and Human Services, 2001).
Cigarette smoking does not seem to affect endogenous levels of the major estrogens in either premenopausal (Lucero et al., 2001; Manson et al., 2001) or postmenopausal women (Baron et al., 1990; Law et al., 1997; Verkasalo et al., 2001) although there are reasons for predicting such effects: nicotine or smoke-associated PAHs may inhibit aromatase, an enzyme that is required for estrogen production, in granulosa cells (Barbieri et al., 1986). This lack of effect indicates that smoking alters estrogen-related processes in women in ways other than through direct modulation of endogenous estrogen levels. Nevertheless, among postmenopausal women who take oral estrogens, cigarette smoking has been shown to reduce circulating levels of estradiol (Jensen et al., 1985; Jensen & Christiansen, 1988; Bjarnason & Christiansen, 2000), unbound estradiol (not bound to sex hormone-binding globulin) (Cassidenti et al., 1990) and estrone (Jensen et al., 1985; Jensen & Christiansen, 1988; Geisler et al., 1999). During oral estrogen therapy, estrone sulfate accounts for a higher proportion of circulating total estrogens in smokers than in nonsmokers (Geisler et al., 1999; Cassidenti et al., 1990). With lower-dose regimens, smokers seem to have higher levels of follicle-stimulating hormone than nonsmokers (Bjarnason & Christiansen, 2000).
On the other hand, smoking does not affect the estradiol or estrone levels in plasma of women treated with parenteral (transdermal) estrogens (Jensen & Christiansen, 1988), at least in the dose-range studied. However, plasma levels of estrone sulfate may be reduced during parenteral estrogen therapy, although not significantly (Geisler et al., 1999). This difference from the findings after oral treatment suggests that cigarette smoking induces the hepatic enzymes that affect hormone metabolism in a ‘first pass’ manner.
The biological mechanisms that may explain an anti-estrogenic effect of smoking are not clear. Changes in body weight and age at menopause caused by smoking do not seem sufficient by themselves to provide an explanation. Some of the potentially anti-estrogenic effects occur in premenopausal women and adjustment for body weight and age at menopause does not greatly affect the observed associations. Smoking-related changes in adrenal androgens are also insufficient to explain the effects observed (Thomas et al., 1993) (see below). A smoking-induced shift in estrogen metabolism towards 2-hydroestrogens and catechol estrogens with weak estrogenic potency may play a role (Michnovicz et al., 1986, 1988). A recently described molecular mechanism may also be relevant. Cigarette smoking is a rich source of PAHs and other ligands for the aryl hydrocarbon receptor. The ligand–receptor complex has the capacity to bind to response elements in the promoter region of estrogen-regulated genes, thereby serving as a transcription repressor. This inhibition of estrogen may explain the lack of estrogen stimulation in women with normal estradiol and estrone levels (Safe et al., 1998, 2001; Safe & McDougal, 2002).
Androgens
In women, the most potent circulating androgen, testosterone, derives both from the ovary (predominantly in premenopausal women) and the adrenal gland. Androstenedione and dihydroepiandrosterone, which have an adrenal origin, are much less potent as androgens, but may serve as precursors for other, more potent hormones, such as dihydroepiandrosterone sulfate (DHEAS). Circulating levels of testosterone, androstenedione and perhaps DHEAS have been associated with cancers of the breast and endometrium (Dorgan et al., 1997; Zeleniuch-Jacquotte et al., 1997; Akhmedkhanov et al., 2001; Endogenous Sex Hormones and Breast Cancer Collaborative Group, 2002).
In postmenopausal women, cigarette smoking clearly increases serum concentrations of the adrenal androgens, androstenedione and DHEAS (Law et al., 1997). In premenopausal women who smoke, DHEAS levels are significantly increased (Manson et al., 2001); for androstenedione, findings are consistent with an increase, but the relevant studies are small and the results of individual studies are not statistically significant (Longcope & Johnston, 1988; Ruiz et al., 1992; Thomas et al., 1993).
The levels of the major circulating androgen, testosterone, seem unaffected by smoking in women. For example, Law et al. (1997) found that testosterone concentrations in postmenopausal women seem not to be substantially affected. Studies in premenopausal women have generally been small and have yielded conflicting results. Nevertheless, one larger investigation (of > 600 women) reported higher testosterone concentrations in the serum of female smokers than in nonsmokers (Sowers et al., 2001), whereas an earlier study had reported a decreased free testosterone index (Ortego-Centeno et al., 1994).
(ii) Diabetes mellitus and insulin resistance
Diabetes mellitus is a risk factor for endometrial and breast cancers and possibly also for colorectal cancer (Giovannucci, 2001; Gupta et al., 2002). Epidemiological data on type 2 diabetes suggest that cigarette smoking is a risk factor for this common disease. Type 2 diabetes, generally incident among children or young adults, has strong immunological and genetic determinants, and has essentially not been studied with respect to possible associations with tobacco use.
Recent cohort studies have shown a modest association of smoking with risk for incident type 2 diabetes mellitus, though typically the increased risks were found only in the heaviest smokers and in those who start smoking at a younger age (Feskens & Kromhout, 1989; Rimm et al., 1993, 1995; Kawakami et al., 1997; Manson et al., 2000). The association was still observed after adjustment for multiple covariates, including body mass index and alcohol intake.
The biological plausibility of these findings is supported by the results from other investigations showing that cigarette smoking can induce insulin resistance (Facchini et al., 1992; Hautanen & Adlercreutz, 1993; Zavaroni et al., 1994; Eliasson et al., 1997). Smokers have also been observed to have higher concentrations of glycosylated haemoglobin (HbA1 and HbA1C) than nonsmokers, indicating a tendency to higher glucose levels (Modan et al., 1988; Simon et al., 1989; Nilsson et al., 1995; Sargeant et al., 2001).
There are some hormonal mechanisms that might explain an association of smoking with an increased risk for diabetes. Smoking increases secretion of catecholamines, glucocorticoids and probably growth hormone; these are all ‘counter-regulatory’ hormones, i.e. they counteract the effects of insulin in glucose metabolism.
(iii) Body weight and obesity
Cigarette smoking distorts the normal association between leanness and health because current cigarette smokers weigh less than never-smokers and have a lower body mass index (Klesges et al., 1989; Perkins, 1993; Flegal et al., 1995; US Department of Health and Human Services, 2001). The relationship is ‘U-shaped’; moderately heavy smokers weigh less than nonsmokers and also less than heavy smokers. The weight differences are larger for women and become more pronounced with age (Klesges et al., 1989; Williamson et al., 1991; Perkins, 1993; Rasky et al., 1996; Molarius et al., 1997; US Department of Health and Human Services, 2001). Initiation of smoking seems not to lead to weight loss — rather, continued smoking probably suppresses age-related weight gain (US Department of Health and Human Services, 2001).
It is clear that smoking cessation leads to weight gain, such that former smokers weigh more than current smokers of the same age (and smoking duration before cessation) (Williamson et al., 1991; Perkins, 1993; Flegal et al., 1995; US Department of Health and Human Services, 2001). The weight gain following cessation has generally been found to be greater in women than in men (Flegal et al., 1995; O'Hara et al., 1998; US Department of Health and Human Services, 2001).
The mechanisms underlying these effects have been studied extensively. The lower body weights of current smokers are not the result of taking more exercise or of lower caloric intake: if anything, smokers tend to consume more calories and be less active than never-smokers (Klesges et al., 1989; Grunberg, 1990; US Department of Health and Human Services, 2001). After smoking cessation, there seems to be a transient increase in caloric intake, but this is not sufficient to explain the weight gain (Perkins, 1993). Metabolic studies, however, have repeatedly shown that smoking acutely increases energy expenditure, particularly in individuals who are not at rest (Perkins, 1993). Animal experiments and studies of individuals who use nicotine replacement as an aid to stop smoking suggest that nicotine is responsible for the weight differences (Grunberg, 1990). This may be due to the release of catecholamine associated with smoking (US Department of Health and Human Services, 1983).
In addition to its effects on body weight, cigarette smoking also affects the distribution of body weight. Although body weight is lower, the waist-to-hip ratio, an index of abdominal obesity associated with an increased risk for cardiovascular disease, is higher in smokers. As for body weight, this association also appears to be stronger among women than among men (Duncan et al., 1995; Croft et al., 1996; Ishizaki et al., 1999; US Department of Health and Human Services, 2001).
(iv) Growth hormone, insulin-like growth factors and insulin-like growth factor-binding proteins
Growth hormone is secreted by the pituitary gland; most of its effects are mediated through insulin-like growth factors (IGFs) that are synthesized under the influence of growth hormone in a variety of tissues. Relatively heavy exposures to tobacco smoke (e.g. smoking of two or more cigarettes in succession) acutely increase the levels of circulating growth hormone, at least in men (Cryer et al., 1976; Winternitz & Quillen, 1977; Wilkins et al., 1982). In women, data are limited, but are consistent with a similar effect. The increase in growth hormone concentration is similar in obese and lean female smokers (Szostak-Wegierek et al., 1996).
Under the control of growth hormone, the liver synthesizes IGFs that are secreted into the circulation. High serum concentrations of IGF1 have been associated with cancers of the breast, prostate, colorectum and lung (Chan et al., 1998; Ma et al., 1999; Yu & Rohan, 2000; Kaaks et al., 2000; Toniolo et al., 2000; Giovannuci, 2001; Fürstenberger & Senn, 2002). The data regarding circulating IGF1 levels and cigarette smoking are conflicting. Although the studies on growth hormone have suggested that IGF1 levels might be increased by smoking (Eliasson et al., 1993; Kaklamani et al., 1999), a few studies have reported decreased IGF1 levels in smokers (Landin-Wilhelmsen et al., 1994; Probst-Hensch et al., 2001) and other investigations have found no association (Goodman-Gruen & Barrett-Connor, 1997; Ma et al., 1999; Lukanova et al., 2001). One study calculated free IGF1 levels and reported lower values in ever-smokers than in nonsmokers, but this result is biased by the inclusion of former smokers with current smokers, and by the apparent lack of adjustment for covariates (Janssen et al., 1998).
Research in this area is complicated by the presence of IGF-binding proteins, which can alter the availability and effects of the IGFs. There are few data regarding the effect of smoking on the binding proteins and some have suggested that smoking may decrease the levels of IGFBP3, the principal binding protein for IGF1 (Kaklamani et al., 1999; Lukanova et al., 2001), although other studies have found no association (Yu et al., 1999; Probst-Hensch et al., 2001).
(v) Vitamin D
Vitamin D is well known as a family of hormones that regulate calcium, magnesium, phosphorus and bone metabolism. Vitamin D also has important effects on cell differentiation and proliferation, and may be inversely related to the risks for cancers of the breast, prostate and colorectum (Schwartz & Hulka, 1990; Martinez & Willett, 1998; Lipkin & Newmark, 1999; Bretherton-Watt et al., 2001; Polek & Weigel, 2002).
Cigarette smoking has repeatedly been associated with reductions in serum levels of 25-hydroxyvitamin D (25-OH vitamin D), the compound that best reflects vitamin D status. A number of studies have reported that smokers have lower serum concentrations of 25-OH vitamin D than nonsmokers (Mellstrom et al., 1993; Brot et al., 1999; Harris et al., 2000; Rapuri et al., 2000; Chapurlat et al., 2001; Need et al., 2002). Most of these investigations adjusted for several covariates such as bone mineral density, vitamin D intake and exposure to sunlight (Brot et al., 1999; Harris et al., 2000; Rapuri et al., 2000). 1,25-Dihydroxyvitamin D (1,25-(OH)2 vitamin D) levels are tightly regulated and do not reflect vitamin D status except under conditions of obvious deficiency or excess. Nevertheless, in some studies (Brot et al., 1999; Need et al., 2002), but not all (Rapuri et al., 2000), serum concentrations of this hormone have been found to be decreased in smokers.
The data reviewed in this section can be summarized as follows:
- Cigarette smoking has widespread and serious effects on reproductive function in women. Some of these effects may have implications for cancer risks in women.
- Cigarette smoking has important hormonal and metabolic effects that may be related to cancer risk at several sites. Smokers have an increased risk for type 2 diabetes, which may, in turn, increase the risk for cancer at several sites. Smokers also have a lower average body weight than nonsmokers, and smoking cessation has been associated with weight gain. Although this weight gain is admittedly a negative outcome, the benefits of smoking cessation still far exceed it.
- The ‘anti-estrogenic’ effect of cigarette smoking may be relevant to the development of cancers at several anatomical sites in women, and the effects of smoking on vitamin D status may be involved in the carcinogenic effects of tobacco.
- The implications of the findings regarding the association between smoking, insulin-like growth factors and adrenal androgens are not clear.
4.3.2. Animals
(a) Reproductive and perinatal effects
(i) Effects on embryonic growth and malformations
A study was conducted to determine whether chronic treatment of gestating monkeys with nicotine alters the concentrations of known regulators of energy balance in the newborn offspring. Gestating rhesus monkeys were treated with nicotine tartrate (1.5 mg/kg bw per day) starting on day 26 of gestation and were maintained until day 160 of gestation. Exposure to nicotine had no significant effect on absolute birth weights of the monkeys, although there was a 10% reduction in birth weights in animals exposed to nicotine when normalized to maternal weight. Plasma leptin concentrations on postnatal day 1 were lower by about 50% in the nicotine-treated group than those in controls, suggesting that the infant monkeys exposed to nicotine may also have lower body-fat levels. These data suggest that exposure to nicotine during gestation may increase energy expenditure in the developing fetus through actions on hypothalamic systems, resulting in lower birth weights and body-fat levels (Grove et al, 2001).
Gestating C57BL or mutant ‘curly tail’ mice were exposed to tobacco smoke in a smoking machine for 10 min, three times a day, either on the day of conception (day 0) and days 1 and 2, on days 3, 4 and 5, or from day 0 until day 17. After the first two treatments, embryonic development was subsequently assessed on day 9. Both these periods of exposure were associated with a dose-related retardation in embryonic growth, but the retardation was more marked in embryos exposed on days 0, 1 and 2. It would seem, therefore, that even brief episodes of maternal smoking are detrimental to the very early embryo, and even if smoking is stopped, the effects persist at least for some days and there is no immediate catch-up growth. In mice exposed continuously for 17 days, the fetuses were studied on day 18: a significant reduction in fetal body weight was observed in both strains of mice. There was also a reduction in the number of skeletal ossification centres, showing that developmental delay also occurred. In C57BL mice, one rib abnormality occurred, but no major congenital malformations were seen. However, in the curly tail mutants, 60% of which normally have a curly tail or an open neural tube defect, there was a modest increase in the frequency of open spina bifida and exencephaly; a few minor rib abnormalities also occurred, and one case of cleft lip with cleft palate. These results indicate that tobacco smoke, although detrimental to the developing fetus, is not a potent teratogen in the mouse, but may have minor effects in those individuals genetically predisposed to an abnormality. These results may explain the generally inconclusive findings regarding congenital malformations in the children of women who smoked during pregnancy. In all experiments, the detrimental effects were seen with both higher-tar cigarettes (tar and nicotine yields, 12.9 and 1.19 mg/cigarette, respectively) and lower-tar cigarettes (4.8 and 0.54 mg/cigarette, respectively), indicating that tobacco modification is not beneficial to the developing fetus (Seller & Bnait, 1995).
(ii) Effects on the fetal respiratory system
Exposure of fetal rats to nicotine gives rise to increased mortality when animals are challenged postnatally with hypoxia. In one study, gestating rats received nicotine infusions simulating the plasma nicotine concentrations of smokers. At 1–2 days postpartum, the nicotine-treated group displayed normal heart rates, electrocardiogram waveforms and respiratory rates under normal oxygen conditions. With hypoxia (5% O2, for 10 min), controls showed initial tachycardia and a subsequent slight decline in heart rate. Atrioventricular conduction was gradually impaired and repolarization abnormalities also appeared. The group exposed to nicotine showed no tachycardia and their heart rate declined rapidly and precipitously within a few minutes after the start of hypoxia. Changes in respiration were identical in the two groups: initial tachypnoea and subsequent decline. These results suggest that prenatal exposure to nicotine affects sinoatrial reactivity to hypoxia without impairing cardiac conduction per se. These mechanisms would explain the increased hypoxia-induced mortality noted in animals exposed to nicotine prenatally, and could contribute to increased morbidity, mortality and sudden infant death syndrome in humans (Slotkin et al., 1997).
Rats were exposed to cigarette smoke or room air from days 2–22 of gestation. Immunoblots of dorsocaudal brainstem lysates at day 2 postpartum revealed no differences in protein kinase C (PKC) (α and β) or endothelial NOS expression. However, the immunoreactivities of PKC-γ, PKC-δ and neuronal NOS were reduced in the smoke-exposed group. The results indicate that smoking during gestation is associated with selective reductions in PKC and NOS isoforms within the dorsocaudal brainstem, which could decrease respiratory drive and lead to enhanced vulnerability to hypoxia in infants of mothers who smoke. These conditions are implicated in sudden infant death syndrome (Hasan et al., 2001).
Gestating rats received either nicotine (6 mg/kg bw per day) or vehicle administered continuously with an osmotic minipump from day 6 of gestation to days 5 or 6 postpartum. On days 5 or 6 postpartum, pups were either exposed to a single period of hypoxia (97% N2; 3% CO2) and their time to last gasp was determined, or exposed repeatedly to hypoxia and their ability to autoresuscitate from primary apnoea was determined. Perinatal exposure to nicotine did not alter the time to last gasp, but it did impair the ability of pups to autoresuscitate from primary apnoea. In the control group, pups were able to autoresuscitate from 18 ± 1 (SD) periods of hypoxia, whereas, after exposure to nicotine, the treated pups were able to autoresuscitate from only 12 ± 2 periods (p < 0.001) of hypoxia. These data provide evidence that perinatal exposure to nicotine impairs the ability of newborn rats to autoresuscitate from primary apnoea during repeated exposure to hypoxia, such as may occur during episodes of prolonged sleep apnoea (Fewell & Smith, 1998).
(iii) Mutagenic effects in the embryo
Long-term chronic exposure to tobacco smoke is believed to be necessary for carcinogenesis. An investigation was conducted into the relationship between short-term exposure to smoke and the frequency of deletions in the mouse embryo. Deletions and other genome rearrangements are associated with carcinogenesis and inheritable diseases. The pink-eyed unstable (pun) mutation in C57BL/6J mice is the result of internal duplication of 70 kb of DNA within the p gene. Spontaneous reversion events in homozygous pun/pun mice occur by deletion of one copy of the duplicated sequence. Reversion events occurring in the embryonic premelanocytes of the developing fetus give rise to black spots on the grey fur of the offspring after birth. The effects of exposure of pregnant pun mice to cigarette smoke and cigarette-smoke condensate on the frequency of black spots occurring in the offspring were investigated. Gestating dams were exposed (whole body) to smoke generated by either filtered or unfiltered cigarettes for 4 h, or alternatively, mice were given a 15-mg/kg bw dose of cigarette-smoke condensate during day 10 of gestation. The concentrations of total particulate matter, CO, plasma nicotine and cotinine were determined to characterize the smoke exposure. There was a significant increase in the number of DNA deletions in the embryo as indicated by spotted offspring in both of the smoke-exposed groups and in the condensate-exposed group. The results suggest that embryos are highly sensitive to the genotoxic activity of cigarette smoke following a single exposure of only 4 h (Jalili et al., 1998).
(iv) Effects on the postnatal brain
The effects of prenatal exposure to CO, a major component of cigarette smoke, on the structural and neurochemical development of the postnatal brain at 1 and 8 weeks were studied alone or in combination with postnatal hyperthermia. Gestating guinea-pigs (n = 11) were exposed to 200 ppm CO for 10 h per day from mid-gestation until term (68 days), whereas control dams (n = 10) breathed room air. On postnatal day 4, neonates from the control and CO-exposed groups were exposed to hyperthermia (35 °C) for 75 min or remained at ambient temperature (23 °C). Semiquantitative immunohistochemical techniques revealed the following neurotransmitter alterations in the medulla after 1 week: a decrease in met-enkephalin-immunoreactivity following postnatal hyperthermia and an increase in 5-hydroxytryptamine immunostaining following a combination of exposure to CO and hyperthermia. No alterations were observed in substance P- or tyrosine-hydroxylase staining in animals subjected to any of the treatments. At 8 weeks of age, the combination of prenatal exposure to CO followed by a brief hyperthermic stress postnatally resulted in lesions throughout the brain and an increase in immunoreactivity of glial fibrillary acidic protein in the medulla. Such effects on brain development could be of relevance in cardiorespiratory control in the neonate and could have implications for the etiology of sudden infant death syndrome, in which smoking and hyperthermia are major risk factors (Tolcos et al., 2000).
Because the identity of the teratogenic agent in cigarette smoke remains controversial, a study was conducted to investigate whether nicotine can cause neural dysmorphology and, hence, act as a nervous system teratogen in cultured rat embryos. This in-vitro study confirmed the conclusion of previous reports on in-utero exposure that nicotine leads to growth retardation and impaired development of the nervous system, particularly of the forebrain and the branchial arches. This could lead to microcephaly and cleft palate in term fetuses. Cellular disruption and necrosis occurred in the neuroepithelium and underlying mesenchyme; the effect was dose-dependent. There was severe disruption of cell and organelle membranes, and many healthy cells were found to contain engulfed, whole condensed or remnants of dead cells. The results show that nicotine acts as a nervous system teratogen leading to gross and cellular dysmorphology. This could be explained by a direct effect of this highly lipid-soluble compound on the membranes, or by an indirect effect through oxidative membrane damage (Joschko et al., 1991).
(b) Hormonal effects
(i) Estrogens
Two groups of female rats (aged 2.5–3 months and 6 months) were exposed to mainstream cigarette smoke for 2 h per day for 3 weeks or 3 months. Exposure to tobacco smoke did not induce any changes in uterine weight or estrous cycle, but led to a decrease in the estradiol (E2) concentration in uterine tissue, in particular in the 6-month-old rats and in the young rats after 3 months of exposure. No signs of aneuploidy were found in the uterus of the smoke-exposed animals. Flow cytometry analysis showed that both the cell proliferation index and the proportion of cells in S-phase were increased by 3 weeks of exposure and both were decreased by 3 months of exposure (Berstein et al., 1999)
(ii) Glucose tolerance, insulin
Cigarette smoking is a major risk factor for coronary heart disease. The effect of nicotine on blood pressure and glucose tolerance was studied in adult male Sprague-Dawley rats randomly assigned to receive either nicotine or placebo pellets as subcutaneous implants. Body-weight gain was controlled by pair-feeding, and was not significantly different between nicotine-treated and placebo-treated animals. Blood pressure increased throughout a 3-week treatment period in nicotine-treated animals and was significantly higher (p < 0.05) than in placebo-treated rats; it returned to normal within 1 week following exhaustion of the pellets. Oral glucose tolerance tests conducted at 2.5 weeks after implantation showed similar glucose, insulin and free fatty acid profiles in both groups. The results show that exposure to nicotine leads to sustained but reversible hypertension in rats without deterioration of glucose tolerance or insulin action, under conditions of controlled body-weight gain. Smokeless nicotine adversely affects the coronary risk profile by increasing blood pressure (Swislocki et al., 1997).
(iii) Vitamin D
The effects of 2 months of nicotine treatment on bone formation and resorption were studied in adult female rats. In addition, the concentrations of calciotropic hormones, including parathyroid hormone, calcitonin, 25-OH vitamin D and 1,25-(OH)2 vitamin D were determined. Groups of seven animals received either saline or nicotine at 3.0 or 4.5 mg/kg bw per day, delivered by subcutaneously implanted osmotic minipumps, for 2 months. Serum, right tibia, left femur and lumbar vertebrae (3–5) were collected to determine hormonal concentrations as well as histomorphometric parameters, bone-mineral density, bone-mineral content and vertebral strength. Although nicotine-treated rats had a lower level of 25-OH vitamin D (54.4 ± 3.1 ng/mL for the lower-dose group and 55.8 ± 2.8 ng/mL for the higher-dose group (mean ± SEM)) than the controls (74.8 ± 2.8 ng/mL) (p < 0.01), no significant difference could be detected between the levels of the remaining hormones. Similarly, no statistical differences were detected in histomorphometric parameters, bone-mineral density, bone-mineral content or vertebral strength between nicotine-treated and control rats. The results indicate that exposure to nicotine for 2 months causes a 30% reduction in serum concentration of 25-OH vitamin D, but no alteration in bone mass, strength or formation and resorption (Fung et al., 1998).
(iv) Effects on the hypothalamus–hypophysis–adrenal hormones
The possible long-term effects of postnatal exposure to cigarette smoke were studied in male Sprague-Dawley rats exposed to the smoke from two Kentucky reference IR-1 type cigarettes every morning from day 1 after birth for a period of 5, 10 or 20 days. The rats were killed 24 h (for exposure periods of 5, 10 and 20 days), 1 week (for 20 days of exposure) or 7 months (for 20 days of exposure) after termination of the last exposure. Catecholamine levels and changes in catecholamine utilization in discrete hypothalamic regions were analysed by quantitative histofluorimetry. Serum prolactin, luteinizing hormone, thyroid-stimulating hormone and corticosterone concentrations were determined by radioimmunoassay. In the postnatal period, serum levels of luteinizing hormone were significantly increased 24 h after a 10-day or 20-day period of exposure to cigarette smoke. A highly significant increase in serum prolactin concentrations was observed in adults who had been exposed postnatally to cigarette smoke for a 20-day period, although the levels had been unaltered by this exposure when measured during the postnatal period. Twenty-four hours following a 20-day postnatal exposure, catecholamine utilization was increased in the medial palisade zone of the median eminence and was substantially reduced in the parvocellular and magnocellular parts of the paraventricular hypothalamic nucleus. Following 20 days of postnatal exposure to cigarette smoke, measurements made 1 week and 7 months later revealed no alterations in levels or utilization of catecholamine in various hypothalamic areas including the median eminence. No alteration in the development of body weight was observed with any of the above changes. The results indicate that marked but temporary increases in the secretion of luteinizing hormone occur 24 h after postnatal exposure to cigarette smoke, whereas increases in prolactin secretion develop only in adult life, when maturation of the brain and/or the anterior pituitary gland is complete. Changes in levels and utilization of catecholamine have been found in discrete hypothalamic nerve terminal networks, but do not play a major role in mediating the above changes in anterior pituitary function and are probably the result of a withdrawal phenomenon (Jansson et al., 1992).
Male rats were exposed to the smoke of 1–4 Kentucky reference IR-1 type cigarettes. Catecholamines in the diencephalon were measured by quantitative histofluorimetry in discrete dopamine (DA) and noradrenaline (NA) nerve terminal systems. Blood concentrations of thyroid-stimulating hormone, prolactin, luteinizing hormone, follicle-stimulating hormone, adrenocorticotropic hormone, vasopressin and corticosterone were determined by radioimmunoassays. Exposure to unfiltered, but not to glass fibre-filtered cigarette smoke resulted in dose-dependent reductions of NA levels in the various hypothalamic NA nerve terminal systems, and in dose-dependent increases of amine turnover in the various DA and NA nerve terminal systems in the hypothalamus. The decrease that was observed in secretion of thyroid-stimulating hormone, luteinizing hormone and prolactin after exposure to unfiltered smoke was probably induced by nicotine activating the lateral and medial tubero-infundibular DA neurons. Furthermore, unfiltered cigarette smoke produced a dose-related increase in corticosterone secretion (Andersson et al., 1985).
4.4 Genetic and related effects
This topic was reviewed previously by DeMarini (1983) and Obe (1984) and summarized in the first IARC Monograph on tobacco smoking (IARC, 1986). The present monograph provides only a brief overview of work prior to 1986 and, instead, summarizes work published since then.
4.4.1. Humans
(a) Mutagenicity, sister chromatid exchange, HPRT mutation and other effects
(i) Urinary mutagenicity
Urinary mutagenicity in smokers was detected first by Yamasaki and Ames (1977) by testing the XAD/acetone-extractable organic compounds from urine in the Salmonella (Ames) mutagenicity assay. Several years later, studies using essentially the same methods confirmed and clarified this original observation (Putzrath et al., 1981; Kriebel et al., 1985). The general approach to these studies has remained similar over the years. It involves concentration of the organic compounds from the urine by means of a solid-phase resin followed by elution with an organic solvent and then testing the resulting concentrate, after its fractional analysis by HPLC, in the Salmonella (Ames) mutagenicity assay in the presence of rat liver S9 mix for metabolic activation.
A comparison of three types of resin (C18, XAD-2 and CN) followed by elution with acetone showed that the highest levels of urinary mutagenicity were detected using C18 resin. This study found that urinary genotoxicity was higher in smokers of black tobacco than in smokers of blond tobacco (Kuenemann-Migeot et al., 1996). A study of the stability of stored urine samples showed that no significant loss of mutagenic activity occurred in urine stored frozen for as long as 175 days, although near significance was reached, as a result of decreasing mutant response as storage time increased, for two of the higher doses tested (Williams et al., 1990). A Salmonella microsuspension assay is generally more sensitive at detecting urinary mutagens from smokers than the standard plate-incorporation assay (Kado et al., 1983; Nylander & Berg, 1991). Peak mutagenic activity of the urine occurred 4–5 h after the beginning of smoking and decreased to pre-smoking levels 12 h after the cessation of smoking in occasional smokers and after 18 h in heavy smokers (Kado et al., 1985). This study suggested that the mutagens are absorbed rapidly (3–5 h) and are eliminated from the body following first-order kinetics; the excretion rate constant for the occasional smoker was ∼0.1 h–1, and the half-life (T1/2) was ∼7 h. A study in which the SOS Chromotest was used as the indicator assay showed that urine from smokers that was mutagenic in Salmonella typhimurium TA98 was not mutagenic in the SOS Chromotest; this test is therefore not suitable for assaying urinary mutagens (De Méo et al., 1988). Urine concentrates from subjects who both smoked tobacco and chewed areca nuts induced sister chromatid exchange and chromosomal aberrations in Chinese hamster ovary cells (Trivedi et al., 1993, 1995).
The urine of smokers who smoked cigarettes that heated, but did not burn tobacco had levels of urinary mutagenicity similar to those of nonsmokers (Doolittle et al., 1989; deBethizy et al., 1990; Smith et al., 1996). However, the urine of occupationally exposed bidi tobacco rollers was mutagenic (Bhisey & Govekar, 1991), suggesting that exposure to tobacco per se rather than tobacco pyrolysate products may be sufficient to produce mutagenic urine. Urinary mutagenicity generally correlated with the number, but not the tar level, of cigarettes smoked (Tuomisto et al., 1986; Kuenemann-Migeot et al., 1996). Interestingly, the urine of smokers of black tobacco was twice as mutagenic as the urine from smokers of blond tobacco, and there was a higher risk for bladder cancer in smokers of black tobacco than in smokers of blond tobacco (Vineis et al., 1984; Bryant et al., 1988; Malaveille et al., 1989; see also Section 2.1.2.(a)(iii)). One study of bladder cancer patients showed that there was no association between levels of urinary mutagenicity and tumour status or recurrence of the bladder tumours (Kanaoka et al., 1990).
Although consumption of fried meat can also produce mutagenic urine, experiments with subjects eating controlled diets showed that the higher urinary mutagenicity in smokers compared with nonsmokers was not the result of enhanced mutagenicity caused by diet-related heterocyclic amine mutagens in their urine (Doolittle et al., 1990a). Although not reviewed exhaustively here, smoking was generally a potential factor in studies investigating urinary mutagenicity of occupational exposures, such as inks and pharmaceuticals (Dolara et al., 1981), tyres (Crebelli et al., 1985), coke or graphite-electrodes (Ferreira et al., 1994), steel and coal processing (De Méo et al., 1987) and benzidine (DeMarini et al., 1987).
Indirect evidence suggests that the chemicals responsible for smoking-related urinary mutagenicity are primarily aromatic amines and/or heterocyclic polyaromatic amino compounds. For example, the urine of smokers was much more mutagenic in strain YG1024 of Salmonella, which overproduces O-acetyltransferase, than in strains with less of this activity (e.g. TA98) or that overproduce nitroreductase (YG1021) (Einistö et al., 1990; Camoirano et al., 2001). One study in which the urine of smokers was fractionated by reversed-phase HPLC and the fractions were then evaluated for their ability to induce chromosomal aberrations in Chinese hamster ovary cells concluded that clastogenic agents were present in the urine of smokers. This clastogenic activity was reduced by the addition of catalase or superoxide dismutase, suggesting that the activity of these agents may result from the production of active oxygen species (Dunn & Curtis, 1985).
Urinary mutagenicity has been shown to correlate with the levels of a 4-ABP–DNA adduct1 in exfoliated urothelial cells from smokers (Talaska et al., 1991b). However, the levels of a 4-ABP-haemoglobin adduct2 showed a more complex association with urinary mutagenicity (Bartsch et al., 1990). Chemical analysis of urine from a smoker with exceptionally high urinary mutagenicity revealed the presence of the mutagen 2-amino-7-naphthol, which is a metabolite of the bladder carcinogen 2-aminonaphthalene (β-naphthylamine), a component of cigarette smoke (Connor et al., 1983). Chemical fractionation of mutagenic urine from smokers indicated that much of the mutagenic activity may be due to PAHs and/or heterocyclic amines (Mure et al., 1997). Although the concentration of urinary nicotine plus its metabolites correlated with urinary mutagenicity in smokers (Rahn et al., 1991; Granella et al., 1996), nicotine and its metabolites were not responsible for the mutagenicity. The absence of urinary mutagens in subjects who smoke cigarettes in which tobacco is heated but not burned has been demonstrated, although large quantities of nicotine and cotinine were found in the urine of such subjects (Curvall et al., 1987; Rahn et al., 1991).
(ii) HPRT mutations
Several reviews (Cole & Skopek, 1994; Robinson et al., 1994; Curry et al., 1999) have noted that smoking generally increases the hypoxanthine-guanine phosphoribosyltransferase (HPRT) (also called HGPRT) mutant frequency in peripheral blood lymphocytes by ∼50%. However, the increases did not reach statistical significance in some studies because of the large interindividual variability of HPRT mutant frequencies. Using the autoradiographic HPRT assay, some elevated portion of the HPRT mutant frequency was shown to reflect recent exposure to tobacco smoke rather than the cumulative effect of past exposure. The two categories of smoker (light and heavy) had frequencies of mutant cells significantly different from each other and both were significantly higher than those of nonsmokers and former smokers (Ammenheuser et al., 1997). Most importantly, HPRT mutant frequencies were similar in patients with lung cancer and in controls with the same smoking status, indicating that lung cancer per se has little if any effect on HPRT mutant frequency in lymphocytes. However, when cases and controls were combined, the HPRT mutant frequency was significantly higher in ever-smokers than in never-smokers (Hou et al., 1999). Although some analyses have found no difference in the mutation spectrum at the HPRT locus between smokers and nonsmokers (Curry et al., 1999), an increase in transversions, in particular, GC→TA, has been frequently noted among smokers (Burkhart-Schultz et al., 1996; Podlutsky et al., 1999; Hackman et al., 2000). This is the primary class of base substitution induced by those PAHs that form bulky DNA adducts giving rise to transversion mutations. An excess of this class of mutation in the HPRT mutation spectrum of smokers would be consistent with exposure to the PAHs in cigarette smoke.
(iii) Genotoxic effects in reproductive tissues/fluids and in children of smokers
Pregnant women who smoke not only had elevated HPRT mutant frequencies themselves, but analysis of cord blood indicated that their children also had elevated frequencies (Ammenheuser et al., 1994, 1998). Sequencing of HPRT mutants in cord blood from mothers who smoke indicated that most (85.7%) of the mutations were illegitimate V(D)J recombinase activity-associated exon 2-3 deletions (Bigbee et al., 1999). V(D)J recombinase is an enzyme system that mediates genomic rearrangements of germline-encoded variable (V), diversity (D) and junctional (J) regions responsible for generating T-cell receptor and immunoglobulin gene diversity for antigen-specific recognition. These genomic HPRT deletions are an illegitimate recombination event associated with haematopoietic malignancies in early childhood (Gu et al., 1992). In-utero exposure to tobacco smoke increased translocation frequencies in the newborn, and a significant association was found between the CYP1A1 MspI polymorphism (variant genotypes of CYP1A1) and frequencies of chromosomal aberration in the newborns (Pluth et al., 2000). Evidence has been obtained to suggest that smoking by the mother may cause detectable DNA strand breaks and alkali-labile damage in the lymphocytes of newborns detected by single-cell gel electrophoresis; the percentage of damaged cells increased with the frequency of smoking (Sardas et al., 1995).
Smoking was shown to induce aneuploidy in sperm for certain chromosomes, including 1, 13, and YY disomy (Rubes et al., 1998; Härkönen et al., 1999; Shi et al., 2001), but not for others, such as XX, XY, 7 or 8 (Robbins et al., 1997; Rubes et al., 1998; Härkönen et al., 1999). Smoking also appeared to induce oxidative damage to sperm DNA as indicated by higher levels of 8-OHdG in sperm DNA of smokers than in that of nonsmokers (Shen et al., 1997). Consistent with this was the finding of higher levels of DNA strand breaks in the sperm from smokers than in that from nonsmokers (Potts et al., 1999).
In investigations on the effects of smoking on oocytes, smokers were found to have fewer retrieved oocytes than nonsmokers, a finding consistent with the known reduction in their fertility (Zenzes et al., 1995). The oocytes with diploid complements of chromosomes were more frequent in smokers than in nonsmokers. This diploidy probably resulted from the prevention of first polar body extrusion, which indicates meiotic immaturity. The proportion of diploid oocytes was strongly associated with the number of cigarettes smoked per day (Zenzes et al., 1995). In addition, triploid zygotes occurred more frequently among smokers than nonsmokers, suggesting that digynic fertilization is an important mechanism leading to triploidy among smokers (Zenzes et al., 1995). These results strongly indicated that cigarette smoking is hazardous to the viability and function of developing oocytes and their resulting embryos, and a recent study suggested that expression of the aromatic hydrocarbon receptor-driven Bax gene is required for premature ovarian failure caused by exposure of mice to PAHs. This ovarian damage caused by PAHs was prevented by aromatic hydrocarbon receptor Ah2 or Bax inactivation. Oocytes in human ovarian biopsies grafted into immunodeficient mice also accumulated Bax and underwent apoptosis after exposure to PAHs in vivo (Matikainen et al., 2001). These data also suggest that the early onset of menopause in women smokers is caused, at least in part, by the pro-apoptotic action of tobacco smoke-derived PAHs in human oocytes.
The cervical mucus of smokers was more mutagenic than that of nonsmokers when tested in the Salmonella (Ames) mutagenicity assay (Holly et al., 1986), and XAD/acetone extracts of amniotic fluid from mothers who smoke induced a higher number of sister chromatid exchanges in Chinese hamster ovary cells than did extracts from nonsmokers (Lähdetie et al., 1993). This is consistent with the finding that cervical epithelial cells from smokers had higher numbers of micronuclei than those from nonsmokers (Cerqueira et al., 1998).
As noted in the reviews by Sastry (1991) and Zenzes (2000), smoking causes numerous reproductive problems, as well as effects on meiotic spindle function, DNA damage (oxidative damage as well as PAH adducts) to spermatozoa and oocytes, in addition to gametic and placental transmission of genetic damage. As discussed above, smoking is also associated with second-generation effects. Together, these data provide suggestive evidence for paternal and possible maternal gametic transmission of genetic damage as well as indirect support for the possibility that tobacco smoke may be a germ-cell mutagen.
(iv) Cytogenetic effects
Micronuclei
Bonassi et al. (2003) reviewed many studies that examined the influence of smoking on the frequency of micronuclei in peripheral lymphocytes. However, mixed results have been obtained. A pooled re-analysis of 24 databases from the Human MicroNucleus (HUMN) international collaborative project showed that smokers did not have an overall increase in the frequency of micronuclei in their lymphocytes. However, when the interaction with occupational exposure was considered, smokers of 30 cigarettes or more per day had a significantly higher frequency of micronuclei than did nonsmokers (Bonassi et al., 2003). An increased frequency of micronuclei in smokers has been observed to occur preferentially in B lymphocytes and suppressor/cytotoxic T8 lymphocytes (Larramendy & Knuutila, 1991). Elevated frequencies of micronuclei have also been found in the tracheobronchial epithelium of smokers (Lippman et al., 1990). Workers exposed to tobacco while making bidi cigarettes also had an elevated frequency of micronuclei in buccal epithelium (Bagwe & Bhisey, 1993).
Sister chromatid exchange
In contrast to micronuclei, the frequency of sister chromatid exchange in peripheral lymphocytes was generally higher in smokers than in nonsmokers. Numerous studies of the frequencies of sister chromatid exchange in peripheral lymphocytes in environmentally or occupationally exposed or unexposed populations have found that cigarette smoking induces sister chromatid exchange and can be a confounding factor in occupational studies (Sarto et al., 1985; Stenstrand, 1985; Nagaya & Toriumi, 1985; Wulf et al., 1986; Husgafvel-Pursiainen, 1987; Perera et al., 1987; Bender et al., 1988; Kelsey et al., 1988; Reidy et al., 1988; Thompson et al., 1989; Nordic Study Group, 1990; Brinkworth et al., 1992; Górecka & Gorski, 1993; Lazutka et al., 1994; Rajah & Ahuja, 1995; Anderson et al., 1997; Lemasters et al., 1997; Bukvic et al., 1998; Cebulska-Wasilewska et al., 1999; Rowland & Harding, 1999). Of all the cytogenetic end-points, sister chromatid exchange is the most sensitive to the effect of smoking. One study even demonstrated that smoking was associated with higher frequency of sister chromatid exchange in peripheral lymphocytes than in bone marrow. An explanation of this finding may be that circulating lymphocytes, with an average lifespan of 4.4 years, may accumulate more DNA damage than bone-marrow cells because they have a shorter lifespan (Kao-Shan et al., 1987). However, among healthy adults, smoking may account for only 19% of the variation in frequencies of sister chromatid exchange between individuals (Husum et al., 1986). There was a decrease in sister chromatid exchange in former smokers during the first 78 days after stopping smoking; the decrease then was much slower from the 78th to the 233rd day after cessation (Sarto et al., 1987). The mechanism by which smoking induced sister chromatid exchange does not appear to involve extracellularly generated free radicals (Lee et al., 1989).
The smoking of various types of tobacco product, including bidis (Ghosh & Ghosh, 1987; Murthy et al., 1997; Yadav & Thakur, 2000a) and hookah (Yadav & Thakur, 2000b) also induced sister chromatid exchange.
Chromosomal aberrations
Studies of large populations using classical cytogenetic banding techniques for chromosomal aberrations have given mixed results. One study found that the frequencies of chromosomal aberrations were not increased by smoking (Bender et al., 1988), whereas another found that smoking caused a 10–20% increase in such aberrations (Nordic Study Group, 1990). The results of smaller studies have also been mixed although several have found significantly higher frequencies of chromosomal aberrations in lymphocytes from smokers than in those from nonsmokers (Littlefield & Joiner, 1986; Sinués et al., 1990; Tawn & Whitehouse, 2001). Molecular cytogenetic techniques, such as fluorescence in-situ hybridization, have also yielded mixed results. Some studies found that smoking:
- -
did not increase the frequency of stable or unstable aberrations, but did increase the frequency of hyperploidy (van Diemen et al., 1995);
- -
produced a marginal increase in translocation frequency (Pressl et al., 1999); or
- -
caused a significant increase in stable aberrations (translocations and insertions) (Ramsey et al., 1995).
Chromosomal aberration frequencies were found to be elevated among workers in a bidi tobacco plant who were exposed to tobacco particles and volatile constituents by the cutaneous and nasopharyngeal routes (Mahimkar & Bhisey, 1995).
Mechanistic considerations include the observation that smokers had lower levels of folic acid in their red cells than nonsmokers and this decrease in folic acid may play a role in the higher levels of chromosomal aberrations detected in smokers relative to those in nonsmokers, probably as a result of induction of common fragile sites (Chen et al., 1989). Various studies have found that exposure of peripheral lymphocytes from smokers to various mutagens such as radiations or chemical compounds in vitro resulted in higher frequencies of chromosomal aberrations than such types of exposure caused in lymphocytes from nonsmokers (Au et al., 1991; Ban et al., 1995; Strom et al., 1995; Wang, L.-E. et al., 2000; Paz-y-Miño et al., 2001). The levels of methylpurine–DNA glycosylase and 2,6-diamino-4-hydroxy-5N formamidopyrimidine (FaPy)–DNA glycosylase were higher in the peripheral blood leukocytes of smokers than in those of nonsmokers (Hall et al., 1993). Collectively, these studies suggested that the cells of smokers, especially those from men, were less able to repair DNA damage and that DNA repair enzyme levels, fragile sites and telomeric associations can be affected by recent exposure.
A large international study showed that an elevated frequency of chromosomal aberrations in lymphocytes predicted the risk for cancer independently of exposure to carcinogens, including cigarette smoke (Bonassi et al., 2000). However, many studies have demonstrated an association between smoking and certain genetic changes specifically predictive of various types of tumour. For example, in comparison with those of nonsmokers, the lymphocytes of smokers had higher frequencies of fragile sites and metaphases with extensive breakage, as well as elevated expression of fragile sites at cancer break-points and oncogene sites (Kao-Shan et al., 1987). Analysis of normal bronchial epithelium from smokers using fluorescence in-situ hybridization found a considerable percentage of cancer-free tobacco smokers with trisomy 7 (Lechner et al., 1997), and the frequence of loss of heterozygosity involving microsatellite DNA at three specific loci-chromosomes was significantly elevated at chromosomal sites containing putative tumour-suppressor genes in histologically normal bronchial epithelium from chronic smokers (Mao et al., 1997). Perhaps most importantly, the fractional allelic loss or gain occurred at a much higher frequency in lung tumours from smokers (48%) than in those from nonsmokers (11%), suggesting that lung cancer in smokers resulted from genetic alterations distinct from those resulting in lung cancer in nonsmokers (Sanchez-Cespedes et al., 2001).
Other genetic changes
Microsatellite instability in colon tumours (Slattery et al., 2000) and chromosome 9 alterations in bladder tumours (Zhang et al., 1997) have been associated with cigarette smoking. Smoking has also been associated with mutagen sensitivity of lymphocytes as a predictor of upper aerodigestive tract cancer (Spitz et al., 1993). In another study, smoking was found not to be significantly associated with mutagen sensitivity of lymphocytes as an indication of predisposition to oral premalignant lesions (Wu et al., 2002). Various cytogenetic changes and smoking have been associated with risk for leukaemia and other myelodysplastic syndromes (Sandler et al., 1993; Davico et al., 1998; Björk et al., 2000, 2001; Moorman et al., 2002).
DNA strand breaks and oxidative damage
A higher frequency of DNA strand breaks detected by the single-cell gel electrophoresis ‘comet’ assay has been found in lymphocytes (Einhaus et al., 1994; Piperakis et al., 1998; Poli et al., 1999), buccal cells (Rojas et al., 1996) and urothelial cells (Gontijo et al., 2001) of cigarette smokers than in those of nonsmokers. Smoking also increased the level of F(2)-isoprostanes (one of the lipid peroxidation biomarkers), an index of oxidant stress (Dietrich et al., 2002).
The role of smoking-induced oxidative DNA damage is discussed elsewhere in this monograph (see Section 4.1.1(c)).
(v) Chemoprevention of the formation of smoking-associated biomarkers of genotoxicity
In humans, the administration of N-acetylcysteine to smokers significantly reduced the level of urinary mutagenicity (De Flora et al., 1996) as well as the frequency of micronuclei in the mouth floor and in the soft palate cells (Van Schooten et al., 2002). Administration of vitamins C and E as antioxidants to smokers also reduced the frequency of micronuclei in blood lymphocytes (Schneider et al., 2001). Higher intakes of vitamin A and selenium were associated with a reduced frequency of sister chromatid exchange in smokers (Cheng et al., 1995). However, administration to smokers of the anti-carcinogenic dithiolethione, oltipraz, had no influence on the levels of urinary mutagenicity (Camoirano et al., 2001). Similarly, supplementary niacin (nicotinic acid precursor of NAD+, a substrate for the DNA repair-related enzyme poly(ADP-ribose)polymerase (PARP)) did not decrease the frequencies of HPRT variants or micronuclei in peripheral blood lymphocytes in smokers. However, sister chromatid exchange was increased in these cells after the same supplementation (Hageman et al., 1998). Thus, the levels of smoking-associated urinary mutagenicity and micronuclei can be reduced by appropriate chemoprevention.
(b) Mutations in TP53, K-RAS and related genes
Smoking is associated with cancer of various organs, and mutations in these smoking-associated tumours have been identified in both oncogenes and tumour-suppressor genes. The gene most frequently found to be mutated in smoking-associated lung tumours is TP53, and the studies of this observation have been extensively reviewed (Hernandez-Boussard & Hainaut, 1998; Hainaut & Pfeifer, 2001; Hussain et al., 2001). Briefly, TP53 mutations are more common in smokers than in nonsmokers, and the frequency of TP53 mutations shows a direct correlation with the number of cigarettes smoked. TP53 mutations are found in preneoplastic lesions of the lung, indicating that they are early events that are linked temporally to DNA damage caused by smoking.
The TP53 mutation spectrum in lung tumours of smokers contains 30% GC→TA transversions, whereas only 10% of the TP53 mutations in nonsmokers or other tumours are of this type. This percentage in lung exceeds 75% only after exposure resulting from the use of a PAH-rich smoky coal in poorly ventilated homes (DeMarini et al., 2001). The elevated frequency of GC→TA transversion in smokers reflects the type of DNA damage and resulting mutations produced by PAHs, which are important carcinogenic components of cigarette smoke. There is a precise correspondence between the mutational hot spots and the sites of DNA adducts remaining after cells exposed to BPDE and other diol epoxides have undergone a period of DNA repair (Denissenko et al., 1996; Smith et al., 2000). These mutations are targeted at methylated CpG sites. There is a bias for most of the mutated guanines of the GC→TA mutations to be in the nontranscribed DNA strand in lung tumours from smokers. This bias results from the preferential binding and slow repair of BPDE adducts formed on the nontranscribed strand at mutational hot spot sites in the TP53 gene and therefore the preferential repair of DNA adducts on the transcribed strand (Denissenko et al., 1998). Taken together, these and other data indicate strongly that the TP53 mutations in lung tumours of smokers are a result of direct DNA damage caused by the carcinogens in cigarette smoke. Although one report argued that TP53 mutations in smoking-associated lung cancer were not induced by mutagens in cigarette smoke, but were pre-existing mutations selected by physiological, non-genotoxic stress (Rodin & Rodin, 2000), subsequent analysis has refuted this proposal (Hainaut & Pfeifer, 2001).
Mutations at the K-RAS or (KRAS 2) gene (codons 12, 13 or 61) occur in ∼30% of the lung adenocarcinomas of smokers and are primarily GC→TA transversions as seen in TP53 (Slebos et al., 1991; Husgafvel-Pursiainen et al., 1993; Westra et al., 1993; Gealy et al., 1999; Ahrendt et al., 2001). These mutations are associated with smoking and occur less frequently in nonsmokers (Marchetti et al., 1998; Gealy et al., 1999).
Mutations in TP53 and other genes, such as FHIT1, BCL-2 and BAX, loss of heterozygosity at specific chromosomal locations and gene overexpression (TP53, MDM2) have also been characterized in smoking-associated tumours, including those of the bladder, oral cavity and breast (Spruck et al., 1993; Schreiber et al., 1997; Sozzi et al., 1997; Baral et al., 1998; Kaur et al., 1998; Dosaka-Akita et al., 1999; Gealy et al., 1999; Tseng et al., 1999; LaRue et al., 2000; Hirao et al., 2001; Conway et al., 2002). Collectively, these observations are consistent with the mutagenic effects of cigarette smoke condensate and tobacco smoke as demonstrated in experimental systems.
(c) Influence of polymorphisms in carcinogen-metabolizing genes on smoking-associated biomarkers
Numerous polymorphisms have been studied for their modulating effect on smoking-associated biomarkers, such as urinary mutagenicity or HPRT mutant frequency. However, much of this literature reports studies using small populations, and many studies have either not been repeated, or conflicting results have been obtained in repeated studies (Vineis et al., 1999). Where effects were observed, they were frequently quite modest. Because of the incomplete and/or conflicting nature of this literature, firm conclusions cannot be drawn regarding the modulating effects of polymorphisms on smoking-associated biomarkers.
4.4.2. Experimental systems
(a) Genotoxic effects, mutagenicity of cigarette-smoke condensate, urinary mutagenicity and mutation spectra
As reviewed previously (IARC, 1986), cigarette-smoke condensate (CSC) is mutagenic in a variety of systems. Most studies have used condensate generated from various reference cigarettes, such as K1R4F, which was developed jointly by the US National Cancer Institute, the US Department of Agriculture and the University of Kentucky Tobacco and Health Research Institute (Steele et al., 1995). The average mutagenicity of cigarette-smoke condensates of mainstream smoke from US commercial brands and K1R4F reference cigarettes in the Salmonella mutagenicity assay (on a revertants/mg condensate or revertants/cigarette basis) was not significantly different between cigarettes representing > 70% of the US cigarette market (Steele et al., 1995). Similar results were recently obtained in the studies of the K1R5F cigarette (Chepiga et al., 2000), indicating that these reference cigarettes are acceptable standards with which to compare mutagenicity of cigarettes typically purchased in the USA.
More recent studies have confirmed and extended the initial observations showing that cigarette-smoke condensate is mutagenic in Salmonella and SOS assays (Morin et al., 1987; Ong et al., 1987; Chen & Lee, 1996), induces micronuclei in Vicia faba root tips (Ji & Chen, 1996), deletions at the Tk+/– locus in mouse lymphoma cells (Cobb et al., 1989), Hprt mutants in Chinese hamster ovary cells (Jongen et al., 1985), and can transform human endocervical cells in culture to a malignant line with up-regulated levels of B-Myb, p53 and WAF1 genes (Yang et al., 1997). Comparisons with wood-smoke condensate or liquid smoke food flavourings have shown that some flavourings are more cytotoxic than cigarette-smoke condensate. Nevertheless none of the liquid smoke food flavourings or wood-smoke condensates was mutagenic in an assay using S. typhimurium TA98 and only some of the liquid smoke food flavourings tested were positive with strain TA100, while cigarette-smoke condensate was mutagenic in both strains (Putnam et al., 1999). However, the levels of exposure must be considered in evaluating the risk posed by such substances (Fitzgerald, 2001; Smith et al., 2001). The relative mutagenic potencies of organic extracts of cigarette-smoke condensate and other combustion emissions rank similarly in Salmonella, mouse lymphoma Tk+/– and mouse skin tumour-initiation assays, suggesting that the results obtained using these different systems are reasonably comparable (Williams & Lewtas, 1985).
Most of the sister chromatid exchange-inducing ability of cigarette-smoke condensate appears to reside in its neutral and highly polar, acidic/neutral fractions (Curvall et al., 1985; Salomaa et al., 1988); the sister chromatid exchange response of the acidic/neutral fraction was observed only after metabolic activation, suggesting that the PAHs and acidic compounds in cigarette-smoke condensate are responsible for this activity. Also, the acidic fraction was the most potent direct-acting fraction that induced mutations involving deletions and/or chromosomal loss by non-disjunction in mammalian AL hybrid cells of CHOK1, a Chinese hamster ovary cell containing one human chromosome 11 (Matsukura et al., 1991). The acid fraction included phenolic compounds such as catechol and hydroquinone (Jansson et al., 1986) that may generate free radicals (quinone and oxygen radicals) that could have produced the clastogenic effects observed. Reconstruction of a mixture containing the primary PAHs in cigarette-smoke condensate failed to reproduce the mutagenic activity of the condensate, suggesting that PAHs were not the primary cause of the activity (Asita et al., 1991).
Nicotine and its metabolites were not mutagenic in Salmonella, did not induce sister chromatid exchange in Chinese hamster ovary cells in a study by Doolittle et al. (1995) and nicotine did not produce mutagenic urine in rats (Doolittle et al., 1991). However, nicotine was reported to induce sister chromatid exchange in Chinese hamster ovary cells (Trivedi et al., 1990). Although cigarette-smoke condensate contains a wide variety of agents of varying toxicity (Smith & Hansch, 2000), varying the amounts of 333 ingredients (e.g. casing materials, volatile top flavourings and menthol) added to typical commercially blended test cigarettes did not add significantly to the overall mutagenicity, cytotoxicity or inhalation toxicity of the resulting cigarettes (Carmines, 2002; Roemer et al., 2002).
Several lines of evidence indicate that the primary source of mutagenic activity of cigarette-smoke condensate in the Salmonella mutagenicity assay is the aromatic amine and heterocyclic amine protein pyrolysate products. As reviewed previously (DeMarini, 1983; Obe, 1984; IARC, 1986), most of the mutagenic activity of this condensate resides in the basic or base/neutral fraction (Austin et al., 1985; Salomaa et al., 1988), which contains the aromatic amines and heterocyclic amines. The removal of protein and peptides from flue-cured or burley tobacco using water extraction followed by protease digestion reduced the mutagenicity of the resultant condensate by ∼80% in S. typhimurium TA98 and ∼50% in S. typhimurium TA100 (Clapp et al., 1999). Condensates produced from tobacco smoke aerosols generated at temperatures below 400 °C or 475 °C were not mutagenic in TA98 or TA100, respectively, but those produced above those temperatures were (White et al., 2001). Heterocyclic amine pyrolysate products from proteins are formed only at high temperatures, and the fact that mutagenic activity is found only in cigarette-smoke condensate produced at high temperatures indicates that such compounds are important contributors to this mutagenic effect. Exposure of hamsters to cigarette smoke enhanced the ability of their livers to convert heterocyclic amines to mutagens, suggesting that cigarette smoke, like heterocyclic amines themselves, induces cytochrome P4501A2. Therefore, this induced isoform metabolizes heterocyclic amines (Mori et al., 1995).
Heterocyclic amines are much more mutagenic in S. typhimurium TA98 than in TA100. This probably explains the greatly reduced mutagenic activity of cigarette-smoke condensate in TA98 relative to TA100 when protein was removed from the tobacco prior to combustion (Clapp et al., 1999). In addition, some of the activity detected in TA100 may be due to PAHs, which are more mutagenic in TA100 than in TA98. At the molecular level, the mutation spectrum of cigarette-smoke condensate in TA98 was identical to that of the heterocyclic amine Glu-P-1 (2-amino-6-methyldipyrido[1,2-a:3′,2′-d]imidazole), suggesting that this class of compound was primarily responsible for the frameshift mutagenic activity of the condensate detected in TA98 (DeMarini et al., 1995). In contrast, most of the mutations (78%) induced by CSC in TA100 were GC→TA transversions; this most closely resembled the mutation spectrum of a model PAH, benzo[a]pyrene (DeMarini et al., 1995). GC→TA transversions are not only the primary class of base-substitution induced by cigarette-smoke condensate in TA100, but are also a primary class of base-substitution found in lung tumours of cigarette smokers (see Section 4.4.1(b)).
(b) Cytogenetic effects of cigarette smoke and its condensate in vitro
Data reviewed previously (IARC, 1986) and confirmed by Rutten and Wilmer (1986) have shown that cigarette smoke and its condensate induced sister chromatid exchange in mammalian cells in culture. Tobacco particulate matter has also been shown to induce structural and numerical chromosomal aberrations (Lafi & Parry, 1988) and micronuclei (Jones et al., 1991) in cultured mammalian cells. Based on the lack of association between micronuclei induction and DNA adducts, Jones et al. (1991) suggested that the micronuclei were not induced by aromatic compounds but, perhaps, by a direct-acting agent that may induce small alkylations or damage caused by radicals in DNA. Jansson et al. (1986, 1988) and Curvall et al. (1985) showed that the neutral fractions and weakly acidic, semi-volatile components of cigarette-smoke condensate were the most potent inducers of sister chromatid exchange in cultured human lymphocytes. They identified a number of potential candidate compounds, most of which were alkylphenols and benzaldehydes (benzenes having vicinal oxygenation or a conjugated double bond), that may be responsible for this activity. Whole smoke also induced micronuclei in Chinese hamster V79 cells without a metabolic activation system (S9) (Massey et al., 1998). In another study based on immunocytochemical antikinetochore staining, which allowed differentiation between the clastogenic and aneugenic action of cigarette smoke, V79 cells exposed to cigarette smoke appeared to be kinetochore-positive, suggesting that the micronuclei formed were due to aneuploidy rather than clastogenicity (Veltel & Hoheneder, 1996).
(c) Cytogenetic effects of cigarette smoke in vivo
As noted in the earlier review (IARC, 1986), exposure of rodents to cigarette smoke has generally produced sister chromatid exchange in the bone marrow. However, studies on the induction of chromosomal aberrations in pulmonary alveolar macrophages by exposure of rodents to cigarette smoke have produced some negative results (Lee, C.K. et al., 1992, 1993) and one positive result (Rithidech et al., 1989). Exposure of rodents to cigarette smoke has consistently produced micronuclei in polychromatic erythrocytes of the bone marrow (Balansky et al., 1987; Mohtashamipur et al., 1987; Balansky et al., 1988; Mohtashamipur et al., 1988; Stoichev et al., 1993; Nersessian & Arutyunyan, 1994; Balansky et al., 1999), in normochromatic erythrocytes in peripheral blood (Balansky et al., 1988, 1999) and in pulmonary alveolar macrophages (Balansky et al., 1999).
(d) DNA strand breaks
Several studies have demonstrated that both cigarette smoke and its condensate can induce DNA strand breaks either in rodents, mammalian cells in culture or in DNA in vitro (Nakayama et al., 1985; Willey et al., 1987; Fielding et al., 1989; Leanderson & Tagesson, 1990; Bermudez et al., 1994; Leanderson & Tagesson, 1994; Spencer et al., 1995; Seree et al., 1996; Yoshie & Ohshima, 1997; Liu et al., 1999; Yang, Q. et al., 1999). Cigarette smoke also induced nuclear accumulation of TP53 protein in mouse cells in culture, providing an indirect indication of DNA damage (Hess & Brandner, 1996). Collectively, these studies are consistent with the demonstrated clastogenicity of cigarette smoke and its condensate in experimental systems and humans. Several of these studies have indicated that reactive oxygen or nitrogen species formed in cigarette smoke and its condensate are the primary cause of the strand breaks.
(e) Studies on cigarettes that primarily heat but not burn tobacco, and modified cigarettes
Although cigarettes that primarily heat rather than burn tobacco (Borgerding et al., 1998) are not generally available commercially, studies generally found that their smoke condensate was not mutagenic in Salmonella and did not induce sister chromatid exchange, chromosomal aberrations or Hprt mutations in Chinese hamster ovary cells or unscheduled DNA synthesis in rat cells (Doolittle et al., 1989; deBethizy et al., 1990; Doolittle et al., 1990b; Bombick, B.R. et al., 1997). Whole smoke from such cigarettes was also either non-mutagenic or only slightly mutagenic in Salmonella and a weak inducer of sister chromatid exchange in Chinese hamster ovary cells, and it was negative in rat bone marrow for chromosomal aberrations, micronuclei and sister chromatid exchange. The relative cytotoxic and genotoxic potential observed in Chinese hamster ovary cells exposed to the whole smoke from these cigarettes is likely to reside in the vapour phase (Lee et al., 1990a,b; Bombick et al., 1998). Humans who smoked these cigarettes produced urine that was ∼70% less mutagenic than the urine of smokers of standard cigarettes (Doolittle et al., 1990b; Smith et al., 1996; Bowman et al., 2002).
In 1990, ∼20% of the ∼5200 deaths from fires in the USA occurred in those started by cigarettes (Brunnemann et al., 1994). The smoke condensate from an experimental cigarette that has low-ignition propensity, and is therefore less likely than regular cigarettes to ignite surrounding material, was as mutagenic as that of reference cigarettes (Brunnemann et al., 1994). An experimental carbon filter that reduces the amounts of certain vapour-phase components of tobacco smoke relative to charcoal filters currently used in commercial cigarettes produced whole smoke that was less mutagenic in Salmonella and induced less sister chromatid exchange in Chinese hamster ovary cells than smoke from cigarettes with the usual type of charcoal filter; however, the genotoxicity of the resulting smoke condensate was not significantly different from that of the condensate from standard cigarettes (Bombick, D.W. et al., 1997).
Masheri, a pyrolysed tobacco product mainly used in India to clean the teeth, induced chromosomal aberrations, sister chromatid exchange and micronuclei in bone-marrow cells of mice and Hprt mutations in Chinese hamster lung fibroblast V79 cells as well as mutations in Salmonella (Kulkarni et al., 1987).
(f) Transplacental effects
Exposure of gestating mice to cigarette smoke showed that fetal liver, and peripheral blood and liver of newborn mice had elevated levels of micronuclei (Balansky & Blagoeva, 1989), and such exposure also induced sister chromatid exchange in mouse fetal liver (Karube et al., 1989). Therefore, tobacco smoke contains clastogens that cross the mouse placental barrier and cause chromosomal damage in erythroblasts in fetal liver. Both tobacco smoke and its condensate have been shown to induce DNA deletions (reversion of the pink-eye unstable mutation) in the embryo of exposed mice (Jalili et al., 1998).
(g) Modulation of genotoxicity of tobacco smoke and cigarette-smoke condensate
Many studies have examined the ability of various agents to modulate the genotoxicity of tobacco smoke or its condensate in vitro or in experimental animals. Studies in rodents exposed to tobacco smoke (De Flora et al., 2003) or those in Salmonella exposed to cigarette-smoke condensate (Camoirano et al., 1994; Romert et al., 1994) have identified a variety of agents that either inhibit or modulate the genotoxicity of tobacco smoke and/or its condensate. Such agents include N-acetylcysteine, chlorophyllin, phenolic compounds, isothiocyanates, dithiocarbamates, indoles, tetrapyroles and flavonoids. As noted in Section 4.4.1(a)(v), some of these agents have been shown to modulate the formation of smoking-associated biomarkers in humans.
4.5. Mechanistic considerations
Classically, tobacco smoke has been described as acting both as a tumour initiator and a tumour promoter (IARC, 1986). In the light of recent developments in the understanding of the molecular effects of tobacco smoke, this knowledge can be extended to describe how genetic and epigenetic changes cooperate in tobacco-induced carcinogenesis.
The modern synthesis of molecular biology and cancer biology identifies at least six major pathways that must be disrupted for a normal cell to become a tumour cell. These include self-sufficiency in growth signals, insensitivity to anti-growth signals, evasion of apoptosis, tissue invasion and metastasis, sustained angiogenesis and limitless replicative potential. Disruption of these major pathways can occur through genetic or genomic alterations in well-defined genes or through a number of epigenetic processes, including methylation of DNA, post-translational modifications of proteins and modification of gene expression patterns (Hanahan & Weinberg, 2000). There is evidence that smoking is associated with some of the genetic and epigenetic changes affecting these major pathways (Heusch & Maneckjee, 1998; Aoshiba et al., 2001; Brandau & Bohle, 2001; Chang et al., 2001; Forgacs et al., 2001; Hittelman, 2001; Jull et al., 2001; Kang & Park, 2001; Kim, D.H. et al., 2001; Wistuba et al., 2001). Some of these genetic and epigenetic changes have also been observed in smoking-associated diseases other than cancer, such as atherosclerosis and rheumatoid arthritis, indicating that these diseases share common pathways with cancer, and that these pathways are disrupted by tobacco smoke (Chen & Loo, 1995; Albano et al., 2001; Wang, H. et al., 2001).
Conceptually, the effects of tobacco smoke as a carcinogen can be viewed as the result of both genetic and epigenetic changes. Although epigenetic changes can trigger a complex, suppressive cellular stress response, genetic changes may endow some cells with a capacity to escape normal immunosuppression. Thus, the changes in the cellular environment inflicted by tobacco smoke may produce a selection procedure that favours the emergence of cells that have acquired the capacity to undergo clonal expansion. This phenomenon can occur simultaneously at several sites within an exposed tissue field, resulting in multifocal lesions, some of which can progress to cancer (field carcinogenesis).
Moreover, there is a remarkable convergence between the molecular changes induced by tobacco smoke summarized here, such as DNA damage and mutation, and recent experimental evidence for mechanisms of carcinogenesis. Collectively, these data support a multistep model of carcinogenesis in which the components of tobacco smoke are the direct cause of the cellular changes that accumulate to drive the carcinogenenic process.
The previous IARC Monographs on tobacco smoking (IARC, 1986) provided convincing evidence of the carcinogenicity of tobacco smoke. The present volume extends this evidence and provides compelling molecular data that explain some of the mechanisms by which tobacco smoke is carcinogenic. Most importantly, these data have largely been obtained from studies in humans, rather than in experimental animals. Thus, their relevance cannot be denied, and their explanatory powers cannot be easily dismissed.
Footnotes
- 1
There is an equilibrium (50–90%) between 8-oxo-dG and 8-OHdG. The compound will be called 8-OHdG throughout this document.
- 1
[Aminobiphenyls in tobacco have been implicated in bladder cancer etiology in smokers. 3- and 4-ABP–haemoglobin adduct levels are considered valid biomarkers of the internal dose of ABP to the bladder (Probst-Hensch et al., 2000).]
- 1
N7-(benzo[a]pyrene-6-yl) adenine
- 1
4-ABP–DNA adduct: N-(deoxyguanosin-8-yl)-4-aminobiphenyl–DNA adduct
- 2
4-ABP–haemoglobin adduct: N-(deoxyguanosin-8-yl)-4-aminobiphenyl–haemoglobin adduct
- 1
FHIT: fragile histidine triad, a gene coding for a dinucleoside 5′,5′″-P1,P3-triphosphate hydrolase, a putative tumour-suppressor protein
References
- Ahrendt S.A., Decker P.A., Alawi E.A., Zhu Y.-R., Sanchez-Cespedes M., Yang S.C., Haasler G.B., Kajdacsy-Balla A., Demeure M.J., Sidransky D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. [PubMed: 11745231]
- Akhmedkhanov A., Zeleniuch-Jacquotte A., Toniolo P. Role of exogenous and endogenous hormones in endometrial cancer: Review of the evidence and research perspectives. Ann. N.Y. Acad. Sci. 2001;943:296–315. [PubMed: 11594550]
- Albano S.A., Santana-Sahagun E., Weisman M.H. Cigarette smoking and rheumatoid arthritis. Semin. Arthritis Rheum. 2001;31:146–159. [PubMed: 11740796]
- Alexandrie A.-K., Warholm M., Carstensen U., Axmon A., Hagmar L., Levin J.O., Östman C., Rannug A. CYP1A1 and GSTM1 polymorphisms affect urinary 1-hydroxypyrene levels after PAH exposure. Carcinogenesis. 2000;21:669–676. [PubMed: 10753202]
- Alexandrov K., Rojas M., Geneste O., Castegnaro M., Camus A.M., Petruzzelli S., Giuntini C., Bartsch H. An improved fluorometric assay for dosimetry of benzo(a)pyrene diolepoxide-DNA adducts in smokers' lung: Comparisons with total bulky adducts and aryl hydrocarbon hydroxylase activity. Cancer Res. 1992;52:6248–6253. [PubMed: 1423269]
- Alexandrov K., Rojas M., Kadlubar F.F., Lang N.P., Bartsch H. Evidence of antibenzo[a]pyrene diolepoxide–DNA adduct formation in human colon mucosa. Carcinogenesis. 1996;17:2081–2083. [PubMed: 8824539]
- Ali S., Astley S.B., Sheldon T.A., Peel K.R., Wells M. Detection and measurement of DNA adducts in the cervix of smokers and nonsmokers. Int. J. Gynecol. Cancer. 1994;4:188–193. [PubMed: 11578405]
- Al-Saleh I.A. Lead exposure in Saudi Arabia and its relationship to smoking. Biometals. 1995;8:243–245. [PubMed: 7647521]
- Al-Saleh I., Shinwari N., Basile P., Al-Dgaither S., Al-Mutairi M. Exposure to cadmium among sheesah smokers and how do they compare to cigarrete smokers. J. trace Elements exp. Med. 2000;13:381–388.
- Ammenheuser M.M., Berenson A.B., Stiglich N.J., Whorton E.B., Ward J.B. Elevated frequencies of hprt mutant lymphocytes in cigarette-smoking mothers and their newborns. Mutat. Res. 1994;304:285–294. [PubMed: 7506372]
- Ammenheuser M.M., Hastings D.A., Whorton E.B., Ward J.B. Frequencies of hprt mutant lymphocytes in smokers, non-smokers, and former smokers. Environ. mol. Mutag. 1997;30:131–138. [PubMed: 9329637]
- Ammenheuser M.M., Berenson A.B., Babiak A.E., Singleton C.R., Whorton E.B. Frequencies of hprt mutant lymphocytes in marijuana-smoking mothers and their newborns. Mutat. Res. 1998;403:55–64. [PubMed: 9726006]
- Anand C.V., Anand U., Agarwal R. Anti-oxidant enzymes, gamma-glutamyl transpeptidase and lipid peroxidation in kidney of rats exposed to cigarette smoke. Indian J. exp. Biol. 1996;34:486–488. [PubMed: 9063083]
- Andersen F.S., Transbol I., Christiansen C. Is cigarette smoking a promotor of the menopause? Acta med. scand. 1982;212:137–139. [PubMed: 7148504]
- Anderson D., Hughes J.A., Nizankowska E., Graca B., Cebulska-Wasilewska A., Wierzewska A., Kasper E. Factors affecting various biomarkers in untreated lung cancer patients and healthy donors. Environ. mol. Mutag. 1997;30:205–216. [PubMed: 9329645]
- Andersson K., Fuxe K., Eneroth P., Mascagni F., Agnati L.F. Effects of acute intermittent exposure to cigarette smoke on catecholamine levels and turnover in various types of hypothalamic DA and NA nerve terminal systems as well as on the secretion of adenohypophyseal hormones and corticosterone. Acta physiol. scand. 1985;124:277–285. [PubMed: 2861716]
- Andreassen Å., Kure E.H., Nielsen P.S., Autrup H., Haugen A. Comparative synchronous fluorescence spectrophotometry and 32P-postlabeling analysis of PAH–DNA adducts in human lung and the relationship to TP53 mutations. Mutat. Res. 1996;368:275–282. [PubMed: 8692233]
- Andreoli R., Manini P., Bergamaschi E., Mutti A., Franchini I., Niessen W.M.A. Determination of naphthalene metabolites in human urine by liquid chromatography-mass spectrometry with electrospray ionization. J. Chromatogr. 1999;A847:9–17. [PubMed: 10431347]
- Andres R.L., Day M.-C. Perinatal complications associated with maternal tobacco use. Semin. Neonatol. 2000;5:231–241. [PubMed: 10956448]
- Angerer J., Mannschreck C., Gündel J. Biological monitoring and biochemical effect monitoring of exposure to polycyclic aromatic hydrocarbons. Int. Arch. occup. environ. Health. 1997;70:365–377. [PubMed: 9439982]
- Anttila S., Hietanen E., Vainio H., Camus A.-M., Helboin H.V., Park S.S., Heikkilä L., Karjalainen A., Bartsch H. Smoking and peripheral type of cancer are related to high levels of pulmonary cytochrome P450IA in lung cancer patients. Int. J. Cancer. 1991;47:681–685. [PubMed: 1848536]
- Aoshiba K., Tamaoki J., Nagai A. Acute cigarette smoke exposure induces apoptosis of alveolar macrophages. Am. J. Physiol. Lung cell. mol. Physiol. 2001;281:L1392–L1401. [PubMed: 11704535]
- Aringer L., Lidums V. Influence of diet and other factors on urinary levels of thioethers. Int. Arch. occup. environ. Health. 1988;61:123–130. [PubMed: 3198277]
- Armstrong, R.N. (1997) Glutathione transferases. In: Guengerich, F.P., ed., Comprehensive Toxicology, Vol. 3, Biotransformation, New York, Elsevier Science, pp. 307–327.
- Arnould J.P., Verhoest P., Bach V., Libert J.P., Belegaud J. Detection of benzo[a]pyrene–DNA adducts in human placenta and umbilical cord blood. Hum. exp. Toxicol. 1997;16:716–721. [PubMed: 9429085]
- Arnould J. P., Hermant A., Levi-Valensi P., Belegaud J. Detection of benzo[a]pyrene-DNA adducts from leukocytes from heavy smokers. Pathol. Biol. 1998;46:787–790. [PubMed: 9922995]
- Arora A., Willhite C.A., Liebler D.C. Interactions of β-carotene and cigarette smoke in human bronchial epithelial cells. Carcinogenesis. 2001;22:1173–1178. [PubMed: 11470745]
- Asami S., Hirano T., Yamaguchi R., Tomioka Y., Itoh H., Kasai H. Increase of a type of oxidative DNA damage, 8-hydroxyguanine, and its repair activity in human leukocytes by cigarette smoking. Cancer Res. 1996;56:2546–2549. [PubMed: 8653695]
- Asami S., Manabe H., Miyake J., Tsurudome Y., Hirano T., Yamaguchi R., Itoh H., Kasai H. Cigarette smoking induces an increase in oxidative DNA damage, 8-hydroxydeoxyguanosine, in a central site of the human lung. Carcinogenesis. 1997;18:1763–1766. [PubMed: 9328173]
- Ashley M.J. Smoking and diseases of the gastrointestinal system: An epidemiological review with special reference to sex differences. Can. J. Gastroenterol. 1997;11:345–352. [PubMed: 9218861]
- Ashley D.L., Bonin M.A., Hamar B., McGeehin M.A. Removing the smoking confounder from blood volatile organic compounds measurements. Environ. Res. 1995;71:39–45. [PubMed: 8757237]
- Ashley D.L., Bonin M.A., Cardinali F.L., McCraw J.M., Wooten J.V. Measurement of volatile organic compounds in human blood. Environ. Health Perspect. 1996;104:871–877. [PMC free article: PMC1469701] [PubMed: 8933028]
- Asita A.O., Matsui M., Nohmi T., Matsuoka A., Hayashi M., Ishidate M. Jr, Sofuni T., Koyano M., Matsushita H. Mutagenicity of wood smoke condensates in the Salmonella/microsome assay. Mutat. Res. 1991;264:7–14. [PubMed: 1881415]
- Atawodi S.E., Lea S., Nyberg F., Mukeria A., Constantinescu V., Ahrens W., Brueske-Hohlfeld I., Fortes C., Boffetta P., Friesen M.D. 4-Hydroxy-1-(3-pyridyl)-1-butanone–hemoglobin adducts as biomarkers of exposure to tobacco smoke: Validation of a method to be used in multicenter studies. Cancer Epidemiol. Biomarkers Prev. 1998;7:817–821. [PubMed: 9752992]
- Attolini I.., Gantenbein M., Villard P.H., Lacarelle B., Catalin J., Bruguerolle B. Effects of different exposure times to tobacco smoke intoxication on carboxyhemoglobin and hepatic enzymate activities in mice. J. pharmacol. toxicol. Meth. 1996;35:211–215. [PubMed: 8823667]
- Au W.W., Walker D.M., Ward J.B., Whorton E., Legator M.S., Singh V. Factors contributing to chromosome damage in lymphocytes of cigarette smokers. Mutat. Res. 1991;260:137–144. [PubMed: 2046694]
- Aubry M.C., Wright J.L., Myers J.L. The pathology of smoking-related lung diseases. Clin. Chest Med. 2000;21:11–35. [PubMed: 10763087]
- Augood C., Duckitt K., Templeton A.A. Smoking and female infertility: A systematic review and meta-analysis. Hum. Reprod. 1998;13:1532–1539. [PubMed: 9688387]
- Austin A.C., Claxton L.D., Lewtas J. Mutagenicity of the fractionated organic emissions from diesel, cigarette smoke condensate, coke oven, and roofing tar in the Ames assay. Environ. Mutag. 1985;7:471–487. [PubMed: 2414094]
- Bader M., Lewalter J., Angerer J. Analysis of N-alkylated amino acids in human hemoglobin: Evidence for elevated N-methylvaline levels in smokers. Int. Arch. occup environ. Health. 1995;67:237–242. [PubMed: 7591184]
- Bagnasco M., Bennicelli C., Camoirano A., Balansky R.M., De Flora S. Metabolic alterations produced by cigarette smoke in rat lung and liver, and their modulation by oral Nacetylcysteine. Mutagenesis. 1992;7:295–301. [PubMed: 1518414]
- Bagwe A.N., Bhisey R.A. Occupational exposure to tobacco and resultant genotoxicity in bidi industry workers. Mutat. Res. 1993;299:103–109. [PubMed: 7680424]
- Bailey E., Brooks A.G.F., Dollery C. T., Farmer P.B., Passingham B.J., Sleightholm M.A., Yates D.W. Hydroxyethylvaline adduct formation in haemoglobin as a biological monitor of cigarette smoke intake. Arch. Toxicol. 1988;62:247–253. [PubMed: 3240090]
- Baird, D.D. (1992) Evidence for reduced fecundity in female smokers. In: Poswillo, D. & Alberman, E., eds, Effects of Smoking on the Fetus, Neonate, and Child, Oxford, Oxford University Press, pp. 5–22.
- Balansky R.M., Blagoeva P.M. Tobacco smoke-induced clastogenicity in mouse fetuses and in newborn mice. Mutat. Res. 1989;223:1–6. [PubMed: 2716759]
- Balansky R.M., Blagoeva P.M., Mircheva Z.I. Investigation of the mutagenic activity of tobacco smoke. Mutat. Res. 1987;188:13–19. [PubMed: 3553921]
- Balansky R.M., Blagoeva P.M., Mircheva Z.I. The mutagenic and clastogenic activity of tobacco smoke. Mutat. Res. 1988;208:237–241. [PubMed: 3041274]
- Balansky R.M., D'Agostini F., De Flora S. Induction, persistence and modulation of cytogenetic alterations in cells of smoke-exposed mice. Carcinogenesis. 1999;20:1491–1497. [PubMed: 10426797]
- Balint B., Donnelly L.E., Hanazawa T., Kharitonow S.A., Barnes P.J. Increased nitric oxide metabolites in exhaled breath condensate after exposure to tobacco smoke. Thorax. 2001;56:456–461. [PMC free article: PMC1746081] [PubMed: 11359961]
- Ban S., Cologne J.B., Neriishi K. Effect of radiation and cigarette smoking on expression of FUdR-inducible common fragile sites in human peripheral lymphocytes. Mutat. Res. 1995;334:197–203. [PubMed: 7885372]
- Banaszewski J., Szmeja Z., Szyfter W., Szyfter K., Baranczewski P., Möller L. Analysis of aromatic DNA adducts in laryngeal biopsies. Eur. Arch. Otorhinolaryngol. 2000;257:149–153. [PubMed: 10839488]
- Baral R.N., Patnaik S., Das B.R. Co-overexpression of p53 and c-myc proteins linked with advanced stages of betel- and tobacco-related oral squamous cell carcinomas from eastern India. Eur. J. oral Sci. 1998;106:907–913. [PubMed: 9786319]
- Barbieri R.L., McShane P.M., Ryan K.J. Constituents of cigarette smoke inhibit human granulosa cell aromatase. Fertil. Steril. 1986;46:232–236. [PubMed: 3732529]
- Baron J.A. Beneficial effects of nicotine and cigarette smoking: The real, the possible and the spurious. Br. med. Bull. 1996;52:58–73. [PubMed: 8746297]
- Baron J.A., La Vecchia C., Levi F. The antiestrogenic effect of cigarette smoking in women. Am. J. Obstet. Gynecol. 1990;162:502–514. [PubMed: 2178432]
- Bartsch H., Ohshima H., Pignatelli B., Calmels S. Human exposure to endogenous Nnitroso compounds: Quantitative estimates in subjects at high risk for cancer of the oral cavity, oesophagus, stomach and urinary bladder. Cancer Surv. 1989;8:335–362. [PubMed: 2696584]
- Bartsch H., Caporaso N., Coda M., Kadlubar F., Malaveille C., Skipper P., Talaska G., Tannenbaum S.R., Vineis P. Carcinogen hemoglobin adducts, urinary mutagenicity, and metabolic phenotype in active and passive cigarette smokers. J. natl Cancer Inst. 1990;82:1826–1831. [PubMed: 2250298]
- Baskaran S., Lakshmi S., Prasad P.R. Effect of cigarette smoke on lipid peroxidation and antioxidant enzymes in albino rat. Indian J. exp. Biol. 1999;37:1196–1200. [PubMed: 10865887]
- Beach A.C., Gupta R.C. Human biomonitoring and the 32P-postlabeling assay. Carcinogenesis. 1992;13:1053–1074. [PubMed: 1638670]
- Becher G., Bjørseth A. Determination of exposure to polycyclic aromatic hydrocarbons by analysis of human urine. Cancer Lett. 1983;17:301–311. [PubMed: 6831387]
- Becher G., Haugen A., Bjørseth A. Multimethod determination of occupational exposure to polycyclic aromatic hydrocarbons in an aluminum plant. Carcinogenesis. 1984;5:647–651. [PubMed: 6722981]
- Bender M.A., Preston R.J., Leonard R.C., Pyatt B.E., Gooch P.C., Shelby M.D. Chromosomal aberration and sister-chromatid exchange frequencies in peripheral blood lymphocytes of a large human population sample. Mutat. Res. 1988;204:421–433. [PubMed: 3347214]
- Bennett C.H., Richardson D.R. Time-dependent changes in cardiovascular regulation caused by chronic tobacco smoke exposure. J. appl. Physiol. 1990;68:248–252. [PubMed: 2312465]
- Benowitz N.L. Pharmacologic aspects of cigarette smoking and nicotine addiction. New Engl. J. Med. 1988;319:1318–1330. [PubMed: 3054551]
- Benowitz N.L. Clinical pharmacology of transdermal nicotine. Eur. J. Pharm. Biopharm. 1995;41:168–174.
- Benowitz N.L. Cotinine as a biomarker of environmental tobacco smoke exposure. Epidemiol. Rev. 1996;18:188–204. [PubMed: 9021312]
- Benowitz N.L. Biomarkers of environmental tobacco smoke exposure. Environ. Health Perspect. 1999a;107 (Suppl. 2):349–355. [PMC free article: PMC1566286] [PubMed: 10350520]
- Benowitz N.L. Nicotine addiction. Prim. Care. 1999b;26:611–631. [PubMed: 10436290]
- Benowitz N.L., Jacob P. Metabolism of nicotine to cotinine studied by a dual stable isotope method. Clin. Pharmacol. Ther. 1994;56:483–493. [PubMed: 7955812]
- Benowitz, N.L. & Jacob, P., III (1997) Individual differences in nicotine kinetics and metabolism in humans. In: Rapaka, R.S., Chiang, N. & Martin, B.R., eds, Pharmacokinetics, Metabolism, and Pharmaceutics of Drugs of Abuse (NIDA Research Monograph No. 173), Rockville, MD, National Institute on Drug Abuse, Division of Clinical Research, pp. 48–64.
- Benowitz N.L., Jacob P. III, Jones R.T., Rosenberg J. Interindividual variability in the metabolism and cardiovascular effects of nicotine in man. J. Pharmacol. exp. Ther. 1982;221:368–372. [PubMed: 7077531]
- Benowitz N.L., Jacob P. III, Kozlowski L.T., Yu L. Influence of smoking fewer cigarettes on exposure to tar, nicotine, and carbon monoxide. New Engl. J. Med. 1986;315:1310–1313. [PubMed: 3773954]
- Benowitz N.L., Jacob P. III, Fong I., Gupta S. Nicotine metabolic profile in man: Comparison of cigarette smoking and transdermal nicotine. J. Pharmacol. exp. Ther. 1994;268:296–303. [PubMed: 8301571]
- Benowitz N.L., Jacob P. III, Sachs D.P. Deficient C-oxidation of nicotine. Clin. Pharmacol. Ther. 1995;57:590–594. [PubMed: 7768082]
- Benowitz N.L., Jacob P. III, Perez-Stable E. CYP2D6 phenotype and the metabolism of nicotine and cotinine. Pharmacogenetics. 1996;6:239–242. [PubMed: 8807663]
- Benowitz N.L., Pérez-Stable E.J., Fong I., Modin G., Herrera B., Jacob P. III. Ethnic differences in N-glucuronidation of nicotine and cotinine. J. Pharmacol. exp. Ther. 1999;291:1196–1203. [PubMed: 10565842]
- Benowitz N.L., Pérez-Stable E.J., Herrera B., Jacob P. III. Slower metabolism and reduced intake of nicotine from cigarette smoking in Chinese-Americans. J. natl Cancer Inst. 2002;94:108–115. [PubMed: 11792749]
- Bergmark E. Hemoglobin adducts of acrylamide and acrylonitrile in laboratory workers, smokers and nonsmokers. Chem. Res. Toxicol. 1997;10:78–84. [PubMed: 9074806]
- Berlin I., Anthenelli R.M. Monoamine oxidases and tobacco smoking. Int. J. Neuropsychopharmacol. 2001;4:33–42. [PubMed: 11343627]
- Bermudez E., Stone K., Carter K.M., Pryor W.A. Environmental tobacco smoke is just as damaging to DNA as mainstream smoke. Environ. Health Perspect. 1994;102:870–874. [PMC free article: PMC1567343] [PubMed: 9644196]
- Berstein L.M., Tsyrlina E.V., Gamajunova V.B., Bychkova N.V., Krjukova O.G., Dzhumasultanova S.V., Kovalenko I.G., Kolesnik O.S. Study of tobacco smoke influence on content of estrogens and DNA flow cytometry data in uterine tissue of rats of different age. Horm. metab. Res. 1999;31:27–30. [PubMed: 10077346]
- Besarati Nia A., Maas L.M., Van Breda S.G.J., Curfs D.M.J., Kleinjans J.C.S., Wouters E.F.M., Van Schooten F.J. Applicability of induced sputum for molecular dosimetry of exposure to inhalatory carcinogens: 32P-Postlabeling of lipophilic DNA adducts in smokers and nonsmokers. Cancer Epidemiol. Biomarkers Prev. 2000a;9:367–372. [PubMed: 10794480]
- Besarati Nia A., Maas L.M., Brouwer E.M.C., Kleinjans J.C.S., Van Schooten F.J. Comparison between smoking-related DNA adduct analysis in induced sputum and peripheral blood lymphocytes. Carcinogenesis. 2000b;21:1335–1340. [PubMed: 10874011]
- Besarati Nia A., Van Straaten H.W.M., Kleinjans J.C.S., Van Schooten F.-J. Immunoperoxidase detection of 4-aminobiphenyl- and polycyclic aromatic hydrocarbons–DNA adducts in induced sputum of smokers and nonsmokers. Mutat. Res. 2000c;468:125–135. [PubMed: 10882891]
- Besarati Nia A., Van Straaten H.W.M., Godschalk R.W.L., Van Zandwijk N., Balm A.J.M., Kleinjans J.C.S., Van Schooten F.J. Immunoperoxidase detection of polycyclic aromatic hydrocarbon–DNA adducts in mouth floor and buccal mucosa cells of smokers and nonsmokers. Environ. mol. Mutag. 2000d;36:127–133. [PubMed: 11013411]
- Besarati Nia A., Van Schooten F.J., Schilderman P.A.E.L., De Kok T.M.C.M., Haenen G.R., Van Herwijnen M.H.M., Van Agen E., Pachen D., Kleinjans J.C.S. A multi-biomarker approach to study the effects of smoking on oxidative DNA damage and repair and antioxidative defense mechanisms. Carcinogenesis. 2001;22:395–401. [PubMed: 11238178]
- deBethizy J.D., Borgerding M.F., Doolittle D.J., Robinson J.H., McManus K.T., Rahn C.A., Davis R.A., Burger G.T., Hayes J.R., Reynolds J.H., Hayes A.W. Chemical and biological studies of a cigarette that heats rather than burns tobacco. J. clin. Pharmacol. 1990;30:755–763. [PubMed: 2401755]
- Bhisey R.A., Govekar R.B. Biological monitoring of bidi rollers with respect to genotoxic hazards of occupational tobacco exposure. Mutat. Res. 1991;261:139–147. [PubMed: 1922157]
- Bielicki J.K., Forte T.M., McCall M.R. Gas-phase cigarette smoke inhibits plasma lecithin-cholesterol acyltransferase activity by modification of the enzyme's free thiols. Biochim. biophys. Acta. 1995;1258:35–40. [PubMed: 7654778]
- Bielicki J.K., Knoff L.J., Tribble D.L., Forte T.M. Relative sensitivities of plasma lecithin:cholesterol acyltransferase, platelet-activating factor acetylhydrolase, and paraoxonase to in vitro gas-phase cigarette smoke exposure. Atherosclerosis. 2001;155:71–78. [PubMed: 11223428]
- Bigbee W.L., Day R.D., Grant S.G., Keohavong P., Xi L., Zhang L., Ness R.B. Impact of maternal lifestyle factors on newborn HPRT mutant frequencies and molecular spectrum — Initial results from the Prenatal Exposures and Preeclampsia Prevention (PEPP) Study. Mutat. Res. 1999;431:279–289. [PubMed: 10635994]
- Binková B., Lewtas J., Míšková I., Lenícek J., Šrám R. DNA adducts and personal air monitoring of carcinogenic polycyclic aromatic hydrocarbons in an environmentally exposed population. Carcinogenesis. 1995;16:1037–1046. [PubMed: 7767962]
- Binková B., Strejc P., Boubelík O., Stávková Z., Chvátalová I., Šrám R.J. DNA adducts and human atherosclerotic lesions. Int. J. Hyg. environ. Health. 2001;204:49–54. [PubMed: 11725345]
- Bjarnason N.H., Christiansen C. The influence of thinness and smoking on bone loss and response to hormone replacement therapy in early postmenopausal women. J. clin. Endocrinol. Metab. 2000;85:590–596. [PubMed: 10690860]
- Bjelogrlic N., Iscan M., Raunio H., Pelkonen O., Vahäkangas K. Benzo[a]pyrene–DNA-adducts and monooxygenase activities in mice treated with benzo[a]pyrene, cigarette smoke or cigarette smoke condensate. Chem.-biol. Interact. 1989;70:51–61. [PubMed: 2786769]
- Björk J., Albin M., Mauritzson N., Stromberg U., Johansson B., Hagmar L. Smoking and myelodysplastic syndromes. Epidemiology. 2000;11:285–291. [PubMed: 10784245]
- Björk J., Albin M., Mauritzson N., Stromberg U., Johansson B., Hagmar L. Smoking and acute myeloid leukemia: Associations with morphology and karyotypic patterns and evaluation of dose–response relations. Leuk. Res. 2001;25:865–872. [PubMed: 11532519]
- Blömeke B., Greenblatt M.J., Doan V.D., Bowman E.D., Murphy S.E., Chen C.C., Kato S., Shields P.G. Distribution of 7-alkyl-2′-deoxyguanosine adduct levels in human lung. Carcinogenesis. 1996;17:741–748. [PubMed: 8625485]
- Bock K.W., Schrenk D., Forster A., Griese E.U., Morike K., Brockmeier D., Eichelbaum M. The influence of environmental and genetic factors on CYP2D6, CYP1A2 and UDPglucuronosyltransferases in man using sparteine, caffeine, and paracetamol as probes. Pharmacogenetics. 1994;4:209–218. [PubMed: 7987405]
- Bohning D.E., Atkins H.L., Cohn S.H. Long-term particle clearance in man: Normal and impaired. Ann. occup. Hyg. 1982;26:259–271. [PubMed: 7181269]
- Bolumar F., Olsen J., Boldsen J., the European Study Group on Infertility and Subfecundity. Smoking reduces fecundity: A European multicenter study on infertility and sub-fecundity. Am. J. Epidemiol. 1996;143:578–587. [PubMed: 8610675]
- Bombick B.R., Murli H., Avalos J.T., Bombick D.W., Morgan W.T., Putnam K.P., Doolittle D.J. Chemical and biological studies of a new cigarette that primarily heats tobacco. Part 2. In vitro toxicology of mainstream smoke condensate. Food chem. Toxicol. 1997;36:183–190. [PubMed: 9609391]
- Bombick D.W., Bombick B.R., Ayres P.H., Putnam K.P., Avalos J., Borgerding M.F., Doolittle D.J. Evaluation of the genotoxic and cytotoxic potential of mainstream whole smoke and smoke condensate from a cigarette containing a novel carbon filter. Fundam. appl. Toxicol. 1997;39:11–17. [PubMed: 9325023]
- Bombick D.W., Ayres P.H., Putnam K., Bombick B.R., Doolittle D.J. Chemical and biological studies of a new cigarette that primarily heats tobacco. Part 3. In vitro toxicity of whole smoke. Food chem. Toxicol. 1998;36:191–197. [PubMed: 9609392]
- Bonassi S., Hagmar L., Strömberg U., Montagud A.H., Tinnerberg H., Forni A., Heikkilä P., Wanders S., Wilhardt P., Hansteen I.-L., Knudsen L.E., Norppa H. Chromosomal aberrations in lymphocytes predict human cancer independently of exposure to carcinogens. European Study Group on Cytogenetic Biomarkers and Health. Cancer Res. 2000;60:1619–1625. [PubMed: 10749131]
- Bonassi S., Neri M., Lando C., Ceppi M., Lin Y.-P., Chang W.P., Holland N., Kirsch-Volders M., Zeiger E., Fenech M., the HUMN Collaborative Group. Effect of smoking habit on the frequency of micronuclei in human lymphocytes: Results from the Human Micro-Nucleus Project. Mutat. Res. 2003;543:155–166. [PubMed: 12644185]
- Bond J.A., Chen B.T., Griffith W.C., Mauderly J.L. Inhaled cigarette smoke induces the formation of DNA adducts in lungs of rats. Toxicol. appl. Pharmacol. 1989;99:161–172. [PubMed: 2727995]
- Bono R., Vincenti M., Meineri V., Pignata C., Saglia U., Giachino O., Scursatone E. Formation of N-(2-hydroxyethyl)valine due to exposure to ethylene oxide via tobacco smoke: A risk factor for onset of cancer. Environ. Res. 1999;81:62–71. [PubMed: 10361027]
- Boogaard P.J., van Sittert N.J. Biological monitoring of exposure to benzene: A comparison between S-phenylmercapturic acid, trans, trans-muconic acid, and phenol. Occup. environ. Med. 1995;52:611–620. [PMC free article: PMC1128315] [PubMed: 7550802]
- Boogaard P.J., van Sittert N.J. Suitability of S-phenyl mercapturic acid and trans-transmuconic acid as biomarkers for exposure to low concentrations of benzene. Environ. Health Perspect. 1996;104 (Suppl. 6):1151–1157. [PMC free article: PMC1469762] [PubMed: 9118886]
- Borgerding M.F., Bodnar J.A., Chung H.L., Mangan P.P., Morrison C.C., Risner C.H., Rogers J.C., Simmons D.F., Uhrig M.S., Wendelboe F.N., Wingate D.E., Winkler L.S. Chemical and biological studies of a new cigarette that primarily heats tobacco. Part 1. Chemical composition of mainstream smoke. Food chem. Toxicol. 1998;36:169–182. [PubMed: 9609390]
- Bowman D.L., Smith C.J., Bombick B.R., Avalos J.T., Davis R.A., Morgan W.T., Doolittle D.J. Relationsip between FTC ‘tar’ and urine mutagenicity in smokers of tobacco-burning or Eclipse cigarettes. Mutat. Res. 2002;521:137–149. [PubMed: 12438011]
- Boyd E.J., Wilson J.A., Wormsley K.G. Smoking impairs therapeutic gastric inhibition. Lancet. 1983;i:95–97. [PubMed: 6129458]
- Brandau S., Bohle A. Bladder cancer. I. Molecular and genetic basis of carcinogenesis. Eur. Urol. 2001;39:491–497. [PubMed: 11464028]
- Branner B., Kutzer C., Zwickenpflug W., Scherer G., Heller W.-D., Richter E. Haemoglobin adducts from aromatic amines and tobacco-specific nitrosamines in pregnant smoking and non-smoking women. Biomarkers. 1998;3:35–47. [PubMed: 23899255]
- Bretherton-Watt D., Given-Wilson R., Mansi J.L., Thomas V., Carter N., Colston K.W. Vitamin D receptor gene polymorphisms are associated with breast cancer risk in a UK Caucasian population. Br. J. Cancer. 2001;85:171–175. [PMC free article: PMC2364044] [PubMed: 11461072]
- Brinkworth M.H., Yardley-Jones A., Edwards A.J., Hughes J.A., Anderson D. A comparison of smokers and nonsmokers with respect to oncogene products and cytogenetic parameters. J. occup. Med. 1992;34:1181–1188. [PubMed: 1464786]
- Brockhaus A., Freier I., Ewers U., Jermann E., Dolgner R. Levels of cadmium and lead in blood in relation to smoking, sex, occupation, and other factors in an adult population of the FRG. Int. Arch. occup. environ. Health. 1983;52:167–175. [PubMed: 6629506]
- Bromberger J.T., Matthews K.A., Kuller L.H., Wing R.R., Meilahn E.N., Plantinga P. Prospective study of the determinants of age at menopause. Am. J. Epidemiol. 1997;145:124–133. [PubMed: 9006309]
- Brot C., Jørgensen N.R., Sørensen O.H. The influence of smoking on vitamin D status and calcium metabolism. Eur. J. clin. Nutr. 1999;53:920–926. [PubMed: 10602348]
- Brown B.G., Lee C.K., Bombick B.R., Ayres P.H., Mosberg A.T., Doolittle D.J. Comparative study of DNA adduct formation in mice following inhalation of smoke from cigarettes that burn or primarily heat tobacco. Environ. mol. Mutag. 1997;29:303–311. [PubMed: 9142174]
- Brown B.G., Chang C.J., Ayres P.H., Lee C.K., Doolittle D.J. The effect of cotinine or cigarette smoke co-administration on the formation of O6-methylguanine adducts in the lung and liver of A/J mice treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Toxicol. Sci. 1999;47:33–39. [PubMed: 10048151]
- Brugnone F., Perbellini L., Romeo L., Cerpelloni M., Bianchin M., Tonello A. Benzene in blood as a biomarker of low level occupational exposure. Sci. total Environ. 1999;235:247–252. [PubMed: 10535123]
- Brunnemann K.D., Hoffmann D., Gairola C.G., Lee B.C. Low ignition propensity cigarettes: Smoke analysis for carcinogens and testing for mutagenic activity of the smoke particulate matter. Food chem. Toxicol. 1994;32:917–922. [PubMed: 7959447]
- Bryant M.S., Skipper P.L., Tannenbaum S.R., Maclure M. Hemoglobin adducts of 4-aminobiphenyl in smokers and nonsmokers. Cancer Res. 1987;47:602–608. [PubMed: 3791245]
- Bryant M.S., Vineis P., Skipper P.L., Tannenbaum S.R. Hemoglobin adducts of aromatic amines: Associations with smoking status and type of tobacco. Proc. natl Acad. Sci. USA. 1988;85:9788–9791. [PMC free article: PMC282866] [PubMed: 3200858]
- Buchhalter A.R., Eissenberg T. Preliminary evaluation of a novel smoking system: Effects on subjective and physiological measures and on smoking behavior. Nicotine Tob. Res. 2000;2:39–43. [PubMed: 11072439]
- Buckley T.J., Waldman J.M., Dhara R., Greenberg A., Ouyang Z., Lioy P.J. An assessment of a urinary biomarker for total human environmental exposure to benzo[a]pyrene. Int. Arch. occup. environ. Health. 1995;67:257–266. [PubMed: 7591187]
- Bukvic N., Bavaro P., Elia G., Cassano F., Fanelli M., Guanti G. Sister chromatid exchange (SCE) and micronucleus (MN) frequencies in lymphocytes of gasoline station attendants. Mutat. Res. 1998;415:25–33. [PubMed: 9711259]
- Buratti M., Fustinoni S., Colombi A. Fast liquid chromatographic determination of urinary trans,trans-muconic acid. J. Chromatogr. 1996;B677:257–263. [PubMed: 8704929]
- Burchell, B., McGurk, K., Brierley, C.H. & Clarke, D.J. (1997) UDP-glucuronosyltransferases. In: Guengerich, F.P., ed., Comprehensive Toxicology, Vol. 3, Biotransformation, New York, Elsevier Science, pp. 401–435.
- Burchell B., Soars M., Monaghan G., Cassidy A., Smith D., Ethell B. Drug-mediated toxicity caused by genetic deficiency of UDP-glucuronosyltransferases. Toxicol. Lett. 2000;112–113:333–340. [PubMed: 10720749]
- Burkhart-Schultz K.J., Thompson C.L., Jones I.M. Spectrum of somatic mutation at the hypoxanthine phosphoribosyltransferase (HPRT) gene of healthy people. Carcinogenesis. 1996;17:1871–1883. [PubMed: 8824508]
- Burns, D.M., Major, J.M., Shanks, T.G., Thun, M.J. & Samet, J.M. (2001) Smoking lower yield cigarettes and disease risks. In: Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine (Smoking and Tobacco Control Monograph No. 13; NIH Publication No. 02-5074), Bethesda, MD, US Department of Health and Human Services, National Institutes of Health, pp. 65–158.
- Butkiewicz D., Cole K.J., Phillips D.H., Harris C.C., Chorazy M. GSTM1, GSTP1, CYP1A1 and CYP2D6 polymorphisms in lung cancer patients from an environmentally polluted region of Poland: Correlation with lung DNA adduct levels. Eur. J. Cancer Prev. 1999;8:315–323. [PubMed: 10493307]
- Byrd G.D., Chang K.M., Green J.M., deBethizy J.D. Evidence for urinary excretion of glucuronide conjugates of nicotine, cotinine and trans-3′-hydroxycotinine in smokers. Drug. Metab. Dispos. 1992;20:192–197. [PubMed: 1352209]
- California Environmental Protection Agency (1997) Field Notes. Our World (http://www.calepa.cahwnet.gov/oehha)
- Camoirano A., Balansky R.M., Bennicelli C., Izzotti A., D'Agostini F., De Flora S. Experimental databases on inhibition of the bacterial mutagenicity of 4-nitroquinoline 1-oxide and cigarette smoke. Mutat. Res. 1994;317:89–109. [PubMed: 7511795]
- Camoirano A., Bagnasco M., Bennicelli C., Cartiglia C., Wang J.-B., Zhang B.-C., Zhu Y.-R., Qian G.-S., Egner P.A., Jacobson L.P., Kensler T.W., De Flora S. Oltipraz chemoprevention trial in Qidong, People's Republic of China: Results of urine genotoxicity assays as related to smoking habits. Cancer Epidemiol. Biomarkers Prev. 2001;10:775–783. [PubMed: 11440963]
- Caraballo R.S., Giovino G.A., Pechacek T.F., Mowery P.D., Richter P.A., Strauss W.J., Sharp D.J., Eriksen M.P., Pirkle J.L., Maurer K.R. Racial and ethnic differences in serum cotinine levels of cigarette smokers. Third National Health and Nutrition Examination Survey, 1988–1981. J. Am. med. Assoc. 1998;280:135–139. [PubMed: 9669785]
- Carmella S., LaVoie E.J., Hecht S.S. Quantitative analysis of catechol and 4-methylcatechol in human urine. Food chem. Toxicol. 1982;20:587–590. [PubMed: 6890513]
- Carmella S.G., Kagan S.S., Kagan M., Foiles P.G., Palladino G., Quart A.M., Quart E., Hecht S.S. Mass spectrometric analysis of tobacco-specific nitrosamine hemoglobin adducts in snuff dippers, smokers, and nonsmokers. Cancer Res. 1990;50:5438–5445. [PubMed: 2386948]
- Carmella S.G., Akerkar S., Hecht S.S. Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers' urine. Cancer Res. 1993;53:721–724. [PubMed: 8428352]
- Carmella S.G., Akerkar S., Richie J.P. Jr, Hecht S.S. Intraindividual and interindividual differences in metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in smokers' urine. Cancer Epidemiol. Biomarkers Prev. 1995;4:635–642. [PubMed: 8547830]
- Carmella S.G., Borukhova A., Akerkar S.A., Hecht S.S. Analysis of human urine for pyridine-N-oxide metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific lung carcinogen. Cancer Epidemiol. Biomarkers Prev. 1997;6:113–120. [PubMed: 9037562]
- Carmella S.G., Ye M., Upadhyaya P., Hecht S.S. Stereochemistry of metabolites of a tobacco-specific lung carcinogen in smokers' urine. Cancer Res. 1999;59:3602–3605. [PubMed: 10446969]
- Carmella S.G., Le K.-A., Upadhyaya P., Hecht S.S. Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem. Res. Toxicol. 2002;15:545–550. [PubMed: 11952341]
- Carmichael, P.L., Hewer, A., Jacob, J., Grimmer, G. & Phillips, D.H. (1993) Comparison of total DNA adduct levels induced in mouse tissues and human skin by mainstream and sidestream cigarette smoke condensates. In: Phillips, D.H., Castegnaro, M. & Bartsch, H., eds, Post-labelling Methods for Detection of DNA Adducts (IARC Scientific Publications No. 124), Lyon, IARCPress, pp. 321–326. [PubMed: 8225501]
- Carmines E.L. Evaluation of the potential effects of ingredients added to cigarettes. Part 1: Cigarette design, testing appraoch, and review of results. Food chem. Toxicol. 2002;40:77–91. [PubMed: 11731038]
- Casale G.P., Singhal M., Bhattacharya S., RamaNathan R., Roberts K.P., Barbacci D.C., Zhao J., Jankowiak R., Gross M.L., Cavalieri E.L., Small G.J., Rennard S.I., Mumford J.L., Shen M. Detection and quantification of depurinated benzo[a]pyrene-adducted DNA bases in the urine of cigarette smokers and women exposed to household coal smoke. Chem. Res. Toxicol. 2001;14:192–201. [PubMed: 11258968]
- Cassidenti D.L., Vijod A.G., Vijod M.A., Stanczyk F.Z., Lobo R.A. Short-term effects of smoking on the pharmacokinetic profiles of micronized estradiol in postmenopausal women. Am. J. Obstet. Gynecol. 1990;163:1953–1960. [PubMed: 2256508]
- Castelao J.E., Tuan J.-M., Skipper P.L., Tannenbaum S.R., Gago-Dominguez M., Crowder J.S., Ross R.K., Yu M.C. Gender and smoking-related bladder cancer risk. J. natl Cancer Inst. 2001;93:538–545. [PubMed: 11287448]
- Castles A., Adams E.K., Melvin C.L., Kelsch C., Boulton M.L. Effects of smoking during pregnancy. Five meta-analyses. Am. J. prev. Med. 1999;16:208–215. [PubMed: 10198660]
- Cebulska-Wasilewska A., Wierzewska A., Nizankowska E., Graca B., Hughes J.A., Anderson D. Cytogenetic damage and ras p21 oncoprotein levels from patients with chronic obstructive pulmonary disease (COPD), untreated lung cancer and healthy controls. Mutat. Res. 1999;431:123–131. [PubMed: 10656491]
- Cerqueira E.M., Santoro C.L., Donozo N.F., Freitas B.A., de Braganca Pereira C.A., Bevilacqua R.G., Machado-Santelli G.M. Genetic damage in exfoliated cells of the uterine cervix. Acta cytol. 1998;42:639–649. [PubMed: 9622681]
- Chacko M., Gupta R.C. Evaluation of DNA damage in the oral mucosa of tobacco users and non-users by 32P-adduct assay. Carcinogenesis. 1988;9:2309–2313. [PubMed: 3191577]
- Chan J.M., Stampfer M.J., Giovannucci E., Gann P.H., Ma J., Wilkinson P., Hennekens C.H., Pollak M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science. 1998;279:563–566. [PubMed: 9438850]
- Chang W.-C., Lee Y.-C., Liu C.-L., Hsu J.-D., Wang H.-C., Chen C.-C., Wang C.-J. Increased expression of iNOS and c-fos via regulation of protein tyrosine phosphorylation and MEK1/ERK2 proteins in terminal bronchiole lesions in the lungs of rats exposed to cigarette smoke. Arch. Toxicol. 2001;75:28–35. [PubMed: 11357518]
- Chapurlat R.D., Ewing S.K., Bauer D.C., Cummings S.R. Influence of smoking on the antiosteoporotic efficacy of raloxifene. J. clin. Endocrinol. Metab. 2001;86:4178–4182. [PubMed: 11549646]
- Chen C.-C., Lee H. Genotoxicity and DNA adduct formation of incense smoke condensates: Comparison with environmental tobacco smoke condensates. Mutat. Res. 1996;367:105–114. [PubMed: 8600366]
- Chen C., Loo G. Inhibition of lecithin:cholesterol acyltransferase activity in human blood plasma by cigarette smoke extract and reactive aldehydes. J. biochem. Toxicol. 1995;10:121–128. [PubMed: 7473602]
- Chen A.T.L., Reidy J.A., Annest J.L., Welty T.K., Zhou H.G. Increased chromosome fragility as a consequence of blood folate levels, smoking status, and coffee consumption. Environ. mol. Mutag. 1989;13:319–324. [PubMed: 2737183]
- Cheng T.-J., Christiani D.C., Xu X., Wain J.C., Wiencke J.K., Kelsey K.T. Glutathione S-transferase µ genotype, diet, and smoking as determinants of sister chromatid exchange frequency in lymphocytes. Cancer Epidemiol. Biomarkers Prev. 1995;4:535–542. [PubMed: 7549811]
- Cheng Y.-W., Chen C.-Y., Lin C.-P., Huang K.H., Lin T.-S., Wu M.-H., Lee H. DNA adduct level in lung tissue may act as a risk biomarker of lung cancer. Eur. J. Cancer. 2000;36:1381–1388. [PubMed: 10899651]
- Chepiga T.A., Morton M.J., Murphy P.A., Avalos J.T., Bombick B.R., Doolittle D.J., Borgerding M.F., Swauger J.E. A comparison of the mainstream smoke chemistry and mutagenicity of a representative sample of the US cigarette market with two Kentucky reference cigarettes (K1R4F and K1R5F). Food chem. Toxicol. 2000;38:949–962. [PubMed: 11039328]
- Clapp W.L., Fagg B.S., Smith C.J. Reduction in Ames Salmonella mutagenicity of mainstream cigarette smoke condensate by tobacco protein removal. Mutat. Res. 1999;446:167–174. [PubMed: 10635338]
- Clark G.C. Comparison of the inhalation toxicity of kretek (clove cigarette) smoke with that of American cigarette smoke. I. One day exposure. Arch. Toxicol. 1989;63:1–6. [PubMed: 2742495]
- Clark G.C. Comparison of the inhalation toxicity of kretek (clove cigarette) smoke with that of American cigarette smoke. II. Fourteen days exposure. Arch. Toxicol. 1990;64:515–521. [PubMed: 2073125]
- Clark P.I., Gautam S., Gerson L.W. Effect of menthol cigarettes on biochemical markers of smoke exposure among black and white smokers. Chest. 1996;110:1194–1198. [PubMed: 8915220]
- Cobb R.R., Martin J., Korytynski E., Monteith L., Hughes T.J. Preliminary molecular analysis of the TK locus in L5178Y large- and small-colony mouse lymphoma cell mutants. Mutat. Res. 1989;226:253–258. [PubMed: 2761566]
- Coghlin J., Gann P.H., Hammond S.K., Skipper P.L., Taghizadeh K., Paul M., Tannenbaum S.R. 4-Aminobiphenyl hemoglobin adducts in fetuses exposed to the tobacco smoke carcinogen in utero. J. natl Cancer Inst. 1991;83:274–280. [PubMed: 1994056]
- Cole J., Skopek T. Somatic mutation frequency, mutation rates and mutation spectra in the human population in vivo. Mutat. Res. 1994;304:33–106. [PubMed: 7506357]
- Conney, A.H., Garland, W.A., Rubio, F., Kornychuk, H., Norkus, E.P. & Kuenzig, W. (1986) Factors influencing the urinary excretion of nitrosodimethylamine and nitrosoproline in human beings. In: Hoffmann, D. & Harris, C.C., eds, Mechanisms in Tobacco Carcinogenesis (Banbury Report 23), Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, pp. 21–32.
- Connor T.H., Ramanujam V.M., Ward J.B., Legator M.S. The identification and characterization of a urinary mutagen resulting form cigarette smoke. Mutat. Res. 1983;113:161–172. [PubMed: 6835243]
- Conway K., Edmiston S.N., Cui L., Drouin S.S., Pang J., He M., Tse C.-K., Geradts J., Dressler L., Liu E.T., Millikan R., Newman B. Prevalance and spectrum of p53 mutations associated with smoking in breast cancer. Cancer Res. 2002;62:1987–1995. [PubMed: 11929815]
- Cooper G.S., Thorp J.M. Jr. FSH levels in relation to hysterectomy and to unilateral oophorectomy. Obstet. Gynecol. 1999;94:969–972. [PubMed: 10576184]
- Cooper, C.S., Grover, P.L. & Sims, P. (1983) The metabolism and activation of benzo[a]pyrene. In: Bridges, J.W. & Chasseaud, L.F., eds, Progress in Drug Metabolism, Vol. 7, London, Wiley & Sons, pp. 295–396.
- Cooper G.S., Baird D.D., Hulka B.S., Weinberg C.R., Savitz D.A., Hughes C.L. Jr. Follicle-stimulating hormone concentrations in relation to active and passive smoking. Obstet. Gynecol. 1995;85:407–411. [PubMed: 7862381]
- Cramer D.W., Barbieri R.L., Fraer A.R., Harlow B.L. Determinants of early follicular phase gonadotrophin and estradiol concentrations in women of late reproductive age. Hum. Reprod. 2002;17:221–227. [PubMed: 11756392]
- Crawford F.G., Mayer J., Santella R.M., Cooper T.B., Ottman R., Tsai W.-Y., Simon-Cereijido G.., Wang M., Tang D., Perera F.P. Biomarkers of environmental tobacco smoke in preschool children and their mothers. J. natl Cancer Inst. 1994;86:1398–1402. [PubMed: 8072033]
- Crebelli R., Paoletti A., Falcone E., Aquilina G., Fabri G., Carere A. Mutagenicity studies in a tyre plant: In vitro activity of workers' urinary concentrates and raw materials. Br. J. ind. Med. 1985;42:481–487. [PMC free article: PMC1007513] [PubMed: 4015996]
- Croft J.B., Freedman D.S., Keenan N.L., Sheridan D.P., Macera C.A., Wheeler F.C. Education, health behaviors, and the black-white difference in waist-to-hip ratio. Obes. Res. 1996;4:505–512. [PubMed: 8946435]
- Cryer P.E., Haymond M.W., Santiago J.V., Shah S.D. Norepinephrine and epinephrine release and adrenergic mediation of smoking-associated hemodynamic and metabolic events. New Engl. J. Med. 1976;295:573–577. [PubMed: 950972]
- Culp S.J., Roberts D.W., Talaska G., Lang N.P., Fu P.P., Lay J.O. Jr, Teitel C.H., Snawder J.E., Von Tungeln L.S., Kadlubar F.F. Immunochemical, 32P-postlabeling, and GC/MS detection of 4-aminobiphenyl-DNA adducts in human peripheral lung in relation to metabolic activation pathways involving pulmonary N-oxidation, conjugation, and peroxidation. Mutat. Res. 1997;378:97–112. [PubMed: 9288889]
- Curigliano G., Zhang Y.-J., Wang L.-Y., Flamini G., Alcini A., Ratto C., Giustacchini M., Alcini E., Cittadini A., Santella R.M. Immunohistochemical quantitation of 4-aminobiphenyl-DNA adducts and p53 nuclear overexpression in T1 bladder cancer of smokers and nonsmokers. Carcinogenesis. 1996;17:911–916. [PubMed: 8640937]
- Curry J., Karnaoukhova L., Guenette G.C., Glickman B.W. Influence of sex, smoking and age on human hprt mutation frequencies and spectra. Genetics. 1999;152:1065–1077. [PMC free article: PMC1460655] [PubMed: 10388825]
- Curtis K.M., Savitz D.A., Arbuckle T.E. Effects of cigarette smoking, caffeine consumption, and alcohol intake on fecundability. Am. J. Epidemiol. 1997;146:32–41. [PubMed: 9215221]
- Curvall M., Enzell C.R. Monitoring absorption by means of determination of nicotine and cotinine. Arch. Toxicol. 1986;9:88–102. [PubMed: 3468933]
- Curvall M., Jansson T., Pettersson B., Hedin A., Enzell C.R. In vitro studies of biological effects of cigarette smoke condensate. I. Genotoxic and cytotoxic effects of neutral, semivolatile constituents. Mutat. Res. 1985;157:169–180. [PubMed: 4022041]
- Curvall M., Romert L, Norlén E., Enzell C.R. Mutagen levels in urine from snuff users, cigarette smokers and non tobacco users — A comparison. Mutat. Res. 1987;188:105–110. [PubMed: 3295535]
- Dallinga J.W., Pachen D.M.F.A., Wijnhoven S.W.P., Breedijk A., van't Veer L., Wigbout G., van Zandwijk N., Maas L.M., van Agen E., Kleinjans J.C., van Schooten F.J. The use of 4-aminobiphenyl hemoglobin adducts and aromatic DNA adducts in lymphocytes of smokers as biomarkers of exposure. Cancer Epidemiol. Biomarkers Prev. 1998;7:571–577. [PubMed: 9681524]
- Daube H., Scherer G., Riedel K., Ruppert T., Tricker A.R., Rosenbaum P., Adlkofer F. DNA adducts in human placenta in relation to tobacco smoke exposure and plasma antioxidant status. J. Cancer Res. clin. Oncol. 1997;123:141–151. [PubMed: 9119879]
- Davico L., Sacerdote C., Ciccone G., Pegorano L., Kerim S., Ponzio G., Vineis P. Chromosome 8, occupational exposures, smoking, and acute nonlymphocytic leukemias: A population-based study. Cancer Epidemiol. Biomarkers Prevent. 1998;7:1123–1125. [PubMed: 9865431]
- Debanne S.M., Rowland D.Y., Riedel T.M., Cleves M.A. Association of Alzheimer's disease and smoking: The case for sibling controls. J. Am. Geriatr. Soc. 2000;48:800–806. [PubMed: 10894320]
- De Flora S., Camoirano A., Bagnasco M., Bennicelli C., van Zandwijk N., Wigbout G., Qian G.S., Zhu Y.R., Kensler T.W. Smokers and urinary genotoxins: Implications for selection of cohorts and modulation of endpoints in chemoprevention trials. J. Cell Biochem. 1996;25 (Suppl.):92–98. [PubMed: 9027604]
- De Flora S., D'Agostini F., Balansky R., Camoirano A., Bennicelli C., Bagnasco M., Cartiglia C., Tampa E., Longobardi M.G., Lubet R.A., Izzotti A. Modulation of cigarette smoke-related end-points in mutagenesis and carcinogenesis. Mutat. Res. 2003;523–524:237–252. [PubMed: 12628522]
- Degawa M., Stern S.J., Martin M.V., Guengerich F.P., Fu P.P., Ilett K.F., Kaderlik R.K., Kadlubar F.F. Metabolic activation and carcinogen–DNA adduct detection in human larynx. Cancer Res. 1994;54:4915–4919. [PubMed: 8069857]
- van Delft J.H.M, Steenwinkel M.-J.S.T., van Asten J.G., de Vogel N., Bruijntjes-Rozier T.C.D.M., Schouten T., Cramers P., Maas L., Van Herwijnen M.H., van Schooten F.-J., Hopmans P.M.J. Biological monitoring the exposure to polycyclic aromatic hydrocarbons of coke oven workers in relation to smoking and genetic polymorphisms for GSTM1 and GSTT1. Ann. occup. Hyg. 2001;45:395–408. [PubMed: 11418090]
- Del Santo P., Moneti G., Salvadori M., Saltutti C., Delle Rose A., Dolara P. Levels of the adducts of 4-aminobiphenyl to hemoglobin in control subjects and bladder carcinoma patients. Cancer Lett. 1991;60:245–251. [PubMed: 1756515]
- DeMarini D.M. Genotoxicity of tobacco smoke and tobacco smoke condensate. Mutat. Res. 1983;114:59–89. [PubMed: 6219288]
- DeMarini D.M., Brooks L.R., Bhatnagar V.K., Hayes R.B., Eischen B.T., Shelton M.L., Zenser T.V., Talaska G., Kashyap S.K., Dosemeci M., Kashyap R., Parikh D.J., Lakshmi V., Hsu F., Davis B.B., Jaeger M., Rothman N. Urinary mutagenicity as a biomarker in workers exposed to benzidine: Correlation with urinary metabolites and urothelial DNA adducts. Carcinogenesis. 1987;18:981–988. [PubMed: 9163684]
- DeMarini D.M., Shelton M.L., Levine J.G. Mutation spectrum of cigarette smoke condensate in Salmonella: Comparison to mutations in smoking-associated tumors. Carcinogenesis. 1995;16:2535–2542. [PubMed: 7586163]
- DeMarini D.M., Landi S., Tian D., Hanley N.M., Li X., Hu F., Roop B.C., Mass M.J., Keohavong P., Gao W., Olivier M., Hainaut P., Mumford J.L. Lung tumor KRAS and TP53 mutations in nonsmokers reflect exposure to PAH-rich coal combustion emissions. Cancer Res. 2001;61:6679–6681. [PubMed: 11559534]
- De Méo M.P., Duménil G., Botta A.H., Laget M., Zabaloueff V., Mathias A. Urine mutagenicity of steel workers exposed to coke oven emissions. Carcinogenesis. 1987;8:363–367. [PubMed: 3545525]
- De Méo M.P., Miribel V., Botta A., Laget M., Duménil G. Applicability of the SOS Chromotest to detect urinary mutagenicity caused by smoking. Mutagenesis. 1988;3:277–283. [PubMed: 3137423]
- Denissenko M.F., Pao A., Tang M.-S., Pfeifer G.P. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–432. [PubMed: 8832894]
- Denissenko M.F., Pao A., Pfeifer G.P., Tang M.-S. Slow repair of bulky DNA adducts along the nontranscribed strand of the human p53 gene may explain the strand bias of trans-version mutations in cancers. Oncogene. 1998;16:1241–1247. [PubMed: 9546425]
- van Diemen P.C., Maasdam D., Vermeulen S., Darroudi F., Natarajan A.T. Influence of smoking habits on the frequencies of structural and numerical chromosomal aberrations in human peripheral blood lymphocytes using the fluoresence in situ hybridization (FISH) technique. Mutagenesis. 1995;10:487–495. [PubMed: 8596467]
- Dietrich M., Block. G., Hudes M., Morrow J.D., Norkus E.P., Traber M.G., Cross C.E., Packer L. Antioxidant supplementation decreases lipid peroxidation biomarker F2-isoprostanes in plasma of smokers. Cancer Epidemiol. Biomarkers Prev. 2002;11:7–13. [PubMed: 11815395]
- Djordjevic M.V., Sigountos C.W., Brunnemann K.D., Hoffmann D. Formation of 4-(methylnitrosamino)-4-(3-pyridyl)butyric acid in vitro and in mainstream cigarette smoke. J. agric. Food Chem. 1991;39:209–213.
- Dolara P., Mazzoli S., Rosi D., Buiatti E., Baccetti S., Turchi A., Vannucci V. Exposure to carcinogenic chemicals and smoking increases urinary excretion of mutagens in humans. J. Toxicol. environ. Health. 1981;8:95–103. [PubMed: 7328717]
- Domino, E.F. (1999) Pharmacological significance of nicotine. In: Gorrod, J.W. & Jacob, P., III, eds, Analytical Determination of Nicotine and Related Compounds and their Metabolites, New York, Elsevier, pp. 1–11.
- Doolittle D.J., Rahn C.A., Burger G.T., Davis R., deBethizy J.D., Howard G., Lee C.K., McKarns S.C., Riccio E., Robinson J., Reynolds J., Hayes A.W. Human urine mutagenicity study comparing cigarettes which burn or only heat tobacco. Mutat. Res. 1989;223:221–232. [PubMed: 2739680]
- Doolittle D.J., Rahn C.A., Riccio E., Passananti G.T., Howard G., Vesell E.S., Burger G.T., Hayes A.W. Comparative studies of the mutagenicity of urine from smokers and nonsmokers on a controlled non-mutagenic diet. Food chem. Toxicol. 1990a;28:639–646. [PubMed: 2272562]
- Doolittle D.J., Lee C.K., Ivett J.L., Mirsalis J.C., Riccio E., Rudd C.J., Burger G.T., Hayes A.W. Comparative studies on the genotoxic activity of mainstream smoke condensate from cigarettes which burn or only heat tobacco. Environ. mol. Mutag. 1990b;15:93–105. [PubMed: 2407532]
- Doolittle D.J., Rahn C.A., Lee C.K. The effect of exposure to nicotine, carbon monoxide, cigarette smoke or cigarette smoke condensate on the mutagenicity of rat urine. Mutat. Res. 1991;260:9–18. [PubMed: 2027346]
- Doolittle D.J., Winegar R., Lee C.K., Caldwell W.S., Hayes A.W., deBethizy J.D. The genotoxic potential of nicotine and its major metabolites. Mutat. Res. 1995;344:95–102. [PubMed: 7491133]
- van Doorn R., Bos R.P., Leijdekkers C.-M., Wagenaas-Zegers M.A.P., Theuws J.L.G., Henderson P.T. Thioether concentration and mutagenicity of urine from cigarette smoker. Int. Arch. occup. environ. Health. 1979;43:159–166. [PubMed: 457288]
- van Doorn R., Leijdekkers C.M., Bos R.P., Brouns R.M.E., Henderson P.T. Detection of human exposure to electrophilic compounds by assay of thioether detoxication products in urine. Ann. occup. Hyg. 1981;24:77–92. [PubMed: 7015964]
- Dor F., Haguenoer J.-M., Zmirou D., Empereur-Bissonnet P., Jongeneelen F.J., Nedellec V., Person A., Ferguson C.C., Dab W. Urinary 1-hydroxypyrene as a biomarker of polycyclic aromatic hydrocarbons exposure of workers on a contaminated site: Influence of exposure conditions. J. occup. environ. Med. 2000;42:391–397. [PubMed: 10774508]
- Dorgan J.F., Stanczyk F.Z., Longcope C., Stephenson H.E. Jr, Chang. L., Miller R., Franz C., Falk R.T., Kahle L. Relationship of serum dehydroepiandrosterone (DHEA), DHEA sulfate, and 5-androstene-3beta,17beta-diol to risk of breast cancer in postmenopausal women. Cancer Epidemiol. Biomarkers Prev. 1997;6:177–181. [PubMed: 9138660]
- Dosaka-Akita H., Katabami M., Hommura H., Fujioka Y., Katoh H., Kawakami Y. Bcl-2 expression in non-small cell lung cancers: Higher frequency of expression in squamous cell carcinomas with earlier pT status. Oncology. 1999;56:259–264. [PubMed: 10202283]
- Duell E.J., Wiencke J.K., Cheng T.-J., Varkonyi A., Zuo Z.F., Ashok T.D.S., Mark E. J., Wain J. C., Christiani D.C., Kelsey K.T. Polymorphisms in the DNA repair genes XRCC1 and ERCC2 and biomarkers of DNA damage in human blood mononuclear cells. Carcinogenesis. 2000;21:965–971. [PubMed: 10783319]
- Duffel, M.W., (1997) Sulfotransferases. In: Guengerich, F.P., ed., Comprehensive Toxicology, Vol. 3, Biotransformation, New York, Elsevier Science, pp. 365–383.
- Duncan B.B., Chambless L.E., Schmidt M.I., Szklo M., Folsom A.R., Carpenter M.A., Crouse J.R. III., for the Atherosclerosis Risk in Communities (ARIC) Study Investigators. Correlates of body fat distribution. Variation across categories of race, sex, and body mass in the atherosclerosis risk in communities study. Ann. Epidemiol. 1995;5:192–200. [PubMed: 7606308]
- Dunn B.P., Curtis J.R. Clastogenic agents in the urine of coffee drinkers and cigarette smokers. Mutat. Res. 1985;147:179–188. [PubMed: 4022032]
- Dunn B.P., Stich H.F. 32P-Postlabelling analysis of aromatic DNA adducts in human oral mucosal cells. Carcinogenesis. 1986;7:1115–1120. [PubMed: 3719906]
- Dunn B.P., Vedal S., San R.H.C., Kwan W.-F., Nelems B., Enarson D.A., Stich H.F. DNA adducts in bronchial biopsies. Int. J. Cancer. 1991;48:485–492. [PubMed: 2045196]
- Dyke G.W., Craven J.L., Hall R., Garner R.C. Smoking-related DNA adducts in human gastric cancers. Int. J. Cancer. 1992;52:847–850. [PubMed: 1459723]
- Einhaus M., Holz O., Meissner R., Krause T., Warncke K., Held I., Scherer G., Tricker A.R., Adlkofer F., Rüdiger H.W. Determination of DNA single-strand breaks in lymphocytes of smokers and nonsmokers exposed to environmental tobacco smoke using the nick translation assay. Clin. Invest. 1994;72:930–936. [PubMed: 7894226]
- Einistö P., Nohmi T., Watanabe M., Ishidate M. Jr. Sensitivity of Salmonella typhimurium YG1024 to urine mutagenicity caused by cigarette smoking. Mutat. Res. 1990;245:87–92. [PubMed: 2215555]
- Ejiofor A., Hutchison D.C.S., Baum H. Inhibition of human leucocyte elastase activity by cigarette smoke. Biosci. Rep. 1981;1:715–720. [PubMed: 6921046]
- Eke B.C., Vural N., Iscan M. Combined effects of ethanol and cigarette smoke on hepatic and pulmonary xenobiotic metabolizing enzymes in rats. Chem.-biol. Interact. 1996;102:155–167. [PubMed: 9021168]
- Eke B.C., Vural N., Iscan M. Age dependent differential effects of cigarette smoke on hepatic and pulmonary xenobiotic metabolizing enzymes in rats. Arch. Toxicol. 1997;71:696–702. [PubMed: 9363843]
- El Bayoumy K., Donahue J.M., Hecht S.S., Hoffmann D. Identification and quantitative determination of aniline and toluidines in human urine. Cancer Res. 1986;46:6064–6067. [PubMed: 3779628]
- Eliasson M., Hägg E., Lundblad D., Karlsson R., Bucht E. Influence of smoking and snuff use on electrolytes, adrenal and calcium regulating hormones. Acta endocrinol. 1993;128:35–40. [PubMed: 8447192]
- Eliasson B., Attwall S., Taskinen M.R., Smith U. Smoking cessation improves insulin sensitivity in health middle-aged men. Eur. J. clin. Invest. 1997;27:450–456. [PubMed: 9179554]
- Ellingsen D.G., Thomassen Y., Aaseth J., Alexander J. Cadmium and selenium in blood and urine related to smoking habits and previous exposure to mercury vapour. J. appl. Toxicol. 1997;17:337–343. [PubMed: 9339747]
- El-Nemr A., Al-Shawaf T., Sabatini L., Wilson C., Lower A.M., Grudzinskas J.G. Effect of smoking on ovarian reserve and ovarian stimulation in in-vitro fertilization and embryo transfer. Hum. Reprod. 1998;13:2192–2198. [PubMed: 9756295]
- Endogenous Sex Hormones and Breast Cancer Collaborative Group. Endogenous sex hormones and breast cancer in postmenopausal women: Reanalysis of nine prospective studies. J. natl Cancer Inst. 2002;94:606–616. [PubMed: 11959894]
- Endoh K., Leung F.W. Effects of smoking and nicotine on the gastric mucosa: A review of clinical and experimental evidence. Gastroenterology. 1994;107:864–878. [PubMed: 7915701]
- US Environmental Protection Agency (1993) Smoking and Tobacco Control. Monograph 4. Respiratory Health Effects of Passive Smoking: Lung Cancer and Other Disorders (NIH Publication No. 93-3605), Washington DC, US Department of Health and Human Services.
- Etter J.F., Vu Duc T., Perneger T.V. Saliva cotinine levels in smokers and nonsmokers. Am. J. Epidemiol. 2000;151:251–258. [PubMed: 10670549]
- Etzel R.A. A review of the use of saliva cotinine as a marker of tobacco smoke exposure. Prev. Med. 1990;19:190–197. [PubMed: 2193308]
- Evans, D.A.P. (1986) Acetylation. In: Kalow, W., Goedde, H.W. & Agarwal, D.P., eds, Ethnic Differences in Reactions to Drugs and Xenobiotics, New York, Alan R. Liss, pp. 209–242.
- Everson R.B., Randerath E., Santella R.M., Cefalo R.C., Avitts T.A., Randerath K. Detection of smoking-related covalent DNA adducts in human placenta. Science. 1986;231:54–57. [PubMed: 3941892]
- Everson R.B., Randerath E., Santella R.M., Avitts T.A., Weinstein I.B., Randerath K. Quantitative associations between DNA damage in human placenta and maternal smoking and birth weight. J. natl Cancer Inst. 1988;80:567–576. [PubMed: 3373547]
- Facchini F.S., Hollenbeck C.B., Jeppesen J., Chen Y.D., Reaven G.M. Insulin resistance and cigarette smoking. Lancet. 1992;339:1128–1130. [PubMed: 1349365]
- Fagerström K.O., Hughes J.R., Rasmussen T., Callas P.W. Randomized trial investigating effect of a novel nicotine delivery device (Eclipse) and a nicotine oral inhaler on smoking behaviour, nicotine and carbon monoxide exposure, and motivation to quit. Tob. Control. 2000;9:327–333. [PMC free article: PMC1748373] [PubMed: 10982578]
- Falter B., Kutzer C., Richter E. Biomonitoring of hemoglobin adducts: Aromatic amines and tobacco-specific nitrosamines. Clin. Invest. 1994;72:364–371. [PubMed: 8086771]
- Farmer P.B., Neumann H.-G., Henschler D. Estimation of exposure of man to substances reacting covalently with macromolecules. Arch. Toxicol. 1987;60:251–260. [PubMed: 3307702]
- Favatier F., Polla B.S. Tobacco-smoke-inducible human haem oxygenase-1 gene expression: Role of distinct transcription factors and reactive oxygen intermediates. Biochem. J. 2001;353:475–482. [PMC free article: PMC1221592] [PubMed: 11171043]
- Fay L.B., Leaf C.D., Gremaud E., Aeschlimann J.-M., Steen C., Shuker D.E.G., Turesky R.J. Urinary excretion of 3-methyladenine after consumption of fish containing high levels of dimethylamine. Carcinogenesis. 1997;18:1039–1044. [PubMed: 9163693]
- Fennell T.R., MacNeela J.P., Morris R.W., Watson M., Thompson C.L., Bell D.A. Hemoglobin adducts from acrylonitrile and ethylene oxide in cigarette smokers: Effects of glutathione S-transferase T1-null and M1-null genotypes. Cancer Epidemiol. Biomarkers Prev. 2000;9:705–712. [PubMed: 10919741]
- Fernandez-Salguero P., Hoffman S.M., Cholerton S., Mohrenweiser H., Raunio H., Rautio A., Pelkonen O., Huang J.-D., Evans W.E., Idle J.R., Gonzalez F.J. A genetic polymorphism in coumarin 7-hydroxylation: Sequence of the human CYP2A genes and identification of variant CYP2A6 alleles. Am. J. hum. Genet. 1995;57:651–660. [PMC free article: PMC1801261] [PubMed: 7668294]
- Ferreira M. Jr, Buchet J.P., Burrion J.B., Moro J., Cupers L., Delavignette J.P., Jacques J., Lauwerys R. Determinants of urinary thioethers, D-glucaric acid and mutagenicity after exposure to polycyclic aromatic hydrocarbons assessed by air monitoring and measurement of 1-hydroxypyrene in urine: A cross-sectional study in workers of coke and graphite-electrode-producing plants. Int. Arch. occup. environ. Health. 1994;65:329–338. [PubMed: 8175189]
- Feskens E.J.M., Kromhout D. Cardiovascular risk factors and the 25-year incidence of diabetes mellitus in middle-aged men. Am. J. Epidemiol. 1989;130:1101–1108. [PubMed: 2589303]
- Fewell J.E., Smith F.G. Perinatal nicotine exposure impairs ability of newborn rats to autoresuscitate from apnea during hypoxia. J. appl. Physiol. 1998;85:2066–2074. [PubMed: 9843527]
- Fielding S., Short C., Davies K., Wald N., Bridges B.A., Waters R. Studies on the ability of smoke from different types of cigarettes to induce single-strand breaks in cultured human cells. Mutat. Res. 1989;214:147–151. [PubMed: 2770760]
- Finch G.L., Nikula K.J., Chen B.T., Barr E.B., Chang I.Y., Hobbs C.H. Effect of chronic cigarette smoke exposure on lung clearance of tracer particles inhaled by rats. Fundam. appl. Toxicol. 1995;24:76–85. [PubMed: 7713345]
- Finch G.L., Lundgren D.L., Barr E.B., Chen B.T., Griffith W.C., Hobbs C.H., Hoover M.D., Nikula K.J., Mauderly J.L. Chronic cigarette smoke exposure increases the pulmonary retention and radiation dose of 239Pu inhaled as 239PuO2 by F344 rats. Health Phys. 1998;75:597–609. [PubMed: 9827506]
- Finkelstein E.I., Nardini M., van der Vliet A. Inhibition of neutrophil apoptosis by acrolein: A mechanism of tobacco-related lung disease? Am. J. Physiol. Lung cell. mol. Physiol. 2001;281:L732–L739. [PubMed: 11504702]
- Fitzgerald J. Cigarette smoke comparative toxicology. Food chem. Toxicol. 2001;39:175–176. [PubMed: 11288744]
- Flamini G., Romano G., Curigliano G., Chiominto A., Capelli G., Boninsegna A., Signorelli C., Ventura L., Santella R.M., Sgambato A., Cittadini A. 4-Aminobiphenyl–DNA adducts in laryngeal tissue and smoking habits: An immunohistochemical study. Carcinogenesis. 1998;19:353–357. [PubMed: 9498288]
- Flaws J.A., Rhodes J.C., Langenberg P., Hirshfield A.N., Kjerulff K., Sharara F.I. Ovarian volume and menopausal status. Menopause. 2000;7:53–61. [PubMed: 10646704]
- Flegal K.M., Troiano R.P., Pamuk E.R., Kuczmarski R.J., Campbell S.M. The influence of smoking cessation on the prevalence of overweight in the United States. New Engl. J. Med. 1995;333:1165–1170. [PubMed: 7565970]
- Foiles P.G., Akerkar S.A., Carmella S.G., Kagan M., Stoner G.D., Resau J.H., Hecht S.S. Mass spectrometric analysis of tobacco-specific nitrosamine–DNA adducts in smokers and nonsmokers. Chem. Res. Toxicol. 1991;4:364–468. [PubMed: 1912321]
- Forgacs E., Zöchbauer-Müller S., Oláh E., Minna J.D. Molecular genetic abnormalities in the pathogenesis of human lung cancer. Pathol. Oncol. Res. 2001;7:6–13. [PubMed: 11349214]
- Fowler J.S., Volkow N.D., Wang G.J., Pappas N., Logan J., MacGregor R., Alexoff D., Shea C., Schlyer D., Wolf A. P., Warner D., Zezulkova I., Cilento R. Inhibition of monoamine oxidase B in the brains of smokers. Nature. 1996a;379:733–736. [PubMed: 8602220]
- Fowler J.S., Volkow N.D., Wang G.J., Pappas N., Logan J., Shea C., Alexoff D., MacGregor R.R., Schlyer D.J., Zezulkova I., Wolf A.P. Brain monoamine oxidase A inhibition in cigarette smokers. Proc. natl Acad. Sci. USA. 1996b;93:14065–14069. [PMC free article: PMC19495] [PubMed: 8943061]
- Fowler J.S., Wang G.J., Volkow N.D., Franceschi D., Logan J., Pappas N., Shea C., MacGregor R.R., Garza V. Smoking a single cigarette does not produce a measurable reduction in brain MAO B in nonsmokers. Nicotine Tob. Res. 1999;1:325–329. [PubMed: 11072429]
- Fraig M., Shreesha U., Savici D., Katzenstein A.L. Respiratory bronchiolitis: A clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am. J. Surg. Pathol. 2002;26:647–653. [PubMed: 11979095]
- Fratiglioni L., Wang H.X. Smoking and Parkinson's and Alzheimer's disease: Review of the epidemiological studies. Behav. Brain Res. 2000;113:117–120. [PubMed: 10942038]
- Fung Y.K., Mendlik M.G., Haven M.C., Akhter M.P., Kimmel D.B. Short-term effects of nicotine on bone and calciotropic hormones in adult female rats. Pharmacol. Toxicol. 1998;82:243–249. [PubMed: 9646330]
- Fürstenberger G., Senn H.-J. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3:298–302. [PubMed: 12067807]
- Gairola C.G. Pulmonary aryl hydrocarbon hydroxylase activity of mice, rats and guinea pigs following long term exposure to mainstream and sidestream cigarette smoke. Toxicology. 1987;45:177–184. [PubMed: 3603583]
- Gairola C.G., Drawdy M.L., Block A.E., Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein E–/– mice. Atherosclerosis. 2001;156:49–55. [PubMed: 11368996]
- Galdston M., Levytska V., Schwartz M.S., Magnusson B. Ceruloplasmin. Increased serum concentration and impaired antioxidant activity in cigarette smokers, and ability to prevent suppression of elastase inhibitory capacity of alpha1-proteinase inhibitor. Am. Rev. respir. Dis. 1984;129:258–263. [PubMed: 6607691]
- Gallagher, J.E., Mumford, J., Li, X., Shank, T., Manchester, D. & Lewtas, J. (1993a) DNA adduct profiles and levels in placenta, blood and lung in relation to cigarette smoking and smoky coal emissions. In: Phillips, D.H., Castegnaro, M. & Bartsch, H., eds, Postlabelling Methods for Detection of DNA Adducts (IARC Scientific Publications No. 124), Lyon, IARCPress, pp. 283–292. [PubMed: 8225497]
- Gallagher J.E., Vine M.F., Schramm M.M., Lewtas J., George M.H., Hulka B.S., Everson R.B. 32P-Postlabeling analysis of DNA adducts in human sperm cells from smokers and nonsmokers. Cancer Epidemiol. Biomarkers Prev. 1993b;2:581–585. [PubMed: 8268777]
- Gealy R., Zhang L., Siegfried J.M., Luketich J.D., Keohavong P. Comparison of mutations in the p53 and K-ras genes in lung carcinomas from smoking and nonsmoking women. Cancer Epidemiol. Biomarkers Prev. 1999;8:297–302. [PubMed: 10207632]
- Geisler J., Omsjo I.H., Helle S.I., Ekse D., Silsand R., Lonning P.E. Plasma oestrogen fractions in postmenopausal women receiving hormone replacement therapy: Influence of route of administration and cigarette smoking. J. Endocrinol. 1999;162:265–270. [PubMed: 10425465]
- Geneste O., Camus A.-M., Castegnaro M., Petruzzelli S., Macchiarini P., Angeletti C.-A., Giuntini C., Bartsch H. Comparison of pulmonary DNA adduct levels, measured by 32P-postlabelling and aryl hydrocarbon hydroxylase activity in lung parenchyma of smokers and ex-smokers. Carcinogenesis. 1991;12:1301–1305. [PubMed: 2070496]
- Georgiadis P., Samoli E., Kaila S., Katsouyanni K., Kyrtopoulos S.A. Ubiquitous presence of O6-methylguanine in human peripheral and cord blood DNA. Cancer Epidemiol. Biomarkers Prev. 2000;9:299–305. [PubMed: 10750669]
- Ghittori S., Maestri L., Fiorentino M.L., Imbriani M. Evaluation of occupational exposure to benzene by urinalysis. Int. Arch. occup. environ. Health. 1995;67:195–200. [PubMed: 7591178]
- Ghittori S., Maestri L., Rolandi L., Lodola L., Fiorentino M.L., Imbriani M. The determination of trans,trans-muconic acid in urine as an indicator of occupational exposure to benzene. Appl. occup. environ. Hyg. 1996;11:187–191.
- Ghosh R., Ghosh P.K. The effect of tobacco smoking on the frequency of sister chromatid exchanges in human lymphocyte chromosomes. Cancer Genet. Cytogenet. 1987;27:15–19. [PubMed: 3581036]
- Giovannucci E. Insulin, insulin-like growth factors and colon cancer: A review of the evidence. J. Nutr. 2001;131:3109S–3120S. [PubMed: 11694656]
- Girre C., Lucas D., Hispard E., Menez C., Dally S., Menez J.-F. Assessment of cytochrome P4502E1 induction in alcoholic patients by chlorzoxazone pharmacokinetics. Biochem. Pharmacol. 1994;47:1503–1508. [PubMed: 7910460]
- Gocze P.M., Freeman D.A. Cytotoxic effects of cigarette smoke alkaloids inhibit the progesterone production and cell growth of cultured MA-10 Leydig tumor cells. Eur. J. Obstet. Gynecol. reprod. Biol. 2000;93:77–83. [PubMed: 11000509]
- Godden P.M., Kass G., Mayer R.T., Burke M.D. The effects of cigarette smoke compared to 3-methylcholanthrene and phenobarbitone on alkoxyresorufin metabolism by lung and liver microsomes from rats. Biochem. Pharmacol. 1987;36:3393–3398. [PubMed: 3675601]
- Godschalk R.W.L., Maas L.M., Van Zandwijk N., van't Veer L.J., Breedijk A., Borm P.J.A., Verhaert J., Kleinjans J.C.S., van Schooten F.-J. Differences in aromatic–DNA adduct levels between alveolar macrophages and subpopulations of white blood cells from smokers. Carcinogenesis. 1998;19:819–825. [PubMed: 9635869]
- Godschalk R.W.L., Dallinga J.W., Wikman H., Risch A., Kleinjans J.C.S., Bartsch H., Van Schooten F.-J. Modulation of DNA and protein adducts in smokers by genetic polymorphisms in GSTM1, GSTT1, NAT1 and NAT2. Pharmacogenetics. 2001;11:389–398. [PubMed: 11470992]
- Gold E.B., Bromberger J., Crawford S., Samuels S., Greendale G.A., Harlow S.D., Skurnick J. Factors associated with age at natural menopause in a multiethnic sample of midlife women. Am. J. Epidemiol. 2001;153:865–874. [PubMed: 11323317]
- Goldman R., Enewold L., Pellizzari E., Beach J.B., Bowman E.D., Krishnan S.S., Shields P.G. Smoking increases carcinogenic polycyclic aromatic hydrocarbons in human lung tissue. Cancer Res. 2001;61:6367–6371. [PubMed: 11522627]
- Gontijo Á.M.deM.C., Elias F.N., Salvadori D.M.F., de Oliveira M.L.C.S., Correa L.A., Goldberg J., Trindade J.C.deS., de Camargo J.L. Single-cell gel (comet) assay detects primary DNA damage in nonneoplastic urothelial cells of smokers and ex-smokers. Cancer Epidemiol. Biomarkers Prev. 2001;10:987–993. [PubMed: 11535552]
- Gonzalez F.J., Gelboin H.V. Role of human cytochromes P450 in the metabolic activation of chemical carcinogens and toxins. Drug Metab. Rev. 1994;26:165–183. [PubMed: 8082563]
- Goodman-Gruen D., Barrett-Connor E. Epidemiology of insulin-like growth factor-I in elderly men and women. The Rancho Bernardo Study. Am. J. Epidemiol. 1997;145:970–976. [PubMed: 9169905]
- Gordon S.M. Identification of exposure markers in smokers' breath. J. Chromatogr. 1990;511:291–302. [PubMed: 2211914]
- Górecka D., Gorski T. The influence of cigarette smoking on sister chromatid exchange frequencies in peripheral lymphocytes among nurses handling cytostatic drugs. Polish J. occup. Med. environ. Health. 1993;6:143–148. [PubMed: 8219905]
- Gorrod, J.W., Schepers, G. (1999) Biotransformation of nicotine in mammalian systems. In: Gorrod, J.W. & Jacob, P., III, eds, Analytical Determination of Nicotine and Related Compounds and their Metabolites, New York, Elsevier, pp. 45–67.
- Granath F., Westerholm R., Peterson A., Törnqvist M., Ehrenberg L. Uptake and metabolism of ethene studied in a smoke-stop experiment. Mutat. Res. 1994;313:285–291. [PubMed: 7523913]
- Granella M., Priante E., Nardini B., Bono R., Clonfero E. Excretion of mutagens, nicotine and its metabolites in urine of cigarette smokers. Mutagenesis. 1996;11:207–211. [PubMed: 8671740]
- Grimmer G., Dettbarn G., Jacob J. Biomonitoring of polycyclic aromatic hydrocarbons in highly exposed coke plant workers by measurement of urinary phenanthrene and pyrene metabolites (phenols and dihydrodiols). Int. Arch. occup. environ. Health. 1993;65:189–199. [PubMed: 8282417]
- Grimmer G., Jacob J., Dettbarn G., Naujack K.-W. Determination of urinary metabolites of polycyclic aromatic hydrocarbons (PAH) for the risk assessment of PAH-exposed workers. Int. Arch. occup. environ. Health. 1997;69:231–239. [PubMed: 9137996]
- Grimmer G., Dettbarn G., Seidel A., Jacob J. Detection of carcinogenic aromatic amines in the urine of nonsmokers. Sci. total Environ. 2000;247:81–90. [PubMed: 10721145]
- Grinberg-Funes R.A., Singh V.N., Perera F.P., Bell D.A., Young T.L., Dickey C., Wang L.W., Santella R.M. Polycyclic aromatic hydrocarbon–DNA adducts in smokers and their relationship to micronutrient levels and the glutathione-S-transferase M1 genotype. Carcinogenesis. 1994;15:2449–2454. [PubMed: 7955090]
- Grove K.L., Sekhon H.S., Brogan R.S., Keller J.A., Smith M.S., Spindel E.R. Chronic maternal nicotine exposure alters neuronal systems in the arcuate nucleus that regulate feeding behavior in the newborn rhesus macaque. J. clin. Endocrinol. Metab. 2001;86:5420–5426. [PubMed: 11701716]
- Grunberg, N.E. (1990) The inverse relationship between tobacco use and body weight. In: Kozlowski, L.T., Annis, H.M., Cappell, H.D., Glaser, F.B., Goodstadt, M.S., Israel, Y., Kalant, H., Sellers, E.M. & Vingilis, E.R., eds, Research Advances in Alcohol and Drug Problems, Vol. 10, New York, Plenum Press, pp. 273–315.
- Gu Y., Cimino G., Alder H., Nakamura T., Prasad R., Canaani O., Moir D.T., Jones C., Nowell P.C., Croce C.M., Canaani O. The (4;11)(q21;q23) chromosome translocations in acute leukemias involve the VDJ recombinase. Proc. natl Acad. Sci. USA. 1992;89:10464–10468. [PMC free article: PMC50359] [PubMed: 1438235]
- Guengerich, F.P. (1997) Cytochrome P450 enzymes. In: Guengerich, F.P., ed., Comprehensive Toxicology, Vol. 3, Biotransformation, New York, Elsevier Science, pp. 37–68.
- Guengerich F.P., Kim D.-H., Iwasaki M. Role of human cytochrome P-450 IIE1 in the oxidation of many low molecular weight cancer suspects. Chem. Res. Toxicol. 1991;4:168–179. [PubMed: 1664256]
- Guidotti T.L. Critique of available studies on the toxicology of kretek smoke and its constituents by routes of entry involving the respiratory tract. Arch. Toxicol. 1989;63:7–12. [PubMed: 2662942]
- Gündel J., Angerer J. High-performance liquid chromatographic method with fluorescence detection for the determination of 3-hydroxybenzo[a]pyrene and 3- hydroxybenz[a]anthracene in the urine of polycyclic aromatic hydrocarbon-exposed workers. J. Chromatogr. 2000;B738:47–55. [PubMed: 10778925]
- Gupta R.C., Sopori M.L., Gairola C.G. Formation of cigarette smoke-induced DNA adducts in the rat lung and nasal mucosa. Cancer Res. 1989;49:1916–1920. [PubMed: 2702635]
- Gupta R.C., Arif J.M., Gairola C.G. Enhancement of pre-existing DNA adducts in rodents exposed to cigarette smoke. Mutat. Res. 1999;424:195–205. [PubMed: 10064861]
- Gupta K., Krishnaswamy G., Karnad A., Peiris A.N. Insulin: A novel factor in carcinogenesis. Am. J. med. Sci. 2002;323:140–145. [PubMed: 11908858]
- Habib M.P., Clements N.C., Garewal H.S. Cigarette smoking and ethane exhalation in humans. Am. J. resp. crit. Care Med. 1995;151:1368–1372. [PubMed: 7735586]
- Hackman P., Hou S.-M., Nyberg F., Pershagen G., Lambert B. Mutational spectra at the hypoxanthine-guanine phosphoribosyltransferase (HPRT) locus in T-lymphocytes of nonsmoking and smoking lung cancer patients. Mutat. Res. 2000;468:45–61. [PubMed: 10863157]
- Hageman G.J., Stierum R.H., van Herwijnen M.H., van der Veer M.S., Kleinjans J.C. Nicotinic acid supplementation: Effects on niacin status, cytogenetic damage, and poly(ADPribosylation) in lymphocytes of smokers. Nutr. Cancer. 1998;32:113–120. [PubMed: 9919621]
- Hainaut P., Pfeifer G.P. Patterns of p53 G→T transversions in lung cancer reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–374. [PubMed: 11238174]
- Hall J., Brésil H., Donato F., Wild C.P., Loktionova N.A., Kazanova O.I., Komyakov I.P., Lemekhov V.G., Likhachev A.J., Montesano R. Alkylation and oxidative-DNA damage repair activity in blood leukocytes of smokers and nonsmokers. Int. J. Cancer. 1993;54:728–733. [PubMed: 8325702]
- Hanahan D., Weinberg R.A. The hallmarks of cancer (Review). Cell. 2000;100:57–70. [PubMed: 10647931]
- Hanawalt P.C. Controlling the efficiency of excision repair. Mutat. Res. 2001;485:3–13. [PubMed: 11341989]
- Hankey G.J. Smoking and risk of stroke. J. cardiovasc. Risk. 1999;6:207–211. [PubMed: 10501270]
- Hansen C., Sørensen L.D., Asmussen I., Autrup H. Transplacental exposure to tobacco smoke in human-adduct formation in placenta and umbilical cord blood vessels. Teratog. Carcinog. Mutag. 1992;12:51–60. [PubMed: 1359662]
- Hansen C., Asmussen I., Autrup H. Detection of carcinogen–DNA adducts in human fetal tissues by the 32P-postlabeling procedure. Environ. Health Perspect. 1993;99:229–231. [PMC free article: PMC1567065] [PubMed: 8319630]
- Hansen Å.M., Poulsen O.M., Sigsgaard T., Christensen J.M. The validity of determination of α-naphthol in urine as a marker for exposure to polycyclic aromatic hydrocarbons. Anal. chim. Acta. 1994;291:341–347.
- Hargreave F.E., Leigh R. Induced sputum, eosinophilic bronchitis, and chronic obstructive pulmonary disease. Am. J. respir. crit. Care Med. 1999;160:S53–S57. [PubMed: 10556171]
- Härkönen K., Viitanen T., Larsen S.B., Bonde J.P., Lahdetie J. Aneuploidy in sperm and exposure to fungicides and lifestyle factors. ASCLEPIOS. A European Concerted Action on Occupational Hazards to Male Reproductive Capability. Environ. mol. Mutag. 1999;34:39–46. [PubMed: 10462722]
- Harris S.S., Soteriades E., Coolidge J.A., Mudgal S., Dawson-Hughes B. Vitamin D insufficiency and hyperparathyroidism in a low income, multiracial, elderly population. J. clin. Endocrinol. Metab. 2000;85:4125–4130. [PubMed: 11095443]
- Hasan S.U., Simakajornboon N., MacKinnon Y., Gozal D. Prenatal cigarette smoke exposure selectively alters protein kinase C and nitric oxide synthase expression within the neonatal rat brainstem. Neurosci. Lett. 2001;301:135–138. [PubMed: 11248441]
- Hatch M.C., Warburton D., Santella R.M. Polycyclic aromatic hydrocarbon–DNA adducts in spontaneously aborted fetal tissue. Carcinogenesis. 1990;11:1673–1675. [PubMed: 2401055]
- Haugen A., Becher G., Benestad C., Vahakangas K., Trivers G.E., Newman M.J., Harris C.C. Determination of polycyclic aromatic hydrocarbons in the urine, benzo[a]pyrene diol epoxide–DNA adducts in lymphocyte DNA, and antibodies to the adducts in sera from coke oven workers exposed to measured amounts of polycyclic aromatic hydrocarbons in the work atmosphere. Cancer Res. 1986;46:4178–4183. [PubMed: 3731085]
- Hautanen A., Adlercreutz H. Hyperinsulinaemia, dyslipidaemia and exaggerated adrenal androgen response to adrenocorticotropin in male smokers. Diabetologia. 1993;36:1275–1281. [PubMed: 8307255]
- Hawkins B.T., Brown R.C., Davis T.P. Smoking and ischemic stroke: A role for nicotine? Trends pharmacol. Sci. 2002;23:78–82. [PubMed: 11830264]
- Hecht S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998;11:559–603. [PubMed: 9625726]
- Hecht S.S. Tobacco smoke carcinogens and lung cancer. J. natl Cancer Inst. 1999;91:1194–1210. [PubMed: 10413421]
- Hecht. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002a;3:461–469. [PubMed: 12147432]
- Hecht. Human urinary carcinogen metabolites: Biomarkers for investigating tobacco and cancer. Carcinogenesis. 2002b;23:907–922. [PubMed: 12082012]
- Hecht, S.S., Peterson, L.A., Spratt, T.E. (1994) Tobacco-specific nitrosamines. In: Hemminki, K., Dipple, A., Shuker, D.E.G., Kadlubar, F.F., Segerbäck, D. Bartsch, H., eds, DNA Adducts: Identification and Biological Significance (IARC Scientific Publications No. 125), Lyon, IARCPress, pp. 91–106.
- Hecht S.S., Chung F.-L., Richie J.P. Jr, Akerkar S.A., Borukhova A., Skowronski L., Carmella S.G. Effects of watercress consumption on metabolism of a tobacco-specific lung carcinogen in smokers. Cancer Epidemiol. Biomarkers Prev. 1995;4:877–884. [PubMed: 8634661]
- Hecht S.S., Carmella S.G., Chen M., Dor Koch J.F., Miller A.T., Murphy S.E., Jensen J.A., Zimmerman C.L., Hatsukami D.K. Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. 1999a;59:590–596. [PubMed: 9973205]
- Hecht S.S., Hatsukami D.K., Bonilla L.E., Hochalter J.B. Quantitation of 4-oxo-4-(3pyridyl)butanoic acid and enantiomers of 4-hydroxy-4-(3-pyridyl)butanoic acid in human urine: A substantial pathway of nicotine metabolism. Chem. Res. Toxicol. 1999b;12:172–179. [PubMed: 10027795]
- Hecht S.S., Hochalter J.B., Villalta P.W., Murphy S.E. 2′-Hydroxylation of nicotine by cytochrome P450 2A6 and human liver microsomes: Formation of a lung carcinogen precursor. Proc. natl Acad. Sci. USA. 2000;97:12493–12497. [PMC free article: PMC18791] [PubMed: 11050152]
- Hecht S.S., Ye M., Carmella S.G., Fredrickson A., Adgate J.L., Greaves I.A., Church T.R., Ryan A.D., Mongin S.J., Sexton K. Metabolites of a tobacco-specific lung carcinogen in the urine of elementary school-aged children. Cancer Epidemiol. Biomarkers Prev. 2001;10:1109–1116. [PubMed: 11700257]
- Hecht S.S., Carmella S.G., Ye M., Le K.-A., Jensen J.A., Zimmerman C.L., Hatsukami D.K. Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res. 2002;62:129–134. [PubMed: 11782369]
- Heikkilä P., Luotamo M., Pyy L., Riihimäki V. Urinary 1-naphthol and 1-pyrenol as indicators of exposure to coal tar products. Int. Arch. occup. environ. Health. 1995;67:211–217. [PubMed: 7591181]
- Helander A., Löwenmo C., Wikström T., Curvall M. Inhibition of human blood aldehyde dehydrogenase activity by cigarette-smoke condensate. Life Sci. 1991;49:1901–1905. [PubMed: 1745106]
- Hernan M.A., Takkouche B., Caamano-Isorna F., Gestal-Otero J.J. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann. Neurol. 2002;52:276–284. [PubMed: 12205639]
- Hernandez-Boussard T.M., Hainaut P. A specific spectrum of p53 mutations in lung cancer from smokers: Review of mutations compiled in the IARC p53 database. Environ. Health Perspect. 1998;106:385–391. [PMC free article: PMC1533115] [PubMed: 9637795]
- Hess R.D., Brandner G. DNA damage by filtered, tar- and aerosol-free cigarette smoke in rodent cells: A novel evaluation. Toxicol. Lett. 1996;88:9–13. [PubMed: 8920710]
- Heudorf U., Angerer J. Urinary monohydroxylated phenanthrenes and hydroxypyrene — The effects of smoking habits and changes induced by smoking on monooxygenasemediated metabolism. Int. Arch. occup. environ. Health. 2001;74:177–183. [PubMed: 11355291]
- Heusch W.L., Maneckjee R. Signalling pathways involved in nicotine regulation of apoptosis of human lung cancer cells. Carcinogenesis. 1998;19:551–556. [PubMed: 9600337]
- Hirao T., Nelson H.H., Ashok T.D., Wain J.C., Mark E.J., Christiani D.C., Wiencke J.K., Kelsey K.T. Tobacco smoke-induced DNA damage and an early age of smoking initation induce chromosome loss at 3p21 in lung cancer. Cancer Res. 2001;61:612–615. [PubMed: 11212258]
- Hittelman W.N. Genetic instability in epithelial tissues at risk for cancer. Ann. N.Y. Acad. Sci. 2001;952:1–12. [PubMed: 11795428]
- Hoffmann K., Becker K., Friedrich C., Helm D., Drause C., Seifert B. The German Environmental Survey 1990/1992 (GerES II): Cadmium in blood, urine and hair of adults and children. J. Expos. Anal. environ. Epidemiol. 2000;10:126–135. [PubMed: 10791594]
- Hoffmann D., Hoffmann I., El-Bayoumy K. The less harmful cigarette: A controversial issue. A tribute to Ernst L. Wynder. Chem. Res. Toxicol. 2001;14:767–790. [PubMed: 11453723]
- Holly E.A., Petrakis N.L., Friend N.F., Sarles D.L., Lee R.E., Flander L.B. Mutagenic mucus in the cervix of smokers. J. natl Cancer Inst. 1986;76:983–986. [PubMed: 3458964]
- Holz O., Krause T., Scherer G., Schmidt-Preuss U., Rüdiger H.W. 32P-Postlabelling analysis of DNA adducts in monocytes of smokers and passive smokers. Int. Arch. occup. environ. Health. 1990;62:299–303. [PubMed: 2379960]
- Holz O., Meissner R., Einhaus M., Koops F., Warncke K., Scherer G., Adlkofer F., Baumgartner E., Rüdiger H.W. Detection of DNA single-strand breaks in lymphocytes of smokers. Int. Arch. occup. environ. Health. 1993;65:83–88. [PubMed: 8253515]
- Hou S.M., Yang K., Nyberg F., Hemminki K., Pershagen G., Lambert B. Hprt mutant frequency and aromatic DNA adduct level in non-smoking and smoking lung cancer patients and population controls. Carcinogenesis. 1999;20:437–444. [PubMed: 10190559]
- Hou S.M., Fält S., Yang K., Nyberg F., Pershagen G., Hemminki K., Lambert B. Differential interactions between GSTM1 and NAT2 genotypes on aromatic DNA adduct level and HPRT mutant frequency in lung cancer patients and population controls. Cancer Epidemiol. Biomarkers Prev. 2001;10:133–140. [PubMed: 11219770]
- Hou S.M., Fält S., Angelini S., Yang K., Nyberg F., Lambert B., Hemminki K. The XPD variant alleles are associated with increased aromatic DNA adduct level and lung cancer risk. Carcinogenesis. 2002;23:599–603. [PubMed: 11960912]
- Howard L.A., Micu A.L., Sellers E.M., Tyndale R.F. Low doses of nicotine and ethanol induce CYP2E1 and chlorzoxazone metabolism in rat liver. J. Pharmacol. exp. Ther. 2001;299:542–550. [PubMed: 11602665]
- Hsu T.M., Zhang Y.-J., Santella R.M. Immunoperoxidase quantitation of 4-aminobiphenyl– and polycyclic aromatic hydrocarbon–DNA adducts in exfoliated oral and urothelial cells of smokers and nonsmokers. Cancer Epidemiol. Biomarkers Prev. 1997;6:193–199. [PubMed: 9138663]
- Huang Y.-L., Chuang I.-C., Pan C.-H., Hsieh C., Shi T.-S., Lin T.-H. Determination of chromium in whole blood and urine by graphite furnace AAS [atomic absorption spectroscopy]. Atom. Spectroscop. 2000;21:10–16.
- Hughes E.G., Brennan B.G. Does cigarette smoking impair natural or assisted fecundity? Fertil. Steril. 1996;66:679–689. [PubMed: 8893667]
- Hull M.G., North K., Taylor H., Farrow A., Ford W.C. Delayed conception and active and passive smoking. The Avon Longitudinal Study of Pregnancy and Childhood Study Team. Fertil. Steril. 2000;74:725–733. [PubMed: 11020514]
- Hurt R.D., Croghan G.A., Wolter T.D., Croghan I.T., Offord K.P., Williams G.M., Djordjevic M.V., Richie J.P. Jr, Jeffrey A.M. Does smoking reduction result in reduction of biomarkers associated with harm? A pilot study using a nicotine inhaler. Nicotine Tob. Res. 2000;2:327–336. [PubMed: 11197312]
- Husgafvel-Pursiainen K. Sister-chromatid exchange and cell proliferation in cultured lymphocytes of passively and actively smoking restaurant personnel. Mutat. Res. 1987;190:211–215. [PubMed: 3821782]
- Husgafvel-Pursiainen K., Hackman P., Ridanpää M., Anttila S., Karjalainen A., Partanen T., Taikina-Aho O., Heikkilä L., Vainio H. K-ras mutations in human adenocarcinoma of the lung: Association with smoking and occupational exposure to asbestos. Int. J. Cancer. 1993;53:250–256. [PubMed: 8425762]
- Hussain S.P., Hofseth L.J., Harris C.C. Tumor suppressor genes: At the crossroads of molecular carcinogenesis, molecular epidemiology and human risk assessment. Lung Cancer. 2001;34 (Suppl. 2):S7–S15. [PubMed: 11720736]
- Husum B, Wulf H.C., Niebuhr E. Sister chromatid exchange frequency correlates with age, sex and cigarette smoking in a 5-year material of 553 healthy adults. Hereditas. 1986;105:17–21. [PubMed: 3793516]
- IARC (1972) IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, Vol. 1, Some Inorganic Substances, Chlorinated Hydrocarbons, Aromatic Amines, N-Nitroso Compounds, and Natural Products, Lyon, IARCPress, pp. 74–79.
- IARC (1974) IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man, Vol. 4, Some Aromatic Amines, Hydrazine and Related Substances, N-Nitroso Compounds and Miscellaneous Alkylating Agents, Lyon, IARCPress, pp. 97–111.
- IARC (1978a) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 17, Some N-Nitroso Compounds, Lyon, IARCPress, pp. 125–175. [PubMed: 355094]
- IARC (1978b) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 17, Some N-Nitroso Compounds, Lyon, IARCPress, pp. 303–311. [PubMed: 355104]
- IARC (1978c) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 17, Some N-Nitroso Compounds, Lyon, IARCPress, pp. 327–335. [PubMed: 680690]
- IARC (1978d) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 17, Some N-Nitroso Compounds, Lyon, IARCPress, pp. 313–326. [PubMed: 355105]
- IARC (1979) IARC Monographs on the Evaluation of the Carcinogenic Risks of Chemicals to Humans, Vol. 19, Some Monomers, Plastics and Synthetic Elastomers, and Acrolein, Lyon, IARCPress, pp. 377–438. [PubMed: 285915]
- IARC (1980) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 23, Some Metals and Metallic Compounds, Lyon, IARCPress, pp. 325–415. [PubMed: 7000667]
- IARC (1982) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 29, Some Industrial Chemicals and Dyestuffs, Lyon, IARCPress, pp. 93–148. [PubMed: 6957390]
- IARC (1983a) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 32, Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data, Lyon, IARCPress, pp. 33–91, 211–224.
- IARC (1983b) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 32, Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data, Lyon, IARCPress, pp. 419–430. [PubMed: 6586639]
- IARC (1983c) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 32, Polynuclear Aromatic Compounds, Part 1, Chemical, Environmental and Experimental Data, Lyon, IARCPress, pp. 431–445. [PubMed: 6586639]
- IARC (1985a) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 37, Tobacco Habits Other than Smoking; Betel-Quid and Areca-Nut Chewing; and Some Related Nitrosamines, Lyon, IARCPress, pp. 209–223. [PubMed: 3866741]
- IARC (1985b) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 37, Tobacco Habits Other than Smoking; Betel-Quid and Areca-Nut Chewing; and Some Related Nitrosamines, Lyon, IARCPress, pp. 241–261. [PubMed: 3866741]
- IARC (1986) IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans, Vol. 38, Tobacco Smoking, Lyon, IARCPress. [PubMed: 3460963]
- IARC (1987) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Suppl. 7, Overall Evaluations of Carcinogenicity: An Updating of IARC Monographs Volumes 1 to 42, Lyon, IARCPress. [PubMed: 3482203]
- IARC (1990a) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 49, Chromium, Nickel and Welding, Lyon, IARCPress, pp. 257–445.
- IARC (1990b) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 49, Chromium, Nickel and Welding, Lyon, IARCPress, pp. 49–256.
- IARC (1993a) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 56, Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins, Lyon, IARCPress, pp. 229–242.
- IARC (1993b) Monographs on the Carcinogenic Risk of Chemicals to Humans, Vol. 58, Beryllium, Cadmium, Mercury, and Exposures in the Glass Manufacturing Industry, Lyon, IARCPress, pp. 119–237. [PMC free article: PMC7681476] [PubMed: 8022054]
- IARC (1994a) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 60, Some Industrial Chemicals, Lyon, IARCPress, pp. 73–159. [PMC free article: PMC7681578] [PubMed: 7869582]
- IARC (1994b) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 60, Some Industrial Chemicals, Lyon, IARCPress, pp. 45–71. [PMC free article: PMC7681339] [PubMed: 7869580]
- IARC (1995a) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 63, Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals, Lyon, IARCPress, pp. 337–372. [PMC free article: PMC7681564] [PubMed: 9139128]
- IARC (1995b) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 62, Wood Dust and Formaldehyde, Lyon, IARCPress, pp. 217–362.
- IARC (1999a) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Reevaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part Three), Lyon, IARCPress, pp. 1037–1047. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999b) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part Two), Lyon, IARCPress, pp. 319–335. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999c) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part Two), Lyon, IARCPress, pp. 749–768. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999d) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part Two), Lyon, IARCPress, pp. 691–719. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999e) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part Two), Lyon, IARCPress, pp. 433–451. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999f) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 71, Reevaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part One), Lyon, IARCPress, pp. 109–225. [PMC free article: PMC7681305] [PubMed: 10507919]
- IARC (1999g) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 73, Some Chemicals that Cause Tumours of the Kidney or Urinary Bladder in Rodents and Some Other Substances, Lyon, IARCPress, pp. 49–58.
- IARC (2000) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 77, Some Industrial Chemicals, Lyon, IARCPress, pp. 267–322. [PMC free article: PMC7197284] [PubMed: 11100404]
- IARC (2002) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 82, Some Traditional Herbal Medicines, Some Mycotoxins, Naphthalene and Styrene, Lyon, IARCPress, pp. 367–435. [PMC free article: PMC4781602] [PubMed: 12687954]
- Ichiba M., Hagmar L., Rannug A., Hogstedt B., Alexandrie A.-K., Carstensen U., Hemminki K. Aromatic DNA adducts, micronuclei and genetic polymorphism for CYP1A1 and GST1 in chimney sweeps. Carcinogenesis. 1994;15:1347–1352. [PubMed: 8033310]
- Inoue O., Seiji K., Kasahara M., Nakatsuka H., Watanabe T., Yin S.-G., Li G.-L., Cai S.-X., Jin C., Ikeda M. Determination of catechol and quinol in the urine of workers exposed to benzene. Br. J. ind. Med. 1988;45:487–492. [PMC free article: PMC1009634] [PubMed: 3395585]
- Inoue O., Seiji K., Nakatsuka H., Watanabe T., Yin S.-N., Li G.-L., Cai S.-X., Jin C., Ikeda M. Excretion of 1,2,4-benzenetriol in the urine of workers exposed to benzene. Br. J. ind. Med. 1989;46:559–565. [PMC free article: PMC1009826] [PubMed: 2775675]
- Ishizaki M., Yamada Y., Morikawa Y., Noborisaka Y., Ishida M., Miura K., Nakagawa H. The relationship between waist-to-hip ratio and occupational status and life-style factors among middle-aged male and female Japanese workers. Occup. Med. 1999;49:177–182. [PubMed: 10451599]
- Izzotti A., Rossi G.A., Bagnasco M., De Flora S. Benzo[a]pyrene diolepoxide–DNA adducts in alveolar macrophages of smokers. Carcinogenesis. 1991;12:1281–1285. [PubMed: 1906379]
- Izzotti A., Balansky R.M., Coscia N., Scatolini L., D'Agostini F., De Flora S. Chemoprevention of smoke-related DNA adduct formation in rat lung and heart. Carcinogenesis. 1992;13:2187–2190. [PubMed: 1423892]
- Izzotti A., De Flora S., Petrilli G.L., Gallagher J., Rojas M., Alexandrov K., Bartsch H., Lewtas J. Cancer biomarkers in human atherosclerotic lesions: Detection of DNA adducts. Cancer Epidemiol. Biomarkers Prev. 1995a;4:105–110. [PubMed: 7742716]
- Izzotti A., Balansky R., Scatolini L., Rovida A., De Flora S. Inhibition by N-acetylcysteine of carcinogen–DNA adducts in the tracheal epithelium of rats exposed to cigarette smoke. Carcinogenesis. 1995b;16:669–672. [PubMed: 7697831]
- Izzotti A., Balansky R.M., Blagoeva P.M., Mircheva Z.I., Tulimiero L., Cartiglia C., De Flora S. DNA alterations in rat organs after chronic exposure to cigarette smoke and/or ethanol ingestion. FASEB J. 1998;12:753–758. [PubMed: 9619454]
- Jacob J., Grimmer G., Dettbarn G. Profile of urinary phenanthrene metabolites in smokers and nonsmokers. Biomarkers. 1999;4:319–327. [PubMed: 23902352]
- Jacobson J.S., Begg M.D., Wang L.W., Wang Q., Agarwal M., Norkus E., Singh V.N., Young T.L., Yang D., Santella R.M. Effects of a 6-month vitamin intervention on DNA damage in heavy smokers. Cancer Epidemiol. Biomarkers Prev. 2000;9:1303–1311. [PubMed: 11142415]
- Jahnke G.D., Thompson C.L., Walker M.P., Gallagher J.E., Lucier G.W., DiAugustine R.P. Multiple DNA adducts in lymphocytes of smokers and nonsmokers determined by 32P-postlabeling analysis. Carcinogenesis. 1990;11:205–211. [PubMed: 2105856]
- Jalili T., Murthy G.G., Schiestl R.H. Cigarette smoke induces DNA deletions in the mouse embryo. Cancer Res. 1998;58:2633–2638. [PubMed: 9635590]
- Jansen E.H.J.M., Schenk E., den Engelsman G., van de Werken G. Use of biomarkers in exposure assessment of polycyclic aromatic hydrocarbons. Clin. Chem. 1995;41:1905–1906.
- Janssen J.A.M.J.L., Stolk R.P., Pols H.A.P., Grobbee D.E., Lamberts S.W.J. Serum total IGF-I, free IGF-I, and IGFBP-1 levels in an elderly population. Relation to cardiovascular risk factors and disease. Arterioscler. Thromb. vasc. Biol. 1998;18:277–282. [PubMed: 9484994]
- Jansson T., Curvall M., Hedin A., Enzell C.R. In vitro studies of biological effects of cigarette smoke condensate. II. Induction of sister-chromatid exchanges in human lymphocytes by weakly acidic, semivolatile contituents. Mutat. Res. 1986;169:129–139. [PubMed: 3951466]
- Jansson T., Curvall M., Hedin A., Enzell C.R. In vitro studies of the biological effects of cigarette smoke condensate. III. Induction of SCE by some phenolic and related constituents derived from cigarette smoke. A study of structure–activity relationships. Mutat. Res. 1988;206:17–24. [PubMed: 3412370]
- Jansson A., Andersson K., Bjelke B., Eneroth P., Fuxe K. Effects of a postnatal exposure to cigarette smoke on hypothalamic catecholamine nerve terminal systems and on neuroendocrine function in the postnatal and adult male rat. Evidence for long-term modulation of anterior pituitary function. Acta physiol. scand. 1992;144:453–462. [PubMed: 1605047]
- Jarvis M.J., Tunstall-Pedoe H., Feyerabend C., Vesey C., Saloojee Y. Comparison of tests used to distinguish smokers from nonsmokers. Am. J. public Health. 1987;77:1435–1438. [PMC free article: PMC1647100] [PubMed: 3661797]
- Jarvis M.J., Feyerabend C., Bryant A., Hedges B., Primatesta P. Passive smoking in the home: Plasma cotinine concentrations in non-smokers with smoking partners. Tob. Control. 2001;10:368–374. [PMC free article: PMC1747624] [PubMed: 11740030]
- Jensen J., Christiansen C. Effects of smoking on serum lipoproteins and bone mineral content during postmenopausal hormone replacement therapy. Am. J. Obstet. Gynecol. 1988;159:820–825. [PubMed: 2972209]
- Jensen J., Christiansen C., Rodbro P. Cigarette smoking, serum estrogens, and bone loss during hormone-replacement therapy early after menopause. New Engl. J. Med. 1985;17:973–975. [PubMed: 4047104]
- Jensen T.K., Henriksen T.B., Hjollund N.H., Scheike T., Kolstad H., Giwercman A., Ernst E., Bonde J.P., Skakkebaek N.E., Olsen J. Adult and prenatal exposures to tobacco smoke as risk indicators of fertility among 430 Danish couples. Am. J. Epidemiol. 1998;148:992–997. [PubMed: 9829871]
- Ji Q., Chen Y. Vicia faba root tip micronucleus test on the mutagenicity of water-soluble contents of cigarette smoke. Mutat. Res. 1996;359:1–6. [PubMed: 8569797]
- Jick H., Porter J., Morrison A.S. Relation between smoking and age of natural menopause. Report from the Boston Collaborative Drug Surveillance Program, Boston University Medical Center. Lancet. 1977;i:1354–1355. [PubMed: 69066]
- Jin Y.P., Kobayashi E., Nogawa K., Kido T., Suwazono Y., Sakurada I., Yin K.O., Nakagawa H., Tsuritani I. Lead levels in 24-h urine in Japanese adults. Toxicol. Lett. 1997;92:173–178. [PubMed: 9334827]
- Jo W.-K., Pack K.-W. Utilization of breath analysis for exposure estimates of benzene associated with active smoking. Environ. Res. 2000;A83:180–187. [PubMed: 10856191]
- Jones N.J., Kadhim M.A., Hoskins P.L., Parry J.M., Waters R. 32P-Postlabelling analysis and micronuclei induction in primary Chinese hamster lung cells exposed to tobacco particulate matter. Carcinogenesis. 1991;12:1507–1514. [PubMed: 1860172]
- Jones N.J., McGregor A.D., Waters R. Detection of DNA adducts in human oral tissue: Correlation of adduct levels with tobacco smoking and differential enhancement of adducts using the butanol extraction and nuclease P1 versions of 32P postlabeling. Cancer Res. 1993;53:1522–1528. [PubMed: 8453617]
- Jongen W.M., Hakkert B.C., van der Hoeven J.C. Genotoxicity testing of cigarette-smoke condensate in the SCE and HGPRT assays with V79 Chinese hamster cells. Food Chem. Toxicol. 1985;23:603–607. [PubMed: 4040105]
- Jongeneelen F.J. Biological monitoring of environmental exposure to polycyclic aromatic hydrocarbons; 1-hydroxypyrene in urine of people. Toxicol. Lett. 1994;72:205–211. [PubMed: 8202933]
- Jongeneelen F.J. Benchmark guideline for urinary 1-hydroxypyrene as biomarker of occupational exposure to polycyclic aromatic hydrocarbons. Ann. occup. Hyg. 2001;45:3–13. [PubMed: 11137694]
- Jongeneelen F.J., Anzion R.B.M., Leijdekkers C.M., Bos R.P., Henderson P.T. 1Hydroxypyrene in human urine after exposure to coal tar and a coal tar derived product. Int. Arch. occup. environ. Health. 1985;57:47–55. [PubMed: 4077281]
- Joschko M.A., Dreosti I.E., Tulsi R.S. The teratogenic effects of nicotine in vitro in rats: A light and electron microscope study. Neurotoxicol. Teratol. 1991;13:307–316. [PubMed: 1886540]
- Jull B.A., Plummer H.K. III, Schuller H.M. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J. Cancer Res. clin. Oncol. 2001;127:707–717. [PubMed: 11768610]
- Kaaks R., Toniolo P., Akhmedkhanov A., Lukanova A., Biessy C., Dechaud H., Rinaldi S., Zeleniuch-Jacquotte A., Shore R.E., Riboli E. Serum C-peptide, insulin-like growth factor (IGF)-I, IGF-binding proteins and colorectal cancer risk in women. J. natl Cancer Inst. 2000;92:1592–1600. [PubMed: 11018095]
- Kadlubar, F.F. (1994) DNA adducts of carcinogenic aromatic amines. In: Hemminki, K., Dipple, A., Shuker, D.E.G., Kadlubar, F.F., Segerbäck, D. & Bartsch, H., eds, DNA Adducts, Identification and Biological Significance (IARC Scientific Publications No. 125), Lyon, IARCPress, pp. 199–216.
- Kadlubar, F.F. & Beland, F.A. (1985) Chemical properties of ultimate carcinogenic metabolites of arylamines and arylamides. In: Harvey, R.G., ed., Polycyclic Hydrocarbons and Carcinogenesis (ACS Symposium Series 283), Washington DC, American Chemical Society, pp. 341–370.
- Kado N.Y., Langley D., Eisenstadt E. A simple modification of the Salmonella, liquid incubation assay. Mutat. Res. 1983;121:25–32. [PubMed: 6306458]
- Kado N.Y., Manson C., Eisenstadt E., Hsieh D.P. The kinetics of mutagen excretion in the urine of cigarette smokers. Mutat. Res. 1985;157:227–233. [PubMed: 3894961]
- Kadohama N., Shintani K., Osawa Y. Tobacco alkaloid derivatives as inhibitors of breast cancer aromatase. Cancer Lett. 1993;75:175–182. [PubMed: 8313352]
- Kaklamani V.G., Linos A., Kaklamani E., Markaki I., Mantzoros C. Age, sex, and smoking are predictors of circulating insulin-like growth factor 1 and insulin-like growth factor-binding protein 3. J. clin. Oncol. 1999;17:813–817. [PubMed: 10071271]
- Kalra R., Singh S.P., Savage S.M., Finch G.L., Sopori M.L. Effects of cigarette smoke on immune response: Chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca2+ stores. J. Pharmacol. exp. Ther. 2000;293:166–171. [PubMed: 10734166]
- Kanaoka T., Miyakawa M., Yoshida O. [Study on influence of cigarette smoking on the mutagenicity of urine. 1. Influence of cigarette smoking on the mutagenicity of urine in health smokers and bladder cancer patients.] Acta. urol. jpn. 1990;36:385–393. (in Japanese) [PubMed: 2378301]
- Kang M.K., Park N.H. Conversion of normal to malignant phenotype: Telomere shortening, telomerase activation, and genomic instability during immortalization of human oral keratinocytes. Crit. Rev. oral Biol. Med. 2001;12:38–54. [PubMed: 11349961]
- Kao-Shan C.-S., Fine R.L., Whang-Peng J., Lee E.C., Chabner B.A. Increased fragile sites and sister chromatid exchanges in bone marrow and peripheral blood of young cigarette smokers. Cancer Res. 1987;47:6278–6282. [PubMed: 3677077]
- Karube T., Odagiri Y., Takemoto K., Watanabe S. Analyses of transplacentally induced sister chromatid exchanges and micronuclei in mouse fetal liver cells following maternal exposure to cigarette smoke. Cancer Res. 1989;49:3550–3552. [PubMed: 2731176]
- Kasai H., Iwamoto-Tanaka N., Miyamoto T., Kawanami K., Kawanami S., Kido R., Ikeda M. Life style and urinary 8-hydroxydeoxyguanosine, a marker of oxidative DNA damage: Effects of exercise, working conditions, meat intake, body mass index, and smoking. Jpn J. Cancer Res. 2001;92:9–15. [PMC free article: PMC5926588] [PubMed: 11173538]
- Kato I., Tominaga S., Suzuki T. Factors related to late menopause and early menarche as risk factors for breast cancer. Jpn. J. Cancer Res. 1988;79:165–172. [PMC free article: PMC5917460] [PubMed: 3130350]
- Kato S., Bowman E.D., Harrington A.M., Blomeke B., Shields P.G. Human lung carcinogen–DNA adduct levels mediated by genetic polymorphisms in vivo. J. natl Cancer Inst. 1995;87:902–907. [PubMed: 7666479]
- Kato I., Toniolo P., Akhmedkhanov A., Koenig K.L., Shore R., Zeleniuch-Jacquotte A. Prospective study of factors influencing the onset of natural menopause. J. clin. Epidemiol. 1998;51:1271–1276. [PubMed: 10086819]
- Kaufman D.W., Slone D., Rosenberg L., Miettinen O.S., Shapiro S. Cigarette smoking and age at natural menopause. Am. J. public Health. 1980;70:420–422. [PMC free article: PMC1619390] [PubMed: 7361965]
- Kaur J., Srivastava A., Ralhan R. Prognostic significance of p53 protein overexpression in betel- and tobacco-related oral oncogenesis. Int. J. Cancer. 1998;79:370–375. [PubMed: 9699529]
- Kawakami N., Takatsuka N., Shimizu H., Ishibashi H. Effects of smoking on the incidence of non-insulin-dependent diabetes mellitus. Replication and extension in a Japanese cohort of male employees. Am. J. Epidemiol. 1997;145:103–109. [PubMed: 9006306]
- Kelsey K.T., Wiencke J.K., Little F.F., Baker E.L. Jr, Little J.B. Effects of cigarette smoking and solvent exposure on sister chromatid exchange frequency in painters. Environ. mol. Mutag. 1988;11:389–399. [PubMed: 3356184]
- Kharitonov S.A., Robbins R.A., Yates D., Keatings V., Barnes P.J. Acute and chronic effects of cigarette smoking on exhaled nitric oxide. Am. J. respir. crit. Care Med. 1995;152:609–612. [PubMed: 7543345]
- Kidd L.C., Stillwell W.G., Yu M.C., Wishnok J.S., Skipper P.L., Ross R.K., Henderson B.E., Tannenbaum S.R. Urinary excretion of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in white, African-American, and Asian-American men in Los Angeles County. Cancer Epidemiol. Biomarkers Prev. 1999;8:439–445. [PubMed: 10350440]
- Kim S.Y., Chung J.H., Kang K.W., Joe C.O., Park K.H. Relationship between activities of cytochrome P-450 monooxygenases in human placental microsomes and binding of benzo(a)pyrene metabolites to calf thymus DNA. Drug chem. Toxicol. 1992;15:313–327. [PubMed: 1459043]
- Kim H., Kim Y.-D., Lee H., Kawamoto T., Yang M., Katoh T. Assay of 2-naphthol in human urine by high-performance liquid chromatography. J. Chromatogr. 1999;B734:211–217. [PubMed: 10595719]
- Kim D.-H., Nelson H.H., Wiencke J.K., Christiani D.C., Wain J.C., Mark E.J., Kelsey K.T. Promoter methylation of DAP-kinase: Association with advanced stage in non-small cell lung cancer. Oncogene. 2001;20:1765–1770. [PubMed: 11313923]
- Kim H., Cho S.-H., Kang J.-W., Kim Y.-D., Nan H.-M., Lee C.-H., Lee H., Kawamoto T. Urinary 1-hydroxypyrene and 2-naphthol concentrations in male Koreans. Int. Arch. occup. environ. Health. 2001;74:59–62. [PubMed: 11196083]
- King M.M., Hollingsworth A., Cuzick J., Garner R.C. The detection of adducts in human cervix tissue DNA using 32P-postlabelling: A study of the relationship with smoking history and oral contraceptive use. Carcinogenesis. 1994;15:1097–1100. [PubMed: 8200076]
- Kitagawa K., Kunugita N., Katoh T., Yang M., Kawamoto T. The significance of the homozygous CYP2A6 deletion on nicotine metabolism: A new genotyping method of CYP2A6 using a single PCR–RFLP. Biochem. biophys. Res. Commun. 1999;262:146–151. [PubMed: 10448083]
- Kivistö H., Pekari K., Peltonen K., Svinhufvud J., Veidebaum T., Sorsa M., Aitio A. Biological monitoring of exposure to benzene in the production of benzene and in a cokery. Sci. total Environ. 1997;199:49–63. [PubMed: 9200847]
- Kiyosawa H., Suko M., Okudaira H., Murata K., Miyamoto T., Chung M.-H., Kasai H., Nishimura S. Cigarette smoking induces formation of 8-hydroxydeoxyguanosine, one of the oxidative DNA damages in human peripheral leukocytes. Free Radic. Res. Commun. 1990;11:23–27. [PubMed: 2074046]
- Klesges R.C., Meyers A.W., Klesges L.M., La Vasque M.E. Smoking, body weight, and their effects on smoking behavior: A comprehensive review of the literature. Psychol. Bull. 1989;106:204–230. [PubMed: 2678202]
- Kopplin A., Eberle-Adamkiewicz G., Glüsenkamp K.-H., Nehls P., Kirstein U. Urinary excretion of 3-methyladenine and 3-ethyladenine after controlled exposure to tobacco smoke. Carcinogenesis. 1995;16:2637–2641. [PubMed: 7586179]
- Kriebel D., Henry J., Gold J.C., Bronsdson A., Commoner B. The mutagenicity of cigarette smokers' urine. J. environ. Pathol. Toxicol. Oncol. 1985;6:157–169. [PubMed: 4078685]
- Kriek E., Rojas M., Alexandrov K., Bartsch H. Polycyclic aromatic hydrocarbon–DNA adducts in humans: Relevance as biomarkers for exposure and cancer risk. Mutat. Res. 1998;400:215–231. [PubMed: 9685648]
- Krokan H., Grafstrom R.C., Sundqvist K., Esterbauer H., Harris C.C. Cytotoxicity, thiol depletion and inhibition of O6-methylguanine–DNA methyltransferase by various aldehydes in cultured human bronchial fibroblasts. Carcinogenesis. 1985;6:1755–1759. [PubMed: 4064250]
- Kuenemann-Migeot C., Callais F., Momas I., Festy B. Urinary promutagens of smokers: Comparison of concentration methods and relation to cigarette consumption. Mutat. Res. 1996;368:141–147. [PubMed: 8684404]
- Kulkarni J.R., Sarkar S., Bhide S.V. Mutagenicity of extracts of brown and black masheri, pyrolysed products of tobacco using short-term tests. Mutagenesis. 1987;2:263–266. [PubMed: 3325755]
- Kulp K.S., Knize M.G., Malfatti M.A., Salmon C.P., Felton J.S. Identification of urine metabolites of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine following consumption of a single cooked chicken meal in humans. Carcinogenesis. 2000;21:2065–2072. [PubMed: 11062169]
- Kwon J.T., Nakajima M., Chai S., Yom Y.K., Kim H.K., Yamazaki H., Sohn D.R., Yamamoto T., Kuroiwa Y., Yokoi T. Nicotine metabolism and CYP2A6 allele frequencies in Koreans. Pharmacogenetics. 2001;11:317–323. [PubMed: 11434509]
- Lafi A., Parry J.M. Cytogenetic activities of tobacco particulate matter (TPM) derived from a low to middle tar British cigarette. Mutat. Res. 1988;201:365–374. [PubMed: 3050488]
- Lähdetie J., Engström K., Husgafvel-Pursiainen K., Nylund L., Vainio H., Sorsa J. Maternal smoking induced cotinine levels and genotoxicity in second trimester amniotic fluid. Mutat. Res. 1993;300:37–43. [PubMed: 7683766]
- Landin-Wilhelmsen K., Wilhelmsen L., Lappas G., Rosen T., Lindstedt G., Lundberg P.A., Bengtsson B.A. Serum insulin-like growth factor I in a random population sample of men and women: Relation to age, sex, smoking habits, coffee consumption and physical activity, blood pressure and concentrations of plasma lipids, fibrinogen, parathyroid hormone and osteocalcin. Clin. Endocrinol. 1994;4:351–357. [PubMed: 7955442]
- Larramendy M.L., Knuutila S. Increased frequency of micronuclei in B and T8 lymphocytes from smokers. Mutat. Res. 1991;259:189–195. [PubMed: 1825233]
- LaRue H., Allard P., Simoneau M., Normand C., Pfister C., Moore L., Meyer F., Têtu B., Fradet Y. P53 point mutations in initial superficial bladder cancer occur only in tumors from current or recent cigarette smokers. Carcinogenesis. 2000;21:101–106. [PubMed: 10607740]
- Lauwerys R.R., Buchet J.P., Andrien F. Muconic acid in urine: A reliable indicator of occupational exposure to benzene. Am. J. ind. Med. 1994;25:297–300. [PubMed: 8147402]
- LaVoie E.J., Adams J.D., Reinhardt J., Rivenson A., Hoffmann D. Toxicity studies on clove cigarette smoke and constituents of clove: Determination of the LD50 of eugenol by intratracheal instillation in rats and hamsters. Arch. Toxicol. 1986;59:78–81. [PubMed: 3753194]
- Law M.R., Hackshaw A.K. A meta-analysis of cigarette smoking, bone mineral density and risk of hip fracture: Recognition of a major effect. Br. med. J. 1997;315:841–846. [PMC free article: PMC2127590] [PubMed: 9353503]
- Law M.R., Cheng R., Hackshaw A.K., Allaway S., Hale A.K. Cigarette smoking, sex hormones and bone density in women. Eur. J. Epidemiol. 1997;13:553–558. [PubMed: 9258568]
- Lazutka J.R., Dedonyte V., Krapavickaite D. Sister-chromatid exchanges and their distribution in human lymphocytes in relation to age, sex and smoking. Mutat. Res. 1994;306:173–180. [PubMed: 7512216]
- Leanderson P. Cigarette smoke-induced DNA damage in cultured human lung cells. Ann. N.Y. Acad. Sci. 1993;686:249–259. 259–261. [PubMed: 8512251]
- Leanderson P., Tagesson C. Cigarette smoke-induced DNA-damage: Role of hydroquinone and catechol in the formation of the oxidative DNA–adduct, 8-hydroxydeoxyguanosine. Chem.-biol. Interact. 1990;75:71–81. [PubMed: 2114224]
- Leanderson P., Tagesson C. Cigarette tar promotes neutrophil-induced DNA damage in cultured lung cells. Environ. Res. 1994;64:103–111. [PubMed: 8306945]
- Lechner J.F., Neft R.E., Gilliland F.D., Crowell R.E., Belinsky S.A. Molecular identification of individuals at high risk for lung cancer. Radiat. oncol. Inves. 1997;5:103–105. [PubMed: 9303064]
- Lee C.K., Brown B.G., Rice W.Y. Jr, Doolittle D.J. Role of oxygen free radicals in the induction of sister chromatid exchanges by cigarette smoke. Environ. mol. Mutag. 1989;13:54–59. [PubMed: 2642805]
- Lee C.K., Brown B.G., Reed E.A., Lowe G.D., McKarns S.C., Fulp C.W., Coggins C.R., Ayres P.H., Doolittle D.J. Analysis of cytogenetic effects in bone-marrow cells of rats subchronically exposed to smoke from cigarettes which burn or only heat tobacco. Mutat. Res. 1990a;240:251–257. [PubMed: 2330011]
- Lee C.K., Doolittle D.J., Burger G.T., Hayes A.W. Comparative genotoxicity testing of mainstream whole smoke from cigarettes which burn or heat tobacco. Mutat. Res. 1990b;242:37–45. [PubMed: 2202896]
- Lee C.K., Brown B.G., Reed B.A., Rahn C.A., Coggins C.R., Doolittle D.J., Hayes A.W. Fourteen-day inhalation study in rats, using aged and diluted sidestream smoke from a reference cigarette. Fundam. appl. Toxicol. 1992;19:141–146. [PubMed: 1397795]
- Lee B.L., New A.L., Kok P.W., Ong H.Y., Shi C.Y., Ong C.N. Urinary trans, transmuconic acid determined by liquid chromatography: Application in biological monitoring of benzene exposure. Clin. Chem. 1993;39:1788–1792. [PubMed: 8375048]
- Lee C.K., Brown B.G., Reed E.A., Coggins C.R., Doolittle D.J., Hayes A.W. Ninety-day inhalation study in rats, using aged and diluted sidestream smoke from a reference cigarette: DNA adducts and alveolar macrophage cytogenetics. Fundam. appl. Toxicol. 1993;20:393–401. [PubMed: 8314456]
- Le Houezec J., Benowitz N.L. Basic and clinical psychopharmacology of nicotine. Clin. Chest. Med. 1991;12:681–699. [PubMed: 1747987]
- Le Houezec J., Martin C., Cohen C., Molimard R. Failure of behavioral dependence induction and oral nicotine biovailability in rats. Physiol. Behav. 1989;45:103–108. [PubMed: 2727123]
- Lemasters G.K., Livingston G.K., Lockey J.E., Olsen D.M., Shukla R., New G., Selevan S.G., Yiin J.H. Genotoxic changes after low-level solvent and fuel exposure on aircraft maintenance personnel. Mutagenesis. 1997;12:237–243. [PubMed: 9237768]
- Levin J.O. First international workshop on hydroxypyrene as a biomarker for PAH exposure in man — Summary and conclusions. Sci. total Environ. 1995;163:165–168. [PubMed: 7716494]
- Leyden, D.E., Leitner, E. & Siegmund, B. (1999) Determination of nicotine in pharmaceutical products and dietary sources. In: Gorrod, J.W. & Jacob, P., III, eds, Analytical Determination of Nicotine and Related Compounds and Their Metabolites, New York, Elsevier.
- Li D., Wang M., Dhingra K., Hittelman W.N. Aromatic DNA adducts in adjacent tissues of breast cancer patients: Clues to breast cancer etiology. Cancer Res. 1996;56:287–293. [PubMed: 8542582]
- Li H., Krieger R.I., Li Q.X. Improved HPLC method for analysis of 1-hydroxypyrene in human urine specimens of cigarette smokers. Sci. total Environ. 2000;257:147–153. [PubMed: 10989924]
- Li L., Ruan Y., Chen Y., Chu Y., Xu X. [The role of transforming growth factor-beta(1) in smoking-induced chronic bronchitis and emphysema in hamsters.] Zhonghua Jie He He Hu Xi Za Zhi. 2002;25:284–286. (in Chinese) [PubMed: 12133321]
- Linert W., Bridge M.H., Huber M., Bjugstad K.B., Grossman S., Arendash G.W. Invitro and in-vivo studies investigating possible antioxidant actions of nicotine: Relevance to Parkinson's and Alzheimer's diseases. Biochim. biophys. Acta. 1999;1454:143–152. [PubMed: 10381559]
- Lipkin M., Newmark H.L. Vitamin D, calcium and prevention of breast cancer: A review. J. Am. Coll. Nutr. 1999;18 (Suppl. 5):392–397. [PubMed: 10511319]
- Lippman S.M., Peters E.J., Wargovich M.J., Stadnyk A.N., Dixon D.O., Dekmezian R.H., Loewy J.W., Morice R.C., Cunningham J.E., Hong W.K. Broncial micronuclei as a marker of an early stage of carcinogenesis in the human tracheobronchial epithelium. Int. J. Cancer. 1990;45:811–815. [PubMed: 2335384]
- Littlefield L.G., Joiner E.E. Analysis of chromosome aberrations in lymphocytes of long-term heavy smokers. Mutat. Res. 1986;170:145–150. [PubMed: 3713724]
- Liu X., Lu J., Liu S. Synergistic induction of hydroxyl radical-induced DNA single-strand breaks by chromium (VI) compound and cigarette smoke solution. Mutat. Res. 1999;440:109–117. [PubMed: 10095134]
- Liu Y., Egyhazi S., Hansson J., Bhide S.V., Kulkarni P.S., Grafström R.C. O6-Methylguanine–DNA methyltransferase activity in human buccal mucosal tissue and cell cultures. Complex mixtures related to habitual use of tobacco and betel quid inhibit the activity in vitro. Carcinogenesis. 1997;18:1889–1895. [PubMed: 9363996]
- Lodovici M., Akpan V., Giovannini L., Migliani F., Dolara P. Benzo[a]pyrene diolepoxide DNA adducts and levels of polycyclic aromatic hydrocarbons in autoptic samples from human lungs. Chem.-biol. Interact. 1998;116:199–212. [PubMed: 9920462]
- Loechler E.L., Green C.L., Essigmann J.M. In-vivo mutagenesis by O6-methylguanine built into a unique site in a viral genome. Proc. natl Acad. Sci. USA. 1984;81:6271–6275. [PMC free article: PMC391905] [PubMed: 6093094]
- Loennechen J.P., Beisvag V., Arbo I., Waldum H.L., Sandvik A.K., Knardahl S., Ellingsen O. Chronic carbon monoxide exposure in vivo induces myocardial endothelin-1 expression and hypertrophy in rat. Pharmacol. Toxicol. 1999;85:192–197. [PubMed: 10563519]
- Loft S., Poulsen H.E. Estimation of oxidative DNA damage in man from urinary excretion of repair products. Acta biochim. polon. 1998;45:133–144. [PubMed: 9701506]
- Loft S., Vistisen K., Ewertz M., Tjonneland A., Overvad K., Poulsen H.E. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: Influence of smoking, gender and body mass index. Carcinogenesis. 1992;13:2241–2247. [PubMed: 1473230]
- Longcope C., Johnston C.C. Jr. Androgen and estrogen dynamics in pre- and postmenopausal women: A comparison between smokers and nonsmokers. J. clin. Endocrinol. Metab. 1988;67:379–383. [PubMed: 3392163]
- Lucero J., Harlow B.L., Barbieri R.L., Sluss P., Cramer D.W. Early follicular phase hormone levels in relation to patterns of alcohol, tobacco, and coffee use. Fertil. Steril. 2001;76:723–729. [PubMed: 11591405]
- Lukanova A., Toniolo P., Akhmedkhanov A., Hunt K., Rinaldi S., Zeleniuch-Jacquotte A., Haley N.J., Riboli E., Stattin P., Lundin E., Kaaks R. A cross-sectional study of IGF-1 determinants in women. Eur. J. Cancer Prev. 2001;10:443–452. [PubMed: 11711759]
- Luoto R., Kaprio J., Uutela A. Age at natural menopause and sociodemographic status in Finland. Am. J. Epidemiol. 1994;139:64–76. [PubMed: 8296776]
- Ma L., Chow J.Y., Cho C.H. Effects of cigarette smoking on gastric ulcer formation and healing: Possible mechanisms of action. J. clin. Gastroenterol. 1998;27 (Suppl. 1):S80–S86. [PubMed: 9872502]
- Ma J., Pollak M.N., Giovannucci E., Chan J.M., Tao Y., Hennekens C.H., Stampfer M.J. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J. natl Cancer Inst. 1999;91:620–625. [PubMed: 10203281]
- van Maanen J.M.S., Welle I.J., Hageman G., Dallinga J.W., Mertens P.L.J.M., Kleinjans J.C.S. Nitrate contamination of drinking water: Relationship with HPRT variant frequency in lymphocyte DNA and urinary excretion of N-nitrosamines. Environ. Health Perspect. 1996;104:522–528. [PMC free article: PMC1469364] [PubMed: 8743440]
- Mackenzie P.I., Miners J.O., McKinnon R.A. Polymorphisms in UDP glucuronosyltransferase genes: Functional consequences and clinical relevance. Clin. Chem. Lab. Med. 2000;38:889–892. [PubMed: 11097345]
- Maclure M., Katz R.B.-A., Bryant M.S., Skipper P.L., Tannenbaum S.R. Elevated blood levels of carcinogens in passive smokers. Am. J. public Health. 1989;79:1381–1384. [PMC free article: PMC1350179] [PubMed: 2782507]
- Maclure M., Bryant M.S., Skipper P.L., Tannenbaum S.R. Decline of the hemoglobin adduct of 4-aminobiphenyl during withdrawal from smoking. Cancer Res. 1990;50:181–184. [PubMed: 2293553]
- Maestrelli P., Saetta M., Mapp C.E., Fabbri L.M. Remodeling in response to infection and injury. Airway inflammation and hypersecretion of mucus in smoking subjects with chronic obstructive pulmonary disease. Am. J. respir. crit. Care Med. 2001;164:S76–S80. [PubMed: 11734472]
- Maggio R., Riva M., Vaglini F., Fornai F., Molteni R., Armogida M., Racagni G., Corsini G.U. Nicotine prevents experimental parkinsonism in rodents and induces striatal increase of neurotrophic factors. J. Neurochem. 1998;71:2439–2446. [PubMed: 9832142]
- Mahimkar M.B., Bhisey R.A. Occupational exposure to bidi tobacco increases chromosomal aberrations in tobacco processors. Mutat. Res. 1995;334:139–144. [PubMed: 7885365]
- Malaveille C., Vineis P., Estève J., Ohshima H., Brun G., Hautefeuille A., Gallet P., Ronco G., Terracini B., Bartsch H. Levels of mutagens in the urine of smokers of black and blond tobacco correlate with their risk of bladder cancer. Carcinogenesis. 1989;10:577–586. [PubMed: 2924402]
- Malin D.H. Nicotine dependence. Studies with a laboratory model. Pharmacol. Biochem. Behav. 2001;70:551–559. [PubMed: 11796153]
- Manchester D.K., Jacoby E.H. Sensitivity of human placental monooxygenase activity to maternal smoking. Clin. Pharmacol. Ther. 1981;30:687–692. [PubMed: 7297026]
- Manchester D.K., Weston A., Choi J.-S., Trivers G.E., Fennessey P.V., Quintana E., Farmer P.B., Mann D.L., Harris C.C. Detection of benzo[a]pyrene diol epoxide–DNA adducts in human placenta. Proc. natl Acad. Sci. USA. 1988;85:9243–9247. [PMC free article: PMC282715] [PubMed: 3143115]
- Manchester D.K., Wilson V.L., Hsu I.-C., Choi J.S., Parker N.B., Mann D.L., Weston A., Harris C.C. Synchronous fluorescence spectroscopic, immunoaffinity chromatographic and 32P-postlabeling analysis of human placental DNA known to contain benzo[a]pyrene diol epoxide adducts. Carcinogenesis. 1990;11:553–559. [PubMed: 2322996]
- Manchester D.K., Bowman E.D., Parker N.B., Caporaso N.E., Weston A. Determinants of polycyclic aromatic hydrocarbon–DNA adducts in human placenta. Cancer Res. 1992;52:1499–1503. [PubMed: 1540958]
- Mancini R., Romano G., Sgambato A., Flamini G., Giovagnoli M.R., Boninsegna A., Carraro C., Vecchione A., Cittadini A. Polycyclic aromatic hydrocarbon–DNA adducts in cervical smears of smokers and nonsmokers. Gynecol. Oncol. 1999;75:68–71. [PubMed: 10502428]
- Manson J.E., Ajani U.A., Liu S., Nathan D.M., Hennekens C.H. A prospective study of cigarette smoking and the incidence of diabetes mellitus among US male physicians. Am. J. Med. 2000;109:538–542. [PubMed: 11063954]
- Manson J.M., Sammel M.D., Freeman E.W., Grisso J.A. Racial differences in sex hormone levels in women approaching the transition to menopause. Fertil. Steril. 2001;75:297–304. [PubMed: 11172830]
- Mao L., Lee J.S., Kurie J.M., Fan Y.H., Lippman S.M., Lee J.J., Ro J.Y., Broxson A., Yu R., Morice R.C., Kemp B.L., Khuri F.R., Walsh G.L., Hittelman W.N., Hong W.K. Clonal genetic alterations in the lungs of current and former smokers. J. natl. Cancer Inst. 1997;89:834–836. [PubMed: 9196251]
- March T.H., Barr E.B., Finch G.L., Hahn F.F., Hobbs C.H., Menache M.G., Nikula K.J. Cigarette smoke exposure produces more evidence of emphysema in B6C3F1 mice than in F344 rats. Toxicol. Sci. 1999;51:289–290. [PubMed: 10543031]
- Marchetti A., Pelligrini S., Sozzi G., Bertacca G., Gaeta P., Buttitta F., Carnicelli V., Griseri P., Chella A., Angeletti C.A., Pierotti M., Bevilacqua G. Genetic analysis of lung tumours of non-smoking subjects: p53 Gene mutations are constantly associated with loss of heterozygosity at the FHIT locus. Br. J. Cancer. 1998;78:73–78. [PMC free article: PMC2062949] [PubMed: 9662254]
- Martinez M.E., Willett W.C. Calcium, vitamin D, and colorectal cancer: A review of the epidemiologic evidence. Cancer Epidemiol. Biomarkers Prev. 1998;7:163–168. [PubMed: 9488592]
- Marubio L.M., Changeux J.-P. Nicotinic acetylcholine receptor knockout mice as animal models for studying receptor function. Eur. J. Pharmacol. 2000;393:113–121. [PubMed: 10771004]
- Mascher D.G., Mascher H.J., Scherer G., Schmid E.R. High-performance liquid chromatographic–tandem mass spectrometric determination of 3-hydroxypropylmercapturic acid in human urine. J. Chromatogr. 2001;B750:163–169. [PubMed: 11204217]
- Massey E., Aufderheide M., Koch W., Lodding H., Pohlmann G., Windt H., Jarck P., Knebel J.W. Micronucleus induction in V79 cells after direct exposure to whole cigarette smoke. Mutagenesis. 1998;13:145–149. [PubMed: 9568586]
- Matikainen T., Perez G.I., Jurisicova A., Pru J.K., Schlezinger J.J., Ryu H.-Y., Laine J., Sakai T., Korsmeyer S.J., Capser R.F., Sherr D.H., Tilly J.L. Aromatic hydrocarbon receptor-driven Bax gene expression is required for premature ovarian failure caused by biohazardous environmental chemicals. Nat. Genet. 2001;28:355–360. [PubMed: 11455387]
- Matsukura N., Willey J., Miyashita M., Taffe B., Hoffmann D., Waldren C., Puck T.T., Harris C.C. Detection of direct mutagenicity of cigarette smoke condensate in mammalian cells. Carcinogenesis. 1991;12:685–689. [PubMed: 2013132]
- Mattison D.R. The effects of smoking on fertility from gametogenesis to implantation. Environ. Res. 1982;28:410–433. [PubMed: 6749489]
- Mattison D.R., Thorgeirsson S.S. Smoking and industrial pollution, and their effects on menopause and ovarian cancer. Lancet. 1978;i:187–188. [PubMed: 74610]
- Matullo G., Palli D., Peluso M., Guarrera S., Carturan S., Celentano E., Krogh V., Munnia A., Tumino R., Polidoro S., Piazza A., Vineis P. XRCC1, XRCC3, XPD gene polymorphisms, smoking and (32)P–DNA adducts in a sample of healthy subjects. Carcinogenesis. 2001;22:1437–1445. [PubMed: 11532866]
- Mauderly, J.L., Chen, B.T., Hahn, F.F., Lundgren, D.L., Cuddihy, R.G., Namenyi, J., Rebar, A.H. (1989) The effect of chronic cigarette smoke inhalation on the long-term pulmonary clearance of inhaled particles in the rat. In: Wehner, A.P., ed., Biological Interaction of Inhaled Mineral Fibers and Cigarette Smoke, Columbus, OH, Battelle Press, pp. 223–239.
- McFadden D., Wright J.L., Wiggs B., Churg A. Smoking inhibits asbestos clearance. Am. Rev. respir. Dis. 1986;133:372–374. [PubMed: 2869726]
- McKinlay S.M., Bifano N.L., McKinlay J.B. Smoking and age at menopause in women. Ann. intern. Med. 1985;103:350–356. [PubMed: 4026083]
- Meger M., Meger-Kossien I., Dietrich M., Tricker A.R., Scherer G., Adlkofer F. Metabolites of 4-(N-methylnitrosamino)-1-(3-pyridyl)-1-butanone in urine of smokers. Eur. J. Cancer Prev. 1996;5 (Suppl. 1):121–124. [PubMed: 8972306]
- Meger M., Meger-Kossien I., Riedel K., Scherer G. Biomonitoring of environmental tobacco smoke (ETS)-related exposure to 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Biomarkers. 2000;5:33–45. [PubMed: 23885904]
- Melikian A.A., Prahalad A.K., Hoffmann D. Urinary trans,trans-muconic acid as an indicator of exposure to benzene in cigarette smokers. Cancer Epidemiol. Biomarkers Prev. 1993;2:47–51. [PubMed: 8420612]
- Melikian A.A., Prahalad A.K., Secker-Walker R.H. Comparison of the levels of the urinary benzene metabolite trans,trans-muconic acid in smokers and nonsmokers, and the effects of pregnancy. Cancer Epidemiol. Biomarkers Prev. 1994;3:239–244. [PubMed: 8019374]
- Melikian A.A., Sun P., Pierpont C., Coleman S., Hecht S.S. Gas chromatographic–mass spectrometric determination of benzo[a]pyrene and chrysene diol epoxide globin adducts in humans. Cancer Epidemiol. Biomarkers Prev. 1997;6:833–839. [PubMed: 9332767]
- Melikian A.A., Sun P., Prokopczyk B., El-Bayoumy K., Hoffmann D., Wang X., Waggoner S. Identification of benzo[a]pyrene metabolites in cervical mucus and DNA adducts in cervical tissues in humans by gas chromatography–mass spectrometry. Cancer Lett. 1999;146:127–134. [PubMed: 10656617]
- Mellstrom D., Johansson C., Johnell O., Lindstedt G., Lundberg P.A., Obrant K., Schoon I.M., Toss G., Ytterberg B.O. Osteoporosis, metabolic aberrations, and increased risk for vertebral fractures after partial gastrectomy. Calcif. Tissue int. 1993;53:370–377. [PubMed: 8293349]
- Memisoglu A., Samson L. Base excision repair in yeast and mammals. Mutat. Res. 2000;451:39–51. [PubMed: 10915864]
- Merchant C., Tang M.X., Albert S., Manly J., Stern Y., Mayeux R. The influence of smoking on the risk of Alzheimer's disease. Neurology. 1999;52:1408–1412. [PubMed: 10227626]
- Merlo F., Andreassen Å., Weston A., Pan C.-F., Haugen A., Valerio F., Reggiardo G., Fontana V., Garte S., Puntoni R., Abbondandolo A. Urinary excretion of 1-hydroxypyrene as a marker for exposure to urban air levels of polycyclic aromatic hydrocarbons. Cancer Epidemiol. Biomarkers Prev. 1998;7:147–155. [PubMed: 9488590]
- Meschia M., Pansini F., Modena A.B., de Aloysio D., Gambacciani M., Parazzini F., Campagnoli C., Maiocchi G., Peruzzi E. Determinants of age at menopause in Italy: Results from a large cross-sectional study. ICARUS Study Group. Italian Climacteric Research Group Study. Maturitas. 2000;34:119–125. [PubMed: 10714906]
- Messina E.S., Tyndale R.F., Sellers E.M. A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J. Pharmacol. exp Ther. 1997;282:1608–1614. [PubMed: 9316878]
- Michnovicz J.J., Hershcopf R.J., Naganuma H., Bradlow H.L., Fishman J. Increased 2-hydroxylation of estradiol as a possible mechanism for the anti-estrogenic effect of cigarette smoking. New Engl. J. Med. 1986;315:1305–1309. [PubMed: 3773953]
- Michnovicz J.J., Naganuma H., Hershcopf R.J., Bradlow H.L., Fishman J. Increased urinary catechol estrogen excretion in female smokers. Steroids. 1988;52:69–83. [PubMed: 2854668]
- Midgette A.S., Baron J.A. Cigarette smoking and the risk of natural menopause. Epidemiology. 1990;1:474–480. [PubMed: 2090286]
- Minoia C., Apostoli P., Maranelli G., Baldi C., Pozzoli L., Capodaglio E. Urinary chromium levels in subjects living in two north Italy regions. Sci. total Environ. 1988;71:527–531. [PubMed: 3406718]
- Miyaura S., Eguchi H., Johnston J.M. Effect of a cigarette smoke extract on the metabolism of the proinflammatory autacoid, platelet-activating factor. Circ. Res. 1992;70:341–347. [PubMed: 1735133]
- Modan M., Meytes D., Rozeman P., Yosef S.B., Sehayek E., Yosef N.B., Lusky A., Halkin H. Significance of high HbA1 levels in normal glucose tolerance. Diabetes Care. 1988;11:422–428. [PubMed: 3391093]
- Mohtashamipur E., Norpoth K., Straeter H. Clastogenic effect of passive smoking on bone marrow polychromatic erythrocytes of NMRI mice. Toxicol. Lett. 1987;35:153–156. [PubMed: 3810675]
- Mohtashamipur E., Steinforth T., Norpoth K. Comparative bone marrow clastogenicity of cigarette sidestream, mainstream and recombined smoke condensates in mice. Mutagenesis. 1988;3:419–422. [PubMed: 3070276]
- Molarius A., Seidell J.C., Kuulasmaa K., Dobsen A.J., Sans S. Smoking and relative body weight: An international perspective from the WHO MONICA Project. J. Epidemiol. Community Health. 1997;51:252–260. [PMC free article: PMC1060469] [PubMed: 9229053]
- Mollerup S., Ryberg D., Hewer A., Phillips D.H., Haugen A. Sex differences in lung CYP1A1 expression and DNA adduct levels among lung cancer patients. Cancer Res. 1999;59:3317–3320. [PubMed: 10416585]
- Mooney L.A., Santella R.M., Covey L., Jeffrey A.M., Bigbee W., Randall M.C., Cooper T.B., Ottman R., Tsai W.Y., Wazneh L., Glassman A.H., Young T.-L., Perera F.P. Decline of DNA damage and other biomarkers in peripheral blood following smoking cessation. Cancer Epidemiol. Biomarkers Prev. 1995;4:627–634. [PubMed: 8547829]
- Mooney L.A., Bell D.A., Santella R.M., Van Bennekum A.M., Ottman R., Paik M., Blaner W.S., Lucier G.W., Covey L., Young T.L., Cooper T.B., Glassman A.H., Perera F.P. Contribution of genetic and nutritional factors to DNA damage in heavy smokers. Carcinogenesis. 1997;18:503–509. [PubMed: 9067549]
- Moorman A.V., Roman E., Cartwright R.A., Morgan G.J. Smoking and the risk of acute myeloid leukaemia in cytogenetic subgroups. Br. J. Cancer. 2002;86:60–62. [PMC free article: PMC2746540] [PubMed: 11857012]
- Morabia A., Bernstein M.S., Curtin F., Berode M. Validation of self-reported smoking status by simultaneous measurement of carbon monoxide and salivary thiocyanate. Prev. Med. 2001;32:82–88. [PubMed: 11162330]
- Mori Y., Iimura K., Furukawa F., Nishikawa A., Takahashi M., Konishi Y. Effect of cigarette smoke on the mutagenic activation of various carcinogens in hamster. Mutat. Res. 1995;346:1–8. [PubMed: 7530323]
- Morimoto T., Nakaya K., Sugitani A., Yamada F., Imai J. Studies on the influence of environmental pollution with heavy metals on human health. IV. Nickel and chromium contents in human urine. Gifu-Ken Eisei Kenkyusho Ho. 1977;22:13–17.
- Morin R.S., Tulis J.J., Claxton L.D. The effect of solvent and extraction methods on the bacterial mutagenicity of sidestream cigarette smoke. Toxicol. Lett. 1987;38:279–290. [PubMed: 3310338]
- Mostafa M.H., Helmi S., Badawi A.F., Tricker A.R., Spiegelhalder B., Preussmann R. Nitrate, nitrite and volatile N-nitroso compounds in the urine of Schistosoma haematobium and Schistosoma mansoni infected patients. Carcinogenesis. 1994;15:619–625. [PubMed: 8149471]
- Motykiewicz G., Malusecka E., Michalska J., Kalinowska E., Wloch J., Butkiewicz D., Mazurek A., Lange D., Perera F.P., Santella R.M. Immunoperoxidase detection of polycyclic aromatic hydrocarbon–DNA adducts in breast tissue sections. Cancer Detect. Prev. 2001;25:328–335. [PubMed: 11531009]
- Moxham J. Nicotine addiction. Br. med. J. 2000;320:391–392. [PMC free article: PMC1117526] [PubMed: 10669423]
- Müller M., Krämer A., Angerer J., Hallier E. Ethylene oxide–protein adduct formation in humans: Influence of glutathione-S-transferase polymorphisms. Int. Arch. occup. environ. Health. 1998;71:499–502. [PubMed: 9826084]
- Mumford J.L., Li X., Hu F., Lu X.B., Chuang J.C. Human exposure and dosimetry of polycyclic aromatic hydrocarbons in urine from Xuan Wei, China with high lung cancer mortality associated with exposure to unvented coal smoke. Carcinogenesis. 1995;16:3031–3036. [PubMed: 8603481]
- Muranaka H., Higashi E., Itani S., Shimizu Y. Evaluation of nicotine, cotinine, thiocyanate, carboxyhemoglobin, and expired carbon monoxide as biochemical tobacco smoke uptake parameters. Int. Arch. occup. environ. Health. 1988;60:37–41. [PubMed: 3258284]
- Mure K., Hayatsu H., Takeuchi T., Takeshita T., Morimoto K. Heavy cigarette smokers show higher mutagenicity in urine. Mutat. Res. 1997;373:107–111. [PubMed: 9015159]
- Murthy M.K., Bhargava M.K., Augustus M. Sister chromatid exchange studies in oral cancer patients. Indian J. Cancer. 1997;34:49–58. [PubMed: 9491662]
- Mustonen R., Hemminki K. 7-Methylguanine levels in DNA of smokers' and nonsmokers' total white blood cells, granulocytes and lymphocytes. Carcinogenesis. 1992;13:1951–1955. [PubMed: 1423861]
- Mustonen R., Schoket B., Hemminki K. Smoking-related DNA adducts: 32P-Postlabeling analysis of 7-methylguanine in human bronchial and lymphocyte DNA. Carcinogenesis. 1993;14:151–154. [PubMed: 8425264]
- Myers S.R., Spinnato J.A., Pinorini-Godly M.T., Cook C., Boles B., Rodgers G.C. Characterization of 4-aminobiphenyl–hemoglobin adducts in maternal and fetal blood samples. J. Toxicol. environ. Health. 1996;47:553–566. [PubMed: 8614023]
- Nagaya T., Toriumi H. Effects of smoking on spontaneous and induced sister chromatid exchanges in lymphocytes. Toxicol. Lett. 1985;25:293–296. [PubMed: 3925600]
- Nakajima M., Yamamoto T., Nunoya K.-I., Yokoi T., Nagashima K., Inoue K., Funae Y., Shimada N., Kamataki T., Kuroiwa Y. Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab. Dispos. 1996;24:1212–1217. [PubMed: 8937855]
- Nakajima M., Yamagishi S.-I., Yamamoto H., Yamamoto T., Kuroiwa Y., Yokoi T. Deficient cotinine formation from nicotine is attributed to the whole deletion of the CYP2A6 gene in humans. Clin. Pharmacol. Ther. 2000;67:57–69. [PubMed: 10668854]
- Nakajima M., Kwon J.-T., Tanaka N., Zenta T., Yamamoto Y., Yamamoto H., Yamazaki H., Yamamoto T., Kuroiwa Y., Yokoi T. Relationship between interindividual differences in nicotine metabolism and CYP2A6 genetic polymorphism in humans. Clin. Pharmacol. Ther. 2001;69:72–78. [PubMed: 11180041]
- Nakayama T., Kaneko M., Kodama M., Nagata C. Cigarette smoke induces DNA single-strand breaks in human cells. Nature. 1985;314:462–464. [PubMed: 2984577]
- Nan H.-M., Kim H., Lim H.-S., Choi J.K., Kawamoto T., Kang J.-W., Lee C.-H., Kim Y.-D., Kwon E.H. Effects of occupation, lifestyle and genetic polymorphisms of CYP1A1, CYP2E1, GSTM1 and GSTT1 on urinary 1-hydroxypyrene and 2-naphthol concentrations. Carcinogenesis. 2001;22:787–793. [PubMed: 11323399]
- Nath R.G., Ocando J.E., Guttenplan J.B., Chung F.L. 1,N2-Propanodeoxyguanosine adducts: Potential new biomarkers of smoking-induced DNA damage in human oral tissue. Cancer Res. 1998;58:581–584. [PubMed: 9485001]
- National Research Council (1986) Environmental Tobacco Smoke. Measuring Exposures and Assessing Health Effects, Washington DC, National Academy Press. [PubMed: 25032469]
- Need A.G., Kemp A., Giles N., Morris H.A., Horowitz M., Nordin B.E. Relationships between intestinal calcium absorption, serum vitamin D metabolites and smoking in postmenopausal women. Osteoporos. int. 2002;13:83–88. [PubMed: 11883410]
- Nersessian A.K., Arutyunyan R.M. The comparative clastogenic activity of mainstream tobacco smoke from cigarettes widely consumed in Armenia. Mutat. Res. 1994;321:89–92. [PubMed: 7510850]
- Nerurkar P.V., Okinaka L., Aoki C., Seifried A., Lum-Jones A., Wilkens L.R., Le Marchand L. CYP1A1, GSTM1, and GSTP1 genetic polymorphisms and urinary 1-hydroxypyrene excretion in non-occupationally exposed individuals. Cancer Epidemiol. Biomarkers Prev. 2000;9:1119–1122. [PubMed: 11045797]
- Newman M.G., Lindsay M.K., Graves W. Cigarette smoking and pre-eclampsia: Their association and effects on clinical outcomes. J. matern. fetal Med. 2001;10:166–170. [PubMed: 11444784]
- Nguyen H., Finkelstein E., Reznick A., Cross C., van der Vliet A. Cigarette smoke impairs neutrophil respiratory burst activation by aldehyde-induced thiol modifications. Toxicology. 2001;160:207–217. [PubMed: 11246141]
- Nilsson P.M., Lind L., Pollare T., Berne C., Lithell H.O. Increased level of hemoglobin A1c, but not impaired insulin sensitivity, found in hypertensive and normotensive smokers. Metabolism. 1995;44:557–561. [PubMed: 7752901]
- Nishio E., Watanabe Y. Cigarette smoke extract inhibits plasma paraoxonase activity by modification of the enzyme's free thiols. Biochem. biophys. Res. Commun. 1997;236:289–293. [PubMed: 9240427]
- Norbury C.J., Hickson I.D. Cellular responses to DNA damage. Ann. Rev. Pharmacol. Toxicol. 2001;41:367–401. [PubMed: 11264462]
- Nordic Study Group on the Health Risk of Chromosome Damage. A Nordic data base on somatic chromosome damage in humans. Mutat. Res. 1990;241:325–337. [PubMed: 2366811]
- Nunoya K., Yokoi T., Takahashi Y., Kimura K., Kinoshita M., Kamataki T. Homologous unequal cross-over within the human CYP2A gene cluster as a mechanism for the deletion of the entire CYP2A6 gene associated with the poor metabolizer phenotype. J. Biochem. (Tokyo). 1999;126:402–407. [PubMed: 10423536]
- Nylander G., Berg K. Mutagenicity study of urine from smoking and non-smoking road tanker drivers. Int. Arch. occup. environ. Health. 1991;63:229–232. [PubMed: 1743763]
- Obe, G., ed. (1984) Mutations in Man, Berlin, Springer.
- Ogushi F., Hubbard R.C., Vogelmeier C., Fells G.A., Crystal R.G. Risk factors for emphysema. Cigarette smoking is associated with a reduction in the association rate constant of lung α1-antitrypsin for neutrophil elastase. J. clin. Invest. 1991;87:1060–1065. [PMC free article: PMC329901] [PubMed: 1999486]
- O'Hara P., Connett J.E., Lee W.W., Nides M., Murray R., Wise R. Early and late weight gain following smoking cessation in the Lung Health Study. Am. J. Epidemiol. 1998;148:821–830. [PubMed: 9801011]
- Ohshima H., Bartsch H. Quantitative estimation of endogenous nitrosation in humans by monitoring N-nitrosoproline excreted in the urine. Cancer Res. 1981;41:3658–3662. [PubMed: 7260920]
- Okonofua F.E., Lawal A., Bamgbose J.K. Features of menopause and menopausal age in Nigerian women. Int. J. Gynecol. Obstet. 1990;31:341–345. [PubMed: 1969819]
- Ong T.-M., Stewart J., Wen Y.-F., Whong W.-Z. Application of SOS umu-test for the detection of genotoxic volatile chemicals and air pollutants. Environ. Mutag. 1987;9:171–176. [PubMed: 2434323]
- Ong C.N., Lee B.L., Shi C.Y., Ong H.Y., Lee H.P. Elevated levels of benzene-related compounds in the urine of cigarette smokers. Int. J. Cancer. 1994;59:177–180. [PubMed: 7927915]
- Ong C.N., Kok P.W., Lee B.L., Shi C.Y., Ong H.Y., Chia K.S., Lee C.S., Luo X.W. Evaluation of biomarkers for occupational exposure to benzene. Occup. environ. Med. 1995;52:528–533. [PMC free article: PMC1128288] [PubMed: 7663638]
- Ong C.N., Kok P.W., Ong H.Y., Shi C.Y., Lee B.L., Phoon W.H., Tan K.T. Biomarkers of exposure to low concentrations of benzene: A field assessment. Occup. environ. Med. 1996;53:328–333. [PMC free article: PMC1128475] [PubMed: 8673180]
- Ortego-Centeno N., Muñoz-Torres M., Hernandez-Quero J., Jurado-Duce A., de la Higuera Torres-Puchol J. Bone mineral density, sex steroids, and mineral metabolism in premenopausal smokers. Calcif. Tissue int. 1994;55:403–407. [PubMed: 7895176]
- Oscarson M., Gullstén H., Rautio A., Bernal M.L., Sinues B., Dahl M.-L., Stengård J.H., Pelkonen O., Raunio H., Ingelman-Sundberg M. Genotyping of human cytochrome P450 2A6 (CYP2A6), a nicotine C-oxidase. FEBS Lett. 1998;438:201–205. [PubMed: 9827545]
- Oscarson M., McLellan R.A., Gullstén H., Agúndez J.A., Benítez J., Rautio A., Raunio H., Pelkonen O., Ingelman-Sundberg M. Identification and characterisation of novel polymorphisms in the CYP2A locus: Implications for nicotine metabolism. FEBS Lett. 1999;460:321–327. [PubMed: 10544257]
- Osterman-Golkar S.M., MacNeela J.P., Turner M.J., Walker V.E., Swenberg J.A., Sumner S.J., Youtsey N., Fennell T.R. Monitoring exposure to acrylonitrile using adducts with N-terminal valine in hemoglobin. Carcinogenesis. 1994;15:2701–2707. [PubMed: 8001224]
- Pachinger A., Begutter H., Ultsch I., Klus H. Determination of 4-(methylnitrosamino)4-(3-pyridyl)-butyric acid in tobacco, tobacco smoke and the urine of rats and smokers. J. Chromatogr. 1993;620:55–60. [PubMed: 8106592]
- Parazzini F., Negri E., La Vecchia C. Reproductive and general lifestyle determinants of age at menopause. Maturitas. 1992;15:141–149. [PubMed: 1470046]
- Parsons W.D., Carmella S.G., Akerkar S., Bonilla L.E., Hecht S.S. A metabolite of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in the urine of hospital workers exposed to environmental tobacco smoke. Cancer Epidemiol. Biomarkers Prev. 1998;7:257–260. [PubMed: 9521443]
- Paschal D.C., Burt V., Caudill S.P., Gunter E.W., Pirkle J.L., Sampson E.J., Miller D.T., Jackson R.J. Exposure of the US population aged 6 years and older to cadmium: 1988–1994. Arch. environ. Contam. Toxicol. 2000;38:377–383. [PubMed: 10667937]
- Pasquini R., Scassellati Sforzolini G., Savino A., Angeli G., Monarca S. Enzyme induction in rat lung and liver by condensates and fractions from main-stream and side-stream cigarette smoke. Environ. Res. 1987;44:302–311. [PubMed: 3691449]
- Pastorelli R., Guanci M., Cerri A., Negri E., La Vecchia C., Fumagalli F., Mezzetti M., Cappelli R., Panigalli T., Fanelli R., Airoldi L. Impact of inherited polymorphisms in glutathione S-transferase M1, microsomal epoxide hydrolase, cytochrome P450 enzymes on DNA, and blood protein adducts of benzo(a)pyrene-diolepoxide. Cancer Epidemiol. Biomarkers Prev. 1998;7:703–709. [PubMed: 9718223]
- Pastorelli R., Guanci M., Restano J., Berri A., Micoli G., Minoia C., Alcini D., Carrer P., Negri E., La Vecchia C., Fanelli R., Airoldi L. Seasonal effect on airborne pyrene, urinary 1-hydroxypyrene, and benzo(a)pyrene diol epoxide–hemoglobin adducts in the general population. Cancer Epidemiol. Biomarkers Prev. 1999;8:561–565. [PubMed: 10385148]
- Pavanello S., Favretto D., Brugnone F., Mastrangelo G., Dal Pra G., Clonfero E. HPLC/fluorescence determination of anti-BPDE–DNA adducts in mononuclear white blood cells from PAH-exposed humans. Carcinogenesis. 1999;20:431–435. [PubMed: 10190558]
- Paz-y-Miño C., Pérez J.C., Dávalos V., Sánchez M.E., Leone P.E. Telomeric associations in cigarette smokers exposed to low levels of X-rays. Mutat. Res. 2001;490:77–80. [PubMed: 11152974]
- Pegg A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000;462:83–100. [PubMed: 10767620]
- Peluso M., Amasio E., Bonassi S., Munnia A., Altrupa F., Parodi S. Detection of DNA adducts in human nasal mucosa tissue by 32P-postlabeling analysis. Carcinogenesis. 1997;18:339–344. [PubMed: 9054626]
- Peluso M., Airoldi L., Magagnotti C., Fiorini L., Munnia A., Hautefeuille A., Malaveille C., Vineis P. White blood cell DNA adducts and fruit and vegetable consumption in bladder cancer. Carcinogenesis. 2000;21:183–187. [PubMed: 10657956]
- Perera F.P., Santella R.M., Brenner D., Poirier M.C., Munshi A.A., Fischman H.K., Van Ryzin J. DNA adducts, protein adducts, and sister chromatid exchange in cigarette smokers and nonsmokers. J. natl Cancer Inst. 1987;79:449–456. [PubMed: 3114532]
- Perera F., Mayer J., Jaretzki A., Hearne S., Brenner D., Young T.L., Fischman H.K., Grimes M., Grantham S., Tang M.X., Tsai W.-Y., Santella R.M. Comparison of DNA adducts and sister chromatid exchange in lung cancer cases and controls. Cancer Res. 1989;49:4446–4451. [PubMed: 2743334]
- Perera F.P., Estabrook A., Hewer A., Channing K., Rundle A., Mooney L.A., Whyatt R., Phillips D.H. Carcinogen–DNA adducts in human breast tissue. Cancer Epidemiol. Biomarkers Prev. 1995;4:233–238. [PubMed: 7606197]
- Pérez-Stable E.J., Herrera B., Jacob P. III, Benowitz N.L. Nicotine metabolism and intake in black and white smokers. J. Am. med. Assoc. 1998;280:152–156. [PubMed: 9669788]
- Perkins K.A. Weight gain following smoking cessation. J. consult. clin. Psychol. 1993;61:768–777. [PubMed: 8245274]
- Persson M.G., Zetterström O., Agrenius V., Ihre E., Gustafsson L.E. Single-breath nitric oxide measurements in asthmatic patients and smokers. Lancet. 1994;343:146–147. [PubMed: 7904005]
- Petruzzelli S., Franchi M., Gronchi L., Janni A., Oesch F., Pacifici G.M., Giuntini C. Cigarette smoke inhibits cytosolic but not microsomal epoxide hydrolase of human lung. Hum. exp. Toxicol. 1992;11:99–103. [PubMed: 1349227]
- Petruzzelli S., Tavanti L.M., Celi A., Giuntini C. Detection of N7-methyldeoxyguanosine adducts in human pulmonary alveolar cells. Am. J. respir. Cell mol. Biol. 1996;15:216–223. [PubMed: 8703477]
- Petruzzelli S., Celi A., Pulerà N., Baliva F., Viegi G., Carrozzi L., Ciacchini G., Bottai M., Di Pede F., Paoletti P., Giuntini C. Serum antibodies to benzo(a)pyrene diol epoxide–DNA adducts in the general population: Effects of air pollution, tobacco smoking, and family history of lung diseases. Cancer Res. 1998;58:4122–4126. [PubMed: 9751623]
- Pezzagno G., Maestri L., Fiorentino M.L. trans,trans-Muconic acid, a biological indicator to low levels of environmental benzene: Some aspects of its specificity. Am. J. ind. Med. 1999;35:511–518. [PubMed: 10212704]
- Phillips D.H. DNA adducts in human tissues: Biomarkers of exposure to carcinogens in tobacco smoke. Environ. Health Perspect. 1996;104 (Suppl 3):453–458. [PMC free article: PMC1469660] [PubMed: 8781363]
- Phillips D.H. Detection of DNA modifications by the 32P-postlabelling assay. Mutat. Res. 1997;378:1–12. [PubMed: 9288880]
- Phillips, D.H., Farmer, P.B., (1995) Protein and DNA adducts as biomarkers of exposure to environmental mutagens. In: Phillips, D.H. & Venitt, S., eds, Environmental Mutagenesis, Oxford, Bioscience, pp. 367–395.
- Phillips D.H., Hewer A. DNA adducts in human urinary bladder and other tissues. Environ. Health Perspect. 1993;99:45–49. [PMC free article: PMC1567015] [PubMed: 8319657]
- Phillips, D.H. & Ni She, M. (1993) Smoking-related DNA adducts in human cervical biopsies. In: Phillips, D.H., Castegnaro, M. & Bartsch, H., eds, Postlabelling Methods for Detection of DNA Adducts (IARC Scientific Publications No. 124), Lyon, IARCPress, pp. 327–330.
- Phillips D. H., Ni She M. DNA adducts in cervical tissue of smokers and nonsmokers. Mutat. Res. 1994;313:277–284. [PubMed: 7523912]
- Phillips D.H., Hewer A., Martin C.N., Garner R.C., King M.M. Correlation of DNA adduct levels in human lung with cigarette smoking. Nature. 1988;336:790–792. [PubMed: 3205307]
- Phillips D.H., Schoket B., Hewer A., Bailey E., Kostic S., Vincze I. Influence of cigarette smoking on the levels of DNA adducts in human bronchial epithelium and white blood cells. Int. J. Cancer. 1990a;46:569–575. [PubMed: 2210880]
- Phillips D.H., Hewer A., Malcolm A.D., Ward P., Coleman D.V. Smoking and DNA damage in cervical cells (Letter to the Editor). Lancet. 1990b;335:417. [PubMed: 1968150]
- Pianezza M.L., Sellers E.M., Tyndale R.F. Nicotine metabolism defect reduces smoking. Nature. 1998;393:750. [PubMed: 9655391]
- Picciotto M.R., Caldarone B.J., King S.L., Zachariou V. Nicotinic receptors in the brain: Links between molecular biology and behavior. Neuropsychopharmacology. 2000;22:451–465. [PubMed: 10731620]
- Pickworth W.B., Moolchan E.T., Berlin I., Murty R. Sensory and physiologic effects of menthol and nonmenthol cigarettes with different nicotine delivery. Pharmacol. Biochem. Behav. 2002;71:55–61. [PubMed: 11812507]
- Pietilä K., Ahtee L. Chronic nicotine administration in the drinking water affects the striatal dopamine in mice. Pharmacol. Biochem. Behav. 2000;66:95–103. [PubMed: 10837848]
- Pignatelli B., Li C.-Q., Boffetta P., Chen Q., Ahrens W., Nyberg F., Mukera A., Bruske-Holfeld I., Fortes C., Constantinescu V., Ischiropoulos H., Oshima H. Nitrated and oxidized plasma proteins in smokers and lung cancer patients. Cancer Res. 2001;61:778–784. [PubMed: 11212282]
- Piipari R., Savela K., Nurminen T., Hukkanen J., Raunio H., Hakkola J., Mantyla T., Beaune P., Edwards R.J., Boobis A.R., Anttila S. Expression of CYP1A1, CYP1B1 and CYP3A, and polycyclic aromatic hydrocarbon–DNA adduct formation in bronchoalveolar macrophages of smokers and nonsmokers. Int. J. Cancer. 2000;86:610–616. [PubMed: 10797280]
- Pilger A., Germadnik D., Riedel K., Meger-Kossien I., Scherer G., Rüdiger H.W. Longitudinal study of urinary 8-hydroxy-2′-deoxyguanosine excretion in healthy adults. Free Radic. Res. 2001;35:273–280. [PubMed: 11697126]
- Piperakis S.M., Visvardis E.-E., Sagnou M., Tassiou A.M. Effects of smoking and aging on oxidative DNA damage of human lymphocytes. Carcinogenesis. 1998;19:695–698. [PubMed: 9600358]
- Pirkle J.L., Flegal K.M., Bernert J.T., Brody D.J., Etzel R.A., Maurer K.R. Exposure of the US population to environmental tobacco smoke: The Third National Health and Nutrition Examination Survey, 1988 to 1991. J. Am. med. Assoc. 1996;275:1233–1240. [PubMed: 8601954]
- Pluth J.M., Ramsey M.J., Tucker J.D. Role of maternal exposures and newborn genotypes on newborn chromosome aberration frequencies. Mutat. Res. 2000;465:101–111. [PubMed: 10708975]
- Podlutsky A., Hou S.-M., Nyberg F., Pershagen G., Lambert B. Influence of smoking and donor age on the spectrum of in vivo mutation at the HPRT-locus in T lymphocytes of healthy adults. Mutat. Res. 1999;431:325–339. [PubMed: 10635998]
- Poirier M.C., Santella R.M., Weston A. Carcinogen macromolecular adducts and their measurement. Carcinogenesis. 2000;21:353–359. [PubMed: 10688855]
- Polek T.C., Weigel N.L. Vitamin D and prostate cancer. J. Androl. 2002;23:9–17. [PubMed: 11780928]
- Poli P., Buschini A., Spaggiari A., Rizzoli V., Carlo-Stella C., Rossi C. DNA damage by tobacco smoke and some antiblastic drugs evaluated using the Comet assay. Toxicol. Lett. 1999;108:267–276. [PubMed: 10511271]
- Popp W., Schell C., Kraus R., Vahrenholz C., Wolf R., Radtke J., Bierwirth K., Norpoth K. DNA strand breakage and DNA adducts in lymphocytes of oral cancer patients. Carcinogenesis. 1993;14:2251–2256. [PubMed: 8242851]
- Potts R.J., Newbury C.J., Smith G., Notarianni L.J., Jefferies T.M. Sperm chromatin damage associated with male smoking. Mutat. Res. 1999;423:103–111. [PubMed: 10029686]
- Pourcelot S., Faure H., Firoozi F., Ducros V., Tripier M., Hee J., Cadet J., Favier A. Urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine and 5-(hydroxymethyl)uracil in smokers. Free Radic. Res. 1999;30:173–180. [PubMed: 10711787]
- Pressl S., Edwards A., Stephan G. The influence of age, sex and smoking habits on the background level of FISH-detected translocations. Mutat. Res. 1999;442:89–95. [PubMed: 10393277]
- Preussmann, R. & Stewart, B.W. (1984) N-Nitroso carcinogens. In: Searle, C.E., ed., Chemical Carcinogens, Vol. 2, 2nd Ed. (ACS Monograph 182), Washington DC, American Chemical Society, pp. 643–828.
- Prévost V., Shuker D.E. Cigarette smoking and urinary 3-alkyladenine excretion in man. Chem. Res. Toxicol. 1996;9:439–444. [PubMed: 8839047]
- Prévost V., Shuker D.E.G., Friesen M.D., Eberle G., Rajewsky M.F., Bartsch H. Immunoaffinity purification and gas chromatography–mass spectrometric quantification of 3alkyladenines in urine: Metabolism studies and basal excretion levels in man. Carcinogenesis. 1993;14:199–204. [PubMed: 8435861]
- Priemé H., Loft S., Kharlund M., Grønbæk K., Tønnesen P., Poulsen H.E. Effect of smoking cessation on oxidative DNA modification estimated by 8-oxo-7,8-dihydro-2′-deoxyguanosine excretion. Carcinogenesis. 1998;19:347–351. [PubMed: 9498287]
- Probst-Hensch N.M., Bell D.A., Watson M.A., Skipper P.L., Tannenbaum S.R., Chan K.K., Ross R.K., Yu M.C. N-Acetyltransferase 2 phenotype but not NAT1*10 genotype affects aminobiphenyl–hemoglobin adduct levels. Cancer Epidemiol. Biomarkers Prev. 2000;9:619–623. [PubMed: 10868698]
- Probst-Hensch N.M., Yuan J.M., Stanczyk F.Z., Gao Y.T., Ross R.K., Yu M.C. IGF1, IGF-2 and IGFBP-3 in prediagnostic serum: Association with colorectal cancer in a cohort of Chinese men in Shanghai. Br. J. Cancer. 2001;85:1695–1699. [PMC free article: PMC2363974] [PubMed: 11742490]
- Prokopczyk B., Cox J.E., Hoffmann D., Waggoner S.E. Identification of tobacco-specific carcinogen in the cervical mucus of smokers and nonsmokers. J. natl Cancer Inst. 1997;89:868–873. [PubMed: 9196253]
- Prokopczyk B., Trushin N., Leszczynska J., Waggoner S.E., El-Bayoumy K. Human cervical tissue metabolizes the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3pyridyl)-1-butanone, via α-hydroxylation and carbonyl reduction pathways. Carcinogenesis. 2001;22:107–114. [PubMed: 11159748]
- Prokopczyk B., Hoffmann D., Bologna M., Cunningham A.J., Trushin N., Akerkar S., Boyiri T., Amin S., Desai D., Colosimo S., Pittman B., Leder G., Ramadani M., Henne-Bruns D., Beger H.G., El-Bayoumy K. Identification of tobacco-derived compounds in human pancreatic juice. Chem. Res. Toxicol. 2002;15:677–685. [PubMed: 12018989]
- Pryor W.A. Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ. Health Perspect. 1997;105 (Suppl. 4):875–882. [PMC free article: PMC1470037] [PubMed: 9255574]
- Pulera N., Petruzzelli S., Celi A., Puntoni R., Fornai E., Sawe U., Paoletti P., Giuntini C. Presence and persistence of serum anti-benzo[a]pyrene diolepoxide–DNA adduct antibodies in smokers: Effects of smoking reduction and cessation. Int. J. Cancer. 1997;70:145–149. [PubMed: 9009151]
- Putnam K.P., Bombick D.W., Avalos J.T., Doolittle D.J. Comparison of the cytotoxic and mutagenic potential of liquid smoke food flavourings, cigarette smoke condensate and wood smoke condensate. Food chem. Toxicol. 1999;37:1113–1118. [PubMed: 10566883]
- Putzrath R.M., Langley D., Eisenstadt E. Analysis of mutagenic activity in cigarette smokers' urine by high performance liquid chromatography. Mutat. Res. 1981;85:97–108. [PubMed: 7022187]
- Qu Q., Melikian A.A., Li G., Shore R., Chen L., Cohen B., Yin S., Kagan M.R., Li H., Meng M., Jin X., Winnik W., Li Y., Mu R., Li K. Validation of biomarkers in humans exposed to benzene: Urine metabolites. Am. J. ind. Med. 2000;37:522–531. [PubMed: 10723046]
- Rahn C.A., Howard G., Riccio E., Doolittle D.J. Correlations between urinary nicotine or cotinine and urinary mutagenicity in smokers on controlled diets. Environ. mol. Mutag. 1991;17:244–252. [PubMed: 2050132]
- Rajah T., Ahuja Y.R. In vivo genotoxic effects of smoking and occupational lead exposure in printing press workers. Toxicol. Lett. 1995;76:71–75. [PubMed: 7701519]
- Ramsey M.J., Moore D.H. II, Briner J.F., Lee D.A., Olsen L.A., Senft J.R., Tucker J.D. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutat. Res. 1995;338:95–106. [PubMed: 7565886]
- Randerath E., Avitts T.A., Reddy M.V., Miller R.H., Everson R.B., Randerath K. Comparative 32P-analysis of cigarette smoke-induced DNA damage in human tissues and mouse skin. Cancer Res. 1986;46:5869–5877. [PubMed: 3756927]
- Randerath E., Miller R.H., Mittal D., Avitts T.A., Dunsford H.A., Randerath K. Covalent DNA damage in tissues of cigarette smokers as determined by 32P-postlabeling assay. J. natl Cancer Inst. 1989;81:341–347. [PubMed: 2915370]
- Rao Y., Hoffmann E., Zia M., Bodin L., Zeman M., Sellers E.M., Tyndale R.F. Duplications and defects in the CYP2A6 gene: Identification, genotyping, and in vivo effects on smoking. Mol. Pharmacol. 2000;58:747–755. [PubMed: 10999944]
- Rapuri P.B., Gallagher J.C., Balhorn K.E., Ryschon K.L. Smoking and bone metabolism in elderly women. Bone. 2000;27:429–436. [PubMed: 10962356]
- Rasky E., Stronegger W.J., Freidi W. The relationship between body weight and patterns of smoking in women and men. Int. J. Epidemiol. 1996;25:1208–1212. [PubMed: 9027526]
- Rauscher, D., Bader, M. & Angerer, J. (1994) The t,t-muconic acid excretion of the general population caused by environmental benzene exposures. In: Proceedings of the 1st International Congress on Environmental Medicine, February 23–26, Duisburg.
- Read G.A., Thornton R.E. Preliminary studies of urinary hydroxyproline levels in rodents and in smokers. Tokai J. exp. clin. Med. 1985;10:445–450. [PubMed: 3836526]
- Reidy J.A., Annest J.L., Chen. A.T.L., Welty T.K. Increased sister chromatid exchange associated with smoking and coffee consumption. Environ. mol. Mutag. 1988;12:311–318. [PubMed: 3169009]
- Renner T., Fechner T., Scherer G. Fast quantification of the urinary marker of oxidative stress 8-hydroxy-2′-deoxyguanosine using solid-phase extraction and high-performance liquid chromatography with triple-stage quadrupole mass detection. J. Chromatogr. 2000;B738:311–317. [PubMed: 10718649]
- Richie J.P. Jr, Carmella S.G., Muscat J.E., Scott D.G., Akerkar S.A., Hecht S.S. Differences in the urinary metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in black and white smokers. Cancer Epidemiol. Biomarkers Prev. 1997;6:783–790. [PubMed: 9332760]
- Riffelmann M., Müller G., Schmieding W., Popp W., Norpoth K. Biomonitoring of urinary aromatic amines and arylamine hemoglobin adducts in exposed workers and nonexposed control persons. Int. Arch. occup. environ. Health. 1995;68:36–43. [PubMed: 8847111]
- Rimm E.B., Manson J.E., Stampfer M.J., Colditz G.A., Willett W.C., Rosner B., Hennekens C.H., Speizer F.E. Cigarette smoking and the risk of diabetes in women. Am. J. public Health. 1993;83:211–214. [PMC free article: PMC1694562] [PubMed: 8427325]
- Rimm E.B., Chan J., Stampfer M.J., Colditz G.A., Willett W.C. Prospective study of cigarette smoking, alcohol use, and the risk of diabetes in men. Br. med. J. 1995;310:555–559. [PMC free article: PMC2548937] [PubMed: 7888928]
- Rioux N., Castonguay A. The induction of cyclooxygenase-1 by a tobacco carcinogen in U937 human macrophages is correlated to the activation of NF-κB. Carcinogenesis. 2000;21:1745–1751. [PubMed: 10964107]
- Rithidech K., Chen B.T., Mauderly J.L., Whorton E.B. Jr, Brooks A.L. Cytogenetic effects of cigarette smoke on pulmonary alveolar macrophages of the rat. Environ. mol. Mutag. 1989;14:27–33. [PubMed: 2753026]
- Robbins W.A., Vine M.F., Truong K.Y., Everson R.B. Use of fluoresence in situ hybridization (FISH) to assess effects of smoking, caffeine, and alcohol on aneuploidy load in sperm of healthy men. Environ. mol. Mutag. 1997;30:175–183. [PubMed: 9329642]
- Robertson A.S., Burge P.S., Cockrill B.L. A study of serum thiocyanate concentrations in office workers as a means of validation of smoking histories and assessing passive exposure to cigarette smoke. Br. J. ind. Med. 1987;44:351–354. [PMC free article: PMC1007833] [PubMed: 3593662]
- Robinson D.R., Goodall K., Albertini R.J., O'Neill J.P., Finette B., Sala-Trepat M., Moustacchi E., Tates A.D., Beare D.M., Green M.H.L., Cole J. An analysis of in vivo hprt mutant frequency in circulating T-lymphocytes in the normal human population: A comparison of four datasets. Mutat. Res. 1994;313:227–247. [PubMed: 7523908]
- Rodin S.N., Rodin A.S. Human lung cancer and p53: The interplay between mutagenesis and selection. Proc. natl Acad. Sci. USA. 2000;97:12244–12249. [PMC free article: PMC17326] [PubMed: 11035769]
- Roemer E., Tewes F.J., Meisgen T.J., Veltel D.J., Carmines E.L. Evaluation of the potential effects of ingredients added to cigarettes. Part 3: In vitro genotoxicity and cytotoxicity. Food chem. Toxicol. 2002;40:105–111. [PubMed: 11731040]
- Roggi C., Minoia C., Sciarra G.F., Apostoli P., Maccarini L., Magnaghi S., Cenni A., Fonte A., Nidasio G.F., Micoli G. Urinary 1-hydroxypyrene as a marker of exposure to pyrene: An epidemiological survey on a general population group. Sci. total Environ. 1997;199:247–254. [PubMed: 9200867]
- Rojas M., Alexandrov K., Van Schooten F.-J., Hillebrand M., Kriek E., Bartsch H. Validation of a new fluorometric assay for benzo[a]pyrene diolepoxide–DNA adducts in human white blood cells: Comparisons with 32P-postlabeling and ELISA. Carcinogenesis. 1994;15:557–560. [PubMed: 8118943]
- Rojas M., Alexandrov K., Auburtin G., Wastiaux-Denamur A., Mayer L., Mahieu B., Sebastien P., Bartsch H. Anti-benzo[a]pyrene diolepoxide–DNA adduct levels in peripheral mononuclear cells from coke oven workers and the enhancing effect of smoking. Carcinogenesis. 1995;16:1373–1376. [PubMed: 7788857]
- Rojas E., Valverde M., Sordo M., Ostrosky-Wegman P. DNA damage in exfoliated buccal cells of smokers assessed by the single cell gel electrophoresis assay. Mutat. Res. 1996;370:115–120. [PubMed: 8879269]
- Rojas M., Alexandrov K., Cascorbi I., Brockmöller J., Likhachev A., Pozharisski K., Bouvier G., Auburtin G., Mayer L., Kopp-Schneider A., Roots I., Bartsch H. High benzo[a]pyrene diol-epoxide DNA adduct levels in lung and blood cells from individuals with combined CYP1A1 MspI/MspI-GSTM1*0/*0 genotypes. Pharmacogenetics. 1998;8:109–118. [PubMed: 10022748]
- Rojas M., Cascorbi I., Alexandrov K., Kriek E., Auburtin G., Mayer L., Kopp-Schneider A., Roots I., Bartsch H. Modulation of benzo[a]pyrene diolepoxide–DNA adduct levels in human white blood cells by CYP1A1, GSTM1 and GSTT1 polymorphism. Carcinogenesis. 2000;21:35–41. [PubMed: 10607731]
- Romano G., Mancini R., Fedele P., Curigliano G., Flamini G., Giovagnoli M.R., Malara N., Boninsegna A., Vecchione A., Santella R.M., Cittadini A. Immunohistochemical analysis of 4-aminobiphenyl–DNA adducts in oral mucosal cells of smokers and nonsmokers. Anticancer Res. 1997;17:2827–2830. [PubMed: 9252724]
- Romano G., Sgambato A., Boninsegna A., Flamini G., Curigliano G., Yang Q., La Gioia V., Signorelli C., Ferro A., Capelli G., Santella R.M., Cittadini A. Evaluation of polycyclic aromatic hydrocarbon–DNA adducts in exfoliated oral cells by an immunohistochemical assay. Cancer Epidemiol. Biomarkers Prev. 1999a;8:91–96. [PubMed: 9950245]
- Romano G., Garagnani L., Boninsegna A., Ferrari P., Flamini G., De Gaetani C., Sgambato A., Giovanni F., Curigliano G., Ferretti G., Cittadini A., Trentini G. Analysis of 4ABP–DNA adducts and p53 alterations in urinary bladder carcinoma. Anticancer Res. 1999b;19:4571–4576. [PubMed: 10650812]
- Romert L., Jansson T., Curvall M., Jenssen D. Screening for agents inhibiting the mutagenicity of extracts and constituents of tobacco products. Mutat. Res. 1994;322:97–110. [PubMed: 7519327]
- Ronco G., Vineis P., Bryant M.S., Skipper P.L., Tannenbaum S.R. Haemoglobin adducts formed by aromatic amines in smokers: Sources of inter-individual variability. Br. J. Cancer. 1990;61:534–537. [PMC free article: PMC1971363] [PubMed: 2331440]
- Rose J.E., Behm F.M., Ramsey C., Ritchie J.C. Jr. Platelet monoamine oxidase, smoking cessation, and tobacco withdrawal symptoms. Nicotine Tob. Res. 2001;3:383–390. [PubMed: 11694206]
- Ross G.W., Petrovitch H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson's disease. Drugs Aging. 2001;18:797–806. [PubMed: 11772120]
- Routledge M.N., Garner R.C., Jenkins D., Cuzick J. 32P-Postlabelling analysis of DNA from human tissues. Mutat. Res. 1992;282:139–145. [PubMed: 1378545]
- Rowland R.E., Harding K.M. Increased sister chromatid exchange in the peripheral blood lymphocytes of young women who smoke cigarettes. Hereditas. 1999;131:143–146. [PubMed: 10680296]
- Rubes J., Lowe X., Moore D., Perreault S., Slott V., Evenson D., Selevan S.G., Wyrobek A.J. Smoking cigarettes is associated with increased sperm disomy in teenage men. Fertil. Steril. 1998;70:715–723. [PubMed: 9797104]
- Rubin D.T., Hanauer S.B. Smoking and inflammatory bowel disease. Eur. J. Gastroenterol. Hepatol. 2000;12:855–862. [PubMed: 10958212]
- Ruiz E., Osorio E., Ortega E. Androgenic status in cyclic and postmenopausal women: A comparison between smokers and nonsmokers. Biochem. int. 1992;27:841–845. [PubMed: 1417917]
- Rundle A., Tang D., Hibshoosh H., Estabrook A., Schnabel F., Cao W., Grumet S., Perera F.P. The relationship between genetic damage from polycyclic aromatic hydrocarbons in breast tissue and breast cancer. Carcinogenesis. 2000;21:1281–1289. [PubMed: 10874004]
- Ruppert T., Scherer G., Tricker A.R., Rauscher D., Adlkofer F. Determination of urinary trans, trans-muconic acid by gas chromatography–mass spectrometry. J. Chromatogr. 1995;B666:71–76. [PubMed: 7655623]
- Ruppert T., Scherer G., Tricker A.R., Adlkofer F. trans, trans-Muconic acid as a biomarker of non-occupational environmental exposure to benzene. Int. Arch. occup. environ. Health. 1997;69:247–251. [PubMed: 9137998]
- Russell M.A.H., Jarvis M.J., Feyerabend C., Saloojee Y. Reduction of tar, nicotine and carbon monoxide intake in low tar smokers. J. Epidemiol. Community Health. 1986;40:80–85. [PMC free article: PMC1052494] [PubMed: 3711773]
- Rutten A.A., Wilmer J.W. Effect of cigarette-smoke condensate and norharman on the induction of SCEs by direct and indirect mutagens in CHO cells. Mutat. Res. 1986;172:61–67. [PubMed: 3762569]
- Ryberg D., Hewer A., Phillips D.H., Haugen A. Different susceptibility to smoking-induced DNA damage among male and female lung cancer patients. Cancer Res. 1994;54:5801–5803. [PubMed: 7954403]
- Ryberg D., Skaug V., Hewer A., Phillips D.H., Harries L.W., Wolf C.R., Øgreid D., Ulvik A., Vu P., Haugen A. Genotypes of glutathione transferase M1 and P1 and their significance for lung DNA adduct levels and cancer risk. Carcinogenesis. 1997;18:1285–1289. [PubMed: 9230269]
- Safe S., McDougal A. Mechanism of action and development of selective aryl hydrocarbon receptor modulators for treatment of hormone-dependent cancers (Review). Int. J. Oncol. 2002;20:1123–1128. [PubMed: 12011988]
- Safe S., Wang F., Porter W., Duan R., McDougal A. Ah receptor agonists as endocrine disruptors: Antiestrogenic activity and mechanisms. Toxicol. Lett. 1998;102–103:343–347. [PubMed: 10022276]
- Safe S.H., Pallaroni L., Yoon K., Gaido K., Ross S., Saville B., McDonnell D. Toxicology of environmental estrogens. Reprod. Fertil. Dev. 2001;13:307–315. [PubMed: 11800169]
- Salomaa S., Tuominen J., Skytta E. Genotoxicity and PAC analysis of particulate and vapour phases of environmental tobacco smoke. Mutat. Res. 1988;204:173–183. [PubMed: 3278208]
- Sanchez-Cespedes M., Ahrendt S.A., Piantadosi S., Rosell R., Monzo M., Wu L., Westra W.H., Yang S.C., Jen J., Sidransky D. Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res. 2001;61:1309–1313. [PubMed: 11245426]
- Sandler D.P., Shore D.L., Anderson J.R., Davey F.R., Arthur D., Mayer R.J., Silver R.T., Weiss R.B., Moore J.O., Schiffer C.A., Wurster-Hill D.H., McIntyre O.R., Bloomfield C.D. Cigarette smoking and risk of acute leukemia: Associations with morphology and cytogenetic abnormalities in bone marrow. J. natl. Cancer Inst. 1993;85:1994–2003. [PubMed: 8246285]
- Santella R.M., Grinberg-Funes R.A., Young T.L., Dickey C., Singh V.N., Wang L.W., Perera F.P. Cigarette smoking related polycyclic aromatic hydrocarbon–DNA adducts in peripheral mononuclear cells. Carcinogenesis. 1992;13:2041–2045. [PubMed: 1423873]
- Santella R.M., Gammon M.D., Zhang Y.J., Motykiewicz G., Young T.L., Hayes S.C., Terry M.B., Schoenberg J.B., Brinton L.A., Bose S., Teitelbaum S.L., Hibshoosh H. Immunohistochemical analysis of polycyclic aromatic hydrocarbon–DNA adducts in breast tumor tissue. Cancer Lett. 2000;154:143–149. [PubMed: 10806302]
- Sanyal M.K., Li Y.L., Belanger K. Metabolism of polynuclear aromatic hydrocarbon in human term placenta influenced by cigarette smoke exposure. Reprod. Toxicol. 1994;8:411–418. [PubMed: 7841660]
- Sardas S., Walker D., Akyol D., Karakaya A.E. Assessment of smoking-induced DNA damage in lymphocytes of smoking mothers of newborn infants using the alkaline single-cell gel electrophoresis technique. Mutat. Res. 1995;335:213–217. [PubMed: 8524335]
- Sargeant L.A., Khaw K.T., Bingham S., Day N.E., Luben R.N., Oakes S., Welch A., Wareham N.J. Cigarette smoking and glycaemia: The EPIC-Norfolk Study. European prospective investigation into cancer. Int. J. Epidemiol. 2001;30:547–554. [PubMed: 11416081]
- Sarto F., Faccioli M.C., Cominato I., Levis A.G. Aging and smoking increase the frequency of sister-chromatid exchanges (SCE) in man. Mutat. Res. 1985;144:183–187. [PubMed: 4058436]
- Sarto F., Mustari L., Mazzotti D., Tomanin R., Levis A.G. Variations of SCE frequencies in peripheral lymphocytes of ex-smokers. Mutat. Res. 1987;192:157–162. [PubMed: 3657845]
- Sastry B.V.R. Placental toxicology: Tobacco smoke, abused drugs, multiple chemical interactions, and placental function. Reprod. Fertil. Dev. 1991;3:355–372. [PubMed: 1957023]
- Savela K., Hemminki K. DNA adducts in lymphocytes and granulocytes of smokers and nonsmokers detected by the 32P-postlabelling assay. Carcinogenesis. 1991;12:503–508. [PubMed: 2009595]
- Schaller K.H., Zober A. [Renal excretion of toxicologically relevant metals in occupationally non-exposed individuals.] Ärztl. Lab. 1982;28:209–214. (in German)
- Schell C., Popp W., Kraus R., Vahrenholz C., Norpoth K. 32P-Postlabeling analysis of DNA adducts in different populations. Toxicol. Lett. 1995;77:299–307. [PubMed: 7618154]
- Scherer G., Conze C., von Meyerinck L., Sorsa M., Adlkover F. Importance of exposure to gaseous and particulate phase components of tobacco smoke in active and passive smokers. Int. Arch. occup. environ. Health. 1990;62:459–466. [PubMed: 2246065]
- Scherer G., Doolittle D.J., Ruppert T., Meger-Kossien I., Riedel K., Tricker A.R., Adlkofer F. Urinary mutagenicity and thioethers in nonsmokers: Role of environmental tobacco smoke (ETS) and diet. Mutat. Res. 1996;368:195–204. [PubMed: 8692225]
- Scherer G., Renner T., Meger M. Analysis and evaluation of trans,trans-muconic acid as a biomarker for benzene exposure. J. Chromatogr. 1998;B717:179–199. [PubMed: 9832246]
- Scherer G., Frank S., Riedel K., Meger-Kossien I., Renner T. Biomonitoring of exposure to polycyclic aromatic hydrocarbons of nonoccupationally exposed persons. Cancer Epidemiol. Biomarkers Prev. 2000;9:373–380. [PubMed: 10794481]
- Scherer G., Urban M., Meger M. Biological monitoring of the tobacco smoke-related exposure to alkylating agents (Abstract No. 802). Proc. Am. Assoc. Cancer Res. 2001a;42:149–150.
- Scherer G., Meger M., Meger-Kossien I., Pachinger A. Biological monitoring of the tobacco-smoke related exposure to benzene (Abstract No. 803). Proc. Am. Assoc. Cancer Res. 2001b;42:150.
- Schilling J., Holzer P., Guggenbach M., Gyurech D., Marathia K., Geroulanos S. Reduced endogenous nitric oxide in the exhaled air of smokers and hypertensives. Eur. respir. J. 1994;7:467–471. [PubMed: 8013603]
- Schneider M., Diemer K., Engelhart K., Zankl H., Trommer W.E., Biesalski H.K. Protective effects of vitamins C and E on the number of micronuclei in lymphocytes in smokers and their role in ascorbate free radical formation in plasma. Free Radic. Res. 2001;34:209–219. [PubMed: 11264897]
- Schoket, B., Kostic, S. & Vincze, I. (1993) Determination of smoking-related DNA adducts in lung-cancer and non-cancer patients. In: Phillips, D.H., Castegnaro, M. & Bartsch, H., eds, Postlabelling Methods for Detection of DNA Adducts (IARC Scientific Publications No. 124), Lyon, IARCPress, pp. 315–319. [PubMed: 8225500]
- Schoket B., Phillips D.H., Kostic S., Vincze I. Smoking-associated bulky DNA adducts in bronchial tissue related to CYP1A1 MspI and GSTM1 genotypes in lung patients. Carcinogenesis. 1998;19:841–846. [PubMed: 9635872]
- Schoket B., Papp G., Lévay K., Mracková G., Kadlubar F.F., Vincze I. Impact of metabolic genotypes on levels of biomarkers of genotoxic exposure. Mutat. Res. 2001;482:57–69. [PubMed: 11535249]
- Schreiber G., Fong K.M., Peterson B., Johnson B.E., O'Briant K.C., Bepler G. Smoking, gender, and survival association with allele loss for the LOH11B lung cancer region on chromosome 11. Cancer Epidemiol. Biomarkers Prev. 1997;6:315–319. [PubMed: 9149890]
- Schwartz G.G., Hulka B.S. Is vitamin D deficiency a risk factor to prostate cancer? (Hypothesis). Anticancer Res. 1990;10:1307–1311. [PubMed: 2241107]
- Seagrave J. Oxidative mechanisms in tobacco smoke-induced emphysema. J. Toxicol. environ. Health. 2000;A61:69–78. [PubMed: 10990164]
- Seidman D.S., Paz I., Merlet-Aharoni I., Vremen H., Stevenson D.K., Gale R. Noninvasive validation of tobacco smoke exposure in late pregnancy using end-tidal carbon monoxide measurements. J. Perinatol. 1999;19:358–361. [PubMed: 10685257]
- Sekido Y., Fong K.M., Minna J.D. Progress in understanding the molecular pathogenesis of human lung cancer. Biochim. biophys. Acta. 1998;1378:F21–F59. [PubMed: 9739759]
- Seller M.J., Bnait K.S. Effects of tobacco smoke inhalation on the developing mouse embryo and fetus. Reprod. Toxicol. 1995;9:449–459. [PubMed: 8563188]
- Sellers, E.M., Kaplan, H.L. & Tyndale, R.F. (2000) Inhibition of Cytochrome P450 2A6 Increases Nicotine's Oral Bioavailability and Decreases Smoking, Toronto, University of Toronto, Departments of Pharmacology, Medicine, and Psychiatry and the Centre for Research in Women's Health.
- Selman M., Montano M., Ramos C., Vanda B., Becerril C., Delgado J., Sansores R., Barrios R., Pardo A. Tobacco smoke-induced lung emphysema in guinea pigs is associated with increased interstitial collagenase. Am. J. Physiol. 1996;271:734–743. [PubMed: 8944716]
- Senthilmohan S.T., McEwan M.J., Wilson P.F., Milligan D.B., Freeman C.G. Real time analysis of breath volatiles using SIFT-MS in cigarette smoking. Redox Rep. 2001;6:185–187. [PubMed: 11523595]
- Seree E.M., Villard P.H., Re J.L., De Meo M., Lacarelle B., Attolini L., Dumenil G., Catalin J., Durand A., Barra Y. High inducibility of mouse renal CYP2E1 gene by tobacco smoke and its possible effect on DNA single strand breaks. Biochem. biophys. Res. Commun. 1996;219:429–434. [PubMed: 8605004]
- Seto H., Ohkubo T., Kanoh T., Koike M., Nakamura K., Kawahara Y. Determination of polycyclic aromatic hydrocarbons in the lung. Arch. environ. Contam. Toxicol. 1993;24:498–503. [PubMed: 8507106]
- Sharara F.I., Beatse S.N., Leonardi M.R., Navot D., Scott R.T. Jr. Cigarette smoking accelerates the development of diminished ovarian reserve as evidenced by the clomiphene citrate challenge test. Fertil. Steril. 1994;62:257–262. [PubMed: 8034069]
- Sherman M.P., Aeberhard E.E., Wong V.Z., Simmons M.S., Roth M.D., Tashkin D.P. Effects of smoking marijuana, tobacco or cocaine alone or in combination on DNA damage in human alveolar macrophages. Life Sci. 1995;56:2201–2207. [PubMed: 7776850]
- Shen H.-M., Chia S.-E., Ni Z.-Y., New A.-L., Lee B.-L., Ong C.-N. Detection of oxidative DNA damage in human sperm and the association with cigarette smoking. Reprod. Toxicol. 1997;11:675–680. [PubMed: 9311575]
- Shen H.M., Chia S.E., Ong C.N. Evaluation of oxidative DNA damage in human sperm and its association with male infertility. J. Androl. 1999;20:718–723. [PubMed: 10591610]
- Shi Q., Ko E., Barclay L., Hoang T., Rademaker A., Martin R. Cigarette smoking and aneuploidy in human sperm. Mol. Reprod. Dev. 2001;59:417–421. [PubMed: 11468778]
- Shinozaki R., Inoue S., Choi K.S., Tatsuno T. Association of benzo[a]pyrene-diolepoxide–deoxyribonucleic acid (BPDE–DNA) adduct level with aging in male smokers and nonsmokers. Arch. environ. Health. 1999;54:79–85. [PubMed: 10094284]
- Shuker, D.E.G. & Bartsch, H. (1994) DNA adducts of nitrosamines. In: Hemminki, K., Dipple, A., Shuker, D.E.G., Kadlubar, F.F., Segerbäck, D. & Bartsch, H., eds, DNA Adducts: Identification and Biological Significance (IARC Scientific Publications No. 125), Lyon, IARCPress, pp. 73–89.
- Shuker, D.E.G., Friesen, M.D., Garren, L. & Prévost, V. (1991) A rapid gas chromatography–mass spectrometry method for the determination of urinary 3-methyladenine: Application in human subjects. In: O'Neill, I.K., Chen, J. & Bartsch, H., eds, Relevance to Human Cancer of N-Nitroso Compounds, Tobacco Smoke and Mycotoxins (IARC Scientific Publications No. 105), Lyon, IARCPress, pp. 102–106. [PubMed: 1855830]
- Simon D., Senan C., Garnier P., Saint-Paul M., Papoz L. Epidemiological features of glycated haemoglobin A1c-distribution in a healthy population. The Telecom Study. Diabetologia. 1989;32:864–869. [PubMed: 2693166]
- Simon P., Lafontaine M., Delsaut P., Morele Y., Nicot T. Trace determination of urinary 3-hydroxybenzo[a]pyrene by automated column-switching high-performance liquid chromatography. J. Chromatogr. 2000;B748:337–348. [PubMed: 11087076]
- Simons A.M., Phillips D.H., Coleman D.V. Damage to DNA in cervical epithelium related to smoking tobacco. Br. med. J. 1993;306:1444–1448. [PMC free article: PMC1677905] [PubMed: 8257490]
- Simons A.M., Mugica van Herckenrode C., Rodriguez J.A., Maitland N., Anderson M., Phillips D.H., Coleman D.V. Demonstration of smoking-related DNA damage in cervical epithelium and correlation with human papillomavirus type 16, using exfoliated cervical cells. Br. J. Cancer. 1995;71:246–249. [PMC free article: PMC2033590] [PubMed: 7841037]
- Simpson C.D., Wu M.-T., Christiani D.C., Santella R.M., Carmella S.G., Hecht S.S. Determination of r-7,t-8,9,c-10-tetrahydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene in human urine by gas chromatography negative ion chemical ionization mass spectrometry. Chem. Res. Toxicol. 2000;13:271–280. [PubMed: 10775327]
- Singer, B. & Grunberger, D. (1983) Molecular Biology of Mutagens and Carcinogens, New York, Plenum Press.
- Sinués B., Izquierdo M., Viguera J.P. Chromosome aberrations and urinary thioethers in smokers. Mutat. Res. 1990;240:289–293. [PubMed: 2330013]
- Sithisarankul P., Vineis P., Kang D., Rothman N., Caporaso N., Strickland P. The association of 1-hydroxypyrene-glucuronide in human urine with cigarette smoking and broiled or roasted meat consumption. Biomarkers. 1997;2:217–221. [PubMed: 23899213]
- van Sittert N.J., Boogaard P.J., Beulink G.D.J. Application of the urinary S-phenylmercapturic acid test as a biomarker for low levels of exposure to benzene in industry. Br. J. ind. Med. 1993;50:460–469. [PMC free article: PMC1012165] [PubMed: 8507599]
- Skipper P.L., Peng X., Soohoo C.K., Tannenbaum S.R. Protein adducts as biomarkers of human carcinogen exposure. Drug Metab. Rev. 1994;26:111–124. [PubMed: 8082561]
- Slattery M.L., Curtin K., Anderson K., Ma K.-N., Ballard L., Edwards S., Schaffer D., Potter J., Leppert M., Samowitz W.S. Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J. natl Cancer Inst. 2000;92:1831–1836. [PubMed: 11078760]
- Slebos R.J., Hruban R.H., Dalesio O., Mooi W.J., Offerhaus G.J., Rodenhuis S. Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J. natl Cancer Inst. 1991;83:1024–1027. [PubMed: 2072410]
- Slotkin T.A., Saleh J.L., McCook E.C., Seidler F.J. Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: Implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology. 1997;55:177–184. [PubMed: 9181671]
- Smith C.J., Fischer T.H. Particulate and vapor phase constituents of cigarette mainstream smoke and risk of myocardial infarction. Atherosclerosis. 2001;158:257–267. [PubMed: 11583703]
- Smith C.J., Hansch C. The relative toxicity of compounds in mainstream cigarette smoke condensate. Food chem. Toxicol. 2000;38:637–646. [PubMed: 10942325]
- Smith C.J., McKarns S.C., Davis R.A., Livingston S.D., Bombick B.R., Avalos J.T., Morgan W.T., Doolittle D.J. Human urine mutagenicity study comparing cigarettes which burn or primarily heat tobacco. Mutat. Res. 1996;361:1–9. [PubMed: 8816936]
- Smith L.E., Denissenko M.F., Bennett W.P., Li H., Amin S., Tang M.-S., Pfeifer G.P. Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. J. natl Cancer Inst. 2000;92:803–811. [PubMed: 10814675]
- Smith C.J., Perfetti T.A., Bombick D.W., Rodgman A., Doolittle D.J. Response to ‘cigarette smoke comparative toxicology’. Food chem. Toxicol. 2001;39:177–180.
- Sopori M.L., Cherian S., Chilukuri R., Shopp G.M. Cigarette smoke causes inhibition of the immune response to intratracheally administered antigens. Toxicol. appl. Pharmacol. 1989;97:489–499. [PubMed: 2609346]
- Sowers M., Beebe J., McConnell D., Randolph J., Jannausch M. Testosterone concentrations in women aged 25–50 years: Associations with lifestyle, body composition, and ovarian status. Am. J. Epidemiol. 2001;153:256–264. [PubMed: 11157413]
- Sozzi G., Sard L., De Gregorio L., Marchetti A., Musso K., Buttitta F., Tornielli S., Pellegrini S., Veronese M.L., Manenti G., Incarbone M., Chella A., Angeletti C.A., Pastorino U., Huebner K., Bevilaqua G., Pilotti S., Croce C.M., Pierotti M.A. Association between cigarette smoking and FHIT gene alterations in lung cancer. Cancer Res. 1997;57:2121–2123. [PubMed: 9187107]
- Spencer J.P., Jenner A., Chimel K., Aruoma O.I., Cross C.E., Wu R., Halliwell B. DNA damage in human respiratory tract epithelial cells: Damage by gas phase cigarette smoke apparently involves attack by reactive nitrogen species in addition to oxygen radicals. FEBS Lett. 1995;375:179–182. [PubMed: 7498494]
- Spitz M.R., Fueger J.J., Halabi S., Schantz S.P., Sample D., Hsu T.C. Mutagen sensitivity in upper aerodigestive tract cancer: A case–control analysis. Cancer Epidemiol. Biomarkers Prev. 1993;2:329–333. [PubMed: 7688625]
- Spruck C.H. III, Rideout W.M. III, Olumi A.F., Ohneseit P.F., Yang A.S., Tsai Y.C., Nichols P.W., Horn T., Hermann G.G., Steven K., Ross R.K., Yu M.C., Jones P.A. Distinct pattern of p53 mutations in bladder cancer: Relationship to tobacco usage. Cancer Res. 1993;53:1162–1166. [PubMed: 8439962]
- Steele R.H., Payne V.M., Fulp C.W., Rees D.C., Lee C.K., Doolittle D.J. A comparison of the mutagenicity of mainstream cigarette smoke condensates from a representative sample of the US cigarette market with a Kentucky reference cigarette (K1R4F). Mutat. Res. 1995;342:179–190. [PubMed: 7715619]
- Stenstrand K. Effects of ionizing radiation on chromosome aberrations, sister chromatid exchanges and micronuclei in lymphocytes of smokers and nonsmokers. Hereditas. 1985;102:71–76. [PubMed: 3988542]
- Stillwell W.G., Glogowski J., Xu H.-X., Wishnok J.S., Zavala D., Montes G., Correa P., Tannenbaum S.R. Urinary excretion of nitrate, N-nitrosoproline, 3-methyladenine, and 7-methylguanine in a Colombian population at high risk for stomach cancer. Cancer Res. 1991;51:190–194. [PubMed: 1988083]
- Stoichev I.I., Todorov D.K., Christova L.T. Dominant-lethal mutations and micronucleus induction in male BALB/c, BDF1 and H mice by tobacco smoke. Mutat. Res. 1993;319:285–292. [PubMed: 7504202]
- Stommel P., Müller G., Stücker W., Verkoyen C., Schöbel S., Norpoth K. Determination of S-phenylmercapturic acid in the urine — An improvement in the biological monitoring of benzene exposure. Carcinogenesis. 1989;10:279–282. [PubMed: 2912579]
- Stone J.G., Jones N.J., McGregor A.D., Waters R. Development of a human biomonitoring assay using buccal mucosa: Comparison of smoking-related DNA adducts in mucosa versus biopsies. Cancer Res. 1995;55:1267–1270. [PubMed: 7882320]
- Stratton, K., Shetty, P., Wallace, R. & Bondurant, S. (2001) Clearing the Smoke: Assessing the Science Base for Tobacco Harm Reduction, Washington DC, National Academy Press. [PubMed: 25057541]
- Strickland P.T., Routledge M.N., Dipple A. Methodologies for measuring carcinogen adducts in humans. Cancer Epidemiol. Biomarkers Prev. 1993;2:607–619. [PubMed: 8268781]
- Strom S.S., Wu X., Sigurdon A.J., Hsu T.C., Fueger J.J., Lopez J., Tee P.G., Spitz M.R. Lung cancer, smoking patterns, and mutagen sensitivity in Mexican-Americans. J. natl Cancer Inst. Monogr. 1995;18:29–33. [PubMed: 8562219]
- Swislocki A.L., Tsuzuki A., Tait M., Khuu D., Fann K. Smokeless nicotine administration is associated with hypertension but not with a deterioration in glucose tolerance in rats. Metabolism. 1997;46:1008–1012. [PubMed: 9284888]
- Syrop C.H., Willhoite A., Van Voorhis B.J. Ovarian volume: A novel outcome predictor for assisted reproduction. Fertil. Steril. 1995;64:1167–1171. [PubMed: 7589671]
- Szaniszló J., Ungváry G. Polycyclic aromatic hydrocarbon exposure and burden of outdoor workers in Budapest. J. Toxicol. environ. Health. 2001;A62:297–306. [PubMed: 11261893]
- Szostak-Wegierek D., Bjorntorp P., Marin P., Lindstedt G., Andersson B. Influence of smoking on hormone secretion in obese and lean female smokers. Obes. Res. 1996;4:321–328. [PubMed: 8822756]
- Szyfter K., Hemminki K., Szyfter W., Szmeja Z., Banaszewski J., Yang K. Aromatic DNA adducts in larynx biopsies and leukocytes. Carcinogenesis. 1994;15:2195–2199. [PubMed: 7955053]
- Szyfter K., Hemminki K., Szyfter W., Szmeja Z., Banaszewski J., Pabiszczak M. Tobacco smoke-associated N7-alkylguanine in DNA of larynx tissue and leucocytes. Carcinogenesis. 1996;17:501–506. [PubMed: 8631136]
- Taioli E., Garbers S., Bradlow H.L., Carmella S.G., Akerkar S., Hecht S.S. Effects of indole-3-carbinol on the metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers. Cancer Epidemiol. Biomarkers Prev. 1997;6:517–522. [PubMed: 9232339]
- Takubo Y., Guerassimov A., Ghezzo H., Triantafillopoulos A., Bates J.H., Hoidal J.R., Cosio M.G. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: Parallels with human disease. Am. J. respir. crit. Care Med. 2002;15:166. [PubMed: 12471075]
- Talaska G., Al-Juburi A.Z.S.S., Kadlubar F.F. Smoking related carcinogen–DNA adducts in biopsy samples of human urinary bladder: Identification of N-(deoxyguanosin-8-yl)-4-aminobiphenyl as a major adduct. Proc. natl Acad. Sci. USA. 1991a;88:5350–5354. [PMC free article: PMC51870] [PubMed: 2052611]
- Talaska G., Schamer M., Skipper P., Tannenbaum S., Caporaso N., Unruh L., Kadlubar F.F., Bartsch H., Malaveille C., Vineis P. Detection of carcinogen–DNA adducts in exfoliated urothelial cells of cigarette smokers: Association with smoking, hemoglobin adducts, and urinary mutagenicity. Cancer Epidemiol. Biomarkers Prev. 1991b;1:61–66. [PubMed: 1845172]
- Talaska G., Schamer M., Casetta G., Tizzani A., Vineis P. Carcinogen–DNA adducts in bladder biopsies and urothelial cells: A risk assessment exercise. Cancer Lett. 1994;84:93–97. [PubMed: 8076366]
- Tang D., Santella R.M., Blackwood A.M., Young T.-L., Mayer J., Jaretzki A., Grantham S., Tsai W.-Y., Perera F.P. A molecular epidemiological case-control study of lung cancer. Cancer Epidemiol. Biomarkers Prev. 1995;4:341–346. [PubMed: 7655328]
- Tang D., Phillips D.H., Stampfer M., Mooney L.A., Hsu Y., Cho S., Tsai W.-Y., Ma J., Cole K.J., Shé M.N., Perera F.P. Association between carcinogen–DNA adducts in white blood cells and lung cancer risk in the Physicians Health Study. Cancer Res. 2001;61:6708–6712. [PubMed: 11559540]
- Tavares R., Ramos P., Palminha J., Bispo M.A., Paz I., Bras A., Rueff J., Farmer P.B., Bailey E. Transplacental exposure to genotoxins. Evaluation in haemoglobin of hydroxyethylvaline adduct levels in smoking and non-smoking mothers and their newborns. Carcinogenesis. 1994;15:1271–1274. [PubMed: 8020166]
- Tavares R., Borba H., Monteiro M., Proenca M.J., Lynce N., Rueff J., Bailey E., Sweetman G.M.A., Lawrence R.M., Farmer P.B. Monitoring of exposure to acrylonitrile by determination of N-(2-cyanoethyl)valine at the N-terminal position of haemoglobin. Carcinogenesis. 1996;17:2655–2660. [PubMed: 9006103]
- Tawn E.J., Whitehouse C.A. Frequencies of chromosome aberrations in a control population determined by G banding. Mutat. Res. 2001;490:171–177. [PubMed: 11342242]
- Thomas E.J., Edridge W., Weddell A., McGill A., McGarrigle H.H. The impact of cigarette smoking on the plasma concentrations of gonadotrophins, ovarian steroids and androgens and upon the metabolism of oestrogens in the human female. Hum. Reprod. 1993;8:1187–1193. [PubMed: 8408515]
- Thompson C.L., McCoy Z., Lambert J.M., Andries M.J., Lucier G.W. Relationships among benzo(a)pyrene metabolism, benzo(a)pyrene-diol-epoxide:DNA adduct formation, and sister chromatid exchanges in human lymphocytes from smokers and nonsmokers. Cancer Res. 1989;49:6503–6511. [PubMed: 2510927]
- Tokiwa H., Sera N., Horikawa K., Nakanishi Y., Shigematu N. The presence of mutagens/carcinogens in the excised lung and analysis of lung cancer induction. Carcinogenesis. 1993;14:1933–1938. [PubMed: 8403221]
- Tolcos M., Mallard C., McGregor H., Walker D., Rees S. Exposure to prenatal carbon monoxide and postnatal hyperthermia: Short and long-term effects on neurochemicals and neuroglia in the developing brain. Exp. Neurol. 2000;162:235–246. [PubMed: 10739630]
- Tomingas R., Pott F., Dehnen W. Polycyclic aromatic hydrocarbons in human bronchial carcinoma. Cancer Lett. 1976;1:189–195. [PubMed: 1016943]
- Toniolo P., Bruning P.F., Akhmedkhanov A., Bonfrer J.M., Koenig K.L., Lukanova A., Shore R.E., Zeleniuch-Jacquotte A. Serum insulin-like growth factor-I and breast cancer. Int. J. Cancer. 2000;88:828–832. [PubMed: 11072255]
- Torgerson D.J., Avenell A., Russell I.T., Reid D.M. Factors associated with onset of menopause in women aged 45–49. Maturitas. 1994;19:83–92. [PubMed: 7968648]
- Törnqvist M., Osterman-Golkar S., Kautiainen A., Jensen S., Farmer P.B., Ehrenberg L. Tissue doses of ethylene oxide in cigarette smokers determined from adduct levels in hemoglobin. Carcinogenesis. 1986;7:1519–1521. [PubMed: 3742724]
- Törnqvist M., Svartengren M., Ericsson C.H. Methylations in hemoglobin from mono-zygotic twins discordant for cigarette smoking: Hereditary and tobacco-related factors. Chem.-biol. Interact. 1992;82:91–98. [PubMed: 1547516]
- Trauth J.A., McCook E.C., Seidler F.J., Slotkin T.A. Modeling adolescent nicotine exposure: Effects on cholinergic systems in rat brain regions. Brain Res. 2000;873:18–25. [PubMed: 10915806]
- Tricker A.R. N-Nitroso compounds and man: Sources of exposure, endogenous formation and occurrence in body fluids. Eur. J. Cancer Prev. 1997;6:226–268. [PubMed: 9306073]
- Tricker A.R., Scherer G., Conze C., Adlkofer F., Pachinger A., Klus H. Evaluation of 4-(N-methylnitrosamino)-4-(3-pyridyl)butyric acid as a potential monitor of endogenous nitrosation of nicotine and its metabolites. Carcinogenesis. 1993;14:1409–1414. [PubMed: 8330358]
- Trivedi A.H., Dave B.J., Adhvaryu S.G. Assessment of genotoxicity of nicotine employing in vitro mammalian test system. Cancer Lett. 1990;54:89–94. [PubMed: 2208095]
- Trivedi A.H., Dave B.J., Adhvaryu S.S. Genotoxic effects of nicotine in combination with arecoline on CHO cells. Cancer Lett. 1993;74:105–110. [PubMed: 8287363]
- Trivedi A.H., Roy S.K., Jaju R.J., Patel R.K., Adhvaryu S.G., Balar D.B. Urine of tobacco/areca nut chewers causes genomic damage in Chinese hamster ovary cells. Carcinogenesis. 1995;16:205–208. [PubMed: 7859349]
- Trummer H., Habermann H., Haas J., Pummer K. The impact of cigarette smoking on human semen parameters and hormones. Hum. Reprod. 2002;17:1554–1559. [PubMed: 12042277]
- Trushin N., Hecht S.S. Stereoselective metabolism of nicotine and tobacco-specific Nnitrosamines to 4-hydroxy-4-(3-pyridyl)butanoic acid in rats. Chem. Res. Toxicol. 1999;12:164–171. [PubMed: 10027794]
- Tseng J.E., Kemp B.L., Khuri F.R., Kurie J.M., Lee J.S., Zhou X., Liu D., Hong W.K., Mao L. Loss of Fhit is frequent in stage I non-small cell lung cancer and in the lungs of chronic smokers. Cancer Res. 1999;59:4798–4803. [PubMed: 10519387]
- Tsuda M., Kurashima Y. Tobacco smoking, chewing, and snuff dipping: Factors contributing to the endogenous formation of N-nitroso compounds. Crit. Rev. Toxicol. 1991;21:243–253. [PubMed: 2069710]
- Tsutsumi M., Lasker J.M., Takahashi T., Lieber C.S. In vivo induction of hepatic P450E1 by ethanol: Role of increased enzyme synthesis. Arch. Biochem. Biophys. 1993;304:209–218. [PubMed: 8323286]
- Tunstall-Pedoe H., Woodward M., Brown C.A. The drinking, passive smoking, smoking deception and serum cotinine in the Scottish Hearth Health Study. J. clin. Epidemiol. 1991;44:1411–1414. [PubMed: 1753272]
- Tuomisto J., Kolonen S., Sorsa M., Einistö P. No difference between urinary mutagenicity in smokers of low-tar and medium-tar cigarettes: A double-blind cross-over study. Arch. Toxicol. 1986;Suppl. 9:115–119. [PubMed: 3468891]
- Ulrik C.S., Lange P. Cigarette smoking and asthma. Monaldi Arch. Chest Dis. 2001;56:349–353. [PubMed: 11770219]
- Upadhyaya P., Kenney P.M.J., Hochalter J.B., Wang M., Hecht S.S. Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis. 1999;20:1577–1582. [PubMed: 10426810]
- Upadhyaya P., McIntee E.J., Hecht S.S. Preparation of pyridine-N-glucuronides of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2001;14:555–561. [PubMed: 11368554]
- US Department of Health and Human Services (DHHS) (1983) The Health Consequences of Smoking. Cardiovascular Disease. A Report of the Surgeon General (DHHS Publ. No. (CDC) 84-50204), Washington DC, US Government Printing Office.
- US Department of Health and Human Services (DHHS) (1984) The Health Consequences of Smoking. Chronic Obstructive Disease. A Report of the Surgeon General (DHHS Publ. No. (PHS) 84-50205), Washington DC, US Government Printing Office.
- US Department of Health and Human Services (DHHS) (1986) The Health Consequences of Involuntary Smoking. A Report of the Surgeon General (US DHHS Pub. No. (PHS) 87-8398), Washington DC, Public Health Service, Office of the Assistant Secretary for health, Office of Smoking and Health.
- US Department of Health and Human Services (DHHS) (1988) The Health Consequences of Smoking: Nicotine Addiction. A Report of the Surgeon General (DHHS Publ. No. (CDC) 888406), Washington DC, US Government Printing Office.
- US Department of Health and Human Services (DHHS) (1989) Reducing the Health Consequences of Smoking: 25 Years of Progress. A Report of the Surgeon General (DHHS Publ. No. (CDC) 89-8411), Washington DC, US Government Printing Office.
- US Department of Health and Human Services (DHHS) (2001) Women and Smoking. A Report of the Surgeon General, Rockville, MD [available at http://www.cdc.gov/tobacco/sgr/sgr_forwomen/index]
- Van Rooij J.G.M., Veeger M.M.S., Bodelier-Bade M.M., Scheepers P.T.J., Jongeneelen F.J. Smoking and dietary intake of polycyclic aromatic hydrocarbons as sources of inter-individual variability in the baseline excretion of 1- hydroxypyrene in urine. Int. Arch. occup. environ. Health. 1994;66:55–65. [PubMed: 7927844]
- Van Schooten F.J., Hillebrand M.J.X., van Leeuwen F.E., Lutgerink J.T., van Zandwijk N., Jansen H.M., Kriek E. Polycyclic aromatic hydrocarbon–DNA adducts in lung tissue from lung cancer patients. Carcinogenesis. 1990a;11:1677–1681. [PubMed: 2119262]
- Van Schooten F.J., van Leeuwen F.E., Hillebrand M.J.X., de Rijke M.E., Hart A.A.M., van Veen H.G., Oosterink S., Kriek E. Determination of benzo[a]pyrene diol epoxide–DNA adducts in white blood cell DNA from coke-oven workers: The impact of smoking. J. natl Cancer Inst. 1990b;82:927–933. [PubMed: 2111410]
- Van Schooten F.J., Hillebrand M.J., van Leeuwen F.E., Van Zandwijk N., Jansen H.M., den Engelse L., Kriek E. Polycyclic aromatic hydrocarbon–DNA adducts in white blood cells from lung cancer patients: No correlation with adduct levels in lung. Carcinogenesis. 1992;13:987–993. [PubMed: 1600621]
- Van Schooten F.J., Godschalk R.W., Breedijk A., Maas L.M., Kriek E., Sakai H., Wigbout G., Baas P., Van't Veer L., Van Zandwijk N. 32P-Postlabelling of aromatic DNA adducts in white blood cells and alveolar macrophages of smokers: Saturation at high exposures. Mutat. Res. 1997;378:65–75. [PubMed: 9288886]
- Van Schooten F.J., Hirvonen A., Maas L.M., De Mol B.A., Kleinjans J.C.S., Bell D.A., Durrer J.D. Putative susceptibility markers of coronary artery disease: Association between VDR genotype, smoking, and aromatic DNA adduct levels in human right atrial tissue. FASEB J. 1998;12:1409–1417. [PubMed: 9761785]
- Van Schooten F.J., Nia A.B., De Flora S., D'Agostini F., Izzotti A., Camoirano A., Balm A.J., Dallinga J.W., Bast A., Haenen G.R., Van't Veer L., Baas P., Sakai H., Van Zandwijk N. Effects of oral administration of N-acetyl-L-cysteine: A multi-biomarker study in smokers. Cancer Epidemiol. Biomarkers Prev. 2002;11:167–175. [PubMed: 11867504]
- Vaught J.B., Gurtoo H.L., Parker N.B., LeBoeuf R., Doctor G. Effect of smoking on benzo(a)pyrene metabolism by human placental microsomes. Cancer Res. 1979;39:3177–3183. [PubMed: 36982]
- Velasco E., Malacara J.M., Cervantes F., de Leon J.D., Davalos G., Castillo J. Gonadotropins and prolactin serum levels during the perimenopausal period: Correlation with diverse factors. Fertil. Steril. 1990;53:56–60. [PubMed: 2104807]
- Veltel D., Hoheneder A. Characterization of cigarette smoke-induced micronuclei in vitro. Exp. Toxicol. Pathol. 1996;48:548–550. [PubMed: 8954346]
- Venier P., Clonfero E., Cottica D., Gava C., Zordan M., Pozzoli L., Levis A.G. Mutagenic activity and polycyclic aromatic hydrocarbon levels in urine of workers exposed to coal tar pitch volatiles in an anode plant. Carcinogenesis. 1985;6:749–752. [PubMed: 4006058]
- Verkasalo P.K., Thomas H.V., Appleby P.N., Davey G.K., Key T.J. Circulating levels of sex hormones and their relation to risk factors for breast cancer: A cross-sectional study in 1092 pre- and postmenopausal women (United Kingdom). Cancer Causes Control. 2001;12:47–59. [PubMed: 11227925]
- Villablanca A.C., McDonald J.M., Rutledge J.C. Smoking and cardiovascular disease. Clin. Chest Med. 2000;21:159–172. [PubMed: 10763097]
- Villard P.H., Seree E., Lacarelle B., Therene-Fenoglio M.C., Barra Y., Attolini L., Bruguerole B., Durand A., Catalin J. Effect of cigarette smoke on hepatic and pulmonary cytochromes P450 in mouse: Evidence for CYP2E1 induction in lung. Biochem. biophys. Res. Commun. 1994;202:1731–1737. [PubMed: 8060364]
- Villard P.H., Herber R., Seree E.M., Attolini L., Magdalou J., Lacarelle B. Effect of cigarette smoke on UDP-glucuronosyltransferase activity and cytochrome P450 content in liver, lung and kidney microsomes in mice. Pharmacol. Toxicol. 1998a;82:74–79. [PubMed: 9498235]
- Villard P.H., Seree E.M., Re J.L., De Meo M., Barra Y., Attolini L., Dumenil G., Catalin J., Durand A., Lacarelle B. Effects of tobacco smoke on the gene expression of the Cyp1a, Cyp2b, Cyp2e, and Cyp3a subfamilies in mouse liver and lung: Relation to single strand breaks of DNA. Toxicol. appl. Pharmacol. 1998b;148:195–204. [PubMed: 9473526]
- Vine M.F. Smoking and male reproduction: A review. Int. J. Androl. 1996;19:323–337. [PubMed: 9051418]
- Vine M.F., Margolin B.H., Morrison H.I., Hulka B.S. Cigarette smoking and sperm density: A meta-analysis. Fertil. Steril. 1994;61:35–43. [PubMed: 8293842]
- Vineis P., Estève J., Terracini B. Bladder cancer and smoking in males: Types of cigarettes, age at start, effect of stopping and interaction with occupation. Int. J. Cancer. 1984;34:165–170. [PubMed: 6469396]
- Vineis P., Caporaso N., Tannenbaum S.R., Skipper P.L., Glogowski J., Bartsch H., Coda M., Talaska G., Kadlubar F. Acetylation phenotype, carcinogen–hemoglobin adducts, and cigarette smoking. Cancer Res. 1990;50:3002–3004. [PubMed: 2334904]
- Vineis P., Talaska G., Malaveille C., Bartsch H., Martone T., Sithisarankul P., Strickland P. DNA adducts in urothelial cells: Relationship with biomarkers of exposure to arylamines and polycyclic aromatic hydrocarbons from tobacco smoke. Int. J. Cancer. 1996;65:314–316. [PubMed: 8575850]
- Vineis, P., d'Errico, A., Malats, N. & Boffetta, P. (1999) Overall evaluation and research perspectives. In: Ryder, W., ed., Metabolic Polymorphisms and Susceptibility in Cancer (IARC Scientific Publications No. 148), Lyon, IARCPress, pp. 403–408. [PubMed: 10493267]
- Voncken P., Rustemeier K., Schepers G. Identification of cis-3′-hydroxycotinine as a urinary nicotine metabolite. Xenobiotica. 1990;20:1353–1356. [PubMed: 2075752]
- Vulimiri S.V., Wu X., Baer-Dubowska W., de Andrade M., Detry M., Spitz M.R., DiGiovanni J. Analysis of aromatic DNA adducts and 7,8-dihydro-8-oxo-2′-deoxyguanosine in lymphocyte DNA from a case–control study of lung cancer involving minority populations. Mol. Carcinog. 2000;27:34–46. [PubMed: 10642435]
- Wagenknecht L.E., Manolio T.A., Sidney S., Burke G.L., Haley N.J. Environmental tobacco smoke exposure as determined by cotinine in black and white young adults: The CARDIA study. Environ. Res. 1993;63:39–46. [PubMed: 8404773]
- Wallace L.A., Pellizzari E.D. Personal air exposures and breath concentrations of benzene and other volatile hydrocarbons for smokers and nonsmokers. Toxicol. Lett. 1986;35:113–116. [PubMed: 3810671]
- Wallace L.A., Pellizzari E.D., Hartwell T.D., Perritt R., Ziegenfus R. Exposures to benzene and volatile compounds from active and passive smoking. Arch. environ. Health, 1987;42:272–279. [PubMed: 3452294]
- Wang M., Dhingra K., Hittelman W.N., Liehr J.G., de Andrade M., Li D. Lipid peroxidation-induced putative malondialdehyde–DNA adducts in human breast tissues. Cancer Epidemiol. Biomarkers Prev. 1996;5:705–710. [PubMed: 8877062]
- Wang Y., Ichiba M., Oishi H., Iyadomi M., Shono N., Tomokuni K. Relationship between plasma concentrations of β-carotene and α-tocopherol and life-style factors and levels of DNA adducts in lymphocytes. Nutr. Cancer. 1997;27:69–73. [PubMed: 8970185]
- Wang M., Abbruzzese J.L., Friess H., Hittelman W.N., Evans D.B., Abbruzzese M.C., Chiao P., Li D. DNA adducts in human pancreatic tissues and their potential role in carcinogenesis. Cancer Res. 1998;58:38–41. [PubMed: 9426054]
- Wang L.-E., Bondy M.L., de Andrade M., Strom S.S., Wang X., Sigurdson A., Spitz M.R., Wei Q. Gender difference in smoking effect on chromosome sensitivity to gamma radiation in a health population. Radiat. Res. 2000;154:20–27. [PubMed: 10856961]
- Wang Q., Fan J., Wang H., Liu S. DNA damage and activation of c-ras in human embryo lung cells exposed to chrysotile and cigarette smoking solution. J. environ. Pathol. Toxicol. Oncol. 2000;19:13–19. [PubMed: 10905503]
- Wang H., Liu X., Umino T., Skold C.M., Zhu Y., Kohyama T., Spurzem J.R., Romberger D.J., Rennard S.I. Cigarette smoke inhibits human bronchial epithelial cell repair processes. Am. J. respir. Cell mol. Biol. 2001;25:772–779. [PubMed: 11726404]
- Wardlaw S.A., Nikula K.J., Kracko D.A., Finch G.L., Thornton-Manning J.R., Dahl A.R. Effect of cigarette smoke on CYP1A1, CYP1A2 and CYP2B1/2 of nasal mucosae in F344 rats. Carcinogenesis. 1998;19:655–662. [PubMed: 9600351]
- Wehner A.P., Olson R.J., Busch R.H. Increased life span and decreased weight in hamsters exposed to cigarette smoke. Arch. environ. Health. 1976;31:146–153. [PubMed: 1275559]
- Wei Q., Cheng L., Amos C.I., Wang L.-E., Guo Z., Hong W.K., Spitz M.R. Repair of tobacco carcinogen-induced DNA adducts and lung cancer risk: A molecular epidemiologic study. J. natl Cancer Inst. 2000;92:1764–1772. [PubMed: 11058619]
- Weitberg A.B., Corvese D. Oxygen radicals potentiate the genetic toxicity of tobacco-specific nitrosamines. Clin. Genet. 1993;43:88–91. [PubMed: 8448908]
- Westhoff C., Murphy P., Heller D. Predictors of ovarian follicle number. Fertil. Steril. 2000;74:624–628. [PubMed: 11020495]
- Westman E.C., Levin E.D., Rose J.E. Nicotine as a therapeutic drug. N.C. med. J. 1995;56:48–51. [PubMed: 7862206]
- Weston A. Physical methods for the detection of carcinogen–DNA adducts in humans. Mutat. Res. 1993;288:19–29. [PubMed: 7686262]
- Weston A., Bowman E.D. Fluorescence detection of benzo[a]pyrene–DNA adducts in human lung. Carcinogenesis. 1991;12:1445–1449. [PubMed: 1860165]
- Weston A., Rowe M.L., Manchester D.K., Farmer P.B., Mann D.L., Harris C.C. Fluorescence and mass spectral evidence for the formation of benzo[a]pyrene anti-diolepoxide–DNA and – hemoglobin adducts in humans. Carcinogenesis. 1989a;10:251–257. [PubMed: 2912575]
- Weston A., Manchester D.K., Poirier M.C., Choi J.S., Trivers G.E., Mann D.L., Harris C.C. Derivative fluorescence spectral analysis of polycyclic aromatic hydrocarbon–DNA adducts in human placenta. Chem. Res. Toxicol. 1989b;2:104–108. [PubMed: 2519708]
- Weston A., Caporaso N.E., Taghizadeh K., Hoover R.N., Tannenbaum S.R., Skipper P.L., Resau J.H., Trump B.F., Harris C.C. Measurement of 4-aminobiphenyl–hemoglobin adducts in lung cancer cases and controls. Cancer Res. 1991;51:5219–5223. [PubMed: 1913645]
- Westra W.H., Slebos R.J.C., Offerbaus G.J.A., Goodman S.N., Evers S.G., Kensler T.W., Askin F.B., Rodenhuis S., Hruban R.H. K-ras oncogene activation in lung adenocarcinomas from former smokers. Cancer. 1993;72:432–438. [PubMed: 8319174]
- White J.L., Conner B.T., Perfetti T.A., Bombick B.R., Avalos J.T., Fowler K.W., Smith C.J., Doolittle D.J. Effect of pyrolysis temperature on the mutagenicity of tobacco smoke condensate. Food chem. Toxicol. 2001;39:499–505. [PubMed: 11313117]
- WHO (1992) Cadmium (Environmental Health Criteria No. 134), Geneva, International Programme on Chemical Safety.
- Whyatt R.M., Bell D.A., Jedrychowski W., Santella R.M., Garte S.J., Cosma G., Manchester D.K., Young T.L., Cooper T.B., Ottman R., Perera F.P. Polycyclic aromatic hydrocarbon–DNA adducts in human placenta and modulation by CYP1A1 induction and genotype. Carcinogenesis. 1998;19:1389–1392. [PubMed: 9744534]
- Whyatt R.M., Jedrychowski W., Hemminki K., Santella R.M., Tsai W.Y., Yang K., Perera F.P. Biomarkers of polycyclic aromatic hydrocarbon–DNA damage and cigarette smoke exposures in paired maternal and newborn blood samples as a measure of differential susceptibility. Cancer Epidemiol. Biomarkers Prev. 2001;10:581–588. [PubMed: 11401906]
- Wiencke J.K., Kelsey K.T., Varkonyi A., Semey K., Wain J.C., Mark E., Christiani D.C. Correlation of DNA adducts in blood mononuclear cells with tobacco carcinogen-induced damage in human lung. Cancer Res. 1995;55:4910–4914. [PubMed: 7585529]
- Wiencke J.K., Thurston S.W., Kelsey K.T., Varkonyi A., Wain J.C., Mark E.J., Christiani D.C. Early age at smoking initiation and tobacco carcinogen DNA damage in the lung. J. natl Cancer Inst. 1999;91:614–619. [PubMed: 10203280]
- Wild C.P., Pisani P. Carcinogen DNA and protein adducts as biomarkers of human exposure in environmental cancer epidemiology. Cancer Detect. Prev. 1998;22:273–283. [PubMed: 9674870]
- Wilkins J.N., Carlson H.E., Van Vunakis H., Hill M.A., Gritz E., Jarvik M.E. Nicotine from cigarette smoking increases circulating levels of cortisol, growth hormone, and prolactin in male chronic smokers. Psychopharmacology. 1982;78:305–308. [PubMed: 6818588]
- Willett W., Stampfer M.J., Bain C., Lipnick R., Speizer F.E., Rosner B., Cramer D., Hennekens C.H. Cigarette smoking, relative weight, and menopause. Am. J. Epidemiol. 1983;117:651–658. [PubMed: 6859020]
- Willey J.C., Grafstrom R.C., Moser C.E. Jr, Ozanne C., Sundqvist K., Harris C.C. Biochemical and morphological effects of cigarette smoke condensate and its fractions on normal human bronchial epithelial cells in vitro. Cancer Res. 1987;47:2045–2049. [PubMed: 3828994]
- Williams K., Lewtas J. Metabolic activation of organic extracts from diesel, coke oven, roofing tar, and cigarette smoke emissions in the Ames assay. Environ. Mutag. 1985;7:489–500. [PubMed: 2414095]
- Williams R.W., Watts R., Inmon J., Pasley T., Claxton L. Stability of the mutagenicity in stored cigarette smokers' urine and extract. Environ. mol. Mutag. 1990;16:246–249. [PubMed: 2253603]
- Williamson D.F., Madans J., Anda R.F., Kleinman J.C., Giovino G.A., Byers T. Smoking cessation and severity of weight gain in a national cohort. New Engl. J. Med. 1991;324:739–745. [PubMed: 1997840]
- Windham G.C., Von Behren J., Waller K., Fenster L. Exposure to environmental and mainstream tobacco smoke and risk of spontaneous abortion. Am. J. Epidemiol. 1999;149:243–247. [PubMed: 9927219]
- Winternitz W.W., Quillen D. Acute hormonal response to cigarette smoking. J. clin. Pharmacol. 1977;17:389–397. [PubMed: 881471]
- Wistuba I.I., Lam S., Behrens C., Virmani A.K., Fong K.M., LeRiche J., Samet J.M., Srivastava S., Minna J.D., Gazdar A.F. Molecular damage in the bronchial epithelium of current and former smokers. J. natl Cancer Inst. 1997;89:1366–1373. [PMC free article: PMC5193483] [PubMed: 9308707]
- Wistuba I.I., Gazdar A.F., Minna J.D. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001;28 (Suppl. 4):3–13. [PubMed: 11479891]
- Wong W.Y., Thomas C.M., Merkus H.M., Zielhuis G.A., Doesburg W.H., Steegers-Theunissen R.P. Cigarette smoking and the risk of male factor subfertility: Minor association between cotinine in seminal plasma and semen morphology. Fertil. Steril. 2000;74:930–935. [PubMed: 11056235]
- Woodward M., Tunstall-Pedoe H. Do smokers of lower tar cigarettes consume lower amounts of smoke components? Results from the Scottish Heart Health Study. Br. J. Addict. 1992;87:921–928. [PubMed: 1326360]
- Wright J.L., Dai J., Zay K., Price K., Gilks C.B., Churg A. Effects of cigarette smoke on nitric oxide synthase expression in the rat lung. Lab. Invest. 1999;79:975–983. [PubMed: 10462035]
- Wu X., Lippman S.M., Lee J.J., Zhu Y., Wei Q.V., Thomas M., Hong W.K., Spitz M.R. Chromosome instability in lymphocytes: A potential indicator of predisposition to oral premalignant lesions. Cancer Res. 2002;62:2813–1818. [PubMed: 12019158]
- Wulf H.C., Kromann N., Kousgaard N., Hansen J.C., Niebuhr E., Albøge K. Sister chromatid exchange (SCE) in Greenlandic Eskimos. Dose–response relationship between SCE and seal diet, smoking, and blood cadmium and mercury concentrations. Sci. total Environ. 1986;48:81–94. [PubMed: 3945798]
- Wurzel H., Yeh C.E., Gairola C.C., Chow C.K. Oxidative damage and antioxidant status in the lungs and bronchoalveolar lavage fluid of rats exposed chronically to cigarette smoke. J. Biochem. Toxicol. 1995;10:11–17. [PubMed: 7595927]
- Xu C., Rao Y.S., Xu B., Hoffmann E., Jones J., Sellers E.M., Tyndale R.F. An in vivo pilot study characterizing the new CYP2A6*7, *8, and *10 alleles. Biochem. biophys. Res. Commun. 2002;290:318–324. [PubMed: 11779172]
- Yadav J.S., Thakur S. Cytogenetic damage in bidi smokers. Nicotine Tob. Res. 2000a;2:97–103. [PubMed: 11072447]
- Yadav J.S., Thakur S. Genetic risk assessment in hookah smokers. Cytobios. 2000b;101:101–113. [PubMed: 10756983]
- Yamasaki E., Ames B.N. Concentration of mutagens from urine by adsorption with the nonpolar resin XAD-2: Cigarette smokers have mutagenic urine. Proc. natl Acad. Sci. USA. 1977;74:3555–3559. [PMC free article: PMC431630] [PubMed: 333441]
- Yamazaki H., Inoue K., Hashimoto M., Shimada T. Roles of CYP2A6 and CYP2B6 in nicotine C-oxidation by human liver microsomes. Arch. Toxicol. 1999;73:65–70. [PubMed: 10350185]
- Yang X., Nakao Y., Pater M.M., Tang S.-C., Pater A. Expression of cellular genes in HPV16-immortalized and cigarette smoke condensate-transformed human endocervical cells. J. cell. Biochem. 1997;66:309–321. [PubMed: 9257188]
- Yang Q., Hergenhahn M., Weninger A., Bartsch H. Cigarette smoke induces direct DNA damage in the human B-lymphoid cell line Raji. Carcinogenesis. 1999;20:1769–1775. [PubMed: 10469623]
- Yang M., Koga M., Katoh T., Kawamoto T. A study for the proper application of urinary naphthols, new biomarkers for airborne polycyclic aromatic hydrocarbons. Arch. environ. Contam. Toxicol. 1999;36:99–108. [PubMed: 9828267]
- Yang M., Kunugita N., Kitagawa K., Kang S.-H., Coles B., Kadlubar F.F., Katoh T., Matsuno K., Kawamoto T. Individual differences in urinary cotinine levels in Japanese smokers: Relation to genetic polymorphism of drug-metabolizing enzymes. Cancer Epidemiol. Biomarkers Prev. 2001;10:589–593. [PubMed: 11401907]
- Yates D.H., Breen H., Thomas P.S. Passive smoke inhalation decreases exhaled nitric oxide in normal subjects. Am. J. respir. crit. Care Med. 2001;164:1043–1046. [PubMed: 11587994]
- Yeowell-O'Connell K., Rothman N., Waidyanatha S., Smith M.T., Hayes R.B., Li G., Bechtold W.E., Dosemeci M., Zhang L., Yin S., Rappaport S.M. Protein adducts of 1,4-benzoquinone and benzene oxide among smokers and nonsmokers exposed to benzene in China. Cancer Epidemiol. Biomarkers Prev. 2001;10:831–838. [PubMed: 11489749]
- Yokoi T., Kamataki T. Genetic polymorphism of drug metabolizing enzymes: New mutations in CYP2D6 and CYP2A6 genes in Japanese. Pharm. Res. 1998;15:517–524. [PubMed: 9587945]
- Yong V.W., Perry T.L. Monoamine oxidase B, smoking, and Parkinson's disease. J. neurol. Sci. 1986;72:265–272. [PubMed: 3486945]
- Yoshie Y., Ohshima H. Synergistic induction of DNA strand breakage by cigarette tar and nitric oxide. Carcinogenesis. 1997;18:1359–1363. [PubMed: 9230280]
- Yu H., Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J. natl Cancer Inst. 2000;92:1472–1489. [PubMed: 10995803]
- Yu M.C., Skipper P.L., Taghizadeh K., Tannenbaum S.R., Chan K.K., Henderson B.E., Ross R.K. Acetylator phenotype, aminobiphenyl–hemoglobin adduct levels, and bladder cancer risk in white, black, and Asian men in Los Angeles, California. J. natl Cancer Inst. 1994;86:712–716. [PubMed: 8158701]
- Yu H., Spitz M.R., Mistry J., Gu J., Hong W.K., Wu X. Plasma levels of insulin-like growth factor-I and lung cancer risk: A case–control analysis. J. natl Cancer Inst. 1999;91:151–156. [PubMed: 9923856]
- Zavaroni I., Bonini L., Gasparini P., Dall'Aglio E., Passeri M., Reaven G.M. Cigarette smokers are relatively glucose intolerant, hyperinsulinemic and dyslipidemic. Am. J. Cardiol. 1994;73:904–905. [PubMed: 8184821]
- Zavos P.M., Correa J.R., Antypas S., Zarmakoupis-Zavos P.N., Zarmakoupis C.N. Effects of seminal plasma from cigarette smokers on sperm viability and longevity. Fertil. Steril. 1998;69:425–429. [PubMed: 9531871]
- van Zeeland A.A., de Groot A.J., Hall J., Donato F. 8-Hydroxydeoxyguanosine in DNA from leukocytes of healthy adults: Relationship with cigarette smoking, environmental tobacco smoke, alcohol and coffee consumption. Mutat. Res. 1999;439:249–257. [PubMed: 10023075]
- Zeleniuch-Jacquotte A., Bruning P.F., Bonfrer J.M., Koenig K.L., Shore R.E., Kim M.Y., Pasternack B.S., Toniolo P. Relation of serum levels of testosterone and dehydroepiandrosterone sulfate to risk of breast cancer in postmenopausal women. Am. J. Epidemiol. 1997;145:1030–1038. [PubMed: 9169912]
- Zeman M.V., Hiraki L., Sellers E.M. Gender differences in tobacco smoking: Higher relative exposure to smoke than nicotine in women. J. Women Health Gender-based Med. 2002;11:147–153. [PubMed: 11975862]
- Zenzes M.T. Smoking and reproduction: Gene damage to human gametes and embryos. Hum. Reprod. Update 2000. 2000;6:122–131. [PubMed: 10782570]
- Zenzes M.T., Wang P., Casper R.F. Cigarette smoking may affect meiotic maturation of human oocytes. Hum. Reprod. 1995;10:3213–3217. [PubMed: 8822447]
- Zenzes M.T., Puy L.A., Bielecki R. Immunodetection of benzo[a]pyrene adducts in ovarian cells of women exposed to cigarette smoke. Mol. hum. Reprod. 1998;4:159–165. [PubMed: 9542974]
- Zenzes M.T., Bielecki R., Reed T.E. Detection of benzo(a)pyrene diol epoxide–DNA adducts in sperm of men exposed to cigarette smoke. Fertil. Steril. 1999a;72:330–335. [PubMed: 10439006]
- Zenzes M.T., Puy L.A., Bielecki R., Reed T.E. Detection of benzo[a]pyrene diol epoxide–DNA adducts in embryos from smoking couples: Evidence for transmission by spermatozoa. Mol. hum. Reprod. 1999b;5:125–131. [PubMed: 10065867]
- Zevin S., Benowitz N.L. Drug interactions with tobacco smoking. An update. Clin. Pharmacokinet. 1999;36:425–438. [PubMed: 10427467]
- Zhang Y.J., Hsu T.M., Santella R.M. Immunoperoxidase detection of polycyclic aromatic hydrocarbon–DNA adducts in oral mucosa cells of smokers and nonsmokers. Cancer Epidemiol. Biomarkers Prev. 1995;4:133–138. [PubMed: 7537994]
- Zhang Z.-F., Shu X.-M., Cordon-Cardo C., Orlow I., Lu M.-L., Millon T.V., Cao P.-Q., Connolly-Jenks C., Dalbagni G., Lianes P., Lacombe L., Reuter V.E., Scher H. Cigarette smoking and chromosome 9 alterations in bladder cancer. Cancer Epidemiol. Biomarkers Prev. 1997;6:321–326. [PubMed: 9149891]
- Zhang Y.J., Weksler B.B., Wang L., Schwartz J., Santella R.M. Immunohistochemical detection of polycyclic aromatic hydrocarbon–DNA damage in human blood vessels of smokers and nonsmokers. Atherosclerosis. 1998;140:325–331. [PubMed: 9862275]
- Zhang Y.J., Chen S.-Y., Hsu T.M., Santella R.M. Immunohistochemical detection of malondialdehyde–DNA adducts in human oral mucosa cells. Carcinogenesis. 2002;23:207–211. [PubMed: 11756243]
- Zhao C., Georgellis A., Flato S., Palmberg L., Thunberg E., Hemminki K. DNA adducts in human nasal mucosa and white blood cells from smokers and nonsmokers. Carcinogenesis. 1997;18:2205–2208. [PubMed: 9395222]
- Other Data Relevant to an Evaluation of Carcinogenicity and its Mechanisms - Tob...Other Data Relevant to an Evaluation of Carcinogenicity and its Mechanisms - Tobacco Smoke and Involuntary Smoking
Your browsing activity is empty.
Activity recording is turned off.
See more...