Rapid progress in understanding glycosylation pathways of eukaryotes came with the application of genetic strategies to isolate mutants of mammalian cells and yeast with alterations in glycan synthesis. This chapter reviews methods used to isolate mammalian cell glycosylation mutants and the diversity of mutants that may be obtained from selections and screens. The applications of glycosylation mutants to address functional roles of glycans and in glycosylation engineering are discussed briefly. Many of the cell lines described in this chapter are available through the American Type Culture Collection.
HISTORY
The success of bacterial and yeast genetics in isolating mutants and using them to define biochemical pathways led in the late 1960s to the development of somatic cell genetics using mammalian cells. Chinese hamster ovary (CHO) cells were selected by two independent groups for initial experiments to isolate stable mutants. Somatic cell genetic strategies were applied early to glycobiology, yielding numerous mutants in glycoprotein biosynthesis and later in proteoglycan, glycosylphosphatidylinositol (GPI) anchor, and glycolipid biosynthesis. The ability to isolate glycosylation mutants in mammalian cells made it possible to unravel pathways of glycan synthesis and degradation and to identify, isolate, and map structural and regulatory genes. CHO cells thus became a focus for experiments to decipher glycosylation pathways and, importantly, provided mutant host cells for the production of viruses and glycoproteins with modified glycans. This proved to be extremely beneficial to the biotechnology industry because most recombinant therapeutics are glycoproteins. CHO cells and CHO glycosylation mutants are now the workhorse of the biotechnology industry. They are particularly useful because they produce only minor, if any, quantities of nonhuman glycans or glycan modifications that give rise to undesirable antibodies. Conserved glycosylation pathways in yeast were delineated by similar approaches (Chapter 23).
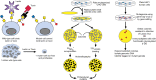
Mutants in any cell type often accumulate the precursor immediately upstream of the block in a pathway and thereby reveal the structure of their substrate(s). Sequencing of mutant alleles reveals specific mutations that may give rise to a glycosylation phenotype. In most cases, mutations are loss-of-function and they reduce or abrogate the activity of an enzyme in a pathway; but there are also gain-of-function mutations that activate a silent glycosylation gene, elevate the expression of an existing activity, or inactivate a negative regulatory factor (Figure ). In nearly all cases, glycosylation mutations lead to the presence of altered glycans on cell-surface glycoconjugates and changes in cell properties that link glycan structure to function. Although gene editing techniques using CRISPR/Cas9 or transcription activator-like effector nucleases (TALENs) are now the method of choice for introducing a mutation that weakens or ablates a glycosylation gene (Chapters 27 and 56), initially such approaches did not allow for the serendipitous findings that often emerge from genetic screens. Subsequently, genetic screens were performed using HAP1 (haploid) human cells mutagenized by retroviral gene trap. Such unbiased screens led to the identification of multiple, previously unknown, glycosylation genes. Lately, however, the evolvement of CRISPR tools and the generation of genome-wide libraries have made it possible to conduct both loss-of-function and gain-of-function screens in nonhaploid cell lines, providing an unbiased strategy to discover new genes that influence cellular glycosylation (Figure ).
Alteration of cell-surface glycans by recessive and dominant glycosylation mutations.
Selections for glycosylation mutants. Cytotoxic lectins or agents that bind to specific sugar residues select for resistant cells (left). Screen for mutants using replica plating. Colonies on plastic are transferred to discs and screened for defects in (more...)
ISOLATION OF GLYCOSYLATION MUTANTS
Cells in culture mutate at a low rate (<10−6 mutations per locus per generation). In CHO cells, some loci are functionally haploid (single copy), and in HAP1 human cells, essentially all loci are haploid, which means that a single hit may generate a recessive mutant. However, typical mammalian cells are diploid and immortalized cells are often hyperploid, so the frequency of finding recessive mutants is low. To greatly increase the probability of finding desirable mutants, mutations may be induced by treating cells with chemical (e.g., alkylating agents), physical (e.g., ionizing radiation), or biological (e.g., a virus) mutagens or, perhaps more likely, by lentiviral transduction with CRIPSR/Cas9 genome-wide or focused libraries. Regardless of the method used to induce mutations, selection or enrichment is usually needed to find rare recessive or dominant mutants bearing a desired glycosylation phenotype (Figure ). For example, direct selection for resistance to cytotoxic plant lectins (Chapters 31 and 32) that bind to cell-surface glycans gives a range of glycosylation mutants. Importantly, many mutants resistant to one or more lectins because of the loss of specific sugars become supersensitive to a different group of lectins that recognize sugar residues exposed by the mutation (Figure ). The latter may be used to select for revertants in the original mutant population. Nontoxic lectins are also useful for enriching lectin-binding mutants (e.g., by flow cytometry). Mutations that affect all stages of glycosylation reactions, including the generation and transport of nucleotide sugars, have been identified using lectins as selective agents.
In principle, any glycan-binding protein (GBP), antibody, or other agent that recognizes cell-surface glycans or a glycoprotein can be used to isolate mutants with a glycosylation defect (Figure ). Conjugation of a GBP or protein domain to a toxin that cannot enter the cell independently, but can kill the cell following entry, can be used to select mutants when cytotoxic lectins are not available. For example, basic fibroblast growth factor FGF2-saporin complexes have been used for the selection of mutants deficient in heparan sulfate (HS). Lectins, antibodies, or ligands that are fluorescently tagged may be used to enrich for mutants that are either deficient in binding or have acquired a novel binding ability because of altered glycosylation or reduced expression of an antigen at the cell surface. Panning or immunodepletion are related techniques. For example, coating a plate with FGF2 allows selection of mutant cells that fail to produce HS proteoglycans and consequently fail to adhere to a FGF2-coated plate. HAP1 cells that fail to glycosylate α-dystroglycan fail to bind a specific monoclonal antibody and may be enriched by immunodepletion (Figure ). Radiation suicide is another direct selection method for obtaining glycosylation mutants. Incubation of cells with a radioactive sugar, sulfate, or other precursor of high radioactivity leads to labeled glycoproteins, glycolipids, or proteoglycans. After prolonged storage of the cells, radiation damage kills wild-type cells, whereas mutants with reduced incorporation of the label survive. Animal cells can also be replica-plated, much like microbial colonies, using porous cloth made of polyester or nylon as the replica (Figure ). Colonies of cells on the disc can be used to identify mutants with reduced incorporation of radioactive precursors or to identify mutants that fail to bind to a lectin, an antibody, or a growth factor. An adaptation of this technique allows detection of mutants affecting a specific enzyme by direct assay for activity in colony lysates generated on a disc. Although this technique has great specificity, its limited capacity makes detection of rare mutants difficult, and mutagenesis before screening is usually a requirement.
The resulting strains must be cloned and carefully characterized for stability and the biochemical and molecular basis of mutation. Additional genetic analyses include somatic cell hybridization for dominance/recessive testing and assigning mutants to different genetic complementation groups. Specifically, when mutations are introduced by CRISPR/Cas9 and single-guide RNAs (sgRNAs), the affected genes can be identified through deep sequencing analysis of enriched sgRNA populations. Regardless of the technique used to isolate mutants, biochemical analysis involves the characterization of glycan structures produced by mutant cells (Chapter 50), the quantitation and analysis of intermediates, and assays for activities thought to be missing or acquired based on the properties of the mutant. Identifying the molecular basis of mutation requires isolation of a complementing cDNA that reverts the mutant phenotype and determining whether the mutation arose from defective transcription, translation, or stability of the gene product or from a missense or nonsense mutation in the coding region of the gene. Targeted gene mutation (Chapter 56) can also be used to validate a phenotype after a gene has been identified in a selected mutant.
CELL LINES FROM MICE OR HUMANS WITH A GLYCOSYLATION MUTATION
Transgenic mice that overexpress a glycosylation gene, or mutant mice that lack a glycosylation activity because of targeted gene inactivation (Chapters 27 and 56), are a source of mutant cells that may be used for glycobiology research. Fibroblasts or lymphoblasts can be obtained readily from humans with a disorder of glycosylation (Chapter 45). Cells may be grown as primary cultures or immortalized by viral transformation. By crossing mutant mice with the Immortomouse, which carries a temperature-sensitive SV40 T antigen in every cell, immortalized mutant cell lines can be derived from essentially any cell type. For mutations that cause embryos to die during gestation, mutant embryonic stem (ES) cells can be derived from blastocysts, provided the mutation does not result in cell-autonomous lethality. The resulting mutant ES cell lines can be used to investigate functions for specific glycans during differentiation in embryoid cell culture, or in vivo in mouse chimeras. A chimera is obtained by injecting wild-type or mutant ES cells into the inner cell mass of a mouse blastocyst. If the ES cells survive, the resulting mouse is a mixture of cells derived from the ES cells and cells derived from the blastocyst and is termed a chimera. Mutant ES cells may not contribute equally well to all tissues. For example, ES cells lacking MGAT1 are unable to make complex or hybrid N-glycans (Chapter 9), but they differentiate normally into many cell types in cultured embryoid bodies. However, following introduction into blastocysts, ES cells lacking MGAT1 do not contribute to the organized layer of bronchial epithelium in chimeric embryos.
Immortalized fibroblasts from patients with defects in glycosylation can be used to study the underlying defect (Chapter 45). Induced pluripotent stem cells derived from fibroblasts from patients with glycosylation disorders provide another approach for obtaining various differentiated cell lines for further study. The use of precise genetic engineering tools allows the correction of a genetic mutation in patient-derived fibroblasts, thereby creating pairs of isogenic cell lines for comparative studies. Alternatively, such isogenic pairs can be generated by introducing the patient-specific mutation in a cell or cell line of choice (Chapter 56).
RECESSIVE GLYCOSYLATION MUTANTS
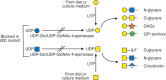
Selection schemes based on isolating rare mutants resistant to cytotoxic plant lectins have yielded a large number of glycosylation mutants affected in diverse aspects of glycan synthesis (Table ). Some mutations affect several types of glycans, such as mutants with reduced nucleotide sugar formation or transport into the Golgi. For example, the UDP-Gal transporter defect in Lec8 mutant cells affects transfer of galactose to O- and N-glycans on glycoproteins, as well as to glycosaminoglycans (GAGs) and glycolipids. The ldlD mutant is particularly interesting in this regard, because it lacks the epimerase responsible for converting UDP-Glc to UDP-Gal and UDP-GlcNAc to UDP-GalNAc (Figure ). Because there are salvage pathways for importing Gal and GalNAc into cells (Chapter 5), the composition of different classes of glycans can be controlled in ldlD cells by nutritional supplementation with either of these two sugars. Mutations in glycosyltransferase genes may ablate activity or affect the kinetic properties of an enzyme (e.g., Lec1A; Table ), or its subcellular localization (e.g., Lec4A; Table ). Sequencing mutant alleles provides leads for further site-directed mutagenesis of the gene in order to define important functional domains of the protein required for catalysis or compartmentalization.
Mutation of UDP-Glc/UDP-GlcNAc-4-epimerase, also called UDP-Gal-4-epimerase or GALE, in ldlD mutant Chinese hamster ovary (CHO) cells prevents the generation of UDP-Gal and UDP-GalNAc preventing addition of Gal and GalNAc to all glycans. Salvage reactions (more...)
Examples of recessive glycosylation mutants
Some lectin-resistant mutants are defective in the formation of dolichol-P-oligosaccharides or in the processing reactions that remove Glc or Man after transfer of the glycan chain to a glycoprotein (Chapter 9). The latter mutants revealed the identity and importance of α-mannosidases in the formation of N-glycans. However, when the α-mannosidase II gene Man2a1 was ablated in mice, no effect was seen in certain tissues because another previously unknown α-mannosidase gene (Man2a2) allowed N-glycans to be synthesized. This finding emphasizes a limitation of somatic cells in that they may not express glycosylation genes that are developmentally regulated in a tissue-specific manner, thereby precluding the isolation of mutants affected in those genes from that cell line.
DOMINANT GLYCOSYLATION MUTANTS
The recessive mutants in Table lack a glycosylation activity or fail to make a precursor. Dominant mutations that activate a silent gene reveal activities that may normally be expressed only in a few, very specialized cells in the body. Therefore, dominant mutants are important in glycosylation gene discovery, for identifying mechanisms of glycosylation gene regulation, and for defining pathways of glycan biosynthesis. The mutants in Table show a gain-of-function, dominant, lectin-resistant phenotype caused by the increased expression of a glycosyltransferase that is normally silent or expressed at very low levels. The activation of a glycosyltransferase gene may reflect a mutation in a regulatory region of the gene or in a trans-acting factor. The genetic bases of the mutants in Table are not known, but their characterization may reveal novel genes or regulatory factors that may not have been previously known to exist.
Examples of dominant mutants expressing a new activity
MUTANTS IN GPI-ANCHOR BIOSYNTHESIS
Glycosylation defects in GPI-anchor biosynthesis reduce expression of GPI-anchored proteins at the cell surface (Chapter 12). Originally, many GPI-anchor mutants were isolated by strategies that took advantage of antibodies to a GPI-anchored glycoprotein. For example, lymphoma cells expressing Thy-1 on their surface were incubated with an antibody to Thy-1 and serum-containing complement, which lysed cells expressing the Thy-1 antigen. Loss of GPI-anchor biosynthesis reduced the expression of Thy-1 on the surface and conferred resistance to the cytolytic effect. Other mutants have been obtained by sorting cells that do not bind to a fluorescent antibody or with bacterial toxins that bind GPI glycans. The GPI-anchor mutants obtained to date fall into many genetic complementation groups, each having a different lesion in GPI-anchor biosynthesis (Chapter 12). These mutants reveal the complexity of GPI-anchor biosynthesis: Multiple gene products are involved in forming the N-acetylglucosamine linkage to phosphatidylinositol, the first committed intermediate in the pathway; dolichol-P-Man is used as the donor of Man; at least three enzymes are involved in the attachment of ethanolamine phosphate residues; and five genes are required for the transfer of the GPI anchor to protein. The available strains show the importance of genetic approaches for identifying genes that might not be obvious from measuring biosynthetic reactions in vitro.
MUTANTS IN PROTEOGLYCAN ASSEMBLY
A large collection of mutants defective in GAG/proteoglycan biosynthesis has been isolated (Table ). Many of these mutants were obtained by replica-plating methods using sulfate incorporation to monitor GAG production in colonies (Figure ). Mutants in the early steps of GAG biosynthesis (complementation groups A, B, and G) lack both CS and HS chains, and enzymatic assays showed that they lack enzymes responsible for the assembly of the core protein linkage tetrasaccharide shared by both these types of GAGs (Chapter 17). Another class of mutants (group D) is defective only in HS biosynthesis. This mutation defines a bifunctional enzyme (EXT1) that catalyzes the alternating addition of GlcNAc and glucuronic acid (GlcA) residues to growing HS chains. Some of the mutant alleles depress both enzyme activities, whereas others only affect the GlcA transfer activity. Thus, the mutants define different functional domains of the protein, which have been mapped by sequencing various mutant alleles. Mutants in another bifunctional enzyme, N-acetylglucosamine N-deacetylase/N-sulfotransferase (NDST1), have only a partial deficiency in N-sulfation of HS chains. Further analysis of the mutant showed that more than one isozyme is present in CHO cells and that the defect affects only one locus. Thus, the mutants revealed early on that the assembly of HS is much more complex than had been appreciated on the basis of known structures, enzyme reactions measured in cell extracts, or intermediates observed in pulse-labeling experiments. The many CHO mutants have recently been complemented with several gene-edited cell libraries that produce and display distinct GAGs with a broad repertoire of modifications.
Examples of mutants defective in proteoglycan assembly
MUTANTS DEFECTIVE IN GLYCOLIPID OR O-GLYCAN SYNTHESIS
Glycolipids and glycans linked by O-GalNAc are often relatively simple in cultured cells. For example, CHO cells synthesize mainly gangliosides GM3 and lactosylceramide with a small amount of glucosylceramide. O-GalNAc glycans contain up to only four sugars in glycoproteins from CHO cells. O-Fuc, O-Glc, and O-Man glycans are expressed on only a small subset of glycoproteins and are generally not detected by glycomic profiling methods (Chapter 50). All of these glycans are affected in the mutants described in Table in which CMP-Neu5Ac, UDP-Gal, UDP-GalNAc, or GDP-Fuc are reduced in the Golgi. Similarly, a defective sialyltransferase or galactosyltransferase may cause these glycans to be truncated. A mutant of B16 melanoma cells that is defective in ceramide glucosyltransferase (glucosylceramide synthase) lacks all glycolipids because this enzyme catalyzes the first step in the synthetic pathway (Chapter 11). However, cultured cell mutants defective in polypeptide O-GalNAc transferases (GALNTs) or protein O-fucosyltransferase 1 (POFUT1) have not been isolated. This may reflect the paucity of cytotoxic lectins or toxins that bind to O-glycans and glycolipids or, in some cases, due to redundancy of enzymes (Chapters 10 and 11). However, cell mutants defective in most of the GALNTs, POFUT1, POGLUT1, and the various O-mannosyl transferases, including POMT1/2 and TMTC1-3, are now made available through precise genetic engineering. The same is the case with mutants in the biosynthesis of glycosphingolipids. In addition, mice lacking specific glycolipid biosynthetic enzymes and glycosyltransferases that transfer GlcNAc or Fuc to protein have been generated and provide a source of mutant cells that may be studied in culture. Interestingly, cells lacking the O-GlcNAc transferase (OGT) that acts in the cytoplasm to transfer GlcNAc to protein have not been obtained, and mouse mutants defective in this transferase become arrested in development at the two-cell-stage embryo, showing that this O-GlcNAc addition is essential for cell viability.
USES OF MAMMALIAN GLYCOSYLATION MUTANTS
Fortunately, the vast majority of glycosylation mutations allow single-cell viability in vitro under ideal culture conditions. Glycosylation mutants of mammalian cells have thus been used to address many questions in glycobiology and for glycosylation engineering of recombinant glycoproteins (Chapter 56). Because mutant selections are broad and often not intentionally biased, they generate mutants defective in both known and novel reactions. Thus, glycosylation mutants play an important role in research to define the pathways and regulation of glycosylation in mammals. In this regard, they are more useful tools than mutant mice because cells in culture are viable in the absence of glycolipids, GPI anchors, proteoglycans, O-GalNAc, O-Fuc, O-Glc, O-Man glycans, and complex or hybrid N-glycans. Glycosylation mutants make truncated or altered glycans and thus provide an opportunity to study functional roles for cell-surface glycans in the context of a living cell. Important insights have been gained into specific sugars required for viral, bacterial, or parasite adhesion and infection, and for leukocyte cell adhesion and motility. In addition, functional roles for glycans in the intracellular sorting and secretion of glycoproteins, in growth factor binding and activation, and in receptor functions have been identified using glycosylation mutants. For example, a panel of CHO glycosylation mutants was used in a coculture assay to show that ligand-induced Notch signaling is reduced when GDP-Fuc levels are low, but is unaffected by reductions in Sia. Similarly, one of the first demonstrations for coreceptor functions for HS used mutant CHO cells defective in HS synthesis and engineered to express the FGF receptor. Most recently, genome-wide gene editing is being combined with screens for an altered phenotype to identify genes that contribute to that particular phenotype. For example, genes that facilitate infection by SARS-CoV-2 were uncovered in a genome-wide genetic screen.
Although glycosylation is in many cases dispensable for survival of isolated cells in a culture dish, it is often crucial in vivo. Gene ablation studies in mice have identified numerous cases in which an intact glycosylation pathway is essential for embryogenesis. Examples include mutants that lack complex and hybrid N-glycans and proteoglycan mutants defective in HS, whereas the corresponding mutants in CHO cells do not cause an obvious growth phenotype. To address the role of glycans in more complex functions, introduction of mutations in stem cells compatible with creation of organoid or even organotypic 3D tissue cultures is used. For example, a human 3D organotypic skin model was used to address the contributions of various glycans on distinct types of glycoconjugates (glycolipids, N-glycans, O-GalNAc, O-Fuc, O-Glc) by targeting of core extension steps. These studies demonstrated distinct tissue phenotypes for each of the mutant lines grown in 3D cultures, whereas no obvious growth phenotype was observed in conventional 2D cultures. Thus, glycosylation is often critical in the context of a multicellular organism but dispensable in isolated cells. This conclusion has been driven home by gene ablation studies in mice and further evidenced by the discovery of human genetic diseases which arise from mutations in genes involved in glycosylation (Chapter 45).
CHO cells have become the cells of choice for the biotechnology industry in the production of recombinant therapeutic glycoproteins and in glycosylation engineering (see Chapter 56). For example, CHO cells lacking FUT8, which adds fucose to the core GlcNAc of complex N-glycans, are used to produce cytotoxic therapeutic antibodies that have a greatly enhanced ability to kill their target cells. In another example, CHO cells with multiple mutations that simplify N- and O-glycans are being used by X-ray crystallographers to produce homogeneous preparations of membrane glycoproteins with highly truncated N- and O-glycans, greatly facilitating their crystallization.
Somatic cell genetics arose from the desire to manipulate the genome of cultured cells in vitro. Today, the availability of genomic sequences from multiple organisms has shifted the emphasis in genetics toward the generation of mutant organisms using the techniques of transgenesis, homologous recombination for gene replacement, conditional gene inactivation, and precise gene editing. However, the study of somatic cell mutants still plays an important role in glycobiology research because it provides a less-expensive and faster method for studying the effects of deleting or expressing particular glycosylation gene products in a cell. Gain-of-function mutants may of course be generated by transfection of cDNAs encoding glycosylation genes, and reduced expression of any gene can be achieved by the use of RNA interference (RNAi), antisense cDNA strategies, or genome-wide gene editing screens. The combination of genome-wide screening and specific agents to select for glycan changes make it possible to discover new genes by screening for phenotypic changes directly related to glycosylation changes. Additionally, cells and mutants with well-characterized glycosylation pathways are ideal hosts for investigating the activity encoded by a putative glycosylation gene identified in genome sequence databases. These mutant cells also provide a platform to test the severity of human mutations in a complementation test: The normal human gene rescues defective glycosylation when transfected into the mutant cell, but the same gene with a pathological mutation does not. Thus, somatic cell mutants provide access to novel genes involved in glycosylation, which in turn guide strategies for sophisticated gene-manipulation experiments in animals. By combining the two approaches, the biological function of a particular glycosyltransferase, sugar residue, or lectin can be defined. Coupled with powerful mass spectrometry techniques for determining glycan structures (Chapter 50) from small samples of tissue or cells, glycosylation mutants of cells and animals provide complementary material for structure/function analyses and identifying mechanistic bases of glycan functions in mammals.
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Carolyn R. Bertozzi and appreciate helpful comments and suggestions from Taroh Kinoshita and Nathan E. Lewis.
FURTHER READING
Stanley P. 1984. Glycosylation mutants of animal cells. Annu Rev Genet
18: 525–552. doi:10.1146/annurev.ge.18.120184.002521 [
PubMed: 6241454] [
CrossRef]
Esko JD. 1989. Replica plating of animal cells. Methods Cell Biol
32: 387–422. doi:10.1016/s0091-679x(08)61183-8 [
PubMed: 2691858] [
CrossRef]
Esko JD. 1991. Genetic analysis of proteoglycan structure, function and metabolism. Curr Opin Cell Biol
3: 805–816. doi:10.1016/0955-0674(91)90054-3 [
PubMed: 1931081] [
CrossRef]
Stanley P. 1992. Glycosylation engineering. Glycobiology
2: 99–107. doi:10.1101/cshperspect.a005199 [
PubMed: 1606361] [
CrossRef]
Stanley P, Raju TS, Bhaumik M. 1996. CHO cells provide access to novel N-glycans and developmentally regulated glycosyltransferases. Glycobiology
6: 695–699. doi:10.1093/glycob/6.7.695 [
PubMed: 8953280] [
CrossRef]
Esko JD, Selleck SB. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem
71: 435–471. doi:10.1146/annurev.biochem.71.110601.135458 [
PubMed: 12045103] [
CrossRef]
Maeda Y, Ashida H, Kinoshita T. 2006. CHO glycosylation mutants: GPI anchor. Methods Enzymol
416: 182–205. doi:10.1016/s0076-6879(06)16012-7 [
PubMed: 17113867] [
CrossRef]
Patnaik SK, Stanley P. 2006. Lectin-resistant CHO glycosylation mutants. Methods Enzymol
416: 159–182. doi:10.1016/s0076-6879(06)16011-5 [
PubMed: 17113866] [
CrossRef]
Zhang L, Lawrence R, Frazier BA, Esko JD. 2006. CHO glycosylation mutants: proteoglycans. Methods Enzymol
416: 205–221. doi:10.1016/s0076-6879(06)16013-9 [
PubMed: 17113868] [
CrossRef]
North SJ, Huang HH, Sundaram S, JangLee J, Etienne AT, Trollope A, Chalabi S, Dell A, Stanley P, Haslam SM. 2010. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals
N-glycans of a novel size and complexity. J Biol Chem
285:
5759–5775. doi:10.1074/jbc.m109.068353 [
PMC free article: PMC2820803] [
PubMed: 19951948] [
CrossRef]
Jae LT, Raaben M, Riemersma M, van Beusekom E, Blomen VA, Velds A, Kerkhoven RM, Carette JE, Topaloglu H, Meinecke P, et al.
2013. Deciphering the glycosylome of dystroglycanopathies using haploid screens for Lassa virus entry. Science
340: 479–483. doi:10.1126/science.1233675 [
PMC free article: PMC3919138] [
PubMed: 23519211] [
CrossRef]
Steentoft C, Bennett EP, Schjoldager KT, Vakhrushev SY, Wandall HH, Clausen H. 2014. Precision genome editing: a small revolution for glycobiology. Glycobiology
24: 663–680. doi:10.1093/glycob/cwu046 [
PubMed: 24861053] [
CrossRef]
Zhu Y, Feng F, Hu G, Wang Y, Yu Y, Zhu Y, Segoe UI, Xu W, Cai X, Sun Z, Han W, Segoe UI, et al.
2021. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat Commun
12:
961. doi:10.1038/s41467-021-21213-4 [
PMC free article: PMC7878750] [
PubMed: 33574281] [
CrossRef]