

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. doi: 10.1101/glycobiology.3e.029


Essentials of Glycobiology [Internet]. 3rd edition.
Show details
Glycans interact with many types of proteins including enzymes, antibodies, and lectins. Protein recognition of glycans represents a major way in which the information contained in glycan structures is deciphered and promotes biological activities. This chapter describes approaches to study the kinetics and thermodynamics of interactions between glycans and glycan-binding proteins (GBPs).
PROTEIN–GLYCAN RECOGNITION
A tremendous variety of GBPs are known and many are discussed in Chapter 28 and elsewhere in this book. GBPs differ in the types of glycans they recognize and in their binding affinity and kinetics. The underlying structural basis by which a GBP binds with specificity and high affinity to a very limited number of glycans (or even a single glycan) among the many thousands that are produced by a cell is discussed in Chapter 30. A wide variety of physical techniques are used to identify and quantify protein–glycan interactions. Differential affinities of glycans for different GBPs revealed by these approaches provide insight into the biological roles of glycans and their cognate GBPs. Characterization of protein–glycan recognition using such techniques, in combination with structural studies by nuclear magnetic resonance (NMR) and crystallography, may be used to identify novel antagonists or inhibitors of GBPs. Such approaches were used, for example, to develop inhibitors of neuraminidases to treat influenza virus infections (Chapter 42) and to screen for high-affinity inhibitors of selectins for the treatment of inflammatory disorders (Chapter 34).
HISTORICAL BACKGROUND
Much of the initial work on understanding protein–glycan interactions came from studies on the combining sites of plant lectins and antibodies against specific blood group antigens. These studies led to the development of quantitative assays using glycans to inhibit binding interactions detected by precipitation and agglutination, which provided early evidence for the importance of specific sugar structures in biological recognition events. Studies of protein–glycan interactions were instrumental in the development of techniques such as equilibrium dialysis and isothermal titration calorimetry, which are now widely used to analyze protein binding to a variety of types of ligands. On the other hand, methods used to study other types of protein–ligand interactions often need to be adapted to accommodate the specific properties of glycans and the proteins that interact with them.
Valency of GBP Interactions
Because many GBPs are oligomeric, with each subunit typically having a single carbohydrate-binding domain (carbohydrate-recognition domain [CRD]), many GBPs show multivalent interactions with glycan ligands. Thus, although the CRD within a GBP may have a particular affinity for a ligand, the multivalent feature enhances binding through increased avidity and allows ligand cross-linking. Although the term affinity in this case, measured at equilibrium as a dissociation constant or affinity Kd, refers to the direct interaction of a single CRD with a monovalent ligand, avidity, measured as an avidity Kd, refers to the overall strength of multivalent interactions (Figure 29.1). Some researchers use the term apparent Kd to denote the nonequilibrium nature of the measurements. Examples of oligomeric and multivalent GBPs include galectins, which are soluble GBPs that associate in dimers and higher oligomers, and soluble C-type lectins, such as serum collectins, which are oligomeric. In fact, some GBPs, such as the mannose receptor and some galectins (Chapters 34 and 36), have multiple CRDs within a single polypeptide and can bind multiple ligands. In such cases, the single protein–glycan interaction may be weak (mm–μm Kd), as for the mannose receptor, which binds α-linked mannose with affinity in the low millimolar range, but which can bind with high avidity to the surface of fungi or microbes rich in mannose-containing ligands.

FIGURE 29.1.
Monovalent and multivalent interactions of a glycan-binding protein (GBP) with monovalent or multivalent glycan ligands. A variety of interactions is possible and these affect the equilibrium constant and contribute to the affinity Kd and the avidity (more...)
Membrane receptors can also function as oligomeric complexes. For example, the C-type lectin P-selectin, which is dimeric, and membrane Siglecs (Sia-binding immunoglobulin-like lectins), both cluster on the cell surface in the presence of glycan receptors on opposing cells. Similarly, influenza virus hemagglutinin is trimeric and is present in multiple copies on a virion, whereas cholera toxin contains a pentamer of glycan-binding B subunits associated with the catalytic and toxic subunit A in an AB5 complex. The glycan of ganglioside GM1 binds strongly to each B subunit of cholera toxin with high affinity because of specific and multiple interactions within each CRD (affinity Kd of ∼40 nm). As a pentamer in the AB5 form, cholera toxin has extremely high avidity (avidity Kd of ∼40 pm) for cells expressing GM1, which clusters at the cell surface.
Most plant lectins are dimers or tetramers and are thus multivalent. Of course, the density of glycans on glycoproteins can also affect the affinity of binding, as some glycoproteins carry multiple multiantennary N-linked chains, each of which may interact with a CRD of the GBP (Figure 29.1).
THERMODYNAMICS OF BINDING
The interaction of glycans with GBPs can be described thermodynamically and kinetically. Consider the simplest case in which a lectin (L) binds with a single site to a glycan (G) with a single binding determinant in a monovalent interaction. The interaction is governed by Equation 1 (Figure 29.2). At equilibrium the affinity constant, K, is defined as an association constant (or Ka) (Equation 2) and is equal to kon/koff, and the inverse of the Ka is the Kd (Equation 3). Like any equilibrium constant, K is related to the standard free-energy change of the binding reaction at pH 7 (ΔGo) in kcal per mole (Equation 4), in which R is the gas constant (0.00198 kcal/mol-K) and T is the absolute temperature (in °K). The affinity constant K is related to the thermodynamic parameters ΔGo, ΔHo, and ΔSo (Equation 4), which represent the changes in free energy, enthalpy, and entropy of binding, respectively.
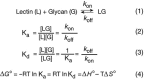
FIGURE 29.2.
Equations governing the interactions of a glycan-binding protein or lectin (L) with a glycan ligand (G). The terms are defined in the text.
The on-rate kon is expressed in units of M−1sec−1 or M−1min−1, whereas koff is expressed in units of sec−1 or min−1. Although it is important to define Ka, kon, koff, ΔGo, ΔHo, and ΔSo for each binding phenomenon under consideration, investigators often discuss data in terms of the Kd (Equation 3), because the units are in concentration (millimolar, micromolar, nanomolar, etc.).
Whereas the binding of a monovalent GBP to a monovalent ligand is easily defined by the equilibrium kinetics described above, binding between multivalent ligands or GBPs involves multiple affinities, and the binding equilibria are more complex and more accurately described by a set of equilibrium constants. Typically, for multivalent ligands and GBPs, the values reported for affinity are apparent affinity constants and usually measure avidity.
TECHNIQUES TO STUDY PROTEIN–GLYCAN INTERACTIONS
There are many different ways to study the binding of glycans to proteins, and each approach has its advantages and disadvantages in terms of thermodynamic rigor, amounts of protein and glycan needed, and speed of analysis. Below is a discussion of some of the major ways in which the binding between a glycan and protein can be studied. Much of the available information about protein–glycan interactions derives from studies of relatively small glycan ligands interacting with a protein. In examining these interactions, two broad categories of techniques have been applied: (1) kinetic and near-equilibrium methods, such as equilibrium dialysis and titration calorimetry; and (2) nonequilibrium methods, such as glycan microarray screening, hapten inhibition, enzyme-linked immunoabsorbent assay (ELISA)-based approaches, and agglutination. In all of these approaches, the concepts of affinity versus avidity must be considered, and because of the multivalency of many GBPs and their ligands, it is difficult to precisely define the kinetic parameters, although the apparent affinity and avidity remain very useful measurements.
Kinetics and Near-Equilibrium Methods
Equilibrium Dialysis for Measuring Kd Values and Interaction Valency
Equilibrium dialysis is one of the earliest and simplest methods to study the binding of a GBP to a glycan. Although the technique is not used that often currently, understanding the principles of equilibrium dialysis helps to clarify the concept of measuring equilibrium constants. A solution of a GBP, such as a lectin or an antibody, is placed in a dialysis chamber of defined volume; the chamber must be permeable to glycans or other small molecules but not to the GBP. The chamber with the GBP inside is then placed in a larger known volume of buffer that contains the glycan in the concentration range of the expected Kd. After equilibrium is achieved, defined as no further changes in concentrations of the glycan either inside or outside the chamber, the total concentration of glycan in the chamber [In] is defined. [In] is a combination of bound glycan (associated with the GBP) plus free glycan versus the concentration of glycan outside the chamber [Out], which is the free glycan only. This difference in glycan [In] and [Out] will depend on the amount and affinity of the GBP. From this information, both the Ka and the valence n can be determined from the relationship
A plot of r/c versus r for different hapten concentrations approximates a straight line with a slope of −Ka. The valence of binding (number of binding sites per mole) is defined by the r intercept at an infinite hapten concentration. If such an analysis were performed with cholera toxin, for example, one would obtain five binding sites per mole of AB5 complex, or 1 mol per mole of B subunit. Nonlinear curve fitting is used to determine equilibrium constants, because older methods using linear conversions have inherent deficiencies and may distort experimental error.
As in any technique for determining binding constants, a number of important assumptions are made and their validity must be considered. These include demonstrating that the protein and glycan(s) are stable and active over the course of the experiment, the glycan is freely diffusible, the complex is at equilibrium, and structurally unrelated small molecules—not expected to bind—show no apparent binding in the experimental setup. If such binding is observed, this may be considered nonspecific and may be subtracted from the specific binding.
There are several advantages to equilibrium dialysis: (1) the approach is relatively easy and sophisticated equipment is not needed; (2) if the affinity is high, then relatively small amounts of protein are needed (typically a few milligrams); (3) if the affinity is high, only small amounts of glycan may be required; (4) if the protein and glycan are very stable, they may be recovered and reused; (5) radioactive or fluorescent-tagged glycans may be used; and (6) reliable equilibrium measurements can be made. Some drawbacks of the approach are that (1) it provides the Ka but not the rate constants (kon or koff); (2) if the affinity of the GBP for the glycan is low, then relatively large amounts of GBP and glycan may be required; and (3) many different measurements must be made if the range of affinity is unknown and this may require many days or weeks to complete.
A variation of this technique is illustrated by the equilibrium gel-filtration method developed by Hummel and Dreyer. In the Hummel–Dreyer method, a GBP is applied to a gel-filtration column that has been preequilibrated with a glycan of interest that is easily detectable (e.g., by radioactive or fluorescent tagging). As the protein binds to the ligand, a complex is formed that emerges from the column as a “peak” above the baseline of ligand alone, followed by a “trough” (where the concentration of ligand is decreased below the baseline) that extends to the included or salt volume of the column. The amount of complex formed is easily determined by the known specific activity of the ligand. Because the amount of complex formed is directly proportional to the amount of protein (or ligand) applied, it is easy to calculate a binding curve from several different Hummel–Dreyer column profiles at different concentrations of either protein or ligand. This binding curve allows the calculation of the equilibrium constant of the interaction. The advantages and drawbacks of this technique are generally the same as for equilibrium dialysis, except that Hummel–Dreyer analyses are often quicker to perform and can be used with ligands of many different sizes. Such an approach has been invaluable in defining the equilibrium binding of selectins to their ligands.
Affinity Chromatography to Assess the Specificity of Binding
In many cases, affinity chromatography is simply used to identify interacting partners. In this technique, a GBP is immobilized to an affinity support, such as Affi-Gel, CNBr-activated Sepharose, Ultralink, or some other activated support. If a glycan binds tightly to an immobilized GBP, a buffer containing a known glycan ligand may be added to force dissociation of the complex. For example, oligomannose-type and hybrid-type N-glycans will bind avidly to an agarose column containing the plant lectin concanavalin A (ConA-agarose) and 10–100 mm α-methyl mannoside is required to elute the bound material efficiently. In contrast, many highly branched complex-type N-glycans will not bind. Biantennary complex-type N-glycans bind to ConA-agarose, but they do not bind as tightly as high-mannose-type N-glycans and their elution can be effected using 10 mm α-methyl glucoside. In this manner, one can assess the binding specificity of a GBP. In practice, this approach is rather crude, and although it gives valuable practical information about the capacity of an immobilized lectin to bind specific glycans, it does not provide quantitative affinity measurements. A variant of this method is to immobilize the glycan ligand through covalent linkage or by capturing a biotinylated glycan on a streptavidin-linked surface and then measuring GBP binding.
A more sophisticated version of this approach, termed frontal affinity chromatography, provides quantitative measurements of the equilibrium binding constants. In this technique, a solution containing a glycan of known concentration is continuously applied to a column of immobilized GBP, and the elution front of the glycan from the column is monitored. Eventually, enough ligand is added through continuous addition that its concentration in the eluant equals that in the starting material. If the glycan has no affinity for the GBP, it will elute in the void volume V0; if, however, the glycan interacts with the GBP, it will elute after the V0 and at a volume Vf (Figure 29.3).
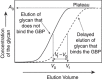
FIGURE 29.3.
Example of frontal affinity chromatography, in which different concentrations of a glycan are applied to a column of immobilized GBP. The profile depicts the elution of one glycan that binds the GBP and the elution of another glycan that does not bind (more...)
The advantages of frontal affinity chromatography are similar to those discussed for equilibrium dialysis: (1) the approach is easy and inexpensive; (2) if the affinity is high, then relatively small amounts of protein are needed (typically a few milligrams), and only a single column is required; (3) correspondingly, small amounts of glycan may be used if the Kd is in the range of 10 nm to 10 mm; (4) if the glycans are stable, they may be recovered and reused; (5) radioactive glycans may be used; and (6) reliable equilibrium measurements can be made. There are several drawbacks to this approach, including (1) only the Kd can be derived, not kon or koff; (2) the conjugation of the GBP to the matrix must be stable and the protein must retain reasonable activity for many different column runs; (3) many different column runs must be made with a single glycan; and (4) if the Kd is high (>1 mm), this approach is typically not feasible. Overall, frontal affinity chromatography is quite useful and has now been automated.
Isothermal Titration Calorimetry to Measure Kd and Binding Enthalpy
Isothermal titration calorimetry (ITC) is one of the most rigorous means of defining the equilibrium binding constant between a glycan and a GBP. The binding of a glycan to the GBP is measured as a change in enthalpy using a commercial microcalorimeter. In this technique, a solution containing a glycan of interest is added in increments into a solution containing a fixed concentration of GBP. The glycan is added at many intervals and the heat evolved from binding is measured relative to a reference cell. Over the course of the experiment, the concentration of glycan is increased in the mixing cell over a glycan-to-GBP molar ratio of 0–10. The heat absorbed or evolved during binding is determined and the data are replotted as kcal/mole of injectant versus the molar ratio (Figure 29.4). These data are then analyzed by replotting data to obtain the Kd. The heat change is directly related to the enthalpy of reaction ΔHo. From knowledge of the Kd and ΔHo, and using Equation 4, it is possible to define the binding entropy ΔSo.
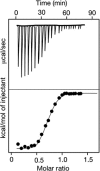
FIGURE 29.4.
Example of isothermal titration calorimetry (ITC). (Top) Increasing amounts of a glycan are injected to a fixed amount of glycan-binding protein (GBP) in a cell, and the heat produced upon binding is measured as μcal/sec. (Bottom) The total kcal/mol (more...)
The major advantage of this approach is that it can provide thermodynamic information about the binding of a glycan to a GBP and is thus superior to equilibrium dialysis and affinity chromatography. The disadvantages of this approach are that (1) relatively large amounts of protein may be required to conduct multiple experiments (>10 mg); (2) relatively large amounts of glycans may be required; and (3) because of the above-mentioned problem, it is not typical for such analyses to use a wide range of different glycans. Nevertheless, this approach is rigorous and if the titration cell dimensions could be decreased in the future, then lower amounts of materials would be required.
Surface Plasmon Resonance to Measure the Kinetics of Binding and the Kd
Surface plasmon resonance (SPR) is used to measure association and dissociation kinetics of ligands (analytes) with a receptor. In SPR, the association of the analyte and receptor, with one or the other immobilized on a sensor chip, which incorporates a critical metal sensing surface, induces a change in total surface plasmon waves resulting in a change in the refractive index of the layer in contact with a gold film (Figure 29.5). This change is recorded as the SPR signal or resonance units (RUs). Binding is measured in real time, and information about the association and dissociation kinetics can thus be obtained, which in turn can be used to obtain Ka and Kd from Equations 2 and 3. There are several instruments available from various companies based on the principle of SPR.
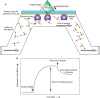
FIGURE 29.5.
Example of surface plasmon resonance (SPR). (A) In SPR, the reflected light is measured and is altered in response to binding of the analyte in the flow cell to the immobilized GBP. (B) An example of a sensorgram showing the binding of the analyte to (more...)
A variety of chemistries are available for coupling ligand or receptor to the surface of the chip, including reaction with amines, thiols, or aldehydes, and noncovalent biotin capture. In some approaches, a glycoprotein ligand for a GBP is immobilized and binding of the GBP is measured directly. It is also possible to degrade the immobilized glycoprotein ligand on the chip sequentially by passing over solutions containing exoglycosidases and reexamining at each step the binding to different GBPs, thereby obtaining structural information about the ligand. The immobilized ligand is usually quite stable and can be used repeatedly for hundreds of runs during a period of months.
The advantages of this approach are that (1) affinities in the range from millimolar to picomolar can be measured; (2) complete measurements of kon and koff are routine (see Equations 2 and 3) making calculations of Kd straightforward; (3) for immobilization of a molecule using amine coupling, only 1–5 µg is normally sufficient; (4) typically, the concentration range of the analyte is 0.1–100×Kd and the typical volumes needed are in the range of 50–150 µL; and (5) measurements are extremely rapid and complete experimental results can be obtained within a few days. The drawbacks of this approach are that (1) analytes must have sufficient mass to cause a significant change in SPR upon binding (thus, the glycan is usually immobilized instead of the protein); (2) coupling of free glycans to the chip surface may be inefficient and thus neoglycoproteins or some other type of large conjugate may be needed; and (3) there may be inhomogeneity in conditions on the SPR instrument because of mass transport effects, which could affect the dissociation rate and thus provide an inaccurate Kd measurement.
Fluorescence Polarization for Measuring Kd
Fluorescence polarization is an old technique but only relatively recently has it been applied to measure the binding constants of glycans to GBPs. This approach is based on the reduced rotational motion of a relatively small glycan when it is bound to a relatively large protein compared with the rotation of the free glycan in solution. Light absorbed by the fluorophore is emitted as fluorescence, but the angle of the emission relative to the incident light is depolarized by rotation of the molecule in solution, as measured through a filter to select molecules oriented close to the plane of incident polarized light. In practice, a fluorescently labeled glycan is incubated with increasing concentrations of a GBP and the fluorescence depolarization is measured. In the absence of the GBP, the fluorescently labeled glycan tumbles randomly and the degree of polarization is low. However, if the fluorescently labeled glycan binds to the GBP, its rate of rotation is diminished and the polarization remains high. By this approach, one can measure directly the Kd of the interaction as a function of the GBP concentration. The advantages of this technique are that (1) it is a homogeneous assay and provides direct measurements of the Kd in solution without derivatization of the GBP; (2) it is relatively simple and can provide rapid measurements of many compounds using microtiter plate–based approaches; (3) it uses relatively small amounts of glycan; (4) the concentrations of all the molecules are known; (5) it avoids complications of multivalent interactions because the glycans are monovalent and free in solution; and (6) it is amenable to inhibition by competitive agents and can be used to determine relative potency of compounds as inhibitors of GBPs. In the latter approach, a single fluorescently labeled glycan is mixed with the GBP, increasing concentrations of inhibitor glycans (which are not fluorescently labeled) are added, and the inhibition of binding is measured. Because the interactions are simple single-site competition, it is possible to use the concentration that causes 50% inhibition (IC50) to derive the approximate Kd for the inhibitor. Some of the disadvantages are that (1) the technique is limited to small glycans (≤2000 Da); (2) it requires fluorescence derivatization of the glycan (the fluorophore may alter the properties of the glycan); and (3) preparation of the glycan and chemical derivatization may be tedious and require large amounts of glycans. However, once the fluorescently labeled glycans are generated, there is usually enough for many assays.
Additional Methods to Define Protein–Glycan Interactions
More complex approaches to measure noncovalent complexes between proteins and glycan ligands include mass spectrometry (MS) and NMR. Soft ionization methods are used to detect protein–glycan complexes in electrospray ionization MS and in matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS. Although such methods have several advantages, including sensitivity and the ability to measure binding to multiple ligands in a single experiment, they may also be complicated to interpret, because the noncovalent complex may dissociate during the measurement, the detection may not be quantitative, and the ionization efficiency of the complex versus the free protein or ligand may differ. Another MS approach is hydrogen deuterium exchange (HDX) mass spectrometry, in which the bound ligand alters the deuterium uptake kinetics on the protein at the binding site of the ligand. Thus, the increased association of ligand decreases deuterium uptake, which can be measured on appropriate peptides proteolytically prepared from the protein after the exchange, and this can be plotted to obtain a binding isotherm. This method has the advantages of sensitivity and ease of use, but it also has drawbacks, including the expense of the approach and instrument time, the problem that relatively small glycans may have limited impact on deuterium exchange, and conformational changes that affect exchange rates in regions outside the CRD.
Several types of NMR spectroscopy measurements are useful for measuring protein–glycan interactions. Interactions can be measured in real time in solution and there is no need to separate the protein–glycan complex from the unassociated GBP or glycan. Both line broadening, when weakly bound ligands are in fast exchange with free ligand, and saturation transfer difference (STD) measurements can be used. However, the protein–glycan complex has to have clear and detectable chemical shifts distinct from the unbound materials (usually 1H-NMR is used to detect glycan only), and often the techniques work best for relatively low affinity interactions.
Another spectroscopic approach to measuring glycan binding to a protein relies on changes in intrinsic fluorescence on interacting with a glycan. Finally, atomic force microscopy (AFM) can also be used to define protein–glycan or glycan–glycan interactions. In this approach, the force required to separate a glycan-coated bead from an immobilized GBP can be measured in highly quantitative methods. Remarkably, single-molecule force measurements can be made by AFM. This method not only gives information about the strength of binding, but also provides insight into the molecular nature of noncovalent bond formation between the protein and its ligand.
Nonequilibrium Methods
ELISA to Measure Specificity and Relative Binding Affinity of Ligands
The conventional ELISA has been adapted for studying glycans and GBPs in a variety of formats. Of course, many glycans are antigens and antibodies to them can be analyzed in the conventional ELISA format. Some of the earliest approaches used biotinylated bacterial polysaccharides captured on streptavidin-coated microtiter plates to measure interactions of antibodies to the polysaccharides. In most types of ELISAs used in glycobiology, either an antibody or a GBP of interest is immobilized and the binding of a glycan to the protein is measured, or the reagents are reversed. In either approach, the glycans are conjugated in some way, such as to biotin or to another protein with an attached reporter group (e.g., a fluorescent moiety or an enzyme such as peroxidase).
Competition ELISA-type assays have also recently been developed to probe the binding site of a GBP. In this approach, a glycan is coupled to a carrier protein (the target), and its binding to an immobilized GBP is detected directly. Competitive glycans are added to the wells and their competition for the GBP is measured as a function of concentration to obtain an IC50. Under appropriate conditions and concentrations of the ligand, the Ki values and Kd values are similar. The major advantages of this approach are that (1) it is relatively easy; (2) it has high-throughput capability and can be used in an automated fashion by robotic handling; (3) it can provide relative Kd values if the GBP concentration is varied appropriately over a large range and binding is saturable; and (4) it has the capacity to define the relative binding activity of a panel of glycoconjugates. The major disadvantages are that (1) it does not provide direct information about affinity constants or other thermodynamic parameters; (2) it can require relatively high amounts of GBP and glycans if used as a general screening array; and (3) it usually requires chemical derivatization of glycans or GBPs.
Glycan Microarrays to Measure Specificity
Glycan microarrays are an extension of both ELISA-type formats and modern DNA and protein microarray technology. In the microarray, glycans are captured, usually covalently, through reaction with N-hydroxysuccinimide (NHS)-esters or epoxide-containing surfaces on a glass slide. Glycans are prepared to contain reactive primary amines at their reducing termini, but other chemical coupling methods are available. In addition, noncovalent immobilization can be used in which lipid-derivatized glycans or glycolipids are deposited onto nitrocellulose-coated slides. The glycans are printed, much like DNA is printed for DNA microarrays, using contact printers or piezoelectric (noncontact) printing (Figure 29.6). Usually, a few nanoliters of solutions containing glycans in concentrations of 1–100 µm are deposited by a robotic printer on the glass surface in ∼100-µm-diameter spots. Slides are incubated for several hours to allow the chemical reactions to covalently fix the samples on the slides. The slides are then blocked to prevent nonspecific binding of reagents, and these microarrays overlaid with a buffer containing the GBP and incubated for several hours to allow equilibrium binding to occur. The slides are washed to remove unbound GBP and then analyzed. Analyses involve fluorescence detection, which means that either the GBP has to be directly fluorescently labeled or a fluorescently tagged antibody to the GBP must be used.
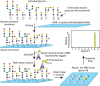
FIGURE 29.6.
Preparation of covalent glycan microarrays printed on N-hydroxysuccinimide (NHS)- or epoxide-activated glass slides. In this example, the glycans have a free amine at the reducing end and are coupled to the glass slide. After washing, the slide is “interrogated” (more...)
The chief feature of the successful microarrays are the variety of glycans they contain and the clustered and high-density presentation of glycans that promotes binding of even relatively low affinity multivalent GBPs. Thus, the density of the ligand should be taken into account when interpreting the results. The type of linker used and the state of the monosaccharide attached to the linked (ring open or closed) can also affect binding. Publicly available glycan microarray binding data repositories are increasingly used in the field. In a typical successful analysis a GBP may bind to several glycans that share structural features, and binding is visualized or imaged as intensely fluorescent spots against a dark background. The data are visually imaged on a scanner and then graphically represented. If desired, the GBP can then be tested for its binding to the identified candidates by other methods such as titration microcalorimetry or fluorescence polarization, to define the Kd as discussed above. The use of microarrays in characterizing GBPs is a central component of functional glycomics (Chapter 51).
Agglutination
Multivalent GBPs interacting with multivalent ligands, as expressed on cells, can cause the cells to agglutinate. This can be exploited by allowing one to measure the ability of a soluble glycan to block the agglutination activity of the GBP. The concentration of the soluble glycan that provides 50% inhibition of agglutination is taken as the inhibitory concentration (IC50). Such approaches have been used for many years in studies on lectin-induced agglutination of cells and were useful in elucidating the nature of the human blood group substances. If a sufficiently large panel of soluble glycans is used, then the relative efficacies of each of these can be measured to help define the specificity of the GBP. A major advantage of this technique is that it does not require tagging of the glycans. Furthermore, polystyrene or dextran beads modified with discrete glycans can be used in lieu of cells. In this case, the glycans on the agglutinating particle are better defined. Usually, the IC50 does not relate directly to the binding affinity, because inhibition is being measured. The actual binding affinity must be defined by other techniques described earlier in this chapter.
Precipitation
The interaction of a multivalent GBP or antibody with a multivalent ligand allows formation of cross-linked complexes in solution. In many cases, these complexes become insoluble and precipitate. Precipitation may be highly specific and reflects the affinity constant of the ligand for the receptor. To quantify this interaction a fixed amount of GBP or antibody is titrated against a glycoprotein or a glycan to which it binds and a precipitate will form at a precise ratio of ligand to receptor. The amount of protein or ligand in the precipitate can be measured directly by chemical means, using assays for glycans or proteins. The technique of precipitation is useful for studying potentially multivalent ligands, and it has been used recently to show that each branch of terminally galactosylated complex-type di-, tri-, and tetra-antennary N-glycans is independently recognized by galactose-binding lectins. Another precipitation approach takes advantage of the fact that a complex between a GBP and a glycan can be “salted out” or precipitated by ammonium sulfate. A variation of this approach was used in early studies on the characterization of the hepatocyte asialoglycoprotein receptor, in which the ligand 125I-labeled asialoorosomucoid was incubated with a preparation of receptor. The sample was treated with sufficient ammonium sulfate to precipitate the complex but not the unbound ligand. The precipitated complex was captured on a filter and the amount of ligand in the complex was determined by γ-counting.
Electrophoresis
In this approach, a glycoprotein (or ligand) is mixed with a GBP or antibody and the mixture is electrophoretically separated in polyacrylamide. For glycosaminoglycans, this technique is termed affinity co-electrophoresis (ACE). This method is particularly useful in defining the apparent Kd of the interaction and allows identification of subpopulations of glycosaminoglycans that differentially interact with the GBP. In another method, termed crossed affinity immunoelectrophoresis, a second step of electrophoresis is conducted in the second perpendicular dimension across an agarose gel that contains precipitating monospecific antibody to the glycoprotein or ligand. The gel is then stained with Coomassie brilliant blue and an immunoelectrophoretogram is obtained. Glycoproteins not interacting with the GBP or antibody have faster mobility than the complex. The amount of glycoprotein or ligand is determined by the area under the curves obtained in the second dimensional analysis. This method is useful for studying glycoforms of proteins and has been particularly valuable in analyzing glycoforms of α1-acid glycoprotein (an acute-phase glycoprotein) in serum and changes in its α1-3 fucosylation.
Expression of Copy DNAs for Ligands and Receptors
An indirect approach to studying protein–glycan interactions is to express a copy DNA (cDNA) encoding a glycosyltransferase in an animal or bacterial cell. The adhesion of the modified cell to a GBP or antibody is then measured and taken to reflect the binding of the GBP or antibody to the new glycans (neoglycans) on the cell surface. Alternatively a cDNA encoding a GBP is expressed in cells and their ability to bind glycan ligands is tested. The expression of selectins, Siglecs, and other GBPs on the cell surface of transfected cells has been helpful in evaluating the roles of GBPs in cell adhesion under physiological flow conditions.
FURTHER READING
- Wu X, Linhardt RJ. 1998. Capillary affinity chromatography and affinity capillary electrophoresis of heparin binding proteins. Electrophoresis 19: 2650–2653. [PubMed: 9848674]
- Leppänen A, Penttilä L, Renkonen O, McEver RP, Cummings RD. 2002. Glycosulfopeptides with O-glycans containing sialylated and polyfucosylated polylactosamine bind with low affinity to P-selectin. J Biol Chem 277: 39749–39759. [PubMed: 12145302]
- Berger G, Girault G. 2003. Macromolecule-ligand binding studied by the Hummel and Dryer method: Current state of the methodology. J Chromatog 797: 51–61. [PubMed: 14630143]
- Duverger E, Frison N, Roche AC, Monsigny M. 2003. Carbohydrate–lectin interactions assessed by surface plasmon resonance. Biochimie 85: 167–179. [PubMed: 12765786]
- Hirabayashi J. 2003. Oligosaccharide microarrays for glycomics. Trends Biotechnol 21: 141–143. [PubMed: 12679056]
- Sorme P, Kahl-Knutson B, Wellmar U, Nilsson UJ, Leffler H. 2003. Fluorescence polarization to study galectin-ligand interactions. Methods Enzymol 362: 504–512. [PubMed: 12968384]
- Bucior I, Burger MM. 2004. Carbohydrate–carbohydrate interactions in cell recognition. Curr Opin Struct Biol 14: 631–637. [PubMed: 15465325]
- Homans SW. 2005. Probing the binding entropy of ligand–protein interactions by NMR. Chembiochem 6: 1585–1591. [PubMed: 16038002]
- Nakamura-Tsuruta S, Uchiyama N, Hirabayashi J. 2006. High-throughput analysis of lectin–oligosaccharide interactions by automated frontal affinity chromatography. Methods Enzymol 415: 311–325. [PubMed: 17116482]
- Paulson JC, Blixt O, Collins BE. 2006. Sweet spots in functional glycomics. Nat Chem Biol 2: 238–248. [PubMed: 16619023]
- Rillahan CD, Paulson JC. 2011. Glycan microarrays for decoding the glycome. Annu Rev Biochem 80: 797–823. [PMC free article: PMC3116967] [PubMed: 21469953]
- de Paz JL, Seeberger PH. 2012. Recent advances and future challenges in glycan microarray technology. Methods Mol Biol 808: 1–12. [PubMed: 22057514]
- Smith DF, Cummings RD. 2013. Application of microarrays for deciphering the structure and function of the human glycome. Mol Cell Proteomics 12: 902–914. [PMC free article: PMC3617337] [PubMed: 23412570]
- Hatakeyama T. 2014. Equilibrium dialysis using chromophoric sugar derivatives. Methods Mol Biol 1200: 165–171. [PubMed: 25117234]
- Dam TK, Brewer CF. 2015. Probing lectin–mucin interactions by isothermal titration microcalorimetry. Meth Mol Biol 1207: 75–90. [PubMed: 25253134]
- Palma AS, Feizi T, Childs RA, Chai W, Liu Y. 2015. The neoglycolipid (NGL)-based oligosaccharide microarray system poised to decipher the meta-glycome. Curr Opin Chem Biol 18: 87–94. [PMC free article: PMC4105633] [PubMed: 24508828]
- Xia L, Gildersleeve JC. 2015. The glycan array platform as a tool to identify carbohydrate antigens. Methods Mol Biol 1331: 27–40. [PMC free article: PMC7714086] [PubMed: 26169733]
- Cockburn D, Wilkens C, Dilokpimol A, Nakai H, Lewinska A, Abou Hachem M, Svensson B. 2017. Using carbohydrate interaction assays to reveal novel binding sites in carbohydrate active enzymes. PLoS ONE 11: e0160112. [PMC free article: PMC4978508] [PubMed: 27504624]
- Review Principles of Glycan Recognition.[Essentials of Glycobiology. 2022]Review Principles of Glycan Recognition.Cummings RD, Schnaar RL, Esko JD, Woods RJ, Drickamer K, Taylor ME. Essentials of Glycobiology. 2022
- Review Principles of Glycan Recognition.[Essentials of Glycobiology. 2009]Review Principles of Glycan Recognition.Cummings RD, Esko JD. Essentials of Glycobiology. 2009
- Fluorescence-based solid-phase assays to study glycan-binding protein interactions with glycoconjugates.[Methods Enzymol. 2010]Fluorescence-based solid-phase assays to study glycan-binding protein interactions with glycoconjugates.Leppänen A, Cummings RD. Methods Enzymol. 2010; 478:241-64.
- Review Glycan Microarrays as Chemical Tools for Identifying Glycan Recognition by Immune Proteins.[Front Chem. 2019]Review Glycan Microarrays as Chemical Tools for Identifying Glycan Recognition by Immune Proteins.Gao C, Wei M, McKitrick TR, McQuillan AM, Heimburg-Molinaro J, Cummings RD. Front Chem. 2019; 7:833. Epub 2019 Dec 13.
- Review Glycans and glycan-binding proteins in immune regulation: A concise introduction to glycobiology for the allergist.[J Allergy Clin Immunol. 2015]Review Glycans and glycan-binding proteins in immune regulation: A concise introduction to glycobiology for the allergist.Schnaar RL. J Allergy Clin Immunol. 2015 Mar; 135(3):609-15. Epub 2015 Jan 30.
- Principles of Glycan Recognition - Essentials of GlycobiologyPrinciples of Glycan Recognition - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...