Summary
Clinical characteristics.
Von Willebrand disease (VWD), a congenital bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF), may only become apparent on hemostatic challenge, and bleeding history may become more apparent with increasing age.
Recent guidelines on VWD have recommended taking a VWF level of 30 or 40 IU/dL as a cutoff for those diagnosed with the disorder. Individuals with VWF levels greater than 30 IU/dL and lower than 50 IU/dL can be described as having a risk factor for bleeding. This change in guidelines significantly alters the proportion of individuals with each disease type.
Type 1 VWD (~30% of VWD) typically manifests as mild mucocutaneous bleeding.
Type 2 VWD accounts for approximately 60% of VWD. Type 2 subtypes include:
Type 2A, which usually manifests as mild-to-moderate mucocutaneous bleeding;
Type 2B, which typically manifests as mild-to-moderate mucocutaneous bleeding that can include thrombocytopenia that worsens in certain circumstances;
Type 2M, which typically manifests as mild-moderate mucocutaneous bleeding;
Type 2N, which can manifest as excessive bleeding with surgery and mimics mild hemophilia A.
Type 3 VWD (<10% of VWD) manifests with severe mucocutaneous and musculoskeletal bleeding.
Management.
Treatment of manifestations: Affected individuals benefit from care in a comprehensive bleeding disorders program. The two main treatments are desmopressin (1-deamino-8-D-arginine vasopressin [DDAVP]) and clotting factor concentrates (recombinant and plasma-derived) containing both VWF and FVIII (VWF/FVIII concentrate). Indirect hemostatic treatments that can reduce symptoms include fibrinolytic inhibitors; hormones for menorrhagia are also beneficial. Individuals with VWD should receive prompt treatment for severe bleeding episodes. Pregnant women with VWD are at increased risk for bleeding complications at or following childbirth.
Prevention of primary manifestations: Prophylactic infusions of VWF/FVIII concentrates in individuals with type 3 VWD to prevent musculoskeletal bleeding and subsequent joint damage.
Prevention of secondary complications: Cautious use of desmopressin (particularly in those age <2 years because of the potential difficulty in restricting fluids in this age group). Vaccination for hepatitis A and B.
Surveillance: Follow up in centers experienced in the management of bleeding disorders. Periodic evaluation by a physiotherapist of those with type 3 VWD to monitor joint mobility.
Agents/circumstances to avoid: Activities involving a high risk of trauma, particularly head injury; medications with effects on platelet function (ASA, clopidogrel, or NSAIDS). Circumcision in infant males should only be considered following consultation with a hematologist.
Evaluation of relatives at risk: If the familial pathogenic variant(s) are known, molecular genetic testing for at-risk relatives to allow early diagnosis and treatment, if needed.
Pregnancy management: As VWF levels increase throughout pregnancy, women with baseline VWF and FVIII levels greater than 30 IU/dL are likely to achieve normal levels by the time of delivery. However, those with a basal level lower than 20 IU/dL and those with baseline VWF:RCo or other VWF activity measurement/VWF:Ag ratio <0.6 are likely to require replacement therapy. Desmopressin has been successfully used to cover delivery in women with type 1 VWD and a proportion of pregnant women with type 2 VWD; delayed, secondary postpartum bleeding may be a problem.
Genetic counseling.
VWD types 2B and 2M are inherited in an autosomal dominant manner. VWD types 1 and 2A are typically inherited in an autosomal dominant manner but may also be inherited in an autosomal recessive manner. VWD types 2N and 3 are inherited in an autosomal recessive manner.
AD inheritance. Most affected individuals have an affected parent. The proportion of cases caused by
de novo pathogenic variants is unknown. Each child of an individual with AD VWD has a 50% chance of inheriting the
pathogenic variant.
AR inheritance. At conception, each sib of an individual with AR VWD has a 25% chance of being affected, a 50% chance of being an asymptomatic
carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for family members at risk for AR VWD is possible once the pathogenic variants have been identified in the family.
Prenatal and preimplantation genetic testing are possible if the pathogenic variant(s) in the family are known.
Diagnosis
Several guidelines and testing algorithms have been published [Keeney et al 2008, Nichols et al 2008, Lassila et al 2011, Laffan et al 2014]. See and .
Initial testing algorithm for von Willebrand disease From Nichols et al [2008]. Reprinted with permission of John Wiley and Sons.
Algorithm for additional testing for von Willebrand disease subtype From Laffan et al [2014]. Reprinted with permission of John Wiley and Sons.
Von Willebrand disease (VWD) is caused by deficient or defective plasma von Willebrand factor (VWF), a large multimeric glycoprotein that plays a pivotal role in primary hemostasis by mediating platelet hemostatic function and stabilizing blood coagulation factor VIII (FVIII).
There are three types of VWD [Sadler et al 2006]:
Type 1. Partial quantitative deficiency of essentially normal VWF
Type 2. Qualitative deficiency of defective VWF; divided into four subtypes depending on VWF function perturbed: 2A, 2B, 2M, 2N
Type 3. Complete quantitative deficiency of (virtually absent) VWF
Suggestive Findings
Von Willebrand disease (VWD) should be suspected in individuals with excessive mucocutaneous bleeding including the following:
Bruising without recognized trauma
Prolonged, recurrent nosebleeds
Bleeding from the gums after brushing or flossing teeth or prolonged bleeding following dental cleaning or dental extractions
Menorrhagia, particularly if occurring since menarche
Prolonged bleeding following surgery, trauma, or childbirth
Gastrointestinal bleeding
The utility of standard clinical assessment tools to score occurrence of symptoms and their severity as part of VWD diagnosis is increasingly recognized [Tosetto et al 2006, Rodeghiero et al 2010, Elbatarny et al 2014, Mittal et al 2015]. These tools can: determine if there is more bleeding than in the general population; justify the diagnosis of a bleeding disorder; quantify the extent of symptoms; indicate situations requiring clinical intervention; and be used to indicate that a bleeding disorder is unlikely [Tosetto et al 2011]. Additionally, bleeding severity assessment correlates with the long-term probability of bleeding [Tosetto 2016].
Establishing the Diagnosis
The diagnosis of VWD is established in a proband with excessive mucocutaneous bleeding and characteristic results of assays of hemostasis factors specific for VWD (see Clinical Laboratory Testing and Table 1) and/or a heterozygous, homozygous, or compound heterozygous pathogenic (or likely pathogenic) variant(s) in VWF identified by molecular genetic testing (see Table 2).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
In addition, the diagnosis requires (in most cases) a positive family history. Note: In those with a risk factor for bleeding (VWF levels >30 and <50 IU/dL), family history may not be positive because of incomplete penetrance and variable expressivity.
Clinical Laboratory Testing
Screening tests
Complete blood count (CBC) may be normal, but could also show a microcytic anemia (if the individual is iron deficient) or a low platelet count (thrombocytopenia), specifically in type 2B VWD.
Activated partial thromboplastin time (aPTT) is often normal, but may be prolonged when the factor VIII (FVIII:C) level is reduced to below 30-40 IU/dL, as can be seen in severe type 1 VWD, type 2N VWD, or type 3 VWD. The normal range for FVIII:C clotting activity is approximately 50-150 IU/dL.
Prothrombin time is normal in VWD.
Other. Although some laboratories may also include a skin bleeding time and platelet function analysis (PFA closure time) in their evaluation of an individual with suspected VWD, these tests lack
sensitivity in persons with mild bleeding disorders.
Hemostasis factor assays. The following specific hemostasis factor assays (see Table 1) should be performed even if the screening tests are normal in an individual in whom VWD is suspected [Budde et al 2006]. Note: Normal ranges are determined by the individual laboratory and thus are indicative only.
The International Society on Thrombosis and Haemostasis has recently published new guidance on assays that measure von Willebrand factor activity (VWF:Act) [Bodó et al 2015]. These tests include:
VWF:RCo. Ristocetin cofactor activity: all assays that use platelets and ristocetin. Ability of VWF to agglutinate platelets, initiated by the antibiotic ristocetin (normal range ~50-200 IU/dL)
VWF:GPIbR. All assays that are based on the ristocetin-induced binding of VWF to a recombinant WT GPIb fragment
VWF:GPIbM. All assays that are based on the spontaneous binding of VWF to a
gain-of-function variant GPIb fragment
VWF:Ab. All assays that are based on the binding of a monoclonal antibody (mAb) to a VWF A1
domain epitope
VWF:Ag. Quantity of VWF protein (antigen) in the plasma, measured antigenically using enzyme-linked immunosorbant assay (ELISA) or by latex immunoassay (LIA) [
Castaman et al 2010a] (normal range ~50-200 IU/dL). A reduced ratio (<0.6) of VWF:Act to VWF:Ag can indicate loss of high-molecular-weight (HMW) multimers.
Factor VIII:C level. Functional FVIII assay (i.e., activity of FVIII in the coagulation cascade) (normal range ~50-150 IU/dL)
If abnormalities in the tests above are identified, specialized coagulation laboratories may also perform the following assays to determine the subtype of VWD:
VWF multimer analysis. SDS-agarose electrophoresis used to determine the complement of VWF oligomers in the plasma. Normal plasma contains VWF ranging from dimers to multimers comprising more than 40 dimers and molecular weight into gigadaltons. Multimers are classified as low (1-5-dimer), intermediate (6-10-dimer), and high (≥10-dimer) molecular weight. HMW multimers are decreased or missing in type 2A VWD and often in 2B VWD; intermediate MW may also be lost in type 2A VWD. Abnormalities in satellite ("triplet") band patterns can give clues as to pathogenesis and help to classify subtypes of type 2 VWD [
Budde et al 2008].
Ristocetin-induced platelet agglutination (RIPA). Ability of VWF to agglutinate platelets at two to three concentrations of ristocetin. Agglutination at a low concentration (~0.5-0.7 mg/mL) is abnormal and may indicate type 2B or platelet-type pseudo VWD (PT-VWD) caused by pathogenic variants in
GP1BA (see
Differential Diagnosis), in which enhanced VWF-platelet binding is present.
Binding of FVIII by VWF (VWF:FVIIIB). Ability of VWF to bind FVIII. Useful, but not widely used to identify type 2N VWD.
Collagen binding assay (VWF:CB). Ability of VWF to bind to collagen (a sub-endothelial matrix component). Used to help define functional VWF discordance (i.e., to help distinguish types 1 and 2 VWD) [
Flood et al 2013]. Collagen I/III mixture is often used, but isolated deficient binding to collagen types IV and VI has recently been recognized [
Flood et al 2012]. Normal range is approximately 50-200 IU/dL. A reduced ratio of VWF:CB/VWF:Ag can indicate loss of HMW multimers.
VWF:GP1BA. Functional tests are used to determine how well VWF binds to GpIbα. Previously, this was assessed using the VWF:RCo assay. Currently, this analysis is undertaken as part of the newer activity assays.
Table 1.
Classification of VWD Based on Specific VWF Tests
View in own window
| VWD Type | VWF:Act 1 | VWF:Ag 1 | Act/Ag | FVIII:C IU/dL 1 | Multimer Pattern 2 | Other |
|---|
| 1 | Low | Low | Equivalent | ~1.5x VWF:Ag | Essentially normal | |
| 2A | Low | Low | VWF:Act < VWF:Ag | Low or normal | Abnormal ↓ HMW | ↓ VWF:GP1BA binding |
| 2B | Low | Low | VWF:Act < VWF:Ag | Low or normal | Often abnormal ↓ HMW | ↑ RIPA 3 (↓ platelet count) |
| 2M | Low | Low | VWF:Act << VWF:Ag | Low or normal | No loss of HMW | ↓ VWF:GP1BA binding
↓VWF:collagen binding 4 |
| 2N | Normal/low | Normal/low | Equivalent | <40 | Normal in most cases | ↓ VWF:FVIIIB 5 |
| 3 | Absent | Absent | NA | <10 | Absent | |
- 1.
Relative to the reference range (approximate values); VWF:Act (50-200 IU/dL), VWF:Ag (50-200 IU/dL), FVIII:C (50-150 IU/dL). VWF activity (VWF:Act) includes VWF:RCo and the newer VWF activity assays in this instance.
- 2.
- 3.
Increased agglutination at low concentrations of ristocetin
- 4.
Reduction in the ability of VWF to bind to collagen. Types I/III are bound by the A3 domain while types IV and VI are bound by the A1 domain. The latter types are not commonly analyzed.
- 5.
Ability of VWF to bind and protect FVIII is reduced. VWF and FVIII levels can look exactly like those in males with mild hemophilia A or in symptomatic hemophilia A carrier females.
Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing, use of a multigene panel, and more comprehensive genomic testing:
A multigene panel that includes
VWF and other genes of interest (see
Differential Diagnosis) may be considered. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Note: Analysis of exons 23 to 34 of
VWF is complicated by the presence of a partial
pseudogene,
VWFP1.
More comprehensive genomic testing (when available) including
exome sequencing and
genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different
gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive
genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
Table 2.
Molecular Genetic Testing Used in von Willebrand Disease (VWD)
View in own window
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
- 8.
In populations with frequent consanguineous partnerships, the rate of recessive forms of VWD may be elevated and type 3 VWD comprises a larger proportion of affected individuals.
Clinical Characteristics
Clinical Description
Von Willebrand disease (VWD) is a congenital bleeding disorder; however, symptoms may only become apparent on hemostatic challenge and bleeding history may become more apparent with increasing age. Thus, it may take some time before a bleeding history becomes apparent.
Recent guidelines on VWD have recommended taking von Willebrand factor (VWF) levels of 30 or 40 IU/dL as a cutoff for those diagnosed with the disorder. Individuals with VWF levels greater than 30 IU/dL and lower than 50 IU/dL can be described as having a risk factor for bleeding. This change in guidelines significantly alters the proportion of individuals with each disease type [Lassila et al 2011, Castaman et al 2013, Laffan et al 2014].
Bleeding history also depends on disease severity; type 3 VWD is often apparent early in life, whereas mild type 1 VWD may not be diagnosed until midlife, despite a history of bleeding episodes.
Individuals with VWD primarily manifest excessive mucocutaneous bleeding (e.g., bruising, epistaxis, menorrhagia) and do not tend to experience musculoskeletal bleeding unless the FVIII:C level is lower than 10 IU/dL, as can be seen in type 2N or type 3 VWD.
Bleeding score. In general, there is an inverse relationship between the VWF level and the severity of bleeding [Tosetto et al 2006]. Bleeding scores (BS) have been documented in several cohort studies and give an indication of the range of bleeding severity associated with different VWD types:
Table 3.
Bleeding Scores (BS) Reported in VWD by Type
View in own window
The higher the bleeding score, the greater the bleeding severity
Note: While the studies reported have all used similar bleeding assessment tools, slight variations in the tools and their application may have contributed to differences in bleeding scores.
Recently established cutoffs for an abnormal BS (≥4 for adult males, ≥6 for adult females, ≥3 for children) can be utilized to objectively assess the affected status of individuals tested using the ISTH-bleeding assessment tool (BAT) in a standard fashion [Elbatarny et al 2014].
BS in adults has also been shown to be a predictor of future bleeding [Federici et al 2014].
Type 1 VWD accounts for approximately 30% of all VWD in populations with infrequent consanguineous partnerships [Batlle et al 2016, Veyradier et al 2016]. It typically manifests as mild mucocutaneous bleeding; however, symptoms can be more severe when VWF levels are lower than 15 IU/dL. Epistaxis and bruising are common symptoms among children. Menorrhagia is the most common finding in women of reproductive age [Ragni et al 2016].
Type 2 VWD accounts for approximately 60% of all VWD. The relative frequency of the subtypes is 2A>2M>2N>2B in European populations [Batlle et al 2016, Veyradier et al 2016].
Type 2A VWD. Individuals with type 2A VWD usually present with mild to moderate mucocutaneous bleeding [
Veyradier et al 2016].
Type 2B VWD. Individuals typically present with mild-moderate mucocutaneous bleeding. Thrombocytopenia may be present. A hallmark of type 2B VWD is a worsening of thrombocytopenia during stressful situations, such as severe infection or during surgery or pregnancy, or if treated with desmopressin [
Federici et al 2009].
Type 2M VWD. Individuals typically present with mild-moderate mucocutaneous bleeding symptoms, but bleeding episodes can be severe, particularly in the presence of very low or absent VWF:RCo [
Castaman et al 2012,
Larsen et al 2013].
Type 2N VWD. Symptoms are essentially the same as those seen in mild hemophilia A and include excessive bleeding at the time of surgery or procedures as both disorders result from reduced FVIII:C [
van Meegeren et al 2015].
Type 3 VWD accounts for up to 10% of VWD (except in areas where consanguineous partnerships are common, where a higher proportion may be found). It manifests with severe bleeding including both excessive mucocutaneous bleeding and musculoskeletal bleeding [Metjian et al 2009, Ahmad et al 2013, Kasatkar et al 2014].
Associated complications
Menorrhagia is experienced by a large proportion of women with VWD.
The development of alloantibodies against VWF is an uncommon but serious complication of VWD treatment. An estimated 5%-10% of individuals with type 3 VWD may experience this complication. Affected individuals present with reduced or absent response to infused VWF concentrates or, in rare cases, with anaphylactic reaction. Individuals who have had multiple transfusions are at highest risk for this complication.
Genotype-Phenotype Correlations
The three phenotypes reflect a partial (type 1 VWD) or complete (type 3 VWD) quantitative deficiency of VWF or qualitative deficits (type 2 VWD) of VWF. See Molecular Genetics, Pathogenic variants for details regarding the genotypes associated with each subtype of VWD.
Individuals with large deletions of VWF are at highest risk for alloantibody development, although some with other null alleles have also been reported to develop this complication [James et al 2013].
Penetrance
Type 1 VWD (AD)
Other AD types (2A, 2B, and 2M). Pathogenic variants are often fully penetrant.
Nomenclature
Changes in nomenclature:
von Willebrand's disease has been replaced by von Willebrand disease.
vWF has been replaced by VWF.
vWD has been replaced by VWD.
FVIII RAg (FVIII related antigen) has been replaced by VWF:Ag.
Platelet-type pseudo von Willebrand (PT-VWD), also called pseudo-VWD, is caused by pathogenic variants in
GP1BA and, thus, is not a form of VWD (see
Differential Diagnosis).
Acquired von Willebrand syndrome (AVWS), previously known as acquired VWD, is the preferred terminology for defects in VWF concentration, structure, or function that are neither inherited nor reflective of pathogenic variants in
VWF, but arise as consequences of other medical conditions (see brief discussion of AVWS under
Differential Diagnosis).
See also Mazurier & Rodeghiero [2001] and Bodó et al [2015].
Prevalence
VWD affects 0.1% to 1% of the population; 1:10,000 seek tertiary care referral.
VWD type 3 affects 0.5:1,000,000-6:1,000,000 population, increasing with the rate of consanguinity.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with von Willebrand disease (VWD), the following evaluations are recommended:
A personal and family history of bleeding to help predict severity and tailor treatment. Use of a bleeding assessment tool can facilitate standardized assessment [
Kaur et al 2016,
Tosetto 2016,
ISTH-BAT].
A joint and muscle evaluation for those with type 3 VWD (Musculoskeletal bleeding is rare in types 1 and 2 VWD.)
Screening for hepatitis B and C as well as HIV if the diagnosis is type 3 VWD or if the individual received blood products or plasma-derived clotting factor concentrates before 1985
Baseline serum concentration of ferritin to assess iron stores, as many individuals with VWD (particularly women with menorrhagia) are iron deficient
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
See Nichols et al [2008] (full text), Castaman et al [2013] (full text), and Laffan et al [2014] (full text) for treatment guidelines.
Individuals with VWD benefit from referral to a comprehensive bleeding disorders program for education, treatment, and genetic counseling.
Medical Treatments
The two main treatments are desmopressin (1-deamino-8-D-arginine vasopressin [DDAVP]) and clotting factor concentrates (recombinant and plasma-derived) containing both VWF and FVIII (VWF/FVIII concentrate). Indirect hemostatic treatments are also beneficial. Individuals with VWD should receive prompt treatment for severe bleeding episodes.
Desmopressin
Most individuals with type 1 VWD and some with type 2 VWD respond to intravenous or subcutaneous treatment with desmopressin [
Castaman et al 2008,
Federici 2008,
Leissinger et al 2014], which promotes release of stored VWF and raises levels three- to fourfold. Intranasal preparations are also available.
Following VWD diagnosis, a desmopressin challenge is advisable to assess VWF response.
Desmopressin is the treatment of choice for acute bleeding episodes or to cover surgery.
It has been used successfully to cover delivery in women with type 1 VWD and also for a proportion of pregnant women with type 2 VWD (where a desmopressin trial has previously proved efficacious) [
Castaman et al 2010b] (see
Pregnancy Management).
Desmopressin is contraindicated in individuals with arteriovascular disease and in those older than age 70 years, for whom VWF/FVIII concentrate is required.
In persons who do not tolerate desmopressin or who have a poor VWF response, clotting factor concentrate is required.
Note: Because desmopressin can cause hyponatremia (which can lead to seizures and coma), fluid intake should be restricted for 24 hours following its administration to minimize this risk.
Intravenous infusion of VWF/FVIII clotting factor concentrates
Indirect treatments. In addition to treatments that directly increase VWF levels, individuals with VWD often benefit from indirect hemostatic treatments, including:
Fibrinolytic inhibitors (i.e., tranexamic acid for treatment or prevention of bleeding episodes);
Hormonal treatments (i.e., the combined oral contraceptive pill for the treatment of menorrhagia).
Combined treatments for menorrhagia. Current treatments and a proposed future treatment trial are described by Ragni et al [2016]. 1,321 women with VWD were assessed between 2011 and 2014, and of these 816 (61.8%) had menorrhagia.
Treatments used most commonly were combined oral contraceptives, tranexamic acid, and desmopressin as first- and second-line therapies, whereas VWF concentrate was the most common third-line therapy used by 13 women (1.6%).
Review of information on 88 women from six published studies showed that a VWF dose of 33-100 IU kg-1 reduced menorrhagia on days one to six of the menstrual cycle in 101 women.
Treatment by VWD Type
Type 1 VWD. Treatments that directly increase VWF levels (e.g., desmopressin or VWF/FVIII clotting factor concentrates) are usually only needed for the treatment or prevention of severe bleeding, as with major trauma or surgery.
Indirect treatment with fibrinolytic inhibitors or hormones is often effective.
Type 2A VWD. Treatment with clotting factor concentrates is usually only required for the treatment or prevention of severe bleeding episodes such as during surgery.
Responsiveness to desmopressin is variable and should be confirmed prior to therapeutic use.
Indirect treatments can be beneficial.
Type 2B VWD. Clotting factor concentrates are usually required to treat severe bleeding or at the time of surgery.
Treatment with desmopressin should be undertaken cautiously as it can precipitate a worsening of any thrombocytopenia. Individuals with mild or atypical type 2B VWD (caused by p.Pro1266Leu, p.Pro1266Glu, and p.Arg1308Leu variants), however, do not appear to develop thrombocytopenia when exposed to desmopressin [Federici et al 2009].
Indirect treatments (i.e., fibrinolytic inhibitors) can be useful.
Type 2M VWD. Because desmopressin response is generally poor, VWF/FVIII concentrate is the treatment of choice.
Type 2N VWD. Desmopressin can be used for minor bleeding, but because the FVIII level will drop rapidly (as FVIII is not protected by VWF), concentrate containing VWF as well as FVIII is required to cover surgical procedures.
Type 3 VWD. Treatment often requires the repeated infusion of VWF/FVIII clotting factor concentrates [Franchini & Mannucci 2016, Lavin & O'Donnell 2016, Lissitchkov et al 2017].
Desmopressin is not effective in type 3 VWD.
Indirect treatments may also be beneficial.
Pediatric Issues
Special considerations for the care of infants and children with VWD include the following:
Infant males should be circumcised only after consultation with a pediatric hemostasis specialist.
Desmopressin should be used with caution, particularly in those younger than age two years, because of the potential difficulty in restricting fluids in this age group.
VWF levels are higher in the neonatal period [
Klarmann et al 2010]; thus, phenotypic testing for milder forms of VWD should be delayed until later in childhood.
Prevention of Primary Manifestations
Individuals with type 3 VWD are often given prophylactic infusions of VWF/FVIII concentrates to prevent musculoskeletal bleeding and subsequent joint damage.
Prevention of Secondary Complications
Desmopressin should be used with caution, particularly in those younger than age two years, because of the potential difficulty in restricting fluids in this age group.
Individuals with VWD should be vaccinated for hepatitis A and B [Nichols et al 2008, Castaman et al 2013].
Prevention of chronic joint disease is a concern for individuals with type 3 VWD. However, controversy exists regarding the specific schedule and dosing of prophylactic regimens. An international trial that investigated prophylactic treatment for symptoms including joint bleeding, nosebleeds, and menorrhagia concluded that rates of bleeding within individuals during prophylaxis were significantly lower than levels prior to prophylaxis [Berntorp et al 2010, Abshire et al 2013]. Additionally, rates of bleeding were also significantly reduced in a trial of the recombinant VWF product Vonvendi®, which was licensed for use in adults in 2015 [Franchini & Mannucci 2016].
Surveillance
Individuals with milder forms of VWD can benefit from being followed by treatment centers with experience in the management of bleeding disorders.
Individuals with type 3 VWD should be followed in experienced centers and should have periodic evaluations by a physiotherapist to monitor joint mobility.
Agents/Circumstances to Avoid
Activities with a high risk of trauma, particularly head injury, should be avoided.
Medications that affect platelet function (ASA, clopidogrel, or NSAIDs) should be avoided as they can worsen bleeding symptoms.
Infant males should be circumcised only after consultation with a pediatric hemostasis specialist.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic at-risk relatives of an affected individual to allow early diagnosis and treatment as needed [Goodeve 2016].
Evaluations can include:
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
VWF levels increase throughout pregnancy with the peak occurring four hours after delivery [James et al 2015]. Nonetheless, pregnant women with VWD are at increased risk for bleeding complications and care should be provided in centers with experience in perinatal management of bleeding disorders [Pacheco et al 2010, Castaman 2013, Biguzzi et la 2015, Reynen & James 2016, Roth & Syed 2016].
Women with baseline VWF and FVIII levels higher than 30 IU/dL are likely to achieve normal levels by the time of delivery, whereas those with a basal level lower than 20 IU/dL and those with baseline VWF:Act/VWF:Ag ratio <0.6 are likely to require replacement therapy [Castaman et al 2010b, James et al 2015, Hawke et al 2016].
Although deliveries should occur based on obstetric indications, instrumentation should be minimized [Demers et al 2005].
Delayed, secondary postpartum bleeding may be a problem. VWF level rapidly returns to pre-pregnancy level following delivery [Castaman et al 2013].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Risk to Family Members – Autosomal Dominant Inheritance
Parents of a proband
Most individuals diagnosed with one of the
autosomal dominant types of VWD have an affected parent.
If the
VWF pathogenic variant causing
autosomal dominant VWD found in the
proband cannot be detected in the leukocyte DNA of either parent, two possible explanations are
germline mosaicism in a parent or
de novo pathogenic variant in the proband. Neither possibility has been sufficiently investigated to comment on relative likelihood of occurrence. Unpublished data on type 1 VWD indicated that approximately 4% of individuals had a
de novo pathogenic variant [Goodeve et al, unpublished data].
The family history of some individuals diagnosed with AD VWD may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the recognition of symptoms, or delayed onset of significant hemostatic challenges in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless appropriate evaluations (e.g., VWD hemostasis factor assays and/or
molecular genetic testing if the
proband's
pathogenic variant is known) have been performed on the parents of the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the
proband is affected, the risk to the sibs is 50%.
The sibs of a
proband with clinically unaffected parents are still at increased risk for the disorder because of the possibility of reduced
penetrance in a parent.
If the
VWF variant found in the
proband cannot be detected in the leukocyte DNA of either parent, the empiric
recurrence risk to sibs is approximately 1% because of the theoretic possibility of parental
germline mosaicism.
Offspring of a proband. Each child of an individual with autosomal dominant VWD has a 50% chance of inheriting the VWF pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, his or her family members may be at risk.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the VWF pathogenic variant(s) in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the VWF pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing for von Willebrand disease are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
von Willebrand Disease: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure.
VWF spans 178 kb of genomic DNA in 52 exons that encode an 8.8-kb mRNA and a 2,813-amino acid protein [Sadler 1998]. For a detailed summary of gene and protein information, see Table A, Gene.
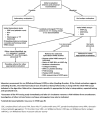
VWF has a partial pseudogene, VWFP1 (exons 23-34), which complicates analysis of these exons. Domain structure and exons encoding each VWF domain are shown in .
VWF protein structure [adapted from Zhou et al 2012] and location of VWF pathogenic variants by VWD type. Bold horizontal lines indicate the approximate position of exons where pathogenic variants are most prevalent; thinner lines indicate exons with (more...)
Pathogenic variants. Most cases of VWD result from single-nucleotide variants; however, large deletions and gene conversion events have been described (Table 4, ) [James & Lillicrap 2006]. Pathogenic variants are catalogued in the EAHAD VWF Database (see Table 5). The types of variants most commonly associated with VWD subtypes are described here.
Type 1 VWD. Partial quantitative deficiency in type 1 VWD is mostly associated with missense variants. Pathogenic variants have been identified in an estimated 60%-65% of individuals with type 1 VWD [Cumming et al 2006, Goodeve et al 2007, James et al 2007a, Yadegari et al 2012]. However, with recent guidelines recommending a VWF level of 30 or 40 IU/dL as a cutoff for VWD, the detection rate is much higher. In a recent Spanish study of individuals with VWD, 133 (27.7%) had type 1 VWD [Batlle et al 2016]; in a French study, 25% had type 1 [Veyradier et al 2016]. Variants associated with type 1 include:
Fully penetrant, dominantly inherited
missense variants that are often identified when VWF:Ag and VWF:RCo levels are lower than 30 IU/dL;
Of note, approximately 50% of pathogenic variants in type 1 VWD are located between exons 18 and 28.
Missense variants, predominantly in the D3 and A1 domains [Haberichter et al 2008, Millar et al 2008, Eikenboom et al 2013], reduce the residence time of VWF in plasma by many fold. The so-called "Vicenza" variant, p.Arg1205His, is the best characterized and the most common of these pathogenic variants. Such pathogenic variants have been referred to as type 1 clearance (1C) [Haberichter et al 2006].
Type 2 VWD. Qualitative deficiency (type 2 VWD) results from pathogenic missense variants in functionally important areas of VWF. Most pathogenic variants seen in types 2A and 2M and all pathogenic missense variants in type 2B are located in exon 28.
Type 2A (AD & AR) VWD. Pathogenic variants are predominantly located in
exon 28, affecting the A2
domain and (to a lesser extent) the A1 domain. Pathogenic variants in the D3 assembly (exons 22 and 25-27) have been reported in dominant disease [
Schneppenheim et al 2010].
In addition to
exon 28, pathogenic
missense variants have also been reported in exons 6-7, 11-16 (AR), and 52 (AD & AR).
Type 3 VWD. Severe quantitative deficiency in type 3 VWD typically results from homozygosity or compound heterozygosity for null alleles, but also a small proportion of missense variants. Pathogenic variants associated with type 3 VWD are found throughout the entire coding region of VWF (). Sequence analysis of the entire coding region plus deletion/duplication analysis identifies two pathogenic variants in approximately 90% of individuals with type 3 VWD and a single heterozygous variant in the remaining approximately 10% [Kasatkar et al 2014, EAHAD VWF Database].
Deletion/duplication analysis of VWF detects approximately 2%-5% of pathogenic variants reported in individuals with VWD [EAHAD VWF Database]. Out-of-frame deletions predominate in type 3 VWD, while in-frame deletions of up to nine exons have also been reported in types 1 and 2 VWD [Sutherland et al 2009, Casari et al 2010]. Duplications of one or two exons have also been reported [Boisseau et al 2013, Obser et al 2016, Veyradier et al 2016].
Gene conversion events with VWFP1 occur from the 3' end of intron 27 into the 5' end of exon 28 [Goodeve 2010]. There are sufficient differences between VWF and VWFP1 to recognize a conversion event through two or more sequential sequence variants from the pseudogene sequence (VWFP1). Conversions of 6 bp to 335 bp are most commonly seen and have been reported in VWD types 1, 2B, 2M, and 3. They have been reported in both multiethnic and Indian populations in higher proportions than often found in other studies [Gupta et al 2005, Kasatkar et al 2014].
Table 4.
Selected VWF Pathogenic Variants
View in own window
| VWD Type 1 | DNA Nucleotide Change | Predicted Protein Change | VWF Exon | Reference Sequences |
|---|
| 1 | c.3614G>A | p.Arg1205His | 27 |
NM_000552.3
NP_000543.2
|
| 1 | c.4751A>G | p.Tyr1584Cys | 28 |
| 2A | c.4517C>T | p.Ser1506Leu | 28 |
| 2A | c.4789C>T | p.Arg1597Trp | 28 |
| 2B | c.3797C>T | p.Pro1266Leu | 28 |
| 2B | c.3797C>A | p.Pro1266Glu | 28 |
| 2B | c.3916C>T | p.Arg1306Trp | 28 |
| 2B | c.3923G>T | p.Arg1308Leu | 28 |
| 2B | c.3946G>A | p.Val1316Met | 28 |
| 2B | c.4022G>A | p.Arg1341Gln | 28 |
| 2B | c.4135C>T | p.Arg1379Cys | 28 |
| 2M | c.3835G>A | p.Val1279Ile | 28 |
| 2M | c.4273A>T | p.Ile1425Phe | 28 |
| 2N | c.2372C>T | p.Thr791Met | 18 |
| 2N | c.2446C>T | p.Arg816Trp | 19 |
| 2N | c.2561G>A | p.Arg854Gln | 20 |
| 3 | c.2435delC | p.Pro812ArgfsTer31 | 18 |
| 3 | c.4975C>T | p.Arg1659Ter | 28 |
| 3 | c.7603C>T | p.Arg2535Ter | 45 |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Examples of the most frequent variants identified in each VWD type are shown. See EAHAD VWF Database for further information on allelic variants and frequencies.
Normal gene product. The 2,813-amino acid VWF protein comprises:
VWF has two key functions:
Binding collagen in the sub-endothelium at sites of vascular damage, which initiates repair through platelet recruitment and clot formation plus binding; and
Protecting FVIII from premature proteolytic degradation and transporting it to sites where fibrin generation is required.
VWF has two sites of synthesis: endothelial cells and megakaryocytes, the precursors of platelets. During synthesis, tail-to-tail disulfide-linked dimers are formed through the CK domains, followed by head-to-head VWF oligomers of up to 40 dimers in length.
During multimer production, the propeptide (VWFpp) is cleaved by furin and is secreted into the plasma along with VWF. The ratio of VWFpp to mature VWF (von Willebrand factor antigen or VWF:Ag) can be used to estimate relative half-life of mature VWF [Haberichter et al 2008] and provide information about the mechanism of pathogenicity [Eikenboom et al 2013].
To render HMW VWF less thrombogenic, it is cleaved by ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 motif) between amino acids 1605 and 1606 following secretion. The pattern of multimer proteolysis products by agarose-SDS gel analysis can be suggestive of VWD subtype [Schneppenheim & Budde 2011].
Abnormal gene product. Abnormalities in VWF depend on the type of pathogenic variant. The protein and nucleotide abnormalities both play a role in determining VWD type:
EAHAD VWF Variant Database
summary. The von Willebrand factor LOVD database currently lists more than 700 unique variants representing more than 1,400 affected individuals. Table 5 summarizes the numbers of affected individuals with propeptide and mature VWF pathogenic variants and the ratio between the two.
Table 5.
EAHAD VWF Mutation Database Summary (January 2017)
View in own window
| VWD Type | Protein Location | All |
|---|
| Propeptide | Mature protein |
|---|
| 1 | 24 | 144 | 168 |
| 2A | 17 | 104 | 121 |
| 2B | 0 | 40 | 40 |
| 2M | 0 | 50 | 50 |
| 2N | 4 | 46 | 50 |
| 3 | 121 | 148 | 269 |
| Total | 166 | 532 | 698 |
| % by VWF location | 24% | 76% | 100% |
Summary of separate patient entries per VWD type. Only a single entry for each patient is included; multiple entries are omitted.




![Figure 3. . VWF protein structure [adapted from Zhou et al 2012] and location of VWF pathogenic variants by VWD type.](/books/NBK7014/bin/von-willebrand-Image003.gif)