Summary
Clinical characteristics.
Hutchinson-Gilford progeria syndrome (HGPS) is characterized by clinical features that typically develop in childhood and resemble some features of accelerated aging. Children with HGPS usually appear normal at birth. Profound failure to thrive occurs during the first year. Characteristic facial features include head that is disproportionately large for the face, narrow nasal ridge, narrow nasal tip, thin vermilion of the upper and lower lips, small mouth, and retro- and micrognathia. Common features include loss of subcutaneous fat, delayed eruption and loss of primary teeth, abnormal skin with small outpouchings over the abdomen and upper thighs, alopecia, nail dystrophy, coxa valga, and progressive joint contractures. Later findings include low-frequency conductive hearing loss, dental crowding, and partial lack of secondary tooth eruption. Motor and mental development is normal. Death occurs as a result of complications of severe atherosclerosis, either cardiac disease (myocardial infarction or heart failure) or cerebrovascular disease (stroke), generally between ages six and 20 years (average 14.5 years) without lonafarnib treatment or cardiac surgery intervention. Average life span is extended to approximately 17-19.5 years with lonafarnib therapy.
Diagnosis/testing.
The diagnosis of classic or nonclassic genotype HGPS is established in a proband with characteristic clinical features, along with identification of a heterozygous pathogenic variant in LMNA that results in production of the abnormal lamin A protein, progerin. Individuals with classic genotype HGPS are heterozygous for pathogenic variant c.1824C>T (~90% of individuals with HGPS). Individuals with nonclassic genotype HGPS have the characteristic clinical features of HGPS and are heterozygous for another LMNA pathogenic variant in exon 11 or intron 11 that results in production of progerin (~10% of individuals with HGPS).
Management.
Targeted therapy: Lonafarnib is an approved medication for HGPS; benefits include improved low-tone hearing, decreased headaches, decreased carotid-femoral pulse wave velocity, decreased arterial wall stiffness, and increased life span.
Supportive care: A regular diet with frequent small meals is recommended. Primary tooth extractions after the secondary tooth has erupted and/or fully descended may be required to avoid dental crowding. Use of sunscreen on all exposed areas of skin, including the head, is recommended for outdoor activities. Hip dislocation is best managed with physical therapy and body bracing; reconstructive hip surgery is possible, but comorbidities of surgery in this high-risk population should be considered. Shoe pads are recommended, as lack of body fat leads to foot discomfort. Routine physical and occupational therapy, active stretching and strengthening exercises, and hydrotherapy are recommended. Maintain optimal hydration, while encouraging physical activity, to minimize stroke risk. Modified transcatheter aortic valve replacement or modified apico-aortic valve replacement are high-risk interventions to treat critical aortic stenosis, yielding improved cardiac status and quality of life and increased life span. Anticoagulation as needed for cardiovascular and neurovascular complications. Medication dosages are based on body weight or body surface area, not age. Nitroglycerin can be beneficial for angina; anticongestive therapy is routine for the treatment of congestive heart failure. General anesthesia and intubation should be performed with extreme caution, ideally with fiberoptic intubation, if possible. Exposure keratopathy can be treated with ocular lubrication. Hearing aids can be used when clinically necessary. Age-appropriate schooling with adaptations for physical needs is usually recommended.
Prevention of secondary complications: Low-dose aspirin (2-3 mg/kg body weight) is recommended for prevention of cardiovascular and stroke complications. Because the stiffened peripheral vasculature may be less tolerant to dehydration, maintaining optimal hydration orally is recommended.
Surveillance: Annual or semiannual electrocardiogram (EKG), annual echocardiogram, carotid duplex ultrasound examination, neurologic examination, MRI/MRA of the head and neck, lipid profile, dental examination, hip x-ray to evaluate for avascular necrosis and progressing coxa valga, dual x-ray absorptiometry and/or peripheral cutaneous computed tomography to measure bone density, physical therapy assessment for joint contractures, ophthalmology examination, audiometry, and assessment of activities of daily living.
Agents/circumstances to avoid: Dehydration; large crowds with taller/larger peers because of the risk of injury, trampolines and bouncy houses due to risk of hip dislocation. Physical activity should be self-limited.
Genetic counseling.
Almost all individuals with HGPS have the disorder as the result of a de novo autosomal dominant pathogenic variant. Recurrence risk to the sibs of a proband is small (as HGPS is typically caused by a de novo pathogenic variant) but greater than that of the general population because of the possibility of parental germline mosaicism. Once the LMNA pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk is possible.
GeneReview Scope
Table
Hutchinson-Gilford progeria syndrome (HGPS), classic Atypical Hutchinson-Gilford progeria syndrome
Diagnosis
Suggestive Findings
Hutchinson-Gilford progeria syndrome (HGPS) should be suspected in individuals with severe growth failure, areas of sclerodermatous skin, partial alopecia that progresses to total alopecia by age two years, generalized lipodystrophy, retrognathia, x-ray findings including distal clavicular and terminal phalangeal resorption as well as coxa valga, and delayed/incomplete primary tooth eruption, all in the setting of normal intellectual development.
Growth deficiency
- Short stature (<3rd percentile)
- Poor weight gain (<3rd percentile), weight distinctly low for height
- Diminished subcutaneous body fat globally
Facial features (See Figure 1.)

Figure 1.
Female age 11 years and male age six years with HGPS displaying classic features Photo courtesy of the Progeria Research Foundation
- Head disproportionately large for face
- Long narrow nose
- Thin vermilion of the upper and lower lips
- Retrognathia and micrognathia
Ectodermal
- Dental. Delayed eruption and delayed loss of primary teeth, partial secondary tooth eruption, dental crowding
- Skin. Taut, variably pigmented, sclerodermatous, skin outpouchings over lower abdomen and/or proximal thighs
- Hair. Total alopecia, sometimes with very sparse downy immature hair remaining; loss of eyebrows
- Dystrophic nails
Musculoskeletal
- Coxa valga with wide-based, shuffling gait, sometimes accompanied by avascular necrosis of the femoral head
- Osteolysis of the distal phalanges
- Short clavicles with distal resorption
- Pear-shaped thorax
Other
- Thin, high-pitched voice
- Low-frequency conductive hearing loss
- Nocturnal lagophthalmos (the inability to fully close the eyes while sleeping)
Establishing the Diagnosis
Five major categories help to define LMNA-related disorders. Categories 1 and 2 define HGPS; categories 3-5 are not considered HGPS:
- 1.
Progerin-producing classic genotype HGPS
- 2.
Progerin-producing nonclassic genotype HGPS
- 3.
Non-progerin-producing progeroid laminopathies (see Differential Diagnosis) due to:
- Heterozygous LMNA pathogenic variant that does not result in progerin production
- Pathogenic variants in other genes (e.g., ZMPSTE24)
- 4.
Non-progeroid laminopathies (See Differential Diagnosis.)
- 5.
Non-laminopathy progeroid syndromes (See Differential Diagnosis.)
The diagnosis of classic genotype HGPS is established in a proband with the above Suggestive Findings and a heterozygous c.1824C>T pathogenic variant in LMNA identified on molecular genetic testing (see Table 1).
The diagnosis of nonclassic genotype HGPS is established in a proband with Suggestive Findings similar to classic genotype HGPS and an autosomal dominant progerin-producing pathogenic variant in either the exon 11 splice junction or intron 11 of LMNA identified on molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing).
Single-gene testing
- Targeted analysis for LMNA pathogenic variants c.1824C>T (identified in 90% of individuals with HGPS), can be performed first in individuals with Suggestive Findings of HGPS.
- Sequence analysis of LMNA can be performed if no pathogenic variant is found on targeted analysis. Sequence analysis of intron 11 should be included if this was not already completed with targeted analysis.Note: LMNA deletions and/or duplications have not been reported in individuals with HGPS.
A multigene panel that includes LMNA, ZMPSTE24, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing. When the phenotype is indistinguishable from many other inherited disorders characterized by progeroid phenotype, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hutchinson-Gilford Progeria Syndrome
Clinical Characteristics
Individuals with classic and nonclassic genotype HGPS have similar clinical features and spectrum of severity.
Clinical Description
Classic and nonclassic genotype Hutchinson-Gilford progeria syndrome (HGPS) are characterized by clinical features that develop in childhood and resemble some features of accelerated aging. Children with progeria usually appear normal at birth and in early infancy.
Growth deficiency. Most infants display normal weight at birth. Profound failure to thrive usually occurs during the first year. Poor weight gain and loss of subcutaneous fat results in weight less than the third percentile for age, and weight that is distinctly low for height. Stature also decreases to below the third percentile for age.
Characteristic facial features (see Figure 1) include a head that appears disproportionately large for face, narrow nasal ridge with a narrow nasal tip, thin vermilion of the upper and lower lips, small mouth, retrognathia, and micrognathia. Ogival (steeple-shaped) palatal vault occurs in 60%-70% of affected individuals. A short, thick lingual frenulum that limits tongue mobility is seen in about 50% of affected individuals. Narrow airway and rigid laryngeal structures cause a high-pitched voice.
Dental. Delayed eruption and delayed loss of primary teeth are common. Dental crowding occurs as a result of a small mouth, lack of primary tooth loss, and secondary tooth eruption behind the primary teeth. Secondary tooth eruption is often partial.
Skin. Skin findings may be evident at birth and are present in all individuals by age two years. "Sclerodermatous" skin changes variably include areas that are described as taut, thickened, fibrotic, indurated, or rippled. In addition, dimpling or irregular small outpouchings can occur over the lower abdomen and proximal thighs. Skin also displays abnormal pigmentation consisting of light or dark macules and patches along with some papules and skin mottling.
Hair. Partial alopecia progresses to total alopecia. Sparse downy hairs may be present on the occiput. Loss of eyebrows is common, and loss of eyelashes occurs in some individuals.
Nails. Fingernails and toenails become dystrophic.
Musculoskeletal. Individuals with HGPS are particularly susceptible to hip dislocation because of the progressive coxa valga malformation, which can be accompanied by avascular necrosis of the hip (osteonecrosis). Avascular necrosis can cause hip pain and is evident on x-ray. The coxa valga causes a wide-based shuffling gait. Additional bone changes include osteolysis of the distal phalanges, short clavicles with distal resorption, a pear-shaped thorax, and mildly low bone density for age. Fractures are not more commonly reported in individuals with HGPS. Extraskeletal calcifications are present in 40% of cases, with unknown clinical significance. Progressive stiffness of the joints due to tightened joint ligaments and osteoarthritis occurs with variable severity.
Endocrine. Affected individuals do not become sexually mature. Females reach Tanner Stage 1 (78%) or 2 (22%) during pubertal years, and approximately 60% of females experience menarche [Greer et al 2018]. No cases of fertility have been described. Serum leptin concentrations are below the limit of detection. Insulin resistance occurs in about 50% of individuals, without the overt development of diabetes mellitus.
Cardiovascular/cerebrovascular. Individuals with HGPS develop severe atherosclerosis, usually without obvious abnormalities in lipid profiles [Gordon et al 2005]. In general, serum cholesterol, LDL, and triglyceride concentrations are not elevated and HDL concentrations may decrease with age. Diastolic dysfunction and cardiac strain are early cardiac abnormalities, usually detected beyond age five years by tissue Doppler echocardiography [Prakash et al 2018, Olsen et al 2023]. Sequential manifestations of cardiovascular decline include impaired relaxation of the heart muscle, followed by ventricular hypertrophy. This may occur in the setting of heart valve thickening or stenosis, or with hypertension that is often labile. Mitral and aortic valve abnormalities, including calcification, stenosis, and regurgitation, usually develop in the second decade of life. Aortic gradient increases exponentially with time at later stages of disease, portending critical aortic stenosis [Gordon et al, unpublished data].
Systolic dysfunction is usually present in the setting of advanced disease, with or without identified coronary vascular insufficiency. Clinical symptoms of angina, dyspnea on exertion, or overt heart failure appear as late findings in the course of disease.
Transient ischemic attacks, silent strokes, or symptomatic strokes have occurred as early as age four years [Silvera et al 2013]. Strokes can occur at any brain site and, therefore can lead to a variety of physical limitations and/or cognitive decline. Partial and complete carotid artery blockages can occur from plaque formation. Despite underlying vascular disease, most children do not have clinically identified strokes.
Raynaud phenomenon in fingers occurs in a minority of affected individuals.
Without lonafarnib treatment, death typically occurs as a result of complications of cardiac or cerebrovascular disease. More than 80% of deaths are due to heart failure and/or myocardial infarction, most often between ages six and 20 years, with an average life span of approximately 14.5 years [Gordon et al 2014, Gordon et al 2018a]. Average life span is extended to approximately 17-19.5 years with lonafarnib therapy, with similar cause of death.
Ophthalmologic. Nocturnal lagophthalmos (the inability to fully close the eyes during sleep) is common. As a result, corneal dryness and clouding can occur. In a minority of individuals, corneal ulceration occurs due to exposure keratitis [Mantagos et al 2017].
Hearing. Conductive hearing loss is highly prevalent at all ages, with low-frequency hearing loss more prevalent than high-frequency [Guardiani et al 2011, Gordon et al 2012].
Other
- Motor and mental development are normal.
- Tumor rate is not increased over that of the general population. One individual died of a chondrosarcoma of the chest wall at age 13 years [King et al 1978].
- Other changes associated with normal aging such as nearsightedness or farsightedness, arcus senilis, senile personality changes, or Alzheimer disease have not been documented.
- Children with HGPS appear to have a normal immune system; they respond as well as the general population when subjected to various infections. Wound healing is normal.
- Liver, kidney, gastrointestinal, neurologic, and cognitive functions are normal.
Genotype-Phenotype Correlations
Table 2.
Classic Genotype HGPS and Nonclassic Genotype HGPS: Causative LMNA Variants and Comparative Clinical Phenotypes
Penetrance
Penetrance is complete.
Nomenclature
HGPS is also referred to as the Hutchinson-Gilford syndrome or progeria.
Prevalence
The prevalence of children with HGPS per total population is one in 20 million [Gordon et al 2014].
The estimated birth incidence for HGPS is one in four million births with no observed differences based on ethnic background [Hennekam 2006].
Genetically Related (Allelic) Disorders
Some 12 distinguishably different genetic conditions with nucleotide variants in LMNA have been identified. See OMIM 150330. In addition, pathogenic variants in ZMPSTE24, which encodes zinc metalloproteinase, an enzyme involved in the post-translational processing of LMNA, can cause excess prelamin A proteins and a related phenotype (OMIM 606480).
Non-progerin-producing progeroid laminopathy can be used to describe phenotypes that overlap with classic and nonclassic genotype HGPS but are obviously different. Various pathogenic variants in LMNA identified through the Progeria Research Foundation Diagnostic Testing Program, International Progeria Registry, and/or Medical and Research Database Program result in a variety of lamin A abnormalities that cause various phenotypes (see Differential Diagnosis).
Non-progeroid laminopathies caused by pathogenic LMNA variants that result in abnormal lamin A protein:
- Autosomal dominant Emery-Dreifuss muscular dystrophy (AD-EDMD)
- Autosomal recessive Emery-Dreifuss muscular dystrophy (AR-EDMD)
- Autosomal dominant familial dilated cardiomyopathy and conduction system defects (See Dilated Cardiomyopathy.)
- Autosomal dominant Dunnigan-type familial partial lipodystrophy (FPLD) (OMIM 151660)
- Autosomal dominant limb-girdle muscular dystrophy 1B (LGMD1B)
- Autosomal recessive axonal neuropathy Charcot-Marie-Tooth disease 2B1 (CMT2B1; see Charcot-Marie-Tooth Hereditary Neuropathy Overview.)
- Autosomal recessive mandibuloacral dysplasia (MAD) [Cao & Hegele 2003]
- Single case reports of individuals with LMNA variants and unique clinical phenotypes [Caux et al 2003, Kirschner et al 2005]
Differential Diagnosis
Non-laminopathy progeroid syndromes. Other syndromes that include some features of premature aging:
- Neonatal progeroid syndrome (Wiedemann-Rautenstrauch syndrome) (OMIM 264090)
- Acrogeria (OMIM 201200)
- Hallermann-Streiff syndrome (OMIM 234100)
- Gerodermia osteodysplastica (OMIM 231070)
- Berardinelli-Seip congenital lipodystrophy (congenital generalized lipodystrophy)
- Petty-Laxova-Weidemann progeroid syndrome (OMIM 612289)
- Ehlers-Danlos syndrome, progeroid form (OMIM 130070)
- Mandibuloacral dysplasia (See Genetically Related Disorders; OMIM 248370.)
- Nestor-Guillermo syndrome (OMIM 614008)
- Penttinen Syndrome (OMIM 601812)
- POLR3A-related Wiedemann-Rautenstrauch syndrome (See POLR3-Related Leukodystrophy; Wambach et al [2018].)
- PYCR1-related Wiedemann-Rautenstrauch-like syndrome [Lessel et al 2018]
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Hutchinson-Gilford progeria syndrome (HGPS), the following evaluations are recommended if they have not already been completed:
- Weight and height plotted on standard growth charts to evaluate growth over time
- Electrocardiogram (EKG) and echocardiogram
- Carotid artery duplex scans to evaluate size of the lumen and intimal thickness in order to establish baseline vascular status
- MRI/MRA of the brain and neck
- Skeletal x-rays to evaluate for characteristic findings: acroosteolysis, clavicular resorption, coxa valga, and extraskeletal soft tissue calcifications [Cleveland et al 2012]
- Orthopedic evaluation for progressive coxa valga and/or avascular necrosis
- Dual-energy x-ray absorptiometry (DXA) to assess bone mineral density. Note: This must be normalized for height-age [Gordon et al 2011].
- Occupational and physical therapy assessments, including six-minute walk test, goniometry to assess joint mobility, and assessment of activities of daily living
- Nutritional assessment, although dietary intake is generally not compromised in these patients
- Audiologic, ophthalmologic, and dental examinations
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
A complete, system-based management guide is available from the Progeria Research Foundation.
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Lonafarnib is a farnesyltransferase inhibitor. For HGPS, its target action is inhibition of post-translational farnesylation of progerin, the active disease-causing protein in HGPS. Lonafarnib is an oral medication administered twice daily at an initial dose of 115 mg/m2 followed by 150 mg/m2 after four months of therapy. Toxicities (with suggested treatments) are mainly diarrhea (loperamide), gastrointestinal upset, nausea (ondansetron), and loss of appetite (cyproheptadine), which usually wane after a few months of therapy. Clinical trial results for lonafarnib have revealed improvement in vascular distensibility as measured via pulse wave velocity and vascular echodensity and increased bone rigidity and neurosensory hearing [Gordon et al 2012], decreased headaches [Ullrich et al 2013], and increased life span by between 2.5-5 years on average (~20%-30%) [Gordon et al 2014, Gordon et al 2023].
Supportive Care
Growth deficiency. Frequent small meals tend to maximize caloric intake.
Dental. Extraction of primary teeth may be required to avoid crowding and development of two rows of teeth. Since secondary teeth may erupt slowly or not at all, pulling primary teeth to make room for secondary teeth should be performed after secondary teeth have fully or almost fully erupted or almost fully descended. Once the primary tooth has been extracted, the secondary tooth often moves into the appropriate position with time.
Skin. Use of sunscreen on all exposed areas of skin, including the head, is recommended for outdoor activities.
Orthopedic. Conservative management of hip dislocation with physical therapy and body bracing when possible; surgical correction if essential, with special attention to intubation and anesthesia guidelines. Reconstructive hip surgery can be performed for cases of repeated dislocations causing pain and significant decrease in quality of life. Surgery should be treated as substantial risk for stroke and/or cardiac events. Podiatric evaluation is indicated to determine if shoe inserts are needed as lack of body fat leads to foot discomfort. Fracture rate is equivalent to the general pediatric population. When children do fracture, treatment and healing are routine.
Routine physical and occupational therapy is recommended to help maintain range of motion in large and small joints; see Physical Therapy and Occupational Therapy in Progeria (pdf). Active stretching and strengthening, along with hydrotherapy, are recommended.
In cases of dysfunctional vaginal bleeding or spotting, short-term (i.e., 3-6 months) provision of low-dose combined oral contraceptive pills can be helpful to stabilize the endometrium.
Cardiovascular. A regular healthy diet is indicated unless the lipid profile becomes abnormal, at which point appropriate treatment includes exercise, diet modification, and medication as warranted. Avoiding anemia, dehydration, and high fever can be important, particularly in individuals with more advanced CV disease. Maintaining optimal hydration orally is recommended.
Prior to decline in cardiovascular or neurologic status (resulting from strokes, angina, or heart attacks), children should be encouraged to be physically active as tolerated, taking into account possible limitations related to restricted range of motion of joints and hip problems including osteoarthritis and hip dislocation.
Critical aortic stenosis has been treated with either modified transcatheter aortic valve replacement (TAVR) or modified apico-aortic valve placement (AAC), yielding improved cardiac status and quality of life and increased life span. However, these are high-risk interventions. When peak aortic gradient reaches 20 mm Hg, more frequent echocardiography and intervention planning should be initiated because aortic gradient increases exponentially with time at later stages of disease. Late accelerated progression of aortic stenosis permits only a small window for optimizing risk-to-benefit ratio before failure ensues. Roughly, peak aortic gradients between 80-90 mm Hg warrant intervention. To date, two individuals have undergone modified TAVR; one was successfully treated and the other experienced intra- and perisurgical cardiac death [Musumeci et al 2020; Gordon et al, unpublished data]. Two individuals have undergone modified AAC surgery successfully [Hoganson et al 2023; Gordon et al, unpublished data].
Anticoagulants other than the routinely recommended aspirin (see Prevention of Secondary Complications) may be warranted if vascular blockage, transient ischemic attacks, stroke, angina, or myocardial infarction occur.
Although lipid profiles are usually normal, dietary therapy ± statin therapy can be implemented if abnormalities occur.
Medications. Dosages should be based on body weight or body surface area and not on age. Anesthetics should be used with particular caution.
- Nitroglycerin is frequently of benefit if angina develops.
- Routine anticongestive therapy is appropriate if congestive heart failure is present.
General anesthesia and intubation should be performed with extreme caution, ideally with fiberoptic intubation. Individuals with HGPS have retrognathia, stiffened laryngeal structures, and a narrow and unusually shaped airway; additionally, they may exhibit an extreme sensitivity to alterations in blood pressure due to vascular stiffness.
Ophthalmologic. Corneal dryness, clouding, or ulceration should be fully evaluated by an ophthalmologist. Exposure keratitis can be treated during daytime with ocular lubrication and during sleep with moisturizing ointment or by closing eyelids with skin tape.
Hearing loss. Low-frequency conductive hearing loss often does not interfere with activities of daily living. Sitting at the front of the classroom can be helpful. Hearing aids can be used, when clinically necessary.
Education. Because intellect and maturity are normal, age-appropriate schooling is usually indicated.
Infections are generally handled as for unaffected children.
Prevention of Secondary Complications
Aspirin. Based on the evidence from adult studies that low doses of aspirin help delay heart attacks and strokes, it is probably appropriate to give children with HGPS low-dose aspirin treatment, at doses of 2-3 mg/kg body weight per day. Note: If chicken pox or influenza is prevalent in the community, it may be advisable to discontinue the aspirin during that time because of the increased risk of Reye syndrome.
Adequate oral hydration is recommended, as the vasculature becomes generally less pliable and the risks of stroke and cardiac complications increase over time due to decreased vascular compensation. This is especially important during hot weather or airplane travel.
Vitamin supplementation. Standard amounts of over-the-counter daily multivitamin tablets are appropriate. Calcium supplementation is not recommended, due to the potential for aggravating extraskeletal calcification formation and hypothetically aggravating vascular plaque status. Maintenance of normal calcium levels from dietary sources is encouraged.
Fluoride supplements are recommended in areas where needed.
Immunizations. The routine doses and administration schedule for all immunizations are recommended. Immunizations are generally handled as for unaffected children.
Surveillance
A complete, system-based management guide is available from the Progeria Research Foundation.
Annually or semiannually. Consistent measurement of blood pressure with the appropriately sized cuff, EKG, echocardiogram, and carotid duplex scans to monitor for cardiovascular disease. Note: Children may experience severe carotid artery atherosclerotic blockage prior to any significant EKG changes.
Annually
- Neurologic assessment for signs and symptoms of headaches and stroke
- MRI/MRA of head and neck to assess for presence of vascular changes and silent strokes and to allow for the assessment of changes (increases) in risk over time
- Lipid profile
- Dental examination, x-ray, and cleaning
- Orthopedic evaluation for avascular necrosis (osteonecrosis) of the hip and progressing coxa valga that result in horse-riding stance and potential hip dislocation
- Occupational and physical therapy assessments including six-minute walk test, goniometry to assess joint mobility, and assessment of activities of daily living
- Ophthalmologic examination with special attention to possible exposure keratopathy
- Audiology evaluation with special attention to possible low-frequency conductive hearing loss
Agents/Circumstances to Avoid
Children should avoid being in the midst of large crowds with much taller and larger peers because of the increased risk of injury. Physical activity should be self-limited. Avoid uneven surfaces that could aggravate hip dysplasia, such as trampolines and bouncy houses. Avoid being carried by underage peers. Avoid dehydration due to increased risk of stroke.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search HGPS or progeria within ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical trials for HGPS.
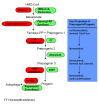
One therapy currently under human clinical trial investigation for HGPS is lonafarnib (see Targeted Therapy) plus everolimus combination therapy (see Figure 2). This study has been completed, and results are pending. Everolimus is a rapalog (rapamycin-like drug) mTOR inhibitor that additionally increases cellular autophagy. It is an oral medication administered once daily. Everolimus is approved as a medication to treat non-HGPS conditions. Rapamycin improved cellular phenotypes in HGPS fibroblasts via increased autophagy [Cao et al 2011, Cenni et al 2011] and extends life span in a lamin A-deficient mouse model. Clinical trial results are not yet known.
Other
- A clinical treatment trial administering lonafarnib in combination with pravastatin and zoledronate demonstrated evidence of increased bone mineral density but no other improvements over that of lonafarnib monotherapy [Gordon et al 2016].
- A clinical treatment trial administering pravastatin and zoledronate as combination therapy has been conducted.
The following proposed treatments have not been tested in humans (see Table 3). For a comprehensive review of evidence for these potential treatments, see Strandgren et al [2017].
Table 3.
Potential Treatments Tested Only In Vitro and/or in Murine Studies
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hutchinson-Gilford progeria syndrome (HGPS) is typically caused by a de novo autosomal dominant pathogenic variant.
Risk to Family Members
Parents of a proband
- Almost all individuals with HGPS have the disorder as the result of a de novo pathogenic variant.
- Approximately 3% of currently living individuals with the classic HGPS genotype identified through the Progeria Research Foundation Diagnostics Program have HGPS as the result of apparent germline (or somatic and germline) mosaicism in a parent [Wuyts et al 2005].
- Parents of probands are not affected.
Sibs of a proband
- Because HGPS is typically caused by a de novo pathogenic variant, the recurrence risk to the sibs of a proband is small.
- Currently there are three non-twin sib cases of HGPS among a total of 113 classic cases identified (2%). The number of unaffected sibs among the 113 total cases is unknown. Thus, the recurrence risk for subsequent pregnancies after one individual has been genetically diagnosed with HGPS is significantly higher than the one in four million incidence for the general population, though still low.
Offspring of a proband. Individuals with classic and nonclassic HGPS are not known to reproduce.
Related Genetic Counseling Issues
Origin of de novo pathogenic variant. Though the number of families evaluated is small, one study identified 4/4 cases as paternal in origin [Eriksson et al 2003]. The pathogenic variant was maternal in origin for the case of mosaicism reported by Wuyts et al [2005]. A study of paternal age effect found that on average the father's age was significantly increased by about five years [Brown et al 1985]. There is no increase in consanguinity.
Prenatal Testing and Preimplantation Genetic Testing
Once the LMNA pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk (because of the rare possibility of germline mosaicism in a parent) and preimplantation genetic testing are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Library of Medicine Genetics Home Reference
- Progeria Research Foundation (PRF)Phone: 978-535-2594Email: info@progeriaresearch.org
- The Progeria Handbook: A Guide for Families & Health Care Providers of Children with Progeria
- Progeria Research Foundation Patient RegistryPO Box 3453 (for mail)Peabody MA 01961-3453Phone: 978-535-2594Fax: 978-535-5849Email: info@progeriaresearch.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hutchinson-Gilford Progeria Syndrome: Genes and Databases
Table B.
OMIM Entries for Hutchinson-Gilford Progeria Syndrome (View All in OMIM)
Molecular Basis of Disease
For the LMNA pathogenic variant c.1824C>T, the C-to-T transition does not change the translated glycine amino acid but activates a cryptic splice site, resulting in a transcript with a deletion of 150 base pairs in the 3' portion of exon 11. Translation followed by post-translational processing of this altered mRNA produces a shortened abnormal prelamin A protein with a 50-amino acid deletion near its C-terminal end, henceforth called "progerin." The 50-amino acid deletion removes the recognition site that leads to proteolytic cleavage of the terminal 18 amino acids of prelamin A, along with the phosphorylation site(s) involved in the dissociation and reassociation of the nuclear membrane at each cell division.
A key component of disease in HGPS is the presumably persistent farnesylation of progerin, which renders it permanently intercalated into the inner nuclear membrane, where it can accumulate and exert progressively more damage to cells as they age. That the failure to remove the farnesyl group is at least in part responsible for the phenotypes observed in HGPS is strongly supported by studies on both cell and mouse models that have either been engineered to produce a nonfarnesylated progerin product or treated with a drug that inhibits farnesylation, rendering a nonfarnesylated progerin product.
Other LMNA variants that do not result in the production of progerin protein result in abnormal lamin A proteins with variable abnormalities in their structure and function (including interactions with the nuclear membrane lamin-associated proteins), all of which produce cellular and organism diseases with varying phenotypes that overlap with HGPS in some aspects.
Gene structure. The coding region of LMNA spans approximately 24 kb and contains 12 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. See Table 4.
Table 4.
LMNA Pathogenic Variants Discussed in This GeneReview
Normal gene product. The nuclear lamina is a protein-containing layer attached to the inner nuclear membrane. In mammals, it is composed of a family of polypeptides, with the major components being the lamins A, B1, B2, and C, with molecular weights ranging from 60,000 to 78,000. Lamins A and C are formed by alternative splicing of the LMNA/C gene transcript. Splicing within exon 10 gives rise to lamin C, whereas transcription of all 12 exons gives rise to lamin A. Lamins B1 and B2 are encoded by separate genes, and there are no known progeroid pathogenic variants within lamins B1 and B2.
Lamin A is normally synthesized as a precursor molecule (prelamin A) and undergoes four major post-translational processing steps. First, because prelamin A contains a CAAX (cysteine / aliphatic / aliphatic / any amino acid) box at its carboxyl terminus, it is modified by farnesylation. Following farnesylation, cleavage of the last three amino acids, methylation of the C-terminus, and internal proteolytic cleavage occur. Removal of the last 15 coding amino acids along with the CAAX box and farnesyl group generates mature lamin A with 646 amino acids.
Abnormal gene product. The HGPS-causing variants in codon 608 of LMNA lead to activation of a cryptic splice site within exon 11, resulting in production of a prelamin A that lacks 50 amino acids near the C terminus [Eriksson et al 2003]. The c.1824C>T pathogenic variant and consequent abnormal splicing produces a prelamin A that still retains the CAAX box and is therefore farnesylated, but is missing the site for endoproteolytic cleavage of the final 16 amino acids along with the farnesyl moiety that normally occurs during the final step in post-translational processing. The resulting protein, named progerin, is shortened and farnesylated. Since the lipophilic farnesyl moiety is utilized to anchor prelamin (and hence progerin) into the inner nuclear membrane, the lack of farnesyl cleavage likely results in long-term progerin association with the inner nuclear membrane.
Chapter Notes
Author Notes
Dr Gordon is an investigator for progeria clinical treatment trials being conducted at Boston Children's Hospital. For more information please contact Dr Gordon at Leslie_Gordon@brown.edu.
Revision History
- 19 October 2023 (lg/sw) Revision: lonafarnib included as a Targeted Therapy
- 17 January 2019 (sw) Comprehensive update posted live
- 8 January 2015 (me) Comprehensive update posted live
- 6 January 2011 (me) Comprehensive update posted live
- 10 August 2006 (me) Comprehensive update posted live
- 12 December 2003 (me) Review posted live
- 31 July 2003 (wtb) Original submission
References
Published Guidelines / Consensus Statements
- Progeria Research Foundation. The Progeria Handbook: A Guide for Families & Health Care Providers of Children with Progeria. Includes genetic testing guidelines. Available online (pdf). Accessed 7-19-22.
Literature Cited
- Bar DZ, Arlt MF, Brazier JF, Norris WE, Campbell SE, Chines P, Larrieu D, Jackson SP, Collins FS, Glover TW, Gordon LB. A novel somatic mutation achieves partial rescue in a child with Hutchinson-Gilford progeria syndrome. J Med Genet 2017; 54: 212-6. [PMC free article: PMC5384422] [PubMed: 27920058]
- Barthélémy F, Navarro C, Fayek R, Da Silva N, Roll P, Sigaudy S, Oshima J, Bonne G, Papadopoulou-Legbelou K, Evangeliou AE, Spilioti M, Lemerrer M, Wevers RA, Morava E, Robaglia-Schlupp A, Lévy N, Bartoli M, De Sandre-Giovannoli A. Truncated prelamin A expression in HGPS-like patients: a transcriptional study. Eur J Hum Genet. 2015;23:1051-61 [PMC free article: PMC4795109] [PubMed: 25649378]
- Brown WT, Zebrower M, Kieras FJ. Progeria, a model disease for the study of accelerated aging. Basic Life Sci. 1985;35:375-96. [PubMed: 4062819]
- Cao H, Hegele RA. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet. 2003;48:271-4. [PubMed: 12768443]
- Cao K, Blair CD, Faddah DA, Kieckhaefer JE, Olive M, Erdos MR, Nabel EG, Collins FS. Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest. 2011;121:2833-44. [PMC free article: PMC3223819] [PubMed: 21670498]
- Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. J Clin Endocrinol Metab. 2003:88:1006-13 [PubMed: 12629077]
- Cenni V, Capanni C, Columbaro M, Ortolani M, D'Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, Squarzoni S, Prencipe S, Lattanzi G. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin-linked progeria. Eur J Histochem. 2011;55:e36. [PMC free article: PMC3284238] [PubMed: 22297442]
- Cleveland RH, Gordon LB, Kleinman ME, Miller DT, Gordon CM, Snyder BD, Nazarian A, Giobbie-Hurder A, Neuberg D, Kieran MW. A prospective study of radiographic manifestations in Hutchinson-Gilford progeria syndrome. Pediatr Radiol. 2012;42:1089-98. [PMC free article: PMC4220680] [PubMed: 22752073]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Lévy N. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003; 300:2055 [PubMed: 12702809]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003; 423:293-8 [PMC free article: PMC10540076] [PubMed: 12714972]
- Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW. Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26:1670-9. [PMC free article: PMC5650062] [PubMed: 21445982]
- Gordon LB, Campbell SE, Massaro JM, D'Agostino RB Sr, Kleinman ME, Kieran MW, Moses MA. Survey of plasma proteins in children with progeria pre-therapy and on-therapy with lonafarnib. Pediatr Res. 2018a. 83: 982-92. [PubMed: 29342131]
- Gordon LB, Harten IA, Patti ME, Lichtenstein AH. Reduced adiponectin and HDL cholesterol without elevated C-reactive protein: clues to the biology of premature atherosclerosis in Hutchinson-Gilford progeria syndrome. J Pediatr. 2005; 146:336-41 [PubMed: 15756215]
- Gordon LB, Kleinman ME, Massaro J, D'Agostino RB Sr, Shappell H, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland RH, Nazarian A, Snyder BD, Ullrich NJ, Silvera VM, Liang MG, Quinn N, Miller DT, Huh SY, Dowton AA, Littlefield K, Greer MM, Kieran MW. Clinical trial of the protein farnesylation inhibitors lonafarnib, pravastatin, and zoledronic acid in children with Hutchinson-Gilford progeria syndrome. Circulation. 2016;134:114-25 [PMC free article: PMC4943677] [PubMed: 27400896]
- Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666-71. [PMC free article: PMC3478615] [PubMed: 23012407]
- Gordon LB, Massaro J, D'Agostino RB Sr, Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130:27-34. [PMC free article: PMC4082404] [PubMed: 24795390]
- Gordon LB, Norris W, Hamren S, Goodson R, LeClair J, Massaro J, Lyass A, D'Agostino RB Sr, Tuminelli K, Kieran MW, Kleinman ME. Plasma progerin in patients with Hutchinson-Gilford progeria syndrome: immunoassay development and clinical evaluation. Circulation. 2023;147:1734-44. [PMC free article: PMC10237348] [PubMed: 36919608]
- Gordon LB, Shappell H, Massaro J, D'Agostino RB Sr, Brazier J, Campbell SE, Kleinman ME, Kieran MW. Association of lonafarnib treatment vs no treatment with mortality rate in patients with Hutchinson-Gilford progeria syndrome. JAMA. 2018b;319:1687-95. [PMC free article: PMC5933395] [PubMed: 29710166]
- Greer MM, Kleinman ME, Gordon LB, Massaro J, D'Agostino RB Sr, Baltrusaitis K, Kieran MW, Gordon CM. Gordon pubertal progression in adolescent females with progeria. J Pediatr Adolesc Gynecol. 2018;31:238-41 [PMC free article: PMC6671321] [PubMed: 29258958]
- Guardiani E, Zalewski C, Brewer C, Merideth M, Introne W, Smith AC, Gordon L, Gahl W, Kim HJ. Otologic and audiologic manifestations of Hutchinson-Gilford progeria syndrome. Laryngoscope. 2011;121:2250-5 [PMC free article: PMC3688450] [PubMed: 21898437]
- Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603-24. [PubMed: 16838330]
- Hisama FM, Lessel D, Leistritz D, Friedrich K, McBride KL, Pastore MT, Gottesman GS, Saha B, Martin GM, Kubisch C, Oshima J. Coronary artery disease in a Werner syndrome-like form of progeria characterized by low levels of progerin, a splice variant of lamin A. Am J Med Genet A. 2011;155A:3002-6. [PMC free article: PMC4679285] [PubMed: 22065502]
- Hoganson DM, Eickhoff ER, Prakash A, Del Nido PJ. Management of severe calcific aortic stenosis in children with progeria syndrome. J Thorac Cardiovasc Surg. 2023. Epub ahead of print. [PubMed: 37236599]
- Iqbal M, Iftikhar A. Progeria - first case report from Pakistan. Rawal Med J. 2008;33:266-7.
- King CR, Lemmer J, Campbell JR, Atkins AR. Osteosarcoma in a patient with Hutchinson-Gilford progeria. J Med Genet. 1978;15:481-4. [PMC free article: PMC1013767] [PubMed: 284134]
- Kirschner J, Brune T, Wehnert M, Denecke J, Wasner C, Feuer A, Marquardt T, Ketelsen UP, Wieacker P, Bonnemann CG, Korinthenberg R. p.S143F mutation in lamin A/C: a new phenotype combining myopathy and progeria. Ann Neurol. 2005; 57:148-51 [PubMed: 15622532]
- Lessel D, Ozel AB, Campbell SE, Saadi A, Arlt MF, McSweeney KM, et al. Analyses of LMNA-negative juvenile progeroid cases confirms biallelic POLR3A mutations in Wiedemann-Rautenstrauch-like syndrome and expands the phenotypic spectrum of PYCR1 mutations. Hum Genet. 2018;137:921-39. [PMC free article: PMC6652186] [PubMed: 30450527]
- Li Y, Zhou G, Bruno IG, Cooke JP. Telomerase mRNA reverses senescence in progeria cells. J Am Coll Cardiol. 2017; 70: 804-5. [PubMed: 28774385]
- Mantagos IS, Kleinman ME, Kieran MW, Gordon LB. Ophthalmologic features of progeria. Am J Ophthalmol. 2017;182:126-32 [PubMed: 28756152]
- Moulson CL, Fong LG, Gardner JM, Farber EA, Go G, Passariello A, Grange DK, Young SG, Miner JH. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum Mutat. 2007; 28:882-9. [PubMed: 17469202]
- Musumeci F, Cammardella AG, Lio A, Musto C, Polizzi V, Buffa V, Montalto A, Comisso M, Ranocchi F, Cassese M. Hutchinson-Gilford progeria syndrome and severe aortic stenosis: a new hope for treatment. Ann Thorac Surg. 2020;110:e365-e367. [PubMed: 32360386]
- Olsen FJ, Gordon LB, Smoot L, Kleinman ME, Gerhard-Herman M, Hegde SM, Mukundan S, Mahoney T, Massaro J, Ha S, Prakash A. Progression of cardiac abnormalities in Hutchinson-Gilford progeria syndrome: a prospective longitudinal study. Circulation. 2023;147:1782-4. [PubMed: 37276254]
- Prakash A, Gordon LB, Kleinman ME, Gurary EB, Massaro J, D'Agostino R Sr, Kieran MW, Gerhard-Herman M, Smoot L. Cardiac abnormalities in patients with Hutchinson-Gilford progeria syndrome. JAMA Cardiol. 2018; 3: 326-34. [PMC free article: PMC5875332] [PubMed: 29466530]
- Reunert J, Wentzell R, Walter M, Jakubiczka S, Zenker M, Brune T, Rust S, Marquardt T. Neonatal progeria: increased ratio of progerin to lamin A leads to progeria of the newborn. Eur J Hum Genet. 2012;20:933-7. [PMC free article: PMC3421121] [PubMed: 22419169]
- Silvera VM, Gordon LB, Orbach DB, Campbell SE, Machan JT, Ullrich NJ. Imaging characteristics of cerebrovascular arteriopathy and stroke in Hutchinson-Gilford progeria syndrome. AJNR Am J Neuroradiol. 2013;34:1091-7. [PMC free article: PMC7964639] [PubMed: 23179651]
- Strandgren C, Revêchon G, Sola-Carvajal A, Eriksson M. Emerging candidate treatment strategies for Hutchinson-Gilford progeria syndrome. Biochem Soc Trans. 2017;45: 1279-93. [PubMed: 29127216]
- Ullrich NJ, Kieran MW, Miller DT, Gordon LB, Cho YJ, Silvera VM, Giobbie-Hurder A, Neuberg D, Kleinman ME. Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment. Neurology. 2013;81:427-30. [PMC free article: PMC3776537] [PubMed: 23897869]
- Wambach JA, Wegner DJ, Patni N, Kircher M, Willing MC, Baldridge D, Xing C, Agarwal AK, Vergano SAS, Patel C, Grange DK, Kenney A, Najaf T, Nickerson DA, Bamshad MJ, Cole FS, Garg A. Bi-allelic POLR3A loss-of-function variants cause autosomal-recessive Wiedemann–Rautenstrauch syndrome. Am J Hum Genet. 2018;103:968-75 [PMC free article: PMC6288318] [PubMed: 30414627]
- Wuyts W, Biervliet M, Reyniers E, D'Apice MR, Novelli G, Storm K. Somatic and gonadal mosaicism in Hutchinson-Gilford progeria. Am J Med Genet A. 2005;135:66-8 [PubMed: 15793835]
Publication Details
Author Information and Affiliations
Alpert Medical School of Brown University
Providence, Rhode Island
Staten Island, New York
Bethesda, Maryland
Publication History
Initial Posting: December 12, 2003; Last Revision: October 19, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford Progeria Syndrome. 2003 Dec 12 [Updated 2023 Oct 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

