

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Reichert WM, editor. Indwelling Neural Implants: Strategies for Contending with the In Vivo Environment. Boca Raton (FL): CRC Press/Taylor & Francis; 2008.

Indwelling Neural Implants: Strategies for Contending with the In Vivo Environment.
Show details6.1. INTRODUCTION
Successfully interfacing the CNS with external electronics holds great potential in improving the quality of life for patients with sensory and motor dysfunctions. The impact is already evident in the profound clinical applications of cochlear implants and deep brain stimulations [1,2]. More recently, the use of an invasive electronic brain implant, also known as a neuromotor prosthesis, to help a patient paralyzed by a tetraplegic spinal cord injury has been reported [3]. In the study, a 96-electrode array was implanted into the patient’s motor cortex to establish a brain–computer interface, where the patient could move a cursor to issue different instructions by thoughts of such motions. Clearly, it adds credibility to the enormous benefits that the interface technology could bring as a potential new therapy to restore independence for those severely disabled patients. Additionally, this interface technology may have significant implications for fundamental studies in neuroscience to understand normal physiology, pathology, or treatment of disorders such as epilepsy.
A key component in such interface technology is the electrode, which is usually placed inside the CNS tissue to record neural impulses. These “spikes” will subsequently be translated into commands for external electronic devices. Currently, several types of electrodes are being used for research purposes, including microwires [4], glass electrodes [5], polymeric electrodes [6], and silicon micromachined implants [7,8]. Among these designs, microwires and silicon electrodes are the most popular. Microwires are well-established, metal-based, tip-recording electrodes. Their features include the ability to record large numbers of single units and ease of fabrication. However, they lack precise positioning inside the tissue. In comparison, silicon micromachined electrodes allow for greater control over electrode placement in vivo, as well as precise and versatile electrode design to accommodate signal recordings at different depths. This, however, comes at the price of a sophisticated multistep fabrication process. There are two prominent silicon electrodes in the field, widely known as the Utah electrode array (UEA) and the Michigan probe. The UEA is a three-dimensional array of needle-like structures with recording sites located at the tips, while the Michigan probe is a thin-film planar array with recording sites spaced out along the electrode shank. Our discussion will mainly refer to the silicon electrode, as such technology has the potential to enable precise dense sampling that will permit detailed mapping of the nervous system and improve the development of prosthetic devices.
Despite the aforementioned therapeutic potentials of electrode interface technology, many obstacles still need to be overcome to make this technology a clinical reality. First, it is not clear for how long these electrodes can record neural activity in vivo. Ideally, the electrode should remain functionally stable in the CNS indefinitely to achieve significant improvements in the lives of disabled patients. In reality, however, the neuronal recording tends to fade over time, for unknown reasons. For example, only 4 out of 11 electrodes remain functioning 6 months after the electrode implantation in cat sensory cortex [9]. Some speculate that the observed loss in recording ability is closely correlated to the adverse tissue response, which is characterized by the formation of glial scar and electrode encapsulation. The biocompatibility of implanted electrodes has been an area of intense research to improve recording reliability [10]. Another challenge for chronic CNS electrode performance is the risk of infection introduced by wires that penetrate the skull and skin to connect the electrode to external hardware. To tackle this problem, effort has been devoted to develop on-chip circuitry and wireless fully implantable micro-systems [11].
Since interactions between the CNS tissue and the electrode are critical in determining the functional performance of the electrode, strategies are being investigated to improve electrode biocompatibility. Several factors must be considered when it comes to addressing the biocompatibility issue. First, the choice of electrode material has an important bearing on its performance and longevity. Ideally, the material should be nontoxic, stable, instigate minimal to no host response, and have the desired electrical properties. Silicon and metals are commonly used for making CNS electrodes. Although they meet the electrical criteria, their mechanical properties are likely to pose a threat to the long-term recording. As we will discuss in a later section, the large mechanical mismatch between the electrode and tissue will create a high strain field at the interface. The high strain field might in turn contribute to the sustained glial response. Therefore, studies are underway to explore alternate softer electrode substrates, such as polyimide [12]. Nevertheless, this will bring new challenges for electrode insertion, as flexible electrodes might lack the stiffness needed to penetrate the pia, resulting in buckling and potentially generating more traumatic injury to the tissue.
The second factor to consider is the size and dimensions of the electrode. Intuitively, the electrode should be made as small as possible, so that tissue damage is minimized. In vivo study has demonstrated that initial glial response decreases as the cross-sectional area of the electrode decreases [13]. The size problem is not insurmountable, as advanced technologies developed in the semiconductor industry could be applied in fabricating smaller electrodes. However, the size of the electrode is closely dependent on the number of recording sites. It is still a matter of debate in the field as to how many neurons should be sampled to produce effective motor outputs. Also, the electrical impedance of the electrode is inversely proportional to the surface area of the recording site, establishing a design constraint in reducing electrode size before losing electrical viability.
The third factor is the surface properties of the electrode. Like many other implants, cell interaction with the electrode is a surface phenomenon; therefore, surface properties play a key role in determining the cellular response. It is well known that cells respond to both topographical and physiochemical cues. For example, a large body of literature has shown that modifying a material surface with extracellular matrix proteins such as fibronectin, collagen, and laminin, or peptide fragment from these proteins such as RGD (Arg-Gly-Asp), IKVAV (Ile-Lys-Val-Ala-Val), or YIGSR (Tyr-Ile-Gly-Ser-Arg), can significantly improve cell adhesion, morphology, and differentiation compared to untreated surfaces. Another example is that cells behave differently on a rough surface compared to a smooth one. Surface features, such as grooves, ridges, pillars, and holes might present physical stimuli to cells in contact with the surface. The critical issue in surface modification for electrode application is to minimize interference with the intrinsic electrical properties while achieving a better cellular interaction. Two critical questions result from a review of this literature: (a) Can these findings translate to the ability to modulate tissue reaction of implantable electrodes? and (b) Can these modifications be conducted without any adverse electrical costs in electrode performance?
6.2. CELLULAR AND MOLECULAR ASPECTS OF BRAIN RESPONSE TO ELECTRODES
Inflammation is generally considered part of the reaction of vascularized living tissue to local injury. It isolates or walls off the injurious substance or process. Although the CNS is considered immune privileged, it follows a similar inflammatory response scenario against biomaterial implants, as shown in Figure 6.1. The response is generally initiated by the implant insertion and sustained by a foreign body reaction against the indwelling implant. It involves several types of cells working in concert by means of molecular mediators. Here we will give a brief over-view of the main cellular and molecular players involved in the brain inflammatory response against the electrode at various stages of the process.
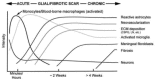
FIGURE 6.1
Hypothesized temporal sequence of inflammation and wound healing response to implanted biomaterials in the CNS. The time and intensity variables are determined by the extent of the injury, as well as the dimension, shape, surface, and bulk properties (more...)
6.2.1. Initial Insertion-Induced Response
6.2.1.1. Vascular damage
The intricate vasculature network in the brain implies that there is a high probability of vascular insult accompanying electrode implantation. En route to the brain parenchyma, the electrode first encounters the meningeal layers (Figure 6.2). The meninges consist of the tough protective outer layer, the dura mater (usually removed prior to electrode insertion), the arachnoid, and the pia mater, the thin innermost layer. Arteries and veins traveling across the pial surface demand extra attention to carefully position the electrode so that large visible surface vessels can be avoided. In addition, the cortex carries the highest vascular density in the brain, up to 160 capillaries per mm2, with shorter and smaller-caliber arteries forming a compact network with the longitudinal vessels [14]. Using brain slices with fluorescently labeled vasculature, Bjornsson and coworkers [15] evaluated vascular damage during electrode insertion. Four general types of vascular damage are commonly observed: (a) fluid displacement; (b) vessel rupture; (c) vessel severing; and (d) dragging of blood vessels by the device [15]. Accompanying such vascular damage is the extravasation of serum proteins and local infiltration of cells such as neutrophils and blood-borne macrophages leading to brain edema formation [16,17].

FIGURE 6.2
Cartoon showing the brain tissue response to an implanted electrode at the cellular level. Microglia activation is demonstrated by the morphological change from highly ramified to amoeboid macrophage-like. Astrocytes become reactive, as shown by the enlargement (more...)
Studies have shown that the extravasated proteins (i.e., thrombin, plasmin, and other proteases) contribute to the inflammatory response. Thrombin is a serine protease known to be an essential component of the coagulation cascade by converting fibrinogen to fibrin. Thrombin has also been suggested as a molecular trigger for astrogliosis, and a microglial activator [18–29]. Thrombin-induced cellular effects have been shown to be mediated by proteinase-activated receptors (PARs) [30,31]. In astrocytes, thrombin has been found to induce morphological changes, proliferation, secretion of endothelin-1, and release of inflammatory mediators [32–34] by activating the thrombin receptors, mainly PAR-1 [19,35,36]. PAR-1-deficient mice (PAR1-1-) showed a reduced astrocytic response to cortical stab, indicating that selective activation of PAR-1 in vivo induces astrogliosis [28]. The molecular mechanism was proposed to be a sustained activation of the extracellular receptor kinase/mitogen-activated protein kinase (ERK/MAPK) signaling pathway [28,35]. Apart from astrocytes, thrombin also takes direct actions on microglial cells. It was demonstrated that thrombin treatment of microglia in vitro induces clear indications of activation including nitric oxide (NO) production, cytokine release, such as tumor necrosis factor-α (TNF-α), and increased cell proliferation [23–26], all of which are clear indications of activation. Moreover, in vivo studies have also shown a thrombin-induced, microglia-mediated inflammatory factor in the CNS [37]. Thrombin receptor PAR-1 contributes primarily to promoting microglial proliferation, while PAR-4 is fully responsible for activating microglia and inducing cytokine production. The signaling pathway of PAR-4 is featured by MAPK activation and subsequent nuclear factor κB (NF-κB) transcription factor activation [22–26]. In summary, plasma enzymes such as thrombin may play an important role in the CNS response to injury. It follows that a better understanding of the molecular mechanisms behind the plasma enzyme–induced CNS inflammation could provide valuable insights for potentially effective therapeutic strategies.
Besides protein extravasation into the CNS parenchyma, early compromise of the microvasculature by the penetrating electrode also leads to the influx of a variety of hematogenous cell types, including blood-borne macrophages, neutrophil granulocytes, and T-lymphocytes. Depending on the extent of vasculature damage, the cell influx could be massive and result in severe inflammatory response. The early work of Fitch and Silver [38] has demonstrated a close correlation between the tissue distribution of the upregulation of chondroitin sulfate proteoglycans (CSPGs) and the presence of activated macrophages as well as a compromised blood–brain barrier (BBB). CSPGs are a family of inhibitory molecules present in the wound healing matrix that may stall axon regeneration in the CNS after trauma. It was also observed that animals with relatively sparse inflammatory cell infiltration but clear evidence of BBB compromise showed no detectable increases in CSPG immunoreactivity. This indicates that qualitatively higher concentrations of activated macrophages are necessary to induce increases in CSPGs, even in the presence of serum components. Clearly, minimizing the access of hematogenous cell types to the CNS will aid in modulating the inflammatory response caused by chronic electrodes.
The electrode will encounter the meningeal surface before descending to the target location. Hence, there is a likelihood that some meningeal cells will be carried into the brain [14], as shown in Figure 6.3, or migrate along the electrode shank in an effort to reform the glia limitans by interacting with local astrocytes [40]. The glia limitans is a structure consisting of layers of astrocytes, layers of meningeal cells, and a layer of basal lamina in between. It is usually located parallel to the surface of the brain and spinal cord. A normal glia limitans functions as a barrier to axonal outgrowth so that axons do not grow out of the brain or spinal cord. The formation of glia limitans between the electrode and the CNS is undesirable, as its inhibitory nature could suppress neurite outgrowth toward the electrode. Moreover, by repelling neurons from the recording sites, the long-term functionality of the electrode (recall that the recording mode is a particular focus in this chapter) would be compromised.
![FIGURE 6.3. A vimentin+/GFAP signal was observed at the recording region of an extracted probe 4 weeks after implantation, shown here with 4,6-diamidine-2-phenylindole (DAPI) (blue) and vimentin (red) overlayed with the bright field image [39].](/books/NBK3930/bin/ch6f3.gif)
FIGURE 6.3
A vimentin+/GFAP signal was observed at the recording region of an extracted probe 4 weeks after implantation, shown here with 4,6-diamidine-2-phenylindole (DAPI) (blue) and vimentin (red) overlayed with the bright field image [39]. Scale bar = 50 μm. (more...)
The contribution of the initial vascular damage to the inflammatory response to implanted electrodes cannot be overemphasized. The extent of damage could be determined by the following factors: the size and geometry of the electrode, the physical insertion parameters, and the location of the insertion site relative to pial structures. Intuitively, the smaller the electrode is, the less damage it causes. While relatively little can be done to change the location of insertion, limited by the intended application, recent findings from the work of Bjornsson and coworkers [14] will help guide the selection of insertion parameters and future design of electrodes. This study evaluated the contribution of tip geometry and insertion speed to the damage caused by insertion. Within the range of speeds and tip geometries studied, faster insertion of sharp electrodes resulted in lower mean effective strain in superficial and middle regions of cortex. Insertion speed seems to play a more important role than tip geometry, with faster insertions (2000 μm s–1) generally resulting in less vascular damage.
6.2.1.2. Mechanical Injury
From the cellular point of view, the initial interaction between the electrode and the brain is relatively traumatic. As the electrode traverses the brain parenchyma, it ruptures the blood vessels, disrupts the integrity of the extracellular matrix, slices the cell bodies, and displaces the tissue. However, the complete cascade of events that occur during the initial insertion, both at the cellular and molecular levels, is not yet fully understood. The consequences of initial damage are very similar to those caused by cortical stab wound. In response to the early mechanical trauma, the wound healing process is initiated. This process is characterized by the early arrival of blood-borne leukocytes, such as macrophages, via the breached vasculature and the activation of microglia, known as the resident immune cells in the nervous system (Figure 6.2). Prior to injury, microglia reside in a resting state, typically characterized as ramified with an elaborate tertiary and quaternary branch structure. The resting microglia take an active role in providing extensive and continuous surveillance of their cellular environment. As they are the first line of defense, microglia quickly become activated after CNS injury. The activation is marked by a number of characteristic events, including contraction of cellular processes and transformation from ramified into a round amoeboid macrophage-like morphology, increased proliferation, induction of immunomolecules such as major histocompatibility complex (MHC) antigen classes I and II, and changes in the pattern of cytokine and growth factor production. Although the precise factors responsible for triggering microglial activation remain to be identified, potential triggers include cellular debris generated by the injury, molecules released from dying neurons, and extravasated plasma constituents. Activated microglia will then engage in phagocytic activity, similar to the role that macrophages play in non-CNS tissue, by engulfing and digesting the debris with lytic enzymes.
The initial inflammatory response also features actions from another type of glial cell, the astrocyte (Figure 6.2). Astrocytes account for 30 to 65% of the glial cells in the CNS [41]. They play an essential role in ensuring normal neuronal activity through uptake and release of glutamate, homeostatic maintenance of extracellular ionic environment and pH, water transport, and preservation of the BBB integrity. Astrocytes are usually characterized by their star-like appearance with fine cellular processes. They respond to injury with a hallmark action, increasing the expression of the glial fibrillary acidic protein (GFAP), a fibrillary intermediate filament that is specific to astrocytes. Functionally, GFAP is essential in long-term maintenance of CNS myelination as well as stabilization of the BBB [42]. The increased expression of GFAP is marked by increases in GFAP mRNA production and is accompanied by an increase in cell volume and the caliber of proximal cell processes (cellular hypertrophy). Furthermore, astrocytes undergo dramatic biochemical and functional transformations upon activation. Collectively, these responses are referred to as reactive astrogliosis. It is generally accepted that reactive astrocytes play a key role in forming a physical barrier, commonly known as the glial scar, in an attempt to prevent injury from spreading to surrounding healthy tissue. However, in relation to its effect on electrode recording, the glial scar is considered undesirable, as it impedes regenerating neural processes coming into contact with the electrode.
6.2.1.3. Molecular Mediators
Accompanying the early cellular response is the release of a variety of molecules that participate actively in orchestrating the tissue response. Some of the molecules are secreted temporarily, while the production of others may extend into the chronic phase of the implant. Although a diverse assortment of molecular mediators is involved in the tissue response, we have chosen to focus our discussion on the expression and effects of cytokines, proteases, and reactive oxygen species.
6.2.1.3.1. Cytokines
Cytokines are low-molecular-weight glycoproteins that function as mediators of intercellular communication. Studies have shown that cytokines are rapidly secreted after CNS injuries and are essential for the initiation, propagation, and termination of the inflammatory response [43]. Cytokines exert their actions through specific cell surface receptors in an autocrine or paracrine fashion. Because of the complexity of the cytokine network induced by injury, our discussion will focus on some of the major players, including TNF-α, interleukin-1 (IL-1), IL-6, and transforming growth factor-beta (TGF-β). The signaling pathways that are closely associated with these cytokines are the ones involving the transcription factor NF-κB and MAPKs such as p38.
6.2.1.3.1.1. TNF-α
TNF-α, a 17-kDa peptide, is one of the prototypic proinflammatory cytokines that are rapidly upregulated in the injured CNS. Activated macrophages, infiltrating through the breached vasculature, are the major cellular source for TNF-α. It has been shown to promote microglia phagocytosis as well as further production of inflammatory cytokines [44]. Activated microglia, which are the resident immune cells in the CNS, are also an early and prominent source of TNF-α. It is likely that the early expression of TNF-α is caused by the initial implantation injury. Staining against TNF-α in a stab wound lesion revealed that the immunoreactivity was predominantly located around the site of the lesion, corresponding to the location of activated microglia and macrophages [45]. Using in situ hybridization, we examined the spatial and temporal profiles of TNF-α expression in rats subjected to Michigan Si electrode implantation. As shown in Figure 6.4, an elevated TNF-α mRNA expression was observed right at the electrode–tissue interface 1 week after implantation, echoing staining results previously reported [45]. However, very little expression was found at the interface 4 weeks postimplantation. Previous studies have demonstrated that the transient expression of proinflammatory cytokine TNF- αis an indication of its key role in controlling the acute inflammatory response and its function in triggering secondary events [46].

FIGURE 6.4
In situ hybridization for mRNA expression of cytokine TNF-α at the electrode –tissue interface. Temporally, the expression was mainly observed in the early time point, 1 week postimplantation (a), as compared to very little expression (more...)
TNF-α serves multiple roles in CNS response to injury. It can activate microglial cells via the autocrine loop to maintain their activated status. TNF-α also induces the proliferation of astrocytes, one of the key features of astrogliosis. In addition, studies have demonstrated that TNF-α can be directly cytotoxic to oligodendrocytes [47] (the myelin-forming cells in the CNS) and neurons [48]. Despite the deleterious role TNF-α plays in CNS tissue response to injury most of the time, studies have shown that under certain situations, TNF-α also plays a beneficial role. It appears that low levels of TNF-α may be neuroprotective [49], and such an effect could be applied indirectly via induction of growth factors such as nerve growth factor (NGF) [50].
6.2.1.3.1.2. IL-1 and IL-6
The interleukin family is composed of a growing list of members, including IL-1, IL-2, IL-3, IL-4, IL-6, IL-8, IL-10, and more. Each of them fulfills a different functional role in the immune response. In the CNS, two widely studied interleukins are IL-1 and IL-6. IL-1 exists in two forms, IL-1α and IL-1β, with the former being mostly membrane associated, while the latter is usually secreted [43]. In normal brain tissue, IL-1 is constitutively expressed at low or undetectable concentrations, at the mRNA and protein level [51]. In response to CNS injury such as electrode implantation, the expression of IL-1α and IL-1β is upregulated. Similar to TNF-α, early expression of IL-1 occurs in cells of monocyte and macrophage lineage, including microglia. Studies have shown that IL-1β is rapidly produced by activated microglia within 15 min following a cortical injury [52]. IL-1 has numerous effects on glial cells and neurons. Of all the glial cells, the effect of IL-1 seems to be strongest on astrocytes. Compelling evidence links IL-1β to the induction and modulation of the astrogliosis process [53,54], featured by a repertoire of astrocyte proliferation, upregulation of GFAP expression, and increase in production of cytokines, growth factors, matrix proteins, and so on. Microglia also express receptors for IL-1β; therefore, via an autocrine feedback loop, the inflammation signal is amplified and further microglial activation is stimulated.
Like TNF-α, the effects of IL-1 in the CNS also come with a positive spin, with several recent studies highlighting the role of IL-1 in the regenerative process in the CNS. It can exert beneficial effects when released in modest concentrations [55]. It is suggested that IL-1 can improve neuronal survival and repair by directly or indirectly inducing a subset of genes primarily associated with neuronal and glial growth and survival, including IL-6, ciliary neurotrophic factor (CNTF), and NGF [56].
Unlike TNF-α and IL-1, which are mostly known for their proinflammatory properties, IL-6 has been observed to act in a proinflammatory and antiinflammatory manner. In the CNS, IL-6 is produced by microglia, astrocytes, and endothelial cells. The production is considered a downstream sequence related to TNF-α or IL-1 stimulation on cells. As IL-6 shares the signaling receptor with several growth factors (e.g., CNTF), studies have shown that IL-6 can promote neuronal survival and neurite growth [57], as well as downregulate the expression of TNF-α [58]. Together, these features are indications of the beneficial roles of IL-6 in repair and modulation of inflammation in the CNS. However, IL-6 also has proinflammatory potential. The cellular action of IL-6 is primarily on reactive astrocytes by promoting cell proliferation [59] and is believed to be involved in astrogliosis.
6.2.1.3.1.3. TGF-β
TGF-β is a multifunctional cytokine with wide-ranging effects on cell proliferation, differentiation, migration, angiogenesis, and extracellular matrix remodeling [56]. In normal CNS, TGF-β is present at low levels, but the expression is upregulated upon injury. Cellular staining for TGF-β reveals that the expression is predominantly around the site of injury and mainly colocalized with GFAP positive reactive astrocytes, indicating that astrocytes are the key cellular source of TGF-β [45]. Additionally, TGF-β can be produced by microglia as a downstream product in response to activation by the IL and TNF families [60]. As is the case with the other cytokines discussed in this section, TGF-β is considered both pro- and antiinflammatory. As a potent antiinflammatory cytokine, TGF-β has immunosuppressive effects on glial cells by inhibiting expression of proinflammatory cytokines such as TNF-α and IL-1, as well as by inhibiting glial cell proliferation [61]. However, TGF-β can also act as a proinflammatory cytokine, especially when expressed at a high concentration, to exacerbate the reactive astrogliosis and scar formation. Studies have demonstrated that, when function-blocking antibodies to TGF-β are administered, the deposition of fibrous scar tissue and the formation of a limiting glial membrane that border the lesion are significantly attenuated [62].
6.2.1.3.2. Proteases
Activated glial cells in the CNS also express proteases that exert both beneficial and harmful effects. These proteases include cathepsins, calpains, plasminogen activators, and matrix metalloproteinases (MMPs). Their physiological roles include orchestrating cell migration as well as extracellular matrix maintenance and remodeling. Here we will take a closer look at the role of MMPs in the CNS inflammatory response. The MMP family consists of over 18 members including gelatinases (MMP-2 and -9), collagenases (MMP-1, -8, -13, -18), and stromelysins (MMP-3, -7, -10, -11) [63]. All cell types in the CNS are potential sources for MMPs. A wealth of data has linked MMPs to CNS injury and inflammation. In cortical stab injury, upregulation of mRNA expression of MMPs is observed within 24 hours. Such elevation has been mainly credited to activated microglia and macrophages and reactive astrocytes. It is likely that MMP elevations postinjury are mediated by MAPK pathways or oxidative stress [64,65]. An overproduction of MMPs could be deleterious, as it contributes to BBB breakdown and therefore results in infiltration of circulating immune cells that will further amplify the inflammatory response. In addition, MMP may also degrade the parenchymal extracellular matrix protein laminin that will disrupt the laminin–neuronal interactions and contribute to neuronal death. Paradoxically, there may be some positive effects. MMPs might play a role in angiogenesis, which is essential in repair. Remodeling of the ECM by MMPs could also facilitate migration of cells to the lesion area to clean up the debris or even assist in axonal elongation.
6.2.1.3.3. Reactive Oxygen Species
Reactive oxygen species (ROS) are a class of oxygen free radicals and related molecules that are capable of exerting oxidative stress on cells when produced in excess. Nitric oxide (NO), a simple yet highly versatile molecule, is a type of ROS frequently studied and involved in a wide range of physiological as well as pathophysiological mechanisms. In normal brain tissue, production of NO is usually neutralized by cellular antioxidants. However, in response to injury, the production of NO can overpower the antioxidants, leading to oxidative stress and cellular damage. Reactive astrocytes and activated microglia have the capacity to generate NO, and the production can be induced by proinflammatory cytokines such as IL-1 and TNF-α. NO is a ubiquitous second messenger [66] that diffuses freely across cell boundaries and can damage neurons by potentiating glutamate excitotoxicity [67].
6.2.2. Tissue Response to the Chronic Presence of Electrodes
To distinguish the impact of initial mechanical injury from the chronic presence of electrodes on tissue response, Biran et al. [68] compared tissue reaction to chronically implanted microelectrodes with time-matched stab wound controls created using identical microelectrodes and an implantation technique. A striking difference was noted, with microelectrode stab wounds eliciting a subtler response that weakened with time, while indwelling microelectrodes generated a glial scar that became more compact and confined to the electrode with time. These results suggest that the initial mechanical-insertion-induced injury is transient, with the persistent presence of the electrode in the tissue accounting for the observed long-term inflammation. To some extent, this finding echoes a previous study by Szarowski et al. [13], which showed that initial injury response was a function of the implant dimensions, while the sustained injury response was independent of the implant size. This suggests that the continuous presence of the implant induces the formation of a sheath composed of reactive glial cells. Such chronic inflammation could be explained from the following perspectives.
6.2.2.1. frustrated phagocytosis
The common mechanism of the body defending against a foreign object is delegating macrophages to interrogate the object. These cells will then either secret lytic enzymes to degrade the object or, in the case of a nondegradable object too large to be phagocytosed, they will fuse into multinucleated foreign body giant cells and continue secretion of degradative agents such as superoxides and free radicals. The latter case is a phenomenon known as “frustrated phagocytosis.” It is likely that the insoluble electrode resists the attempted removal action from activated microglia and macrophages, which means the stimulant will linger and the microglial activation will remain, leading to increased secretion of inflammatory products that further exacerbate neuronal damage.
6.2.2.2. Mechanical strain
Chronic inflammation and glial scarring could also be instigated by the micromotion surrounding the electrode. In contrast to the softness of brain tissue, electrodes are considerably hard and rigid. There is a drastic mechanical mismatch between the electrode material, usually silicon, and the brain tissue, as the Young’s moduli of bulk silicon and brain are ~200 GPa [69] and 6 kPa [70], respectively. Because of this large mismatch, brain micromotion that arises from physiological sources such as cardiac rhythm and fluctuation in respiratory pressure [71], behavioral sources such as spontaneous head movements [72], and mechanical sources such as disturbances of the implanted devices [73] could be translated into mechanical stresses and strains that impose on the tissue adjacent to the electrode, leading to compression, expansion, and even tearing of the tissue.
Recently, several groups including our own have examined the magnitude of tissue micromotion relative to a stationary implant [74] and the interfacial strains induced around the implant by micromotion [75,76]. The surface micromotion in the rodent somatosensory cortex was quantified to be on the order of 10 to 30 μm owing to pressure changes during respiration and 2 to 4 μm owing to vascular pulsatility [74]. Using finite-element modeling (FEM), the interfacial strain profiles were analyzed around a single-shank Michigan type Si microelectrode (Figure 6.5). The maximum strain occurred at the tip of the electrode in the tissue, and the strain regions could extend up to 100 μm away from the implant interface and approximately 70 μm beyond the electrode tip, with strain values decreasing exponentially as a function of distance from the interface. The strain field located around the electrode track is attributed to frictional shear stresses induced by the longitudinal displacement or the “poking” action of the electrode, which also could lead to extensive compression and tearing of the tissue. The simulation results further suggest that induced strain is affected by the extent of adhesion between the electrode and tissue. In the poor adhesion condition where the friction coefficient is finite, high strains resulted (10−1 to 10−2) and were concentrated in a narrow region around the tip. Since the recording sites are usually located in areas toward the tip, such a strain profile is obviously undesirable, as it could cause an elevated tissue response in that region. In contrast, good adhesion representing no slipping between the electrode and tissue led to lower-magnitude strain (10−3 to 10−4) distributed more uniformly along the entire electrode track. Although the results from these simulated studies have yet to be experimentally validated, they provide insight into the events caused by the mechanical mismatch at the interface and a basis for future improvements to the electrode bulk as well as surface properties to minimize mechanically induced strain that will subsequently translate into less scarring in the long term.
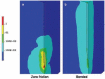
FIGURE 6.5
Von Mises strain contours (logarithmic scale) of the brain tissue on the surfaces of symmetry planes for longitudinal micromotion. (a) The zero friction coefficient case, where the significant high strain is located at the tip of the electrode. (b) The (more...)
Thus, at the cellular level, how are these mechanical signals being transduced to cells, to what cell type(s), and with what consequences? Although there is no direct report on the effect of electrode-related mechanical strain injury on cells in the nervous system, mechanotransduction has been studied in another injury model: traumatic brain injury (TBI). Using an in vitro stretch model, the effects of mechanical strain injury on various brain cell types, especially astrocytes, were studied [77–84]. Astrocytes sense mechanical stress and translate it into chemical messages, such as rapid increase in intracellular free sodium and calcium, activation of phospholipases, free radical formation, and secretion of the potent astrocyte antigen endothelin, that have been implicated in the induction of gliosis. In addition, stretch-induced injury also causes delayed neuronal depolarization, which may be related to the transient neuronal dysfunction observed in vivo [85].
The hypothesis that continual physical insults resulting from micromotion at the electrode–brain interface could exacerbate glial scar formation, which in turn will severely compromise the recording stability of the electrode, is currently being investigated. Utilizing the in vitro stretch model, our group is studying the effects of sustained, low-frequency and low-magnitude strain, mimicking the actual in vivo situation, on astrocytes and microglia [86]. After subjecting an astrocyte culture to 24 hours of low-frequency (30 cycles/min), low-magnitude strain (5%), real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis indicated that there are mild increases in mRNA expression in the following markers: TGF-β1, a cytokine that is known to enhance glial scarring and extracellular matrix deposition at high level [62]; GFAP, a characteristic indicator of astrocyte activation; and neurocan, a member of the CSPG family that is known for its role in blocking axon regeneration and is a major component of the glial scar [87]. As for microglia, although sustained strain had little effect on the gene expression of proinflammatory cytokine TNF-α, it did induce a mild upregulation in the expression of IL-1β, a very potent signaling molecule that modulates the inflammatory response. The in vitro results suggest that micromotion-induced mechanical strain could be perceived as a continuous stimulus and lead to glial scarring by direct stimulation of the astrocytic response or indirectly through IL-1β modulation. Future studies include investigating the mechanical strain effect in an astrocyte–microglia coculture system and exposing the culture to a longer period of strain. Additionally, it would be interesting to further elucidate the mechanical effects using organotypic slice cultures with electrodes implanted to more closely mimic the actual in vivo scenario.
6.2.2.3. Molecular Mediators
The chronic phase of CNS tissue response to electrodes shares a cast of molecular mediators similar to those aforementioned in the initial phase (i.e. cytokines, ROS, proteases, etc.). However, the focus of their actions might shift from engaging the defense system against the insult to restoration of tissue homeostasis and recovery and repair of the injured tissue. The cytokine balance might also tilt toward the side of antiinflammatory cytokines as opposed to initially favoring proinflammatory cytokines.
6.3. MOLECULAR STRATEGIES TO MINIMIZE THE ABOVE RESPONSES
To fully translate the enormous potential of CNS electrodes into clinical realities, one has to overcome several challenges, including the issue of long-term biocompatibility of the electrode. Clearly, a thorough understanding of the underlying biological processes behind CNS tissue response to the electrode, at the cellular and molecular level, will provide valuable implications for the development of therapeutic interventions to improve its chronic CNS performance. Although the details of such complex biological responses to CNS electrodes remain to be delineated, a consensus has been reached among the scientific community that the lack of long-term stability could be attributed to a myriad of responses that the tissue mounts against the invading electrode. As noted above, the prominent aftermath is set with the opening act of electrode insertion, which causes traumatic injury and vessel damage, followed by transition into the acute phase as the tissue attempts to repair the initial injury and remove the electrode, and eventually phased into the chronic response to the persistent inflammatory stimuli, that is, the physical presence of the indwelling electrode and micromotion-induced mechanical strain. We will briefly discuss some of the potential strategies to control CNS tissue response against the electrode. The overarching objective is to minimize the adverse inflammatory response and glial scar formation and to promote neuronal survival and outgrowth toward the electrode. The proposed strategies could potentially intervene at a molecular level.
6.3.1. Strategy I: Blocking Proteinase-Activated Inhibitors (Par) Receptors
Even though the complete consequences of vascular damage by the initial electrode implantation on chronic electrode performance are not yet clear, one cannot overlook the fact that serum components that are extravasated via the compromised BBB play an indispensable role in triggering the inflammatory response. Serine proteases such as thrombin are powerful activators of glial cells in the CNS and can induce reactive gliosis in vivo. However, it would be unwise to completely inhibit thrombin activity, as it could trade dangerously excessive bleeding for its potential therapeutic effect. Thus, it may be beneficial to identify specific molecular targets mediating thrombin effects. A potential approach could be inactivating or blocking the receptors for thrombin expressed by the glial cells. As studies have demonstrated the involvement of the receptors PAR-1 and PAR-4 in thrombin actions on microglia and astrocytes, it is possible to employ anti-PAR1 and anti-PAR4 tactics to subdue the effects of thrombin. The available PAR-1 antagonists include peptide mimetics such as BMS-200261, RWJ-56110, RWJ-53052, and RWJ-58259 and peptidic antagonists such as RPPGF [27]. The selectivity of these antagonists has been characterized in tissues outside the CNS, but their effectiveness has not been evaluated in the CNS except for RPPGF. In comparison with PAR-1, PAR-4 is a relatively newly identified receptor for thrombin; thus, there is very limited information available for selective PAR-4 antagonists, except a synthetic compound YD-3 [88] and a pepducin type antagonist P4pal-10 [89].
Besides identifying the specific molecular targets to combat thrombin-triggered CNS inflammation to the electrode, several other issues need to be addressed as well. In particular, questions such as how to deliver the antagonists, when to release them, at what concentration, and for how long demand careful consideration. As vessel rupture takes place simultaneously with initial electrode insertion, it would be favorable to release the PAR antagonists early on. Also, as studies have suggested the biphasic impact of thrombin in the CNS (low concentrations of thrombin can be neuroprotective, while high concentrations can be deleterious), partially blocking PAR-1 and PAR-4 in the CNS may hold more promising therapeutic value than completely diminishing the PAR effects.
6.3.2. Strategy II: Attenuating Inflammation
One major consequence of the inflammatory response is the formation of glial scar, which not only physically blocks neurons away from the electrode, but also electrically isolates the electrode from surrounding neurons by increased impedance. Considerable effort is being directed toward minimizing, or better, eliminating, the formation of such a barrier. The key members of the inflammation cast are microglia and astrocytes, with a supporting cast of molecular mediators such as cytokines, growth factors, and reactive oxygen species. Also, NF-κB is a pivotal transcription factor for genes that encode proinflammatory cytokines such as IL-1 and TNF-α [90]. Several antiinflammatory agents have been shown to exert powerful effects through inhibition of NF-κB activation, including the neuroimmunomodulatory peptide α-melanocyte-stimulating hormone (α-MSH) and synthetic glucocorticoids such as methylprednisolone and dexamethasone. Spataro et al. [91] found that peripheral injections of dexamethasone beginning at the day of electrode insertion and continuing for 6 days profoundly affected the early (1 week after insertion) and sustained (6 weeks after insertion) reactive responses. However, the success of systemic delivery of glucocorticoids to suppress inflammation is likely to be shadowed by the known adverse side effects. To circumvent this problem, our group is employing a nitrocellulose-based delivery approach for the local release of dexamethasone [92]. Local release of dexamethasone significantly reduced the reactivity of microglia and macrophages, as well as the expression of the inhibitory molecule CSPG 1 week postimplantation (Figure 6.6). Furthermore, such treatment greatly attenuated astroglial response and reduced neuronal loss in the vicinity of the electrode at 1 and 4 weeks postimplantation, implying that such an approach could potentially contribute to improving chronic neuronal recording.
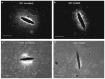
FIGURE 6.6
Representative images of horizontal brain sections inserted with control silicon electrode (a and c) and dexamethasone-coated electrode (b and d) 1 week postimplantation. ED1 stains for reactive microglia and macrophages (a and b) and CS56 stains chondroitin (more...)
Unlike the synthetic glucocorticoid dexamethasone, α-MSH is endogenously expressed in cells of the immune system as well as in cells of the nervous and endocrine systems [93]. The neuropeptide has remarkable antiinflammatory properties, acting directly upon peripheral host cells to modulate the release of inflammatory substances and acting on receptors within the brain that drive descending antiinflammatory pathways [94]. Even though the strategy of delivering antiinflammatory agents can modulate the tissue response to electrodes, it presents the difficulty of sustaining the delivery over long implantation periods (months or even years). To complement this strategy, we proposed intrinsically modifying the electrode surface property by chemically immobilizing the α-MSH to the surface [95]. The peptide is grafted onto the surface using conventional silane chemistry. An in vitro cell study indicates that the immobilized peptide retained its biological property, making the electrode surface inherently antiinflammatory. In rats, the peptide-modified electrode reduced the microglial response at 1 and 4 weeks postimplantation, compared to the untreated control. As microglia are the frontline cells in direct contact with the electrode surface, their response is directly affected by the tethered α-MSH peptide. The astrocytic response was attenuated at the 4-week time point. It will be important to examine how long the tethered peptide will remain on the surface inside the tissue and whether it will be sufficient to modulate the gliosis response induced by “frustrated” phagocytosis as well as micromotion-induced mechanical strain. It is indisputable that to achieve maximum long-term improvement in tissue response, such an immobilization strategy should be applied in combination with other molecular interventions and serve as a foundation for a multilayered combinatorial treatment strategy.
Besides directly delivering antiinflammatory chemical drugs to influence cellular response, sustained release of antisense oligodeoxynucleotides (ODNs) against NF-κB mRNA would also be an effective strategy to diminish inflammation at the electrode implantation site. NF-κB antisense ODN is anticipated to selectively block proinflammatory gene expression, thus regulating cellular response at the mRNA level. Alternatively, strategies using antibodies against major initiators of gliosis, for example, TGF-β, could potentially mitigate the formation of glial scar. Given the complexity of molecules involved in the inflammatory response, many other potential molecular targets could be identified for therapeutic purposes. However, one has to be careful when employing such strategies, as increasing evidence has shown that many inflammatory molecular mediators in the CNS act like double-edged swords. Their effects do not occur in an all-or-none fashion; rather, they are concentration dependant. Therefore, it should be kept in mind that the goal is to contain any excessive activation rather than to remove all activity.
6.3.3. Strategy III: Promoting Migration and Survival of Neurons and Neurites
The electrode’s main point of contact with the CNS is the neuron. Therefore, electrode performance is directly associated with the density of neurons and their proximity to the electrode sites. Theoretically, a recording electrode can detect action potentials from neurons within a radius of approximately 130 μm [96]. However, studies have suggested that the maximum recording distance is between 50 and 100 μm [97–100]. Accompanying brain tissue response to the implanted electrode is a reported neuronal cell loss. About a 40% decrease in neuronal cell bodies within a 100-μm radius of the electrode–tissue interface was observed at 2 and 4 weeks postimplantation [68]. The loss is mainly attributed to the production of neurotoxic factors such as proinflammatory cytokines and free oxygen radicals that are produced during persistent inflammation, rather than to direct mechanical trauma during electrode implantation [68]. This indicates that neuronal survival can be indirectly enhanced by attenuating the inflammatory response using the strategies suggested above.
A more active approach would be to directly promote neuronal survival and growth toward the electrode by releasing neurotrophic factors. A classical example is the study conducted by Kennedy [101] in which glass cone electrodes seeded with pieces of sciatic nerve recorded neural signals with signal to noise ratios that were 5 to 10 times better than those obtained with wire and silicon electrode arrays for over 12 months. It is likely that such improvement was related to the growth factors or chemoattractants released from the sciatic nerve. The neurotrophin family, a subgroup of the neurotrophic factors family, contains members such as NGF, brainderived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4/5. These neurotrophins are widely expressed in nearly all neuronal populations in the CNS and peripheral nervous system (PNS) and are well known for their role in neuronal survival, process outgrowth, and regulation of synaptic plasticity [102]. Additionally, the chemotactic guidance of various migrating neurons is potentially mediated by the neurotrophins. In fact, delivery of growth factors has been proposed and is currently being studied as a potential therapeutic treatment for spinal cord injury [103]. A similar approach can be applied to electrodes implanted in the CNS: nourishing neurons with growth factors so that the number of recordable neurons will be increased. The challenges will be determining the parameters for delivery, specifically, the onset point, the dosage, the duration, and the delivery vehicle. Putatively, such strategic neurotrophin release could be an adjunctive component of therapeutics for achieving stable, long-term electrode performance.
Besides delivering exogenous growth factors, ideas of making the electrode surface more neural adhesive have been tested. In vitro studies have demonstrated that modifying the substrate surface with bioactive molecules such as laminin [104,105], the peptide fragment from laminin YIGSR [106], and cell adhesion molecule L-1 [107] significantly promoted neuron growth on the surface. However, the efficacy of these approaches in vivo is yet to be determined.
The other strategy to increase neuron density in the proximity of the electrode is to clear the path for neurites toward the interface. It is well known that the glial scar is highly inhibitory for neurite outgrowth, because of the presence of inhibitory components such as myelin-associated molecules (myeline-associated glycoprotein [MAG], Nogo, tenascin R) and CSPGs [87]. To encourage neurites to interact intimately with the electrode, this inhibitory environment has to be cleared. At least four possible approaches have been suggested [87], including (a) removing the cells that make inhibitory molecules, mostly astrocytes; (b) preventing the synthesis of inhibitory molecules, such as TGF-β; (c) blocking inhibitory molecules with antibodies that are specific to certain epitopes; and (d) degrading inhibitory molecules, such as using the bacterial enzyme chondroitinase ABC to digest away the glycosaminoglycan side chains of CSPG while leaving the core protein intact.
6.4. CONCLUSION
In this chapter, we have attempted to outline the differing molecular responses so that attempts to modulate tissue response to electrodes can be rationally pursued. Overall, the complex nature of CNS tissue response to chronic electrodes suggests that no single cellular or molecular targeting strategy will prove sufficient for achieving stable and long-term functional outcomes. Instead, a combinatorial strategy that is rationally designed and tailored, aided by a better understanding of underlying biological processes, will lead to cumulative improvements in interfacing technology that will one day be translated into real clinical applications.
ACKNOWLEDGMENTS
Funding support from National Institutes of Health, 1R01 DC06849 and 1R01 NS043486, is gratefully acknowledged (RVB). Special gratitude is expressed to Yinghui Zhong, George C. McConnell, and Thomas Schneider for their illuminating discussions and help in preparation of this manuscript.
REFERENCES
- 1.
- Spelman FA. The past, present, and future of cochlear prostheses. IEEE Eng. Med. Biol. Mag. 1999;18:27. [PubMed: 10337561]
- 2.
- Lozano AM, et al. Deep brain stimulation for Parkinson’s disease: Disrupting the disruption. Lancet Neurol. 2002;1:225. [PubMed: 12849455]
- 3.
- Hochberg LR, et al. Neuronal ensemble control of prosthetic devices by a human with tetraplegia. Nature. 2006;442:164. [PubMed: 16838014]
- 4.
- Nicolelis MA, et al. Chronic, multisite, multielectrode recordings in macaque monkeys. Proc. Natl. Acad. Sci. USA. 2003;100:11041. [PMC free article: PMC196923] [PubMed: 12960378]
- 5.
- Kennedy PR, Bakay RA, Sharpe SM. The cone electrode: Ultrastructural studies following long-term recording in rat and monkey cortex. Neurosci. Lett. 1992;142:89. [PubMed: 1407726]
- 6.
- Sachs HG, et al. Transscleral implantation and neurophysiological testing of subretinal polyimide film electrodes in the domestic pig in visual prosthesis development. J. Neural. Eng. 2005;2:S57. [PubMed: 15876656]
- 7.
- Drake KL, et al. Performance of planar multisite microprobes in recording extracellular single-unit intracortical activity. IEEE Trans. Biomed. Eng. 1998;35:719. [PubMed: 3169824]
- 8.
- Campbell PK, et al. A silicon-based, three-dimensional neural interface: Manufacturing processes for an intracortical electrode array. IEEE Trans. Biomed. Eng. 1991;38:758. [PubMed: 1937509]
- 9.
- Rousche PJ, Normann RA. Chronic recording capability of the Utah intracortical electrode array in cat sensory cortex. J. Neurosci. Methods. 1998;82:1. [PubMed: 10223510]
- 10.
- Polikov VS, Tresco PA, Reichert WM. Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods. 2005;148:1. [PubMed: 16198003]
- 11.
- Wise KD. Silicon microsystems for neuroscience and neural prostheses. IEEE Eng. Med. Biol. Mag. 2005;24:22. [PubMed: 16248114]
- 12.
- Rousche PJ, et al. Flexible polyimide-based intracortical electrode arrays with bioactive capability. IEEE Trans. Biomed. Eng. 2001;48:361. [PubMed: 11327505]
- 13.
- Szarowski DH, et al. Brain response to micro-machined silicon devices. Brain Res. 2003;983:23. [PubMed: 12914963]
- 14.
- Cavaglia M, et al. Regional variation in brain capillary density and vascular response to ischemia. Brain Res. 2001;910:81. [PubMed: 11489257]
- 15.
- Bjornsson CS, et al. Effects of insertion conditions on tissue strain and vascular damage during neuroprosthetic device insertion. J. Neural Eng. 2006;3:196. [PubMed: 16921203]
- 16.
- Klatzo I. Pathophysiological aspects of brain edema. Acta Neuropathol. 1987;72:236. [PubMed: 3564903]
- 17.
- Schilling L, Wahl M. Mediators of cerebral edema. Adv. Exp. Med. Biol. 1999;474:123. [PubMed: 10634998]
- 18.
- Nishino A, et al. Thrombin may contribute to the pathophysiology of central nervous system injury. J. Neurotrauma. 1993;10:167. [PubMed: 7692071]
- 19.
- Pindon A, Berry M, Hantai D. Thrombomodulin as a new marker of lesion-induced astrogliosis: involvement of thrombin through the G-protein-coupled protease- activated receptor-1. J. Neurosci. 2000;20:2543. [PMC free article: PMC6772242] [PubMed: 10729334]
- 20.
- Kubo Y, et al. Thrombin inhibitor ameliorates secondary damage in rat brain injury: Suppression of inflammatory cells and vimentin-positive astrocytes. J. Neurotrauma. 2000;17:163. [PubMed: 10709874]
- 21.
- Xue M, Del Bigio MR. Acute tissue damage after injections of thrombin and plasmin into rat striatum. Stroke. 2001;32:2164. [PubMed: 11546912]
- 22.
- Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J. Neurochemistry. 2003;84:3. [PubMed: 12485396]
- 23.
- Ryu J, et al. Thrombin induces NO release from cultured rat microglia via protein kinase C, mitogen-acivated protein kinase, and NF-kappa B. J. Biol. Chem. 2000;275:29955. [PubMed: 10893407]
- 24.
- Möller T, Hanisch UK, Ransom BR. Thrombin-induced activation of cultured rodent microglia. J. Neurochem. 2000;75:1539. [PubMed: 10987834]
- 25.
- Suo Z, et al. Participation of protease-activated receptor-1 in thrombin-induced microglial activation. J. Neurochem. 2002;80:655. [PubMed: 11841573]
- 26.
- Suo Z, et al. Persistent protease-activated receptor 4 signaling mediates thrombin-induced microglial activation. J. Biol. Chem. 2003;278:31177. [PubMed: 12775717]
- 27.
- Suo Z, Citron BA, Festoff BW. Thrombin: a potential proinflammatory mediator in neurotrauma and neurodegenerative disorders. Curr. Drug Targets Inflamm. Allergy. 2004;3:103. [PubMed: 15032647]
- 28.
- Nicole O, et al. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 2005;25:4319. [PMC free article: PMC6725104] [PubMed: 15858058]
- 29.
- Möller T, Weinstein JR, Hanisch UK. Activation of microglial cells by thrombin: past, present, and future. Semin. Thromb. Hemost. 2006;32(Suppl 1):69. [PubMed: 16673268]
- 30.
- Coughlin SR. Thrombin signaling and protease-activated receptors. Nature. 2000;407:258. [PubMed: 11001069]
- 31.
- Noorbakhsh F, et al. Proteinase-activated receptors in the nervous system. Nat. Rev. Neurosci. 2003;4:981. [PubMed: 14682360]
- 32.
- Beecher KL, et al. Thrombin receptor peptides induce shape change in neonatal murine astrocytes in culture. J. Neurosci. Res. 1994;37:108. [PubMed: 8145298]
- 33.
- Ehrenreich H, et al. Thrombin is a regulator of astrocyte endothelin-1. Brain Res. 1993;600:201. [PubMed: 7679602]
- 34.
- Grabham P, Cunningham DD. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: involvement of tyrosine phosphorylation. J. Neurochem. 1995;64:583. [PubMed: 7830051]
- 35.
- Wang H, et al. Thrombin (PAR-1)-induced proliferation in astrocytes via MAPK involves multiple signaling pathways. Am. J. Physiol. Cell Physiol. 2002;283:C1351. [PubMed: 12372796]
- 36.
- Wang H, Ubl JJ, Reiser G. The four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia. 2002;37:53. [PubMed: 11746783]
- 37.
- Xue M, Del Bigio MR. Acute tissue damage after injections of thrombin and plasmin into rat striatum. Stroke. 2001;32:2164. [PubMed: 11546912]
- 38.
- Fitch MT, Silver J. Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp. Neurol. 1997;148:587. [PubMed: 9417835]
- 39.
- McConnell GC, Bellamkonda RV. Extraction force and cortical tissue reaction of silicon microelectrode arrays implanted in the rat brain. Unpublished manuscript. [PubMed: 17554828]
- 40.
- Krueger S, et al. Three morphologically distinct types of interface develop between adult host and fetal brain transplants: Implications for scar formation in the adult central nervous system. J. Comp. Neurol. 1986;249:103. [PubMed: 3755447]
- 41.
- Nathaniel EJH, Nathaniel DR. The reactive astrocyte. In: Federoff S, editor. Advances in Cellular Neurobiology. Orlando, FL: Academic Press; 1981. pp. 249–301.
- 42.
- Chen Y, Swanson RA. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003;23:137. [PubMed: 12571445]
- 43.
- Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog. Neurobiol. 2002;67:161. [PubMed: 12126659]
- 44.
- Aloisi F. Immune function of microglia. Glia. 2001;36:165. [PubMed: 11596125]
- 45.
- Ghirnikar RS, Lee YL, Eng LF. Inflammation in traumatic brain injury: Role of cytokines and chemokines. Neurochem. Res. 1998;23:329. [PubMed: 9482245]
- 46.
- Streit WJ, et al. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp. Neurol. 1998;152:74. [PubMed: 9682014]
- 47.
- Louis JC, et al. CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science. 1993;259:689. [PubMed: 8430320]
- 48.
- Downen M, et al. Neuronal death in cytokine-activated primary human brain cell culture: Role of tumor necrosis factor-alpha. Glia. 1999;28:114. [PubMed: 10533055]
- 49.
- Scherbel U, Raghupathi R, Nakamura M. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury chemical messengers. Proc. Natl. Acad. Sci. USA. 1999;96:8721. [PMC free article: PMC17583] [PubMed: 10411942]
- 50.
- Baird A, Gage FH. Cytokine regulation of nerve growth factor-mediated cholinergic neurotrophic activity synthesized by astrocytes and fibroblast. J. Neurochem. 1992;59:919. [PubMed: 1494917]
- 51.
- Vitkovic L, Bockaert J, Jacque C. Inflammatory” cytokines: Neuromodulators in normal brain? J. Neurochem. 2000;74:457. [PubMed: 10646496]
- 52.
- Herx LM, Rivest S, Yong VW. Central nervous system-initiated inflammation and neurotrophin in trauma: IL-1 beta is required for the production of ciliary neurotrophis factor. J. Immunol. 2000;165:2232. [PubMed: 10925311]
- 53.
- Herx LM, Yong VW. Interleukin-1 beta is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 2001;60:961. [PubMed: 11589427]
- 54.
- Allan SM, Tyrrell PJ, Rothwell NJ. Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 2005;5:629. [PubMed: 16034365]
- 55.
- Basu A, Krady JK, Levison SW. Interleukin-1: A master regulator of neuroinflammation. J. Neurosci. Res. 2004;78:151. [PubMed: 15378607]
- 56.
- John GR, Lee SC, Brosnan CF. Cytokines: Powerful regulation of glial cell activation. Neuroscientist. 2003;9:10. [PubMed: 12580336]
- 57.
- John GR, et al. IL-1-regulated responses in astrocytes: Relevance to injury and recorvery. Glia. 2005;49:161. [PubMed: 15472994]
- 58.
- Shrikant P, Benveniste EN. The central nervous system as an immunocompetent organ: role of glia cells in antigen presentation. J. Immunol. 1996;157:1819. [PubMed: 8757296]
- 59.
- Selmaj KW, et al. Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J. Immunol. 1990;144:129. [PubMed: 2104886]
- 60.
- Unsicker K, Strelau J. Functions of transforming growth factor-beta isoforms in the nervous system. Cues based on localization and experimental in vitro and in vivo evidence. Eur. J. Biochem. 2000;26:6972. [PubMed: 11106405]
- 61.
- Benveniste EN, Nguyen VT, O’Keefe GM. Immunological aspects of microglia: relevance to Alzheimer’s disease. Neurochem. Int. 2001;39:381. [PubMed: 11578773]
- 62.
- Logan A, et al. Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur. J. Neurosci. 1994;6:355. [PubMed: 8019673]
- 63.
- Yong VW, et al. Matrix metalloproteinases and diseases of the CNS. Trends Neurosci. 1998;21:75. [PubMed: 9498303]
- 64.
- Wang X, et al. Mechanical injury in rat cortical cultures activates MAPK signaling pathways and induces secretion of matrix metalloproteinase-2 and –9. J. Cereb. Blood Flow Metab. 2001;21:s264.
- 65.
- Morita-Fujimura Y, et al. Overexpression of copper and zinc superoxide dismutase in transgenic mice prevents the induction and activation of matrix metalloproteinases after cold injury induced brain trauma. J. Cereb. Blood Flow Metab. 1999;20:130. [PubMed: 10616801]
- 66.
- Denninger JW, Marletta MA. Guanylate cyclase and the NO/cGMP signaling pathway. Biochem. Biophys. Acta. 1999;1411:334. [PubMed: 10320667]
- 67.
- Hewett SJ, Csernansky CA, Choi DW. Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron. 1994;13:487. [PubMed: 7520256]
- 68.
- Biran R, Martin DC, Tresco PA. Neuronal cell loss accompanies the brain tissue response to chronically implanted silicon microelectrode arrays. Exp. Neurol. 2005;195:115. [PubMed: 16045910]
- 69.
- Pearson GL, Read WT, Feldman WL. Deformation and fracture of small silicon crystals. Acta. Metall. 1957;5:181.
- 70.
- Ommaya AK. Mechanical properties of tissues of the nervous system. J. Biomech. 1967;1:127. [PubMed: 16329300]
- 71.
- Britt RH, Rossi GT. Quantitative analysis of methods for reducing physiological brain pulsations. J. Neurosci. Methods. 1982;6:219. [PubMed: 7144235]
- 72.
- Fee MS. Active stabilization of electrodes for intracellular recording in awake behaving animals. Neuron. 2000;27:461. [PubMed: 11055429]
- 73.
- Goldstein SR, Salcman M. Mechanical factors in the design of chronic recording intracortical microelectrodes. IEEE Trans. Biomed. Eng. 1973;20:260. [PubMed: 4196687]
- 74.
- Gilletti A, Muthuswamy J. Brain micromotion around implants in the rodent somatosensory cortex. J. Neural Eng. 2006;3:189. [PubMed: 16921202]
- 75.
- Lee H, et al. Biomechanical analysis of silicon microelectrode-induced strain in the brain. J. Neural Eng. 2005;2:81. [PubMed: 16317231]
- 76.
- Subbaroyan J, Martin DC, Kipke DR. A finite-element model of the mechanical effects of implantable microelectrodes in the cerebral cortex. J. Neural Eng. 2005;2:103. [PubMed: 16317234]
- 77.
- Ostrow LW, Sachs F. Mechanosensation and endothelin in astrocytes: Hypothetical roles in CNS pathophysiology. Brain Res. Rev. 2005;48:488. [PubMed: 15914254]
- 78.
- Floyd CL, Gorin FA, Lyeth BG. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia. 2005;51:35. [PMC free article: PMC2996279] [PubMed: 15779085]
- 79.
- Floyd CL, et al. Traumatic injury of cultured astrocytes alters inositol (1,4,5)-trisphophate- medicated signaling. Glia. 2001;33:12. [PubMed: 11169788]
- 80.
- Neary JT, et al. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic recepters. J. Neurosci. 2003;23:2348. [PMC free article: PMC6742014] [PubMed: 12657694]
- 81.
- Rzigalinski BA, et al. Effect of Ca2+ on in vitro astrocyte injury. J. Neurochem. 1997;68:289. [PubMed: 8978737]
- 82.
- Lamb RG, et al. Alterations in phosphatidylcholine metabolism of stretch-injured cultured rat astrocytes. J. Neurochem. 1997;68:1904. [PubMed: 9109516]
- 83.
- Willoughby KA, et al. S100B protein is released by in vitro trauma and reduces delayed neuronal injury. J. Neurochem. 2004;91:1284. [PubMed: 15584905]
- 84.
- Ahmed SM, et al. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J. Neurochem. 2000;74:1951. [PubMed: 10800938]
- 85.
- Tavalin SJ, Ellis EF, Satin LS. Mechanical perturbation of cultured cortical neurons reveals a stretch-induced delayed depolarization. J. Neurophysiol. 1995;74:2767. [PubMed: 8747234]
- 86.
- Zhong Y, Bellamkonda RV. Response of glial cells to cyclic strain. 2007 In preparation.
- 87.
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res. Bull. 1999;49:377. [PubMed: 10483914]
- 88.
- Wu CC, et al. Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb. Haemost. 2002;87:1026. [PubMed: 12083482]
- 89.
- Covic L, et al. Pepducin-based intervention of thrombin-receptor signaling and systemic platelet activation. Nat. Med. 2002;8:1161. [PubMed: 12357249]
- 90.
- Ichiyama T, et al. Systemically administered α-melanocyte-stimulating peptide inhibit NF-κB activation in experimental brain inflammation. Brain Res. 1991;836:31. [PubMed: 10415402]
- 91.
- Spataro L, et al. Dexamethasone treatment reduces astroglia responses to inserted neuroprosthetic devices in rat neocortex. Exp. Neurol. 2005;194:289. [PubMed: 16022859]
- 92.
- Zhong Y, Bellamkonda RV. Dexamethasone coated neural probes elicit attenuated inflammatory response and neuronal loss compared to uncoated neural probes. Brain Res. 2007;1148:15. [PMC free article: PMC1950487] [PubMed: 17376408]
- 93.
- Rajora N, et al. α-MSH production, receptors, and influence on neopterin in a human monocyte/macrophage cell line. J. Leukoc. Biol. 1996;59:248. [PubMed: 8603997]
- 94.
- Lipton JM, et al. Mechanisms of anti-inflammatory action of α-MSH peptides. Ann. NY Acad. Sci. 1999;885:173. [PubMed: 10816650]
- 95.
- He W, et al. A novel anti-inflammatory surface for neural electrodes. Adv Nat. [(Accepted, 2007)];
- 96.
- Eaton KP, Henriquez CS. Confounded spikes generated by synchrony within neural tissue models. Neurocomputing. 2005;65:851.
- 97.
- Mountcastle VB. Modality and topographic properties of single neurons of cat’s somatic sensory cortex. J. Neurophysiol. 1957;20:408. [PubMed: 13439410]
- 98.
- Rall W. Electrophysiology of a dendritic neuron model. Biophy. J. 1962;2(2 Pt 2):145. [PMC free article: PMC1366481] [PubMed: 14490040]
- 99.
- Rosenthal F. Extracellular potential fields of single PT-neurons. Brain Res. 1972;36:251. [PubMed: 5009638]
- 100.
- Henze DA, et al. Intracellular features predicted by extracellular recordings in the hippocampus in vivo. J. Neurophysiol. 2000;84:390. [PubMed: 10899213]
- 101.
- Kennedy PR. The cone electrode—A long-term electrode that records from neurites grown onto its recording surface. J. Neurosci. Methods. 1989;29:181. [PubMed: 2796391]
- 102.
- Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: Current facts and future prospects. Prog. Neurobiol. 2003;69:341. [PubMed: 12787574]
- 103.
- Thuret S, Moon LDF, Gage FH. Therapeutic interventions after spinal cord injury. Nat. Rev. Neurosci. 2006;7:628. [PubMed: 16858391]
- 104.
- He W, Bellamkonda RV. Nanoscale neuro-integrative coatings for neural implants. Biomaterials. 2005;26:2983. [PubMed: 15603793]
- 105.
- Ignatius MJ, et al. Bioactive surface coatings for nanoscale instruments: effects on CNS neurons. J. Biomed. Mater. Res. 1998;40:264. [PubMed: 9549621]
- 106.
- Cui XY, et al. Surface modification of neural recording electrodes with conducting polymer/biomolecule blends. J. Biomed. Mater. Res. 2001;56:261. [PubMed: 11340598]
- 107.
- Azemi E, et al. Improving the biocompatibility of neural probes by surface immobilization of L1. Poster presented at Neural Interfaces Workshop; Bethesda, MD. August 21–23, 2006.
- Ultrasoft microwire neural electrodes improve chronic tissue integration.[Acta Biomater. 2017]Ultrasoft microwire neural electrodes improve chronic tissue integration.Du ZJ, Kolarcik CL, Kozai TDY, Luebben SD, Sapp SA, Zheng XS, Nabity JA, Cui XT. Acta Biomater. 2017 Apr 15; 53:46-58. Epub 2017 Feb 6.
- Penetrating multichannel stimulation and recording electrodes in auditory prosthesis research.[Hear Res. 2008]Penetrating multichannel stimulation and recording electrodes in auditory prosthesis research.Anderson DJ. Hear Res. 2008 Aug; 242(1-2):31-41. Epub 2008 Jan 31.
- Reliability of signals from a chronically implanted, silicon-based electrode array in non-human primate primary motor cortex.[IEEE Trans Neural Syst Rehabil...]Reliability of signals from a chronically implanted, silicon-based electrode array in non-human primate primary motor cortex.Suner S, Fellows MR, Vargas-Irwin C, Nakata GK, Donoghue JP. IEEE Trans Neural Syst Rehabil Eng. 2005 Dec; 13(4):524-41.
- Review Artificial vision: needs, functioning, and testing of a retinal electronic prosthesis.[Prog Brain Res. 2009]Review Artificial vision: needs, functioning, and testing of a retinal electronic prosthesis.Chader GJ, Weiland J, Humayun MS. Prog Brain Res. 2009; 175:317-32.
- Review Conceptual and Technical Approaches to Human Neural Ensemble Recordings.[Methods for Neural Ensemble Re...]Review Conceptual and Technical Approaches to Human Neural Ensemble Recordings.Turner DA, Patil PG, Nicolelis MAL. Methods for Neural Ensemble Recordings. 2008
- A Molecular Perspective on Understanding and Modulating the Performance of Chron...A Molecular Perspective on Understanding and Modulating the Performance of Chronic Central Nervous System (CNS) Recording Electrodes - Indwelling Neural Implants
Your browsing activity is empty.
Activity recording is turned off.
See more...