

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis; 2006.

A disease phenotype may be modulated by genetic and non-genetic modifiers. The correlation between genotype and phenotype is a statistical relationship that predicts a physical trait in a person or abnormality in a patient with a given mutation or a group of similar mutations. Analysis of genotype–phenotype correlations in Fabry disease is complicated by a number of factors, such as the high proportion of private mutations, the large phenotypic heterogeneity associated with the same mutation – both among patients from the same family and among those from unrelated families – and the fact that patients with Fabry disease may develop disease-related complications that are observed with high prevalence in the general population. Genotype-related information about the enzyme structure derived from crystallographic analysis, together with measurement of residual enzyme activity, can be of assistance in predicting the likelihood of a severe or attenuated phenotype. Individual genotypes may have pharmacogenomic implications; for example, the maximum possible clinical response to molecular chaperone therapy might be predicted from the correlation between the amount of residual enzyme activity and the associated clinical phenotype. Female heterozygotes exhibit significant phenotypic variability, which could be better understood given more comprehensive epidemiological data.
Introduction
The correlation between genotype (from the Greek genos, meaning race, offspring) and phenotype (from the Greek phaino-, from phainein, meaning to show) is defined as an above-chance probability of a distinct mutation being associated with a particular physical feature or abnormality. The genotype and phenotype share a statistical relationship. The more frequently a specific phenotype is observed in association with a certain genotype, the higher the likelihood that an unrelated person with the same genotype will show the traits or abnormalities observed in the population carrying the same allele(s). This relationship could be expressed as the positive predictive value (PPV) of a given genotype for a particular phenotype, as follows:

Gaucher disease (OMIM 230800 type I; OMIM 231000 type III) provides a classic example of an established genotype–phenotype correlation in a lysosomal storage disorder. In this condition, homozygosity for the p.Leu483Pro (traditional designation, Leu444Pro) allele has a high PPV for the clinically well-defined neuronopathic disease phenotype. On the other hand, the p.Asn409Ser (traditional designation, Asn370Ser) allele has a high PPV for a non-neuronopathic phenotype of the same disorder.
General and epidemiological considerations
Genotype–phenotype analysis in Gaucher disease is facilitated by the high prevalence of a small number of individual mutations, representing founder mutations, in the population. Thus, the phenotype of a large number of patients can be evaluated in a cohort that carries the same mutation. Furthermore, the phenotype of neuronopathic Gaucher disease consists of distinct symptoms rarely found in the general population; for example, supra-nuclear gaze palsy, ataxia, dementia and myoclonic epilepsy.
Analysis of genotype–phenotype correlations in Fabry disease is complicated for a number of reasons. First, unlike Gaucher disease, there is a high proportion of private mutations; that is, most of the families carry different mutations [1]. Secondly, there is a high degree of clinical variability both among patients from the same family and among those from unrelated families with the same mutation. Thirdly, many of the clinical features of Fabry disease are frequently observed in the general population, such as neuropathic and abdominal pain, headache, tinnitus, hearing loss, diarrhoea and cardiovascular disease. In this context, Fabry disease can be seen as a risk factor for commonly encountered pathology [2]. Fourthly, it is possible that part of the phenotype may be due to non-genetic factors, such as slowly progressive and/or focal accumulation of the misfolded mutant proteins, which may lead to formation of insoluble aggregates, as has been hypothesized to explain the increased frequency of Parkinsonism among patients who are heterozygous for Gaucher disease [3].
A crystallographic model of the structure of α-galactosidase A has provided new insights into genotype–phenotype correlations in Fabry disease. Garman and Garboczi [4] mapped GLA mutations onto the three-dimensional structural model of the enzyme and correlated genotype and phenotype by a meta-analysis of the phenotypes reported in the literature. It appears that missense mutations that cause a mild phenotype affect residues that tend to be on the surface of the protein or are easily accessible from the surface. In contrast, missense mutations associated with severe Fabry disease more frequently affect residues that are near the active site or disrupt the hydrophobic core of the enzyme [4].
Residual enzyme activity and phenotypic variants
A premature stop codon or mutations affecting the active site of the enzyme usually result in a non-functional protein or a complete absence of the gene product (functional null allele). In contrast, a missense mutation may result in a considerable loss of metabolic activity of α-galactosidase A, although the protein may still retain some residual enzyme activity [5]. Thus, genotype–phenotype analysis in Fabry disease can be performed by relating the very variable and individual genotype with a quantifiable, intermediate phenotype, such as residual activity of α-galactosidase A. For this purpose, patients can be categorized as having or not having residual enzyme activity. For quality assurance, it is imperative that the determination of α-galactosidase A activity in vitro be conducted in the presence of N-acetylgalactosamine, an inhibitor of α-galactosidase B, in order to suppress the background noise in the assay and avoid falsely elevated results. Patients with residual enzyme activity have been described as exhibiting a later onset of renal involvement, a decreased prevalence of neuropathic pain, fewer dys-morphic features and a lower incidence of hearing loss compared with those individuals with no residual enzyme activity [5–8]. In a natural history study of 25 children, three boys with residual enzyme activity had normal globotriaosylceramide concentrations in blood and urine, and were free of cornea verticillata [9]. Occasionally, patients (usually with residual enzyme activity) are reported as having cardiac or renal variants of Fabry disease. We think that these terms reflect a phenotypic 'snap shot' at a given moment in time and should be used with caution, because they do not take into consideration the fact that Fabry disease is a progressive condition. As such, patients showing the so-called cardiac variant may develop proteinuria, which is, by definition, an involvement of the renal system. Cardiac or renal variants therefore reflect the large spectrum of clinical heterogeneity in Fabry disease and may theoretically exist for any given sign or symptom at a given age (Figure 1). Thus, in male patients it appears that the presence of residual activity shifts the curve representing the burden of disease to the right (Figure 2). Further natural history studies of individuals with residual enzyme activity may reveal a continuous spectrum of phenotypes. In addition to gaining a better understanding of the disorder, such studies could potentially help in the prediction of the outcome of treatment with molecular chaperones (see below).
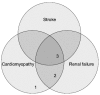
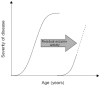
Figure 2
A sufficient level of residual enzyme activity may shift the curve representing the burden of Fabry disease to the right. Data on time and severity are incomplete (indicated by the broken line).
Pharmacogenomics
Enzyme activities in patients who have specific mutations, such as p.Arg301Gln and p.Gln279Glu, can be pharmacologically enhanced in the presence of the amino sugar deoxygalactonojirimycin (DGJ) [10]. Drugs of this class are commonly referred to as molecular chaperones (from the French chaperone, literally 'small hood', meaning to accompany). At low concentrations, these small molecules have been shown to interact with the enzyme and increase its activity, whereas they have inhibitory properties at higher concentrations. In order to assess whether, and to what extent, DGJ has the potential to enhance enzyme activity, in-vitro studies are required to compare the activities of the mutant enzymes in the presence and absence of the chaperone. The disease phenotype of pro-bands with 10% residual enzyme activity, for example, might indicate the maximum achievable level of improvement of the condition in a patient with low residual enzyme activity, that has been therapeutically elevated to 10%. Provided that the in-vitro results translate into clinical benefit, which must be confirmed in clinical trials, this kind of therapy may be useful for subsets of patients with Fabry disease associated with specific genotypes.
Female heterozygotes
In X-linked diseases, heterozygous females are commonly referred to as carriers. This term describes the genetic state of heterozygosity and, consequently, the likelihood of transmitting the given X-linked condition. However, the word 'carrier', by definition, does not provide any information on the female's disease phenotype in X-linked disorders. By reviewing published cases, Dobyns et al. estimated that, in Fabry disease, the penetrance in female heterozygotes is 70% (meaning that 70 out of 100 females have clinical manifestations). In the same study, the clinical severity of the disease in females was estimated as being, on average 4, on a scale from 0 (free from signs or symptoms) to 100 (full disease expression) [11]. However, it is difficult to define penetrance and expressivity reliably in females who are heterozygous for Fabry disease, because, at the time of writing, epidemiological data are incomplete.
Some authors have raised the question of whether Fabry disease is an X-chromosomal dominant or recessive condition. According to the principles summarized by Rimoin and colleagues, an X-chromosomal condition is dominant if the phenotype is similar in both genders and there is an excess of affected females compared with males. By contrast, in a recessive trait, males are affected almost exclusively [12]. In disease registries, females and males are represented at a ratio of approximately 1:1, whereas, statistically, there should be twice as many female heterozygotes as male hemizygotes. Investigators proposing that the condition is dominant will claim that females with Fabry disease are under-diagnosed, whereas those suggesting that the condition is recessive will contend that cases of symptomatic females are over-reported. It is possible that the assessment of female heterozygotes with a mild phenotype is subject to ascertainment bias; that is, the signs and symptoms are not recognized as manifestations of Fabry disease and are consequently not referred to tertiary centres. Both groups agree that, in general, the onset of disease is later and the signs and symptoms tend to be milder in females than in males. Nevertheless, there is increasing evidence documenting high penetrance of Fabry disease in female carriers, with variable phenotypic expression, ranging from no clinical manifestations to the classic disease phenotype, including stroke and renal failure, as seen in hemizygous males [13–19].
In order to reflect the prevalence of symptomatic X-linked conditions in females more appropriately, Dobyns et al. proposed using only the term 'X-linked' trait, because the definitions 'X-linked recessive' or 'X-linked dominant' do not capture the wide spectrum of penetrance and expressivity [11]. The phenotype in heterozygotes may depend on a combination of factors, as is the case in their male relatives, such as the particular GLA mutation or autosomal and/or X-chromosomal modifier genes (see below). In addition, there are a number of issues specific to heterozygotes, such as the pattern of Lyonization in different organ systems, and sequence variants present on the wild-type GLA allele that may also modify the overall phenotype. For the clinical management and counselling of female heterozygotes, it is important to recognize that there is no surrogate marker available to predict the risk of complications, such as arrhythmias, stroke or renal failure.
Genetic modifiers
From the point of view of molecular pathology, Fabry disease may be considered as a metabolic vasculopathy beyond a single gene defect. Therefore, risk factors for cardiovascular and cerebrovascular disease, such as high blood pressure, elevated cholesterol levels and nicotine abuse, will adversely affect the phenotype in a given patient. Thus, the phenotype is probably also modified both by genetic factors unrelated to α-galactosidase A and by environmental factors. In a recent study, the presence of various DNA polymorphisms of genes encoding proteins of the inflammatory and coagulation system, such as interleukin 6 (c.−174G>C), p.Glu298Asp of endothelial nitric oxide synthase, the factor V p.Arg506Gln mutation, and the c.−13A>G and the IVS6 (intron F) +79G>A variants of the gene (PROZ) encoding the vitamin-K-dependent protein Z, were associated with an increased risk of cerebral lesions and stroke in patients with Fabry disease [20]. One might hypothesize that the relative influence of modifier genes may be greater in patients with residual enzyme activity than in those with no enzyme activity. In probands of the former group, GLA-unrelated factors may still have a significant effect on the extent of the GLA-mutation-defined residual enzyme activity and, consequently, on the overall phenotype, which would not be the case in the absence of residual enzyme activity.
Conclusions
Multivariate models based on large-scale systematic data collection are required to derive meaningful associations between genotype and phenotype in Fabry disease. In this context, FOS is likely to prove a valuable resource, providing important information for future efforts aimed at defining the different facets of the genotype–phenotype correlation.
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (National Institutes of Health, Bethesda, MD, USA).
References
- 1.
- Schäfer E, Baron K, Widmer U, Deegan P, Neumann HP, Sunder-Plassmann G. et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat. 2005;25:412. [PubMed: 15776423]
- 2.
- Schiffmann R, Ries M. Fabry's disease – an important risk factor for stroke. Lancet. 2005;366:1754–6. [PubMed: 16298202]
- 3.
- Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–40. [PMC free article: PMC1735652] [PubMed: 15591280]
- 4.
- Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human α-galactosidase. J Mol Biol. 2004;337:319–35. [PubMed: 15003450]
- 5.
- Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G. et al. Natural history of Fabry renal disease: influence of α-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81:122–38. [PubMed: 11889412]
- 6.
- Altarescu GM, Goldfarb LG, Park KY, Kaneski C, Jeffries N, Litvak S. et al. Identification of fifteen novel mutations and genotype–phenotype relationship in Fabry disease. Clin Genet. 2001;60:46–51. [PubMed: 11531969]
- 7.
- Ries M. Quantitative analysis of the neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. School of Medicine. Durham, NC: Duke University; 2005.
- 8.
- Ries M, Moore DF, Robinson CJ, Tifft CJ, Rosenbaum KN, Brady RO. et al. Quantitative dysmorphology assessment in Fabry disease. Genet Med. 2006;8:96–101. [PubMed: 16481892]
- 9.
- Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ. et al. Pediatric Fabry disease. Pediatrics. 2005;115:e344–55. [PubMed: 15713906]
- 10.
- Fan JQ, Ishii S, Asano N, Suzuki Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med. 1999;5:112–5. [PubMed: 9883849]
- 11.
- Dobyns WB, Filauro A, Tomson BN, Chan AS, Ho AW, Ting NT. et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004;129:136–43. [PubMed: 15316978]
- 12.
- Rimoin D, Connor J, Pyeritz RE. Emery and Rimoin's principles and practice of medical genetics. New York: Churchill Livingstone; 1997, p. 93–94.
- 13.
- Guffon N. Clinical presentation in female patients with Fabry disease. J Med Genet. 2003;40:e38. [PMC free article: PMC1735431] [PubMed: 12676911]
- 14.
- Gupta S, Ries M, Kotsopoulos S, Schiffmann R. The relationship of vascular glycolipid storage to clinical manifestations of Fabry disease: a cross-sectional study of a large cohort of clinically affected heterozygous women. Medicine (Baltimore). 2005;84:261–8. [PubMed: 16148726]
- 15.
- MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38:769–75. [PMC free article: PMC1734754] [PubMed: 11732485]
- 16.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C. et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–42. [PubMed: 15025684]
- 17.
- Ries M, Ramaswami U, Parini R, Lindblad B, Whybra C, Willers I. et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767–72. [PubMed: 14505049]
- 18.
- Ries M, Schiffmann R. Fabry disease: angiokeratoma, biomarker, and the effect of enzyme replacement therapy on kidney function. Arch Dermatol. 2005;141:904–5. author reply 905–6. [PubMed: 16027312]
- 19.
- Whybra C, Kampmann C, Willers I, Davies J, Winchester B, Kriegsmann J. et al. Anderson–Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24:715–24. [PubMed: 11804208]
- 20.
- Altarescu G, Moore DF, Schiffmann R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology. 2005;64:2148–50. [PubMed: 15985593]
- Fabry disease: twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes.[J Investig Med. 2000]Fabry disease: twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes.Ashton-Prolla P, Tong B, Shabbeer J, Astrin KH, Eng CM, Desnick RJ. J Investig Med. 2000 Jul; 48(4):227-35.
- Genetic epidemiology of Charcot-Marie-Tooth disease.[Acta Neurol Scand Suppl. 2012]Genetic epidemiology of Charcot-Marie-Tooth disease.Braathen GJ. Acta Neurol Scand Suppl. 2012; (193):iv-22.
- Review Cardiovascular characteristics in Marfan syndrome and their relation to the genotype.[Verh K Acad Geneeskd Belg. 2009]Review Cardiovascular characteristics in Marfan syndrome and their relation to the genotype.De Backer J. Verh K Acad Geneeskd Belg. 2009; 71(6):335-71.
- Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population.[Mol Genet Metab. 2011]Molecular genetics and impact of residual in vitro phenylalanine hydroxylase activity on tetrahydrobiopterin responsiveness in Turkish PKU population.Dobrowolski SF, Heintz C, Miller T, Ellingson C, Ellingson C, Ozer I, Gökçay G, Baykal T, Thöny B, Demirkol M, et al. Mol Genet Metab. 2011 Feb; 102(2):116-21. Epub 2010 Nov 18.
- Review Mucopolysaccharidosis type II: an update on mutation spectrum.[Acta Paediatr. 2007]Review Mucopolysaccharidosis type II: an update on mutation spectrum.Froissart R, Da Silva IM, Maire I. Acta Paediatr. 2007 Apr; 96(455):71-7.
- Genotype–phenotype correlation in Fabry disease - Fabry DiseaseGenotype–phenotype correlation in Fabry disease - Fabry Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...