

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

Loss of telomere homeostasis via chromosome-genomic instability might effectively promote tumour progression. Telomere function may have contrasting roles: inducing replicative senescence and promoting tumourigenesis and these roles may vary between cell types depending on the expression of telomerase enzyme, the level of mutations induced, and deficiency of related DNA repair pathways. Earlier studies in yeast and their recent extension to mammalian systems have convincingly indicated a role for DNA repair proteins in telomere maintenance. An alternative telomere maintenance mechanism has been identified in mouse embryonic stem cells lacking the telomerase RNA unit (mTERC) in which nontelomeric sequences adjacent to existing short stretches of telomere repeats are amplified. Our quest for identifying telomerase-independent or alternative mechanisms for telomere maintenance in mammalian cells has identified the involvement of potential DNA repair factors in such pathways. Studies by us and others have shown the association between the DNA repair factors and telomere function in mammalian cells. Mice deficient in a DNA-break sensing molecule, PARP-1 (poly (ADP)-ribopolymerase), have increased levels of chromosomal instability associated with extensive telomere shortening. Ku80 null cells showed telomere shortening associated with extensive chromosome end fusions whereas Ku80+/- cells exhibited an intermediate level of telomere shortening. This overview will focus mainly on the role of DNA repair/recombination and DNA damage signalling molecules such as PARP-1, DNA-PKcs, Ku70/80, XRCC4 and ATM, which we have been studying for quite sometime. As the maintenance of telomere function is crucial for genomic stability, our results are likely to provide new insights into the telomere regulatory mechanisms and their impact on chromosome instability, ageing and tumour formation.
Telomeres and Telomerase
Telomeres are situated at the ends of linear chromosomes. In most eukaryotes, telomeres consist of non-coding (TTAGGG)n repeats that are associated with an array of proteins.1 Vertebrate telomeres contain such repeated sequences thought to be folded by telomere binding proteins into a duplex T loop structure.2,3 They cap the chromosomes to prevent DNA repair pathways to be triggered for the repair of spontaneous or induced breaks, 4-6 which may result in chromosomal end-to-end fusion. Fusions can be induced through perturbation of the telomeric DNA sequence or depletion of telomere-associated proteins, such that the fundamental structure of the telomere is altered or telomere homeostasis is lost. Specific DNA-binding proteins recognise and associate with either double-strand or single-strand telomeric repeat sequences providing further protection for the chromosome termini.
Telomere repeats are synthesised by the reverse transcriptase enzyme telomerase.7,8 Telomerase is required for the stable maintenance of telomere repeats in vivo and in vitro.9 The appropriate complement of telomere-associated proteins must be present to ensure that the telomere assumes a fully capped configuration and is excluded from processing by DNA damage repair systems.10 Telomeres are believed to serve a protective function in the maintenance of chromosomal genome stability.11 Chromosomes with defective telomere length and/or structure are more susceptible in forming end-to-end fusions that fail to segregate properly in mitosis, resulting in chromosomal breakage accumulation and DNA damage checkpoint activation. The addition of telomeric tracts by telomerase compensates for the progressive loss of telomeric sequences from successive rounds of DNA replication.
Telomeres and Ageing
In most somatic cells, telomeric DNA is lost every time a cell divides.12,13 As a result of this progressive telomere shortening, somatic cells cease to proliferate and become senescent after finite divisions (˜60 population doublings).14 Telomere shortening occurs rapidly in certain cell lines derived from premature ageing disorders, i.e., Werner Syndrome (WS) and Ataxia telangiectasia (AT) leading to premature senescence as compared to age-matched control cell lines15-17 Additionally, cells derived from AT patients show chromosome abnormalities in the form of end-to-end fusions involving telomeres One characteristic feature of Werner Syndrome cells is the “variegated translocation mosaicism” which could be due to shortened telomeres. Thus, telomere shortening is directly related to ageing and senescence in in vitro model systems. However, a recent study has proposed that telomere dynamics at the single cell level in WS fibroblasts are not significantly different from those in normal fibroblasts, and suggest that the accelerated replicative decline seen in WS fibroblasts does not result from accelerated telomere erosion.18
Telomeres and Cancer
The loss of telomeres with age and the resulting genomic instability coupled with onset of telomerase activity are of critical importance in tumourigenesis and in tumour progression.19 Telomere shortening in somatic cells and the subsequent replicative senescence may prevent uncontrolled cell proliferation and thereby malignant transformation. The tumour suppressor function of telomeres has also been suggested.19,20 However, extensive proliferation and the subsequent telomere shortening/lengthening may also result in telomere dysfunction. Chromosome fusions and breaks resulting from telomere dysfunction may facilitate the loss of heterozygosity (LOH) of tumour suppressor genes. Preferential loss of telomere repeats from the ends of a particular chromosome harbouring tumour suppressor genes or oncogenes may result either in their inactivation or activation, repsectively, due to chromosome translocation events and may also facilitate tumour progression. Collectively, telomere dysfunction (see later) and associated chromosome/ genetic instability might effectively promote tumour progression. Thus telomere function may have contrasting roles: inducing replicative senescence and promoting tumourigenesis and these roles may vary between cell types depending on the expression of the enzyme telomerase, the level of mutations induced, and efficiency/deficiency of related DNA repair pathways.
Telomeres and DNA Repair Factors
The question of whether or not telomere shortening triggers double strand DNA break response is still largely unanswered. Telomerase knockout mice showed progressive shortening of telomeres up to the 6th generation after which they became infertile.9,21 However, embryonic fibroblasts obtained from mTER-/- (mouse telomerase RNA or mTERC; telomerase negative) mice could be immortalized in vitro and displayed telomere maintenance with associated chromosomal defects at later passages in culture.22 This study has implicated the existence of telomerase-independent mechanisms in the telomere maintenance. Similarly, we have identified an alternative telomere maintenance mechanism in mouse embryonic stem cells lacking telomerase RNA unit (mTER) with amplification of nontelomeric sequences adjacent to existing short stretches of telomere repeats.23 Recombination of sub-telomeric sequences has been implicated in the telomere maintenance mechanisms in the telomerase negative mouse embryonic stem cells.23 Our quest for identifying telomerase-independent or alternative mechanisms involved in telomere maintenance has implicated the involvement of potential DNA repair factors in such pathways. Several studies have shown the association between the DNA repair factors and telomere function. Extensive studies in yeast have linked the role of DNA repair/recombination and damage signalling molecules in telomere maintenance mechanisms. However, only recently, such roles for these proteins in mammalian cells have been uncovered. This overview will focus mainly on the role of DNA repair /recombination and DNA damage signalling molecules such as DNA-PKcs, ATM, Ku complex, XRCC4 and PARP in telomere chromosome integrity in mammalian cells for which we have sufficient knowledge and data.
Scid (Severe Combined Immuno Deficiency) and Telomeres
In one of our earlier studies, perhaps one of the first in mammalian models, we have shown that mouse scid cells possess abnormal telomere lengths24 being ten times longer than their CB17 parental cells. Besides having abnormally long telomeres, the scid cell line showed unusual telomeric associations. Several chromosomes in the scid cell line were associated with their p-arms to form the so called multi-branched chromosome configuration. The frequency of such multi-branched chromosomes in the scid cell line was about 10%.24 A puzzling observation that longer telomeres and telomere fusions occur in the same cell line has prompted the speculation that probably the telomere telomerase complex may not be efficient in preventing end-to-end fusions in this particular cell line.24 Telomere elongation was seen at both p- and q-arms of the chromosomes suggesting aberrant recombination or defective telomere capping function24,25 (Hande, unpublished results).
To further investigate whether such a difference in telomere length also exists under in vivo condition, the telomere length was analysed in primary cells for different strains of scid mice. In all the strains of scid mice with their parental strains we have studied, 1.5 to 2 times longer telomeres could be detected.25 In contrast to scid cell lines, bone marrow cells from scid mice did not exhibit telomere fusions (Hande, unpublished observation). It should also be noted that the difference in telomere length is wider in the cell lines (ten times) compared to the primary cells (1.5 to 2 times). Bailey et al26 reported a similar occurrence of telomere fusions in a scid cell line though telomere length was not measured in that study. scid mice are deficient in the enzyme DNA-PK (DNA-dependent protein kinase) as a result of the mutation in the gene encoding the catalytic subunit (DNA-PKcs) of this enzyme. Our results on scid cell lines and primary cells from scid mice pointed to the possibility that DNA-PKcs either alone or in complex with other proteins in the nonhomologous end joining (NHEJ) pathway (see later) may, directly or indirectly, be involved in telomere length regulation in mammalian cells.
Nonhomologous End Joining (NHEJ) Complex and Telomeres
Eukaryotic cells use two different pathways for repairing DNA double strand breaks: homologous recombination (HR) and nonhomologous end joining (NHEJ). A key component of the homologous recombination process is Rad51,27 a 50-kDa protein whose expression is cell-cycle dependent, peaking at the S/G2 boundary.28 The NHEJ pathway requires the activity of DNA-PK, a multimeric serine-threonine kinase composed of a catalytic subunit (DNA-PKcs) and two regulatory subunits (Ku70 and Ku80). Ku70 and Ku80 are able to recognise and bind to DNA DSB and then activate DNA-PKcs.29 Another protein that plays a key role in NHEJ is XRCC4,30 which interacts with, and probably controls the function of ligase IV.
DNA repair by NHEJ relies on the Ku70:Ku80 heterodimer in species ranging from yeast to man. In Saccharomyces cerevisiae and Schizosaccharomyces pombe, Ku also controls telomere functions. Based on our observation in scid mouse cells and on the studies in yeast, an analogous role for mammalian Ku complex in telomere maintenance can not be ruled out. Ku70, Ku80, and DNA-PKcs, with which Ku interacts, are associated in vivo with telomeric DNA in several human cell types and we have shown earlier that these associations are not significantly affected by DNA-damaging agents.31 It was also demonstrated that inactivation of Ku80 or Ku70 in the mouse yields telomeric shortening in various primary cell types at different developmental stages (Fig. 1).31 By contrast, telomere length is not altered in cells impaired in XRCC4 or DNA ligase IV, two other NHEJ components (Fig. 1). We also observe higher genomic instability in Ku-deficient cells than in XRCC4-null cells (Fig. 2). This study has suggested that chromosomal instability of Ku-deficient cells results from a combination of compromised telomere stability and defective NHEJ.31
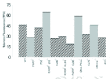
Figure 1
Telomere length dynamics in primary cells from DNA repair deficient mice. Reprinted from Hande with permission; ©2004 S. Karger AG, Basel..

Myung et al32 have demonstrated that the functional inactivation of even a single allele of Ku86 in human somatic cells results in a profound telomere loss, which is accompanied by an increase in chromosomal fusions, translocations, and genomic instability measured by telomere FISH and spectral karyotyping. Taken together, the data available till now support the concept that Ku86, separate from its role in nonhomologous end joining, performs the additional function in human somatic cells of suppressing genomic instability through the regulation of telomere length.
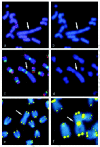
The telomere repeat binding protein, TRF1 and Ku form a complex at the telomere sites.33 The Ku and TRF1 complex determined by coimmunoprecipitation experiments is found to have a specific high-affinity interaction in human cells. Ku does not bind to telomeric DNA directly but localises to telomeric repeats via its interaction with TRF1. Primary mouse embryonic fibroblasts, deficient for Ku80, accumulated a large percentage of telomere fusions (Fig. 2), demonstrating that Ku plays a critical role in telomere capping in mammalian cells.31,33 It was proposed that Ku localizes to internal regions of the telomere via a high-affinity interaction with TRF1.
The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is critical for DNA repair via the nonhomologous end joining pathway. As explained above, bone marrow cells and spontaneously transformed fibroblasts from scid mice have defects in telomere maintenance.24,25 The genetically defective scid mouse arose spontaneously from its parental strain CB17. One known genomic alteration in scid mice is a truncation of the extreme carboxyl terminus of DNA-PKcs, but other as yet unidentified alterations may also exist. In another study, we used a defined system, the DNA-PKcs knockout mouse, to investigate specifically the role played by DNA-PKcs in telomere maintenance. Primary mouse embryonic fibroblasts (MEFs) and primary cultured kidney cells from 6-8 month-old DNA-PKcs-deficient mice accumulated a large number of telomere fusions, yet still retain wild-type telomere length (Figs. 1,2).34 Thus, the phenotype of this defect separates the two-telomere related phenotypes, capping, and length maintenance. DNA-PKcs-deficient MEFs also exhibited elevated levels of chromosome fragments and breaks, which correlated with increased telomere fusions. Based on the high levels of telomere fusions observed in DNA-PKcs deficient cells (e.g., Fig. 3f ), it was concluded that DNA-PKcs plays an important capping role at the mammalian telomere.34 Recently, Espejel et al,35 in a life-long follow-up study of DNA-PKcs-defective mice, observed that DNA-PKcs-defective mice had a shorter life span and showed an earlier onset of ageing-related pathologies than the corresponding wild-type littermates. In addition, DNA-PKcs ablation was associated with a markedly higher incidence of T-lymphomas and infections.35 Therefore, these data link the dual role of DNA-PKcs in DNA repair and telomere length maintenance to organismal ageing and cancer.
PARP-1 and Telomeres
In most eukaryotes, poly(ADP-ribose) polymerase (PARP) recognises DNA strand interruptions generated in vivo. DNA binding by PARP triggers primarily its own modification by the sequential addition of ADP-ribose units to form polymers; this modification, in turn, causes the release of PARP from DNA ends.36 Studies on the effects of the disruption of the gene encoding PARP-1 (Adprt1) in mice have demonstrated roles for PARP in recovery from DNA damage and in suppressing recombination processes involving DNA ends.37-41
Telomeres situated at the natural termini of chromosomes are therefore potential targets for PARP. Telomere shortening was seen in different genetic backgrounds and in different tissues, both from embryos and adult mice (Fig. 1)42 without any change in the in vitro telomerase activity. Furthermore, cytogenetic analysis of mouse embryonic fibroblasts has revealed that lack of PARP-1 is associated with severe chromosomal instability, characterised by increased frequencies of chromosome fusions and aneuploidy (Figs. 2,3). The absence of PARP-1 did not affect the presence of single-strand overhangs, naturally present at the ends of telomeres. The above study has therefore revealed an unanticipated role for PARP-1 in telomere length regulation; though a later study using PARP-1 knockout mice with a different genetic background43 did not reveal any significant change in telomere length. However, it cannot be ruled out that different genetic background mice and different mutations of the same gene might yield different results. Studies are presently in progress in my laboratory to evaluate the role of PARP-1 to address this issue. It will also be interesting to study the importance of other PARP molecules in genome stability.
Ataxia Telangiectasia Mutated (ATM) and Telomeres
Ataxia-telangiectasia (AT) is an autosomally recessive human genetic disease with pleiotropic defects such as neurological degeneration, immunodeficiency, chromosomal instability, cancer susceptibility and premature aging. Cells derived from AT patients and ataxia-telangiectasia mutated (ATM)-deficient mice show slow growth in culture and premature senescence. ATM, which belongs to the PI3 (phosphatidylinositol 3) kinase family along with DNA-PK, plays a major role in signalling the p53 response to DNA strand breaks. Telomere maintenance is perturbed in yeast strains lacking genes homologous to ATM and cells from patients with AT have short telomeres.16 We examined the length of individual telomeres in cells from Atm-/- mice by fluorescence in situ hybridisation techniques. Telomeres were extensively shortened in multiple tissues of Atm-/- mice44 (Fig. 1). More than the expected number of telomere signals was observed in interphase nuclei of Atm-/- mouse fibroblasts. Signals corresponding to 5-25 kb of telomeric DNA that were not associated with chromosomes were also noticed in Atm-/- metaphase spreads. Extrachromosomal telomeric DNA was also detected in fibroblasts from AT patients and may represent fragmented telomeres or by-products of defective replication of telomeric DNA. These results suggested a role for ATM in telomere maintenance and replication, which may contribute to the poor growth of Atm-/- cells and increased tumour incidence in both AT patients and Atm-/- mice.44 Lustig45 has suggested that formation of circular telomeric fragments could be due to the telomere rapid deletion as was seen in yeast. Recently, Herbig et al46 looked at the association of ATM with the DNA damage foci induced by dysfunctional telomere (telomere induced foci) in senescent cells47,48 and found that ATM colocalises with most senescent telomere induced foci.46 Oxidative damage has been suggested to be one of the factors that influences the telomere status in AT cells. We are currently studying the precise role of chronic oxidative damage in normal and ATM-/- cells and in Atm-/- mice with special emphasis on genome stability and ageing.
Concerted Roles of PARP-1 and p53 on Telomeres
Genomic instability is often caused by mutations in genes that are involved in DNA repair and/or cell cycle checkpoints, and it plays an important role in tumourigenesis. To confirm our previous observation of telomere shortening in PARP-1 mice, we took advantage of PARP-1 and p53 double knockout mice to study the telomere-related chromosome instability and tumourigenesis in these mice. As PARP is thought to protect genomic stability,38 its functional interaction with the guardian of the genome, p53 was tested using PARP-1-/-p53-/- mice. Compared to single-mutant cells, PARP-1 and p53 double-mutant cells exhibit many severe chromosome aberrations, including a high degree of aneuploidy, fragmentations, and end-to-end fusions, which to a large extent is attributable to telomere dysfunction.49 While PARP-/- cells showed telomere shortening and p53-/- cells showed normal telomere length, inactivation of PARP-1 in p53-/- cells surprisingly resulted in very long and heterogeneous telomeres, suggesting a functional interplay between PARP-1 and p53 at the telomeres (Fig. 1). Strikingly, PARP-1 deficiency widens the tumour spectrum in mice deficient in p53, resulting in a high frequency of carcinomas in the mammary gland, lung, prostate, and skin, as well as brain tumours. The enhanced tumourigenesis is likely to be caused by PARP-1 deficiency, which facilitates the loss of function of tumour suppressor genes as demonstrated by a high rate of loss of heterozygosity at the p53 locus in these tumours.49 These results indicated that PARP-1 and p53 interact to maintain genome integrity and identify PARP as a cofactor for suppressing tumourigenesis. It is however not clear whether the long and heterogeneous telomeres are responsible for the wider tumour spectrum in these double knockout mice.
Interplay between PARP and Ku80 on Telomere-Chromosome Integrity
PARP-1 and Ku80 null cells showed telomere shortening associated with chromosome instability. Interestingly, haplo-insufficiency of PARP-1 in Ku80 null cells caused more severe telomere shortening and accumulation of chromosome abnormalities compared to either PARP-1 or Ku80 single null cells. Cytogenetic analysis of these cells revealed that many chromosome ends lack detectable telomeres as well. These results demonstrate that DNA break-sensing molecules, PARP-1 and Ku80, synergistically function at telomeres and play an important role in the maintenance of chromosome integrity. More importantly haplo-insufficiency of Ku80 in PARP-1-/- mice promoted the development of hepatocellular adenoma and hepatocellular carcinoma (HCC).50 These tumours exhibited a multistage tumour progression associated with the loss of E-cadherin expression and the mutation of beta-catenin. Cytogenetic analysis revealed that Ku80 heterozygosity elevated chromosomal instability in PARP-1-/- cells and that these liver tumours harboured a high degree of chromosomal aberrations including fragmentations, end-to-end fusions, and recurrent nonreciprocal translocations. These features are reminiscent of human HCC. Taken together, these data implicate a synergistic function of Ku80 and PARP-1 in minimising chromosome aberrations and cancer development.
Homologous Recombination Proteins and Telomeres
The homologous recombination (HR) DNA repair pathway participates in telomere length maintenance in yeast but its putative role at mammalian telomeres is currently being studied. Strong evidence for telomere loss in the murine hypomorphic Rad50(S/S) homozygous mutant is shown by the frequent formation of fusions between chromosomes with short telomeres, which is consistent with a rapid loss of the originally elongated telomere.51 Another component of homologous recombination pathway, NBS1 has been implicated in telomere maintenance. It has been shown that cells from NBS patients have strong telomere phenotypes. NBS cells have significantly shorter telomeres than their normal counterparts.52 A rapid rate of telomere loss in cell lines from Nijmegen breakage syndrome (NBS) patients (who carry a nonnull NBS1 allele), might be caused by a combination of replicative loss, other mechanisms of attrition and an elevated rate of telomeric (end to end) fusions53 (Hande et al unpublished results; (Fig. 3a,b). Introduction of both NBS1 and hTERT (telomerase catalytic subunit) genes was necessary to rescue the telomere shortening phenotypes in fibroblasts from NBS patients52 indicating a strong interaction between NBS1 and telomerase complex. NBS1 is present at meiotic telomeres in mammalian cells.54
Jaco et al55 have shown that Rad54-deficient mice show significantly shorter telomeres than wild-type controls indicating that Rad54 activity plays an essential role in telomere length maintenance in mammals.55 Recently, Tarsounas et al56 have reported that Rad51D is also involved in telomere maintenance. RAD51D was shown to localize to the telomeres of both meiotic and somatic cells. Rad51d-/-Trp53-/- double knockout mouse embryonic fibroblasts exhibited telomere DNA repeat shortening compared to Trp53-/- or wild-type controls accompanied by elevated levels of chromosomal aberrations.
Concluding Remarks
It is evident from the telomere-chromosome data and associated genome instability data from DNA repair deficient mouse cells that the DNA damage/repair and signalling molecules play a vital role in the protection of telomeres and chromosomes and thereby maintain the genome integrity. As indicated in Figure 1, there seems to be a modest loss of telomeres in the cells lacking some of the DNA repair factors. This loss is approximately 30 to 40% of the original telomeres. Data on telomerase negative embryonic stem cells and embryonic fibroblasts lacking telomerase RNA indicated that a loss of approximately 60 to 70% of telomeric repeats was needed for the cells to become fusigenic and to induce the chromosome end-to-end fusion events in these cells. The telomeres are maintained by telomerase-independent mechanisms in mTER-/- mouse cells at later passages. Based on the above observations, it is tempting to speculate that at least in mice, a fraction of telomeres are also maintained by the factors involved in DNA repair/damage response and DNA damage signalling molecules. However, both telomerase-dependent and telomerase-independent mechanisms coexist to maintain telomeres in the mouse cells. It is plausible that telomere maintenance will be compromised by one pathway in the absence of another pathway. More interestingly, loss of DNA repair factors will render the cells to lose telomeres and a majority of these cells exhibit premature senescence. On the other hand, telomerase negative mouse cells escape senescence and could be immortalised in vitro through accumulation of chromosome instability. These two pathways interact with each other very efficiently and loss of either will lead to severe telomere mediated chromosome instability leading to tumourigenesis in mice (Fig. 4). Mice lacking genes involved in either DNA repair complex and/or telomerase complex develop spontaneous tumours and cells from these mice show telomere related chromosome abnormalities. Loss of telomere homeostasis might have contributed to the occurrence of severe chromosome instability in these mice, which led to the spontaneous development of tumours. However, given the many pathways in which DNA damage/repair and signalling molecules are involved, telomeric loss defects might represent a direct or indirect effect of the primary defect.

Figure 4
Role(s) of DNA damage signalling molecules and DNA repair factors in telomere mediated chromosome-genomic instability and tumourigenesis. Modified rom Hande with permission; ©2004 S. Karger AG, Basel.
A study by d'Adda di Fagagna et al47 documented the appearance of DNA damage-inducible foci in senescent primary fibroblasts and provided the functional evidence that these foci contribute directly to the senescent phenotype. This study has established the molecular and mechanistic similarities between critically shortened and uncapped telomeres and that the telomere attrition does induce a DNA damage response in primary human cells.47 Components of the DNA damage response associated with DNA end-to-end joining or homologous recombination have been proposed to mediate alterations in telomere structure believed to be required for telomerase access.57 These activities have also been linked to ‘telomere rapid deletion’ in yeast, which has been proposed to reset telomere size during meiosis.45 Telomere rapid deletion has been defined as an end-mediated intrachromatid homologous-recombination event that results in a deleted telomere and a linear or circular by-product.45 Catastrophic loss of telomere sequences in mouse cells lacking key DNA repair proteins, as observed in our studies with DNA repair deficient mouse cells, might reflect the above mentioned telomere rapid deletion process. However, this process needs to be thoroughly investigated in defined mammalian systems. Many of the DNA repair deficient mice or mouse cells (e.g., Ku80-/- and Atm-/-) do exhibit premature ageing phenotype and cancer susceptibility.58 Therefore, telomere rapid deletion and subsequent defects in genome maintenance systems in the repair deficient mice might contribute to the observed tumour susceptibility phenotype and accelerated ageing.
Acknowledgements
I would like to express my gratitude to Dr. Adayabalam S. Balajee, Centre for Radiological Research, Columbia University, New York, USA for his thoughtful suggestions and criticisms. Current research in my laboratory (Genome Stability Laboratory) is supported by grants from Academic Research Fund, National University of Singapore and Oncology Research Institute, Office of Life Sciences, National University Medical Institutes, Singapore. Dr. Anuradha Poonepalli and Mr. Aik Kia Khaw are acknowledged for their help during the manuscript preparation. Thanks are also due to Dr. Veena Hande for critically reading the manuscript.
References
- 1.
- McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34:331–358. [PubMed: 11092831]
- 2.
- Griffith JD, Comeau L, Rosenfield S. et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97(4):503–14. [PubMed: 10338214]
- 3.
- Moyzis RK, Buckingham JM, Cram LS. et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988;85(18):6622–6. [PMC free article: PMC282029] [PubMed: 3413114]
- 4.
- Blackburn EH. Structure and function of telomeres. Nature. 1991;350(6319):569–73. [PubMed: 1708110]
- 5.
- Greider CW. Telomere length regulation. Annu Rev Biochem. 1996;65:337–65. [PubMed: 8811183]
- 6.
- Zakian VA. Telomeres: Beginning to understand the end. Science. 1995;270(5242):1601–7. [PubMed: 7502069]
- 7.
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43(2 Pt 1):405–13. [PubMed: 3907856]
- 8.
- Lingner J, Hughes TR, Shevchenko A. et al. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science. 1997;276(5312):561–7. [PubMed: 9110970]
- 9.
- Blasco MA, Lee HW, Hande MP. et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25–34. [PubMed: 9335332]
- 10.
- Heacock M, Spangler E, Riha K. et al. Molecular analysis of telomere fusions in Arabidopsis: Multiple pathways for chromosome end-joining. EMBO J. 2004;23(11):2304–2313. [PMC free article: PMC419913] [PubMed: 15141167]
- 11.
- Greider CW. Telomeres. Curr Opin Cell Biol. 1991;3(3):444–51. [PubMed: 1892656]
- 12.
- Allsopp RC, Vaziri H, Patterson C. et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89(21):10114–8. [PMC free article: PMC50288] [PubMed: 1438199]
- 13.
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–60. [PubMed: 2342578]
- 14.
- Granger MP, Wright WE, Shay JW. Telomerase in cancer and aging. Crit Rev Oncol Hematol. 2002;41(1):29–40. [PubMed: 11796230]
- 15.
- Kruk PA, Rampino NJ, Bohr VA. DNA damage and repair in telomeres: Relation to aging. Proc Natl Acad Sci USA. 1995;92(1):258–262. [PMC free article: PMC42857] [PubMed: 7816828]
- 16.
- Metcalfe JA, Parkhill J, Campbell L. et al. Accelerated telomere shortening in ataxia telangiectasia. Nat Genet. 1996;13(3):350–3. [PubMed: 8673136]
- 17.
- Schulz VP, Zakian VA, Ogburn CE. et al. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum Genet. 1996;97(6):750–4. [PubMed: 8641691]
- 18.
- Baird DM, Davis T, Rowson J. et al. Normal telomere erosion rates at the single cell level in Werner syndrome fibroblast cells. Hum Mol Genet. 2004 [PubMed: 15150162]
- 19.
- de Lange T, DePinho RA. Unlimited mileage from telomerase? Science. 1999;283(5404):947–9. [PubMed: 10075559]
- 20.
- Artandi SE, DePinho RA. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr Opin Genet Dev. 2000;10(1):39–46. [PubMed: 10679392]
- 21.
- Lee HW, Blasco MA, Gottlieb GJ. et al. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569–74. [PubMed: 9560153]
- 22.
- Hande MP, Samper E, Lansdorp P. et al. Telomere length dynamics and chromosomal instability in cells derived from telomerase null mice. J Cell Biol. 1999;144(4):589–601. [PMC free article: PMC2132934] [PubMed: 10037783]
- 23.
- Niida H, Shinkai Y, Hande MP. et al. Telomere maintenance in telomerase-deficient mouse embryonic stem cells: Characterization of an amplified telomeric DNA. Mol Cell Biol. 2000;20(11):4115–27. [PMC free article: PMC85781] [PubMed: 10805753]
- 24.
- Slijepcevic P, Hande MP, Bouffler SD. et al. Telomere length, chromatin structure and chromosome fusigenic potential. Chromosoma. 1997;106(7):413–21. [PubMed: 9391214]
- 25.
- Hande P, Slijepcevic P, Silver A. et al. Elongated telomeres in scid mice. Genomics. 1999;56(2):221–3. [PubMed: 10051409]
- 26.
- Bailey SM, Meyne J, Chen DJ. et al. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci USA. 1999;96(26):14899–904. [PMC free article: PMC24745] [PubMed: 10611310]
- 27.
- Baumann P, Benson FE, West SC. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell. 1996;87(4):757–766. [PubMed: 8929543]
- 28.
- Yamamoto A, Taki T, Yagi H. et al. Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol Gen Genet. 1996;251(1):1–12. [PubMed: 8628240]
- 29.
- Critchlow SE, Jackson SP. DNA end-joining: From yeast to man. Trends Biochem Sci. 1998;23(10):394–8. [PubMed: 9810228]
- 30.
- Critchlow SE, Bowater RP, Jackson SP. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr Biol. 1997;7(8):588–98. [PubMed: 9259561]
- 31.
- d'Adda di Fagagna F, Hande MP, Tong WM. et al. Effects of DNA nonhomologous end-joining factors on telomere length and chromosomal stability in mammalian cells. Curr Biol. 2001;11(15):1192–6. [PubMed: 11516951]
- 32.
- Myung K, Ghosh G, Fattah FJ. et al. Regulation of telomere length and suppression of genomic instability in human somatic cells by Ku86. Mol Cell Biol. 2004;24(11):5050–5059. [PMC free article: PMC416406] [PubMed: 15143195]
- 33.
- Hsu HL, Gilley D, Galande SA. et al. Ku acts in a unique way at the mammalian telomere to prevent end joining. Genes Dev. 2000;14(22):2807–12. [PMC free article: PMC317061] [PubMed: 11090128]
- 34.
- Gilley D, Tanaka H, Hande MP. et al. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci USA. 2001;98(26):15084–8. [PMC free article: PMC64987] [PubMed: 11742099]
- 35.
- Espejel S, Martin M, Klatt P. et al. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004;5(5):503–509. [PMC free article: PMC1299048] [PubMed: 15105825]
- 36.
- Lindahl T. Recognition and processing of damaged DNA. J Cell Sci Suppl. 1995;19:73–7. [PubMed: 8655650]
- 37.
- de Murcia JM, Niedergang C, Trucco C. et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA. 1997;94(14):7303–7. [PMC free article: PMC23816] [PubMed: 9207086]
- 38.
- Jeggo PA. DNA repair: PARP - another guardian angel? Curr Biol. 1998;8(2):49–51. [PubMed: 9427640]
- 39.
- Lindahl T, Satoh MS, Dianov G. Enzymes acting at strand interruptions in DNA. Philos Trans R Soc Lond B Biol Sci. 1995;347(1319):57–62. [PubMed: 7746855]
- 40.
- Morrison C, Smith GC, Stingl L. et al. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat Genet. 1997;17(4):479–82. [PubMed: 9398855]
- 41.
- Wang ZQ, Stingl L, Morrison C. et al. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997;11(18):2347–58. [PMC free article: PMC316515] [PubMed: 9308963]
- 42.
- d'Adda di Fagagna F, Hande MP, Tong WM. et al. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet. 1999;23(1):76–80. [PubMed: 10471503]
- 43.
- Samper E, Goytisolo FA, Menissier-de Murcia J. et al. Normal telomere length and chromosomal end capping in poly(ADP-ribose) polymerase-deficient mice and primary cells despite increased chromosomal instability. J Cell Biol. 2001;154(1):49–60. [PMC free article: PMC2196874] [PubMed: 11448989]
- 44.
- Hande MP, Balajee AS, Tchirkov A. et al. Extra-chromosomal telomeric DNA in cells from Atm(-/-) mice and patients with ataxia-telangiectasia. Hum Mol Genet. 2001;10(5):519–28. [PubMed: 11181576]
- 45.
- Lustig AJ. Clues to catastrophic telomere loss in mammals from yeast telomere rapid deletion. Nat Rev Genet. 2003;4(11):916–923. [PubMed: 14634639]
- 46.
- Herbig U, Jobling WA, Chen BP. et al. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004;14(4):501–513. [PubMed: 15149599]
- 47.
- d'Adda dF, Reaper PM, Clay-Farrace L. et al. A DNA damage checkpoint response in telomere initiated senescence. Nature. 2003;426(6963):194–198. [PubMed: 14608368]
- 48.
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13(17):1549–1556. [PubMed: 12956959]
- 49.
- Tong WM, Hande MP, Lansdorp PM. et al. DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol. 2001;21(12):4046–54. [PMC free article: PMC87066] [PubMed: 11359911]
- 50.
- Tong WM, Cortes U, Hande MP. et al. Synergistic role of Ku80 and poly(ADP-ribose) polymerase in suppressing chromosomal aberrations and liver cancer formation. Cancer Res. 2002;62(23):6990–6. [PubMed: 12460917]
- 51.
- Bender CF, Sikes ML, Sullivan R. et al. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002;16(17):2237–2251. [PMC free article: PMC186667] [PubMed: 12208847]
- 52.
- Ranganathan V, Heine WF, Ciccone DN. et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires NBS and telomerase catalytic subunit. Curr Biol. 2001;11(12):962–6. [PubMed: 11448772]
- 53.
- Tauchi H, Matsuura S, Kobayashi J. et al. Nijmegen breakage syndrome gene, NBS1, and molecular links to factors for genome stability. Oncogene. 2002;21(58):8967–8980. [PubMed: 12483513]
- 54.
- Lombard DB, Guarente L. Nijmegen breakage syndrome disease protein and MRE11 at PML nuclear bodies and meiotic telomeres. Cancer Res. 2000;60(9):2331–4. [PubMed: 10811102]
- 55.
- Jaco I, Munoz P, Goytisolo F. et al. Role of mammalian Rad54 in telomere length maintenance. Mol Cell Biol. 2003;23(16):5572–80. [PMC free article: PMC166323] [PubMed: 12897131]
- 56.
- Tarsounas M, Munoz P, Claas A. et al. Telomere maintenance requires the RAD51D recombination/ repair protein. Cell. 2004;117(3):337–347. [PubMed: 15109494]
- 57.
- Blackburn EH. Telomere states and cell fates. Nature. 2000;408(6808):53–56. [PubMed: 11081503]
- 58.
- Hasty P, Campisi J, Hoeijmakers J. et al. Aging and genome maintenance: Lessons from the mouse? Science. 2003;299(5611):1355–1359. [PubMed: 12610296]
- 59.
- Hande MP. DNA repair factors and telomere-chromosome integrity in mammalian cells. Cytogenet Genome Res. 2004;104(1-4):116–122. [PubMed: 15162024]
- Telomeres and Telomerase
- Telomeres and Ageing
- Telomeres and Cancer
- Telomeres and DNA Repair Factors
- Scid (Severe Combined Immuno Deficiency) and Telomeres
- Nonhomologous End Joining (NHEJ) Complex and Telomeres
- PARP-1 and Telomeres
- Ataxia Telangiectasia Mutated (ATM) and Telomeres
- Concerted Roles of PARP-1 and p53 on Telomeres
- Interplay between PARP and Ku80 on Telomere-Chromosome Integrity
- Homologous Recombination Proteins and Telomeres
- Concluding Remarks
- Acknowledgements
- References
- Orchestration of Telomeres and DNA Repair Factors in Mammalian Cells: Implicatio...Orchestration of Telomeres and DNA Repair Factors in Mammalian Cells: Implications for Cancer and Ageing - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...