IGFs (Insulin-like Growth Factors) are peptides known to stimulate a wide range of actions on different tissues. Indeed, IGFs can stimulate anabolism, acute metabolic effects as well as enhancing more chronic effects such as cell proliferation and differentiation together with protecting cells from apoptosis. Recently, it was shown that IGFs induce an epithelial to mesenchymal transition (EMT), a crucial morphogenic event during development and transformation. Here, the cellular and molecular aspects of IGF-induced EMT are reviewed. Major signalling pathways downstream of IGFs are described in order to introduce molecules that are believed to convey the EMT signal. The roles and targets of these molecules are analysed. The importance of IGFs in cellular events which when dysregulated lead to neoplasia is discussed in this review.
Introduction
The insulin-like growth factor (IGF) family contains two highly similar, single-chain ligands of ∼7.5 kDa termed IGF-I and IGF-II. In most species, the major forms of IGF-I and IGF-II are molecules of 70 and 67 amino acids, respectively. However, there are additional isoforms of IGF-I and IGF-II which arise from differential promoter usage, variable transcription initiation sites, differential RNA splicing and alternative polyadenylation site usage.
Both mature proteins have 4 domains: B (N-terminal), C, A and D (C-terminal). This organization is similar to that of pro-insulin which also contains domains B, C and A. The amino acid similarity between insulin (Ins) and IGFs is restricted to the A and B domains but nevertheless insulin, IGF-I and IGF-II are believed to have evolved from a single common ancestor. The primary sequences of both IGF-I and IGF-II have been highly conserved in Mammals (for review see 1).
IGFs are ligands which bind to the Insulin Receptor (IR), the IGF-I Receptor (IGF-1R) and the IGF-II Receptor (IGF-2R). IR and IGF-1R both are Receptor Protein Tyrosine Kinases (RPTK) with intrinsic tyrosine kinase activity. Their overall structure resembles that of the archetypal epidermal growth factor receptor. The mature IR and IGF-1R are large transmembrane glycoproteins (∼300-350kDa) composed of two α and two β subunits. The α and β subunits are proteolytically cleaved from a single common proreceptor and covalently linked into dimers by disulphide bonds. The two α subunits are held together by at least two disulphide bonds whereas only a single -S-S- bond is responsible for the α-β linkage. These dimers then form a functional α2β2 heterotetrameric complex. The putative ligand binding pockets are located in the cysteine rich portion of the extracellular α subunits. About one third of the β subunit is extracellular and the remainder spans the membrane and contains tyrosine kinase domain, ATP and substrate binding pockets and autophosphorylation sites. In sharp contrast, IGF-2R is a single protein of ∼300kDa, devoid of any apparent intrinsic catalytic activity. The same molecule has also been identified as a cation independent receptor for mannose 6-phosphate, being involved in the transport of enzymes linked with phosphorylated mannose to the lysosome. It is believed that IGF-2R is a multifunctional protein binding both IGF-II and proteins tagged with a phosphorylated mannose recognition marker (for review see 1). It is unclear, however, whether any signal transduction pathway relevant to Ins or IGFs is activated either upon ligand binding at the cell surface or upon ligand-induced receptor internalization.
Any study of Ins, IGF-I and IGF-II ligands and their receptor is made infinitely more complicated by the fact that both the receptors and ligands are promiscuous; practically all of the three receptors bind heterologous ligands, but in a concentration-dependent manner. For example, IGF-1R binds these ligands with the following relative affinities: IGF-I > IGF-II >> Insulin; and IGF-2R: IGF-II> IGF-I, but Ins does not bind at physiological concentrations to IGF-1R or IGF-2R (fig. 1). Obviously, each receptor has the highest affinity for its cognate ligand but typically a 100 to 1000 times lower affinity for any of the others. The most crucial point to note is that the affinities of either IGF-I or IGF-II for the IGF-1R are so favourable that in mammals many of the biological effects attributed to IGF-I and IGF-II are believed to be mediated exclusively via IGF-1R.

The biological effects of IGF ligands (but not Ins) are further modulated by a family of structurally related, secreted proteins termed IGF Binding Proteins (IGFBP). They are present ubiquitously in the circulation and in the extracellular space. IGFBP bind to the IGFs with affinities comparable to those for the IGFRs. IGFBP regulate IGFs bioavaibility by (i) protecting them from degradation, (ii) facilitating their transport and (iii) promoting or inhibiting IGF binding to their receptors.
IGFs are produced during both prenatal and postnatal phases where they exert a general anabolic or growth promoting effect. During this time, endocrine IGFs are mainly synthesized by hepatocytes. In human, the serum concentrations of IGF-I and IGF-II increase with age. At 30 weeks of gestation, the serum levels of IGF-I and IGF-II are 20 ng/mL and 100 ng/mL, respectively; at birth, they are 100 ng/mL and 300 ng/mL, respectively; and after birth, they are 200 ng/mL and 700 ng/mL, respectively. IGF-1R is broadly expressed in various tissues during embryonic development. Postnatally, in addition, local production of IGFs begins and continues in numerous cell types including the ovary, testis, nervous system, bones, skeletal muscles. When locally synthesized IGFs have paracrine and autocrine functions.1
IGFs are believed to be important for growth and development and their role has been examined genetically in the mouse. Somatic undergrowth is a typical characteristic of the mutant mice. If the IGF-II gene is disrupted, for example, birth weight is 61.5 % of the weight of wild-type mice. When IGF-1R gene is disrupted, embryo birth weight is 46 % the weight of wild-type littermates. Mutation of both the IGF-II and IGF-1R genes gives an embryo birth weight which is 34 % of that of wild-type littermates. The comparison of these phenotypes suggests that, in vivo, IGF-II signals through another receptor which was identified as IR.
The powerful mitogenic effects of IGFs on many cell types partially explain these phenotypes. Indeed, IGFs induce cell growth and cell division in vivo and in vitro: (1) IGF-I stimulates DNA synthesis within granulosa and granulosa-luteal cells; (2) IGF-I mediates proliferative effects of oestradiol during the endometrial proliferative phase; (3) IGFs stimulate spermatogonial DNA synthesis and Sertoli and Leydig cell division; (4) IGFs regulate proliferation of prostate epithelial cells and of bladder urothelial and smooth cells; (5) IGFs promote axonal growth and partial regeneration even in adult animals; (6) IGFs promote astrocyte proliferation after physical injury in central nervous system; (7) IGFs are key regulators of bone formation by stimulating osteoblast proliferation; (8) IGFs induce proliferation of skeletal muscle cells.1 IGFs also regulate cell number via regulation of apoptosis. In most cases, IGFs are anti-apoptotic and can therefore be classified as survival factors. The overall mitogenic effect of IGFs suggests that abnormal activation of the IGF pathway could be a crucial event in the formation of many cancers. Indeed, IGF signalling is dysregulated in certain pancreatic cancers.
Moreover, IGF participates in physiological invasion mechanisms occurring during development, such as trophoblast invasion of the endometrium during implantation.1
Surprisingly, IGF promotes the acquisition or maintenance of the differentiated status of certain cell types: (1) IGF stimulates differentiation of skeletal muscle cells and Leydig cells; (2) IGF promotes oestradiol production by granulosa and granulosa-luteal cells. IGF-I also synergises with FSH (Follicle Stimulating Hormne) and hCG (human Chorionic Gonadotropin) by increasing oestradiol and progesterone production; (3) IGF modulates androgen production by Sertoli and Leydig cells; (4) IGF may also be important in neuronal differentiation, and promotes myelination in central and peripheral nervous systems.1 Therefore, IGF stimulates both proliferation and differentiation, commonly accepted as being two mutually exclusive cellular events.
This chapter will describe a recently identified biological effect of IGF peptides namely their role in the Epithelial to Mesenchymal Transition (EMT). First, the cellular and molecular events induced by IGFs and associated with EMT will be described. As the induction of EMT by IGFs must proceed through the activation of IGF-1R, the main signalling cascades induced by IGF-1R will be described. Finally, we will explain how the molecular events of this EMT are initiated in the light of the known signalling pathways downstream from IGFRs.
IGFs and Regulation of Cell-cell Adhesion: Cellular and Molecular Aspects
The effects of IGF-I and IGF-II on cell-cell adhesion have been studied using in vitro models including the epithelial cell line NBT-II (= Nara Bladder Tumor II).2 The NBT-II cell line is derived from a chemically induced rat bladder carcinoma. This cell line possesses a typical epithelial cell morphology with extensive cell-cell contacts and has also been used to study the epithelial to mesenchymal transition induced by various factors.3>-5 Importantly, cell-cell adhesion between NBT-II cells is mainly mediated by E-cadherin.
IGFs Induce EMT
In the absence of IGFs peptides and Ins, NBT-II cells have the classical morphology of epithelial cells: they are polarized and tightly attached to one another. Upon either IGF-I or IGF-II treatment (subsequently referred to as IGF), NBT-II cells lose their cell-cell contacts, flatten and spread (fig. 2). Other epithelial cells, such as MCF7 and MDCK cells, or embryonic stem (ES) cells undergo the same morphological changes upon IGF treatment but the mesenchymal NIH-3T3 cells do not. Importantly, these morphological modifications appear very quickly after IGF addition (typically within one hour of application) and are not associated with cell division.
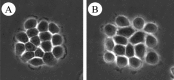
Figure 2
Phase contrast microscopy of NBT-II cells. The NBT-II cells were treated (B) or not (A) with 100 ng/mL of IGF-II for 1 hour. Note that the cells located at the periphery of the colony spread and loose the cell-cell contacts.
The visible loss of cell-cell contacts is associated with various molecular events: (i) the rapid internalisation of E-cadherin and desmoplakin leading to the disruption of junctional complexes such as adherens junctions, desmosomes and certainly gap junctions; and (ii) after 4 days of treatment with IGF the neoexpression of the mesenchymal-specific marker vimentin. Therefore, IGF induces a series of cellular and molecular modifications characteristic of standard EMT. This transition is reversible. After removing IGF from the medium, NBT-II cells revert to an epithelial morphology within 24 hours, and E-cadherin is relocalized at cell-cell contacts.3
A basic EMT can be defined as the transition between tightly attached and polarized epithelial cells to a set of loosely attached and nonpolarized mesenchymal cells. In addition to the characteristics cited above, a full EMT is associated with cellular motility. IGF is able to induce the migration of certain epithelial cells such as MCF76 and melanoma cells7 but not all (e.g., NBT-II cells).3 epithelial cells. In this respect, IGFs induce a basic or a full EMT according to the cell type.
In conclusion, IGF affects cell-cell adhesion. The cadherin/catenin complex is certainly one of the most important complexes involved in cell-cell adhesion. Consequently, the signalling link between IGF-1R and the cadherin/catenin complex has been studied in further detail.
IGF-1R Interacts Indirectly With E-cadherin and β-Catenin
The cadherin cell adhesion molecules bind to one another homophilically at the cell surface in a Ca2+-dependent manner. By binding cells opposing each other they physically adhere cells to each other forming large networks or even tissues. The cytoplasmic domain of cadherin binds to β-catenin, which in turn binds to α-catenin, which connects the cadherin/catenin complex to the actin-based cytoskeleton. In a large majority of epithelial cells, IGF-1R and E-cadherin are coexpressed. They can be coimmunoprecipitated and so constitute a membranous complex.3,8 According to the results of immunoprecipitations, the interaction of IGF-1R with E-cadherin, β-catenin or α-catenin does not appear to dissociate cadherins from catenins.3,8 Presumably, a IGF-1R/E-cadherin/β-catenin/α-catenin supra-molecular complex is present at the cell surface of various cells.
The cytoplasmic domain of E-cadherin is sufficient to interact with the cytoplasmic domain of IGF-1Rβ subunit.3 However, according to protein-protein interaction assays, IGF-1R does not interact directly either with E-cadherin or β-catenin.3 The nature of the molecules linking members of the complex together has yet to be determined.
IGFs Redistribute Proteins of Adherens Junctions
In epithelial cells non exposed to IGF, E-cadherin and β-catenin are mainly concentrated at cell-cell contacts and IGF-1R is present both at cell-cell contacts and in the cytoplasm. Exposure to IGF induces the redistribution of E-cadherin and IGF-1R from the membrane to the cytoplasm. After IGF treatment, E-cadherin is concentrated in a ring around the nucleus. The constant cycling of E-cadherin between the cytoplasm and the cell membrane has been demonstrated.9,10 Large amounts of IGF could affect this equilibrium: E-cadherin could be internalised faster than it is readdressed to the membrane.
These results obtained in vitro are consistent with a correlation of expression of IGF-II, IGF-1R and E-cadherin at the time of gastrulation (fig. 3). At this time, IGF-II is mainly expressed by mesenchymal cells (mesoderm), E-cadherin is mainly absent in murine mesodermal cells11 and IGF-1R is located at the membrane of epithelial cells (ectoderm and endoderm) and in the cytoplasm for mesodermal cells.

Moreover, the degradation, albeit minor, of E-cadherin has been reported as being associated with IGF treatment3 and this is supported by the partial colocalization of a subset of E-cadherin molecules with LAMP1 (lysosomal associated protein), an endosomal and lysosomal marker. Thus, E-cadherin may be degradated in LAMP1-positive organelles.
In conclusion, IGF induces rapid internalization of E-cadherin, associated with both slight degradation and sequestration in vesicles located around the nucleus.3 The reversibility of the EMT can now be explained by a simple hypothesis: the stored E-cadherin is rapidly readdressed to the membrane once IGF is removed from the medium.
Distribution of β-catenin is also affected by IGF. β-catenin is important for cell-cell adhesion and also in signal transduction via the Wnt signalling pathway. In the absence of Wnt, β-catenin is part of a complex containing GSK-3β (Glycogen Synthase Kinase-3β), APC (Adenomatous Polyposis Coli), and axin. GSK-3β is a serine-threonine kinase which phosphorylates β-catenin. β-catenin is then ubiquitinated and degradated by proteasomes.12,13 Upon binding of Wnt to its cell surface receptor, termed Frizzled, the serine-threonine kinase Dishevelled (Dsh) is activated.14-16 Dsh then inhibits the catalytic activity of GSK-3β by phosphorylation. Unphosphorylated and hence undegradable β-catenin accumulates in the cytoplasm and is translocated with TCF/LEF (T Cell Factor/Lymphoïd Enhancer Factor) factors - DNA bending proteins- into the nucleus (for review see 17). β-catenin/TCF complex can induce or repress the expression of a variety of target genes such as Cyclin D1, c-Myc, T-Brachyury, c-Jun, Fra-1, Matrix-Metalloprotease-7 (MMP-7), Fibronectin, Cyclo-oxygenase-2, m-Mitf, EphB2 and EphB3 receptors, and ephrin-B1.18-27> Some of these genes, such as cyclin D1, are expressed ubiquitously and some, such as m-Mitf in the melanocyte lineage, are cell specific.28>,29> IGF redistributes β-catenin from the cell membrane to the nucleus, and induces the translocation of TCF3 from the cytoplasm to the nucleus.3> None of the genes known to be activated/repressed by β-catenin is, alone, able to induce an EMT. As not all β-catenin target genes have been identified, some could be involved in EMT or/and the classical IGF signalling pathway may induce expression of genes involved in EMT.
In summary, IGFs induce a reversible EMT which is based on the reduction of the cell-cell adhesive properties of E-cadherin and the redistribution of the proteins associated with it. Indeed, IGF (i) rapidly delocalizes E-cadherin from cell-cell contacts, (ii) disrupts the interaction of E-cadherin with β-catenin, by phosphorylation, (iii) induces a limited degradation of E-cadherin, (iv) activates genes via β-catenin/TCF and (v) obviously induces the IGF signalling pathway.
Signalling Pathways Activated by IGF-1R
IGF-1R is a RPTK (for reviews see 30-32) and IGF binding leads to the subsequent activation of multiple signalling pathways, which are described below. This signalling multiplicity could explain the diversity of biological effects elicited by IGFs.
Activation of IGF-1R by IGF
IGFs bind to the extracellular domain of IGF-1R and initiate a conformational change in the quiescent receptor that is transmitted to the intracellular β subunit. The tyrosine kinase activity of IGF-1R is thereby activated and results in trans-autophosphorylation of the β-subunit on three tyrosines within the catalytic core: tyrosine residues 1161, 1165 and 1166 according to the numbering of the IGF-1R precursor, tyrosine residues 1131, 1135 and 1136 in α+β IGF-1R protein or tyrosine residues 421, 425 and 426 in the IGF-1R β-subunit, see GenBank accession number P08069. In the following sections, we will refer to the IGF-1R β-subunit to position specific amino acids. This autophosphorylation enhances the capability of the kinase to phosphorylate other substrates. These three phosphotyrosines participate in the activation of cytoplasmic signalling pathways.
Intracellular Signalling Pathways
The three phosphotyrosines located in the IGF-1R β-subunit at positions 421, 425 and 426 participate in the activation of cytoplasmic signalling pathways by two different processes:
- the tyrosine-phosphorylated IGF-1R acts only as a platform for the recruitment of specific cytoplasmic proteins, such as Grb (Growth factor Receptor-Bound).
- the tyrosine-phosphorylated IGF-1R recruits and phosphorylates various particular cytoplasmic proteins, such as PI-3K (PhosphoInositol 3-Kinase), Shc (SH2 Containing proto-oncogene), Crk (Chicken tumor virus CT10 Regulator of Kinase) and IRS (Insulin Receptor Substrate). The recruitment and tyrosine phosphorylation of these proteins by IGF-1R either leads to the activation of the enzymatic activity of PI-3K or creates new docking sites for the recruitment of cytoplasmic molecules interacting with Shc, Crk or IRS.
Thus, five cytoplasmic protein families interact with IGF-1R and propagate the signal from the exterior to the cytoplasm of the cells:
- >Grb is a large family of adaptor proteins containing SH2 (Src Homology 2) and SH3 domains. Some of its members, including various Grb10 isoforms, interact via their SH2 domain with a tyrosine-phosphorylated form of IGF-1R. Tyrosines 425 and 426 of β-subunit are implicated in the binding of Grb10 isoforms.33 When Grb10 isoforms bind to IGF-1R in a similar manner as to IR, it will involve tyrosine residues from the catalytic domain (implicating tyrosines 425 and 426 in the IGF-1R β-subunit).34 The role of Grb10 in IGF signalling is controversial. As a function of the cell type, the overexpression of Grb10 can inhibit or increase the mitogenic effects of IGF.35,36 Nevertheless, recent data suggest that Grb10 inhibits substrate phosphorylation by active IGF-1R.37
- Class I PI-3K is a heterodimer composed of a regulatory subunit, p85, and a tyrosine kinase catalytic subunit, p110. Different classes of PI-3K were characterized. In this chapter, we will focus on class I PI-3K and will just refer to PI-3K. Both subunits contain SH2 domains. The YXXM motif (implicating amino-acids (AA) 633-636) of IGF-1R β-subunit is necessary to interact with SH2 domains of p85.1 p85 is phosphorylated by activated IGF-1R. This phosphorylation induces a conformational change of p85 that drives the activation of the catalytic subunit, p110. The PI-3K pathway can lead to activation of proteins containing a PH (Pleckstrin Homology) domain such as PDK1 (Phosphoinositide Dependent Kinase) and Akt (fig.4A1,A2).
- SHC is also an adaptor protein that contains SH2 domains and also numerous tyrosine residues which can be phosphorylated by IGF-1R. The NPXY motif (AA237-240) of the β-subunit of IGF-1R is necessary for the interaction of IGF-1R with SH2 domains of SHC.38 The tyrosine-phosphorylated SHC mainly binds to the Grb2/Sos complex which drives the activation of the MAPK pathway.39-41 (fig.4B1, B2)
- The Crk adaptor protein family includes three members (Crk-I, Crk-II and Crk-L) containing both SH2 and SH3 domains. Tyrosine phosphorylation of Crk-II is rapid following its interaction with IGF-1R. The tyrosines 233, 240 and 540 of the β-subunit of IGF-1R are required for the interaction with SH2 domains of Crk-II. The tyrosine-phosphorylated Crk-II in turn interacts with two GEF (Guanine nucleotide-Exchange Factor) proteins, C3G and Sos42 and these factors drive the activation of the MAPK pathway.43,44 (fig. 4C1, C2)
- Members of the IRS adaptor protein family contain a PTB (PhosphoTyrosine Binding) domain, a C-terminal region rich in tyrosine residues and a PH domain, but no SH2 domain. IRS-1 and IRS-2 are rapidly phosphorylated by activated IGF-1R. The NPXY motif (AA237-240) of the β-subunit of IGF-1R is necessary for the interaction with PTB domain of IRS-1 and IRS-2.45 IRS-1 and IRS-2 are phosphorylated on various tyrosines by activated IGF-1R. The various phosphorylated residues on IRS are the docking sites for (i) p85 (ii) the three adaptor proteins Grb2, Crk-II and Nck (Non catalytic region of tyrosine kinase), (iii) SHP-2 (Src Homology 2 domain-containing protein tyrosine Phosphatase, also known as Syp or PTP1D), which is a tyrosine phosphatase46,47 and (iv) Fyn and Csk (C-terminal Src Kinase), which are Src Family Kinases (SFK).48
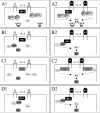
The formation of the IRS-1/Grb2 complex propagates the signal induced initially by the IGF ligand through Sos (Son of sevenless). Before IGF induction, Sos and Grb2 interact and are localized in the cytoplasm, where Sos is inactive. The recruitment, mediated by IRS-1, of Grb2, and therefore Sos, at the membrane activates Sos. Sos substrates, such as Ras, are also present at the membrane. Sos is a GEF protein activating the MAPK pathway (Mitogen Activated Protein Kinase (fig. 4D1,D2). Thus, the MAPK pathway is induced but will be described later. As mentioned above, Crk proteins also activate the MAPK pathway.
Nck- and Csk-activated signalling pathways lead to cytoskeletal reorganization. Csk activates FAK (Focal Adhesion Kinase) increasing the turnover of focal contacts.49> Nck targets members of the Pak (P21rac/cdc42>-activated serine/threonine kinase) family to the plasma membrane. Then, Pak interacts with the GTP-bound Rac/Cdc42 and is activated. Activated Pak1 phosphorylates MLCK (Myosin Light Chain Kinase) promoting actin polymerisation, a crucial event during physiological or pathological migration of cells.50>
Independently, it was shown that the protein Src is associated with the IGF signalling pathway. 51> Currently, however, the molecular link between IGF-1R and Src is unclear.
In summary, IGFs cause: (i) the reorganization of cytoskeleton through Csk-FAK and NCK-PAK; (ii) the activation of MAPK pathway through Grb2-Sos-Ras; and (iii) the activation of pathways downstream from PI-3K, such as the AKT (or PKB) pathway. The MAPK, AKT and Src pathways will be described in more detail below.
MAPK Signalling Pathway
MAPK pathways can be described as a successive activation of three kinase families (i) MAPKKK (MAP Kinase Kinase Kinase) phosphorylates and activates MAPKK, (ii) MAPKK then activates by phosphorylation MAPK and (iii) MAPK phosphorylates numerous proteins driving the biological effects of the pathway. Cytokines, stress and growth factors activate the MAPK pathway.
IGF-activation of the MAPK pathway is initiated by the activation of Ras. Ras is a GTP-binding protein kinase that alternates between an active state when bound to GTP and an inactive state when bound to GDP. Ras activation is accelerated by GEFs which catalyse the GDP/GTP exchange. Downstream from IGF, Sos and C3G activate Ras. One of the main targets of Ras is the serine/threonine kinase Raf, which is a MAPKKK. In Mammals, three Raf proteins have been isolated: A-Raf, B-Raf and C-Raf (or Raf-1). The affinity of Raf for Ras-GTP is greater than its affinity for Ras-GDP. Activated Ras recruits Raf to the plasma membrane where it is phosphorylated and activated. Full activation of A-Raf and C-Raf requires the phosphorylation of two residues, serine 338 and tyrosine 341. Ras phosphorylates the serine 338 whereas Src phosphorylates the tyrosine 341.52 The full activation of B-Raf requires only the phosphorylation of the serine 338.
Activated Raf phosphorylates MAPKKs, termed MEK1 and MEK2 (Map/Erk Kinase 1 and 2), in the growth factor-activated MAPK pathway. This phosphorylation occurs on the tyrosine residue contained in the TXY motifs.
Activated threonine/tyrosine kinases MEKs phosphorylate MAPKs named ERK1 and 2 (Extracellular signal-Regulated Kinases 1 and 2) in the growth factor-activated MAPK pathway. Serine/threonine kinases ERKs phosphorylate target proteins on serine or threonine residues of PX(T/S)P motifs. Some transcription factors are phosphorylated by ERKs.53
Moderate activation of MAPK pathway induces cell division. Indeed, active ERKs enhance the expression of Cyclin D1 and inhibit the expression of p27. Various findings suggest that Shc mediates most of mitogenic effects of IGFs. Strong activation of the MAPK pathway induces cell division arrest and, in some cell types, cell differentiation. These biological effects explain the important role of the MAPK pathway during development. Interestingly, the MAPK pathway is implicated in the loss of E-cadherin dependent adherens junctions upon HGF/SF induction and Src activation.54
PI-3K and AKT Signalling Pathway
PI-3K are heterodimers composed of a regulatory subunit, p85, and a catalytic subunit, p110. p85 binds directly to the tyrosine phosphorylated forms of IGF-1R and IRS-1. These interactions trigger tyrosine phosphorylation of p85. This phosphorylation induces a conformational change of p85 that activates the catalytic subunit, p110. Moreover, Ras-GTP can directly activate p110.55,56
Active PI-3K phosphorylates PtdIns (Phosphatidylinositol), PtdIns(4)P and Ptd(4,5)P on the 3'-OH group of the inositol ring producing Ptd(3)P, PtdIns(3,4)P2 and PtdIns(3,4,5)P3 (subsequently referred to as D3-phosphorylated phosphoinositides) respectively. (see fig. 5). Amphipathic PtdIns(3,4)P2 and PtdIns(3,4,5)P3 molecules bind to proteins containing a PH domain. Serine/Threonine kinases, such as AKT (= RAC = PKB) and PDK1 (Phosphatidylinositol Dependent Kinase 1), are relocalized to cell membrane upon their binding to these phosphorylated PtdIns. AKT is then appropriately localized to be phosphorylated on threonine 308 by PDK1 and on serine 473 by PDK2. More controversial is the identity of PDK2, the kinase(s) responsible for Ser-473/474 phosphorylation.57 AKT phosphorylated on T308 and S473 is fully active. It phosphorylates threonine or serine residues of RXRXX(T/S) motifs in numerous molecules. AKT regulates the activity of several transcription factors: CREB (cAMP Responsive Element Binding protein), members of ForkHead family and Ets-2. Phosphorylation of CREB by AKT stimulates CREB-dependent transcription.58 FKHR (ForKHead in Rhabdomyosarcoma) and FKHRL1 (ForKHead in Rhabdomyosarcoma Like 1) are phosphorylated and inhibited by AKT.59,60 Ets-2 is phosphorylated by JNK-2 in cells in which AKT is also activated, leading to the activation of Ets-2-dependent transcription.61

AKT promotes cell cycle progression, cell survival, and tumour cell invasion.62 Interestingly, AKT phosphorylates and inhibits GSK-3β thereby, presumably, linking IGF and Wnt pathways.
Moreover, Ras-mediated reorganisation of the actin cytoskeleton and cell migration depend on PI-3K. Indeed, some membrane lipid targets of PI-3K regulate: (i) the activity and structure of proteins that bind to actin; (ii) the GTPase Rac thereby inducing the formation of membrane folds.
Src Signalling Pathway
Src is the prototype SFK protein. It contains: (1) an amino-terminal myristylation sequence, (2) a single U specific region, (3) a SH2 domain, (4) a SH3 domain, (5) a tyrosine kinase domain that contains tyrosine-416, (6) a carboxy-terminal domain that contains tyrosine-537 (for review see 63). Autophosphorylation of Src on tyrosine-416 is required for optimal activity of the enzyme.
The activity of Src depends on its phosphorylation and two main intramolecular interactions, involving the SH2 and SH3 domains. Indeed, Src is inactive (i) when the carboxy-terminal part of Src interacts by appropriate folding of the protein with its own SH2 domain and (ii) possibly when the catalytic domain interacts with the SH3 domain. The interaction between the carboxy-terminal part and the SH2 domain is only possible when the tyrosine-537 is phosphorylated. Inactive Src is found in the perinuclear region of the cell and is most likely associated with endosomal membranes.64> The activation of Src induces SH3-dependent association with actin and peripheral targeting.65>,66> Src can be activated in several ways. First, dephosphorylation of tyrosine-537 by PTP-α, PTP1, SHP-1, or SHP-2 phosphatases activates Src. Also, Src-interacting partners can compete with intramolecular interactions between Src domains: PDGFR (Platelet-Derived Growth Factor Receptor) and FAK can bind to the SH2 or SH3 domains of Src and, thereby, activate it (for review see 63).
Activation of Src by IGF-I has been shown in neuroblastoma and in 3T3-L1 preadipocyte cells.51>,67> Interestingly, inactivation of Src by IGF-I has also been shown in NIH-3T3 cells.68> Therefore, the activation/repression of Src by IGF-I is cell type-specific. The mechanisms by which IGF-I activates/represses Src still remain unknown.
Src participates in mitogenic signalling of IGF-I.51> More precisely, Src participates in the activation of the MAPK pathway that is downstream from IGF. As concerns EMT, Src regulates cell-cell adhesion:
In conclusion, IGF is implicated in cell differentiation, division and survival, and also in the regulation of cell-cell adhesion and in cytoskeletal remodelling. It is now well established that IGFs activate MAPK, FAK, Pak and PI-3K pathways50>,76-80> and may activate Src.51>,67> A series of apparently unrelated observations could link these molecular pathways to adhesion and cytoskeletal cellular events:
- MAPK is involved in the disruption of E-cadherin-dependent adherens junctions upon HGF/SF induction.54
- FAK is involved in the turn-over of focal contacts.81
- Pak stimulates actin polymerisation, a crucial molecular event during cell migration.50
- PI-3K is involved in Ras-induced actin reorganization and cell migration. PI-3K also activates AKT, which inhibits GSK-3β, a regulator of β-catenin degradation.
The mechanism by which IGFs induce an EMT is still poorly understood and involves at least the disruption of cell-cell adhesion via the disorganisation of components present in adherens junctions and desmosomes. The next paragraph describes various evidence identifying the molecules involved in the induction of EMT by IGF.
Pathways Activated by IGF and Implicated in the Induction of the EMT
IGF induces an EMT associated with various molecular events: (i) the redistribution of E-cadherin, β-catenin and desmoplakin from plasma membrane to cytoplasm; (ii) the cytoplasmic sequestration and slight degradation of E-cadherin and the translocation of β-catenin to the nucleus; (iii) the neoexpression of mesenchymal markers. How do these molecular events are linked to the signalling pathways induced by IGF?
Effectors Implicated in Redistribution of Proteins
E-cadherin and β-catenin localize at the surface of epithelial cells. After IGF induction, they are internalized, and no longer colocalized.
Internalization of E-cadherin and β-catenin
IGF-1R forms a complex with E-cadherin and β-catenin. Following ligand binding, IGF-1R aggregates in coated pits and the aggregates are quickly internalised. Subsequently, IGF-1R is recycled to the cell surface. The mechanisms of internalisation of E-cadherin and β-catenin are not yet known, but various models can be suggested:
- (i) E-cadherin and β-catenin may be internalised passively, following the sorting of IGF-1R to which they are bound.
- Recently, it was shown that HGF (Hepatocyte Growth Factor) induces endocytosis of E-cadherin after tyrosine-phosphorylation and ubiquitination of this protein.82 A similar mechanism is plausible for IGF. E-cadherin becomes phosphorylated on tyrosines 755 and 756 (numbered according to the murine E-cadherin sequence) upon HGF induction. This form of E-cadherin can interact with Hakai.82 Hakai is a E3 ubiquitin ligase that induces E2-dependent ubiquitination of E-cadherin in vivo and in vitro. Ubiquitination of membrane proteins triggers their internalisation and targeting to lysosomes for degradation. The mechanism responsible for this internalisation has not been determined.. However, membrane proteins that have to be endocytosed are classically recognized by AP (Adaptor Proteins). When membrane-docked AP is bound to the protein destined to be internalised it can recruit clathrin. Polymerisation of clathrin induces the formation of a endocytotic vesicle (for reviews see 83, 84). Therefore, internalisation of phosphorylated and ubiquitinated E-cadherin could be driven by the interaction of E-cadherin with proteins of endocytotic machinery.
- Moreover, IGFs activate AKT.80 This serine-threonine kinase activates Rab proteins. The Rab proteins regulate the rate of vesicle fusion involved in protein trafficking.85 Interestingly, the expression of a constitutively active form of AKT mimics IGF induction by inducing an EMT.86 This transition is associated with the internalisation of E-cadherin and β-catenin. In conclusion, AKT may be an important effector of IGF during EMT and is a candidate for the stimulation of endocytosis.
Regulation of E-cadherin/β-catenin Interaction
After IGF-induction, the localization of IGF-1R, E-cadherin and β-catenin changes dramatically; IGF-1R is recycled, E-cadherin is trapped in organelles surrounding the nucleus and β-catenin is concentrated within the nucleus. The molecular mechanisms responsible for the partition of these three proteins are not known. After their internalisation, E-cadherin and β-catenin no longer colocalize. Post-translational modifications, in particular phosphorylation, are involved in the regulation of protein-protein interactions, and this type of mechanism may be involved in the association/dissociation of cadherin/catenins.
As mentioned earlier, E-cadherin can be tyrosine-phosphorylated. The tyrosine-phosphorylation of E-cadherin decreases its binding to p120ctn and increases its binding to Hakaï.82 The residues implicated are tyrosines 755 and 756, numbered according to the murine E-cadherin sequence. IGF can activate the Src and Fyn tyrosine kinases.51,67,87 Src was first implicated in regulating adherens junctions at the end of the 1980s.88 Also, Src and Yes are abundant at adherens junctions of epithelial cells89 and Src induces tyrosine phosphorylation of E-cadherin.82 So, Src could participate in the regulation of the interaction between E-cadherin and its partners. This leads to various possibilities. One of them is that Src induces the tyrosine-phosphorylation of E-cadherin after IGF induction, and as a result the interaction of the phosphorylated form of E-cadherin with p120 is inhibited and the interaction with Hakai is favoured. The interaction of E-cadherin with Hakai could lead to the internalisation of this new complex. In addition to activating tyrosine kinases, IGF can also activate SHP-2 tyrosine phosphatase. SHP2, PTPμ and PTP1B, can interact with cadherins in vivo, suggesting that cadherins were previously tyrosine-phosphorylated.90 The apparent discrepancy of the kinase and phosphatase inductor effect of IGF could be better understood when it was shown that SHP-2 phosphatase can activate Src by tyrosine-537 dephosphorylation. The tyrosine-phosphorylation status of E-cadherin seems tightly regulated and IGF could be involved in this process. Consequently, IGF may contribute to the maintenance/formation of the adherens junctions.
Tyrosine-phosphorylation of other classical cadherins, including N- and VE-cadherin, has been described.91,92 It appears that this process induces cellular events typically associated with the particular cadherin involved.82,93,94 Increased confluence of endothelial cells in culture is accompanied by a decrease in the tyrosine phosphorylation of VE-cadherin.92 VE-PTP (Vascular Endothelial-Protein Tyrosine Phosphatase), an endothelial receptor-type phosphatase, is a transmembrane binding partner of VE-cadherin that associates through an extracellular domain and reduces the tyrosine phosphorylation of VE-cadherin.95
Moreover, E-cadherin can be serine-phosphorylated.96 The serine-phosphorylation of E-cadherin increases the affinity of its binding to β-catenin. The serine residues implicated map to a region from amino-acid 833 to 862 (numbered according to the murine E-cadherin sequence). This region of E-cadherin is also necessary for the interaction with β-catenin. Of the eight serines in this region, Ser-853 and Ser-855 seem to be particularly important96 and are phosphorylated by Casein Kinase II.96 Another serine, Ser-849, is phosphorylated by GSK-3β. The phosphorylation status of these serine residues in E-cadherin upon IGF induction has, unfortunately, not been determined by phosphopeptide mapping. As E-cadherin does not bind to β-catenin after IGF induction, it seems likely that the serine phosphorylation of E-cadherin is abolished.
In contrast, tyrosine-phosphorylation of β-catenin decreases its affinity for E-cadherin.97-99 Tyrosine-654 of β-catenin seems to be the main residue involved in the modulation of β-catenin binding to E-cadherin.97 Src phosphorylates β-catenin on tyrosine 65497 and IGFs activate Src in certain cell types (see above). Thus, Src could act downstream from IGF to tyrosine-phosphorylate β-catenin and to disrupt the interaction between molecules involved in cell-cell adhesion.
β-catenin is not only involved in cell-cell adhesion when associated to cadherins but is also involved in cell signalling when associated to APC, axin and GSK-3β, and in regulation of gene transcription when associated to TCF/LEF proteins. The status of β-catenin phosphorylation is critical for its various protein-protein interactions.100,101 To elucidate the molecular details of the interactions of β-catenin with its partners, the crystal structure of the central core region of β-catenin has been solved.102 Armadillo repeats of the core region of β-catenin interact with E-cadherin, APC and many other proteins. The three dimensional structure of this region reveals a superhelix that forms a long positively-charged groove. This structure suggests that this segment can interact with acidic, negatively charged regions. Indeed, the region of E-cadherin which binds to β-catenin is highly acidic. One can imagine that addition of a phosphate group to E-cadherin reinforces the acidic properties of the β-catenin-binding domain. In contrast, addition of phosphate groups to β-catenin would diminish the positive nature of the groove and thereby perturb interaction with E-cadherin.103,104
Effectors Implicated in the Stability of E-cadherin and β-catenin Proteins
IGF induces a slight degradation of the extracellular part of E-cadherin.3> This may occur in lysosomes as E-cadherin is partially colocalized with LAMP1, an endosome-lysosome marker.105> The full process of E-cadherin degradation is still unknown although a parallel between IGF and HGF may exists. In the presence of HGF, the tyrosine phosphorylated form of E-cadherin becomes ubiquitinated by Hakaï and consequently targeted to lysosomes.82> The level of expression and the degradation of E-cadherin has not been studied after long periods of IGF induction. Stable expression of active AKT is associated with a low level of E-cadherin protein, mainly due to repression of the E-cadherin promoter.86> The repression of E-cadherin expression is certainly required to “lock” the cells in a mesenchymal state. During embryonic development this process must be rapid and strictly timed, but during transformation it can occur more gradually.
IGF and Wnt pathways both result in nuclear translocation of β-catenin and transcriptional regulation of gene expression.3>,17> The IGF pathway leads to an increase of the β-catenin half-life: IGF induces tyrosine-phosphorylation of β-catenin and doubles the half-life of this protein from 3 to 6 hours98>,106> and AKT serine phosphorylates and inhibits GSK-3β.107> So, AKT may participate in the stabilisation of β-catenin upon IGF induction. Interestingly, the expression of active AKT results in a reduction in the amount of β-catenin protein without any modification of the transcriptional level of the gene.86> Therefore, there is an apparent discrepancy between the effect of IGF and AKT. This discrepancy can be explained by the following reasons:
- The timing of induction is certainly very important. The effect of IGF on the amount of cytoplasmic β-catenin was studied after 17 h of IGF induction whereas studies on the effect of AKT have been performed on cells constitutively expressing an active form of AKT. The long-term regulation of the amount of β-catenin could be different from the short-term regulation.
- β-catenin can be located in three distinct areas of the cell: plasma membrane, cytoplasm and nucleus. In epithelial cells, which do not express an exogenous active form of AKT, the vast majority of β-catenin is present at the plasma membrane. β-catenin is mainly present in the cytoplasm of cells expressing an active exogenous form of AKT.86 Even though the total (plasma membrane + cytoplasm + nucleus) amount of β-catenin is lower in cells expressing exogenous AKT than in parental cells, quantitatively the amount of β-catenin in the cytoplasm is certainly higher and much lower at the plasma membrane in comparison.
- The mechanism of GSK-3β regulation revealed by recent crystallographic and biochemical studies is another important issue.110,111 Wnt and IGF/AKT signalling pathways affect two distinct pools of GSK-3β that in turn target different substrates. AKT can potentiate the Wnt pathway but AKT signalling alone cannot initiate the Wnt pathway.112-114 Consequently, AKT signalling is not the only pathway induced by IGF that is implicated in the regulation of the level of β-catenin. Finally, even though two pools of GSK-3β exist, one associated to IGF and the other to the Wnt, the barrier between the two may be not total.
Effectors Implicated in Transcriptional Regulations
During EMT, there is a massive shift of gene expression. We will focus this paragraph on the regulation of the E-cadherin gene and the role of β-catenin as a transcription factor.
Activation of Genes by the β-catenin Transcription Factor
The role of β-catenin/TCF as a bipartite transcription factor was first shown in 1996.115,116,117 Nevertheless, the induction of the β-catenin/TCF pathway by IGF is still controversial: Playford et al (2000) reported that IGF alone cannot induce β-catenin/TCF-dependent transcriptional activation; in contrast, two groups3,106 have reported that it could. This obvious discrepancy may be explained by the nature of the experiments and that the conclusions cannot be generalized. Indeed, the different conditions of induction may explain the conflicting findings:
- Playford and colleagues (2000) induced cells for no more than 6 hours with 0.65 x 10-8 M of IGF-I;
- Morali and colleagues (2001) and Desbois-Mouthon and colleagues (2001) induced their cellular systems for 16 hours with 1.3 x 10-8 M of IGF-II or 17 hours with 10-8 M of IGF-I, respectively. The activation of β-catenin/TCF by IGF may possibly require a period of greater than 6 hours.
The target genes of the β-catenin/TCF complex isolated from different systems include Cyclin D1, c-Myc, T-Brachyury, c-Jun, Fra-1, Matrix-Metalloprotease-7 (MMP-7), Fibronectin, Cyclo-oxygenase-2, m-Mitf, EphB2 and EphB3 receptors and ephrin-B1.18-27
T-Brachyury belongs to T-box genes family. In mouse, zebrafish and frogs, T-box genes are expressed in mesoderm or mesoderm precursors around the time of gastrulation. In Xenopus laevis, Brat induces mesodermal markers such as XmyoD, Xwnt8, goosecoid and XFKH-1.118-122 MMP-7 (matrilysin), a metalloproteinase, degrades extracellular matrix components and facilitates cell migration/invasion.123,124 Thus, these three proteins could be related to EMT, but none of them is involved in EMT per se.
The expression of Myc and Cyclin D1 genes has been determined after IGF-II treatment, in epithelial and ES cells. In epithelial cells, Myc, but not cyclin D1 expression, increased; in ES cells, the opposite was observed.3,125 Thus such induction seems to be cell specific. This leads to complexity, but also to cell specificity, of each combination of molecular pathways. Obviously, the combination of a series of molecular events (effectors, signalling pathways, transcription factors) will lead to the appropriate associated cellular event, in our case EMT. IGF alone cannot recapitulate EMT completely or prefectly; IGF needs to be associated with other factors to induce the molecular events correctly (such as the full downregulation of E-cadherin expression) and to assure cell specificity. This complexity leads to the specificity observed during development but not during oncogenesis.
Regulation of E-cadherin Expression
Downregulation of E-cadherin seems to be intimately involved in physiological and pathological EMT. In most cases, the downregulation of E-cadherin occurs by transcriptional repression. This repression can be due to methylation of the promoter and/or binding of repressors to the promoter (for review see 126). Snail, SIP1, E47 and Ets-1 transcription factors have all recently been independently identified as strong repressors of the E-cadherin promoter.127-131
The direct effect of IGFs on the activity of the E-cadherin promoter has not been studied. However, in stable transfectant cells, it was shown that active AKT represses the E-cadherin promoter activity86 and Snail mRNA is more abundant.86 The connection between AKT and Snail may be due to the presence of putative binding sites for CREB, members of ForkHead family and Ets-2, in the Snail promoter, because all these transcription factors are direct targets of AKT. So, AKT could indirectly repress E-cadherin promoter via Snail. This does not exclude a possible role for SIP1, E47, Ets or other transcription factors associated or potentially associated with AKT.
In conclusion, the major roles of IGF peptides are associated with cell proliferation and differentiation and as anti-apoptotic proteins. Recent work has lead to a variety of new insights into the biological effects of IGF factors. In particular, it is now clear that IGF is involved in EMT, a crucial cellular mechanism. Our understanding of the molecular mechanisms involved in the interaction between IGF and the IGF-1R/cadherin/catenin system is not clearly yet complete. The in vivo relevance of IGF to correct cell-cell adherens regulation and to EMT also require further elucidation.132
Mitogenicity, apoptosis and cell-cell interactions must be strictly regulated to maintain tissue homeostasis. Indeed, dysregulation of IGF signalling can lead to cancer, and IGF-1R is overexpressed in many types of tumour.133-135 Previously, the presence of this receptor was mainly considered in the light of cell proliferation. We must now consider the role of IGF in EMT. This step forward may well open new possibilities for cancer therapeutics and for gene therapy.
Acknowledgments
This work was supported by grants from Ligue Nationale contre le Cancer, ARC, Gefluc and Fondation de France. We are grateful to Silvia Martinozzi and Patrick Pla for their comments on the manuscript. We thank Pennsylvanie de la Corbeille de Venus for providing additionnal information and cesfo for providing various supplies.
References
- 1.
- Ron G, Rosenfeld CTR. The IGF systemTotowa, New Jersey: Humana Press,1999 .
- 2.
- Toyoshima K, Ito N, Hiasa Y. et al. Tissue culture of urinary bladder tumor induced in a rat by N-butyl-N-(-4-hydroxybutyl)nitrosamine: Establishment of cell line, Nara Bladder Tumor II. J Natl Cancer Inst. 1971;47(5):979–985. [PubMed: 4941696]
- 3.
- Morali OG, Delmas V, Moore R. et al. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene. 2001;20(36):4942–4950. [PubMed: 11526479]
- 4.
- Valles AM, Boyer B, Badet J. et al. Acidic fibroblast growth factor is a modulator of epithelial plasticity in a rat bladder carcinoma cell line. Proc Natl Acad Sci USA. 1990;87(3):1124–1128. [PMC free article: PMC53423] [PubMed: 2153969]
- 5.
- Gavrilovic J, Moens G, Thiery JP. et al. Expression of transfected transforming growth factor alpha induces a motile fibroblast-like phenotype with extracellular matrix-degrading potential in a rat bladder carcinoma cell line. Cell Regul. 1990;1(13):1003–1014. [PMC free article: PMC361698] [PubMed: 2134746]
- 6.
- Guvakova MA, Adams JC, Boettiger D. Functional role of alpha-actinin, PI 3-kinase and MEK1/ 2 in insulin-like growth factor I receptor kinase regulated motility of human breast carcinoma cells. J Cell Sci. 2002;115(Pt 21):4149–4165. [PubMed: 12356918]
- 7.
- Li Y, Bhargava MM, Joseph A. et al. Goldberg ID. Effect of hepatocyte growth factor/scatter factor and other growth factors on motility and morphology of nontumorigenic and tumor cells. In Vitro Cell Dev Biol Anim. 1994;30A(2):105–110. [PubMed: 7516797]
- 8.
- Guvakova MA, Surmacz E. Overexpressed IGF-I receptors reduce estrogen growth requirements, enhance survival, and promote E-cadherin-mediated cell-cell adhesion in human breast cancer cells. Exp Cell Res. 1997;231(1):149–162. [PubMed: 9056422]
- 9.
- Bauer A, Lickert H, Kemler R. et al. Modification of the E-cadherin-catenin complex in mitotic Madin-Darby canine kidney epithelial cells. J Biol Chem. 1998;273(43):28314–28321. [PubMed: 9774455]
- 10.
- Le TL, Yap AS, Stow JL. Recycling of E-cadherin: A potential mechanism for regulating cadherin dynamics. J Cell Biol. 1999;146(1):219–232. [PMC free article: PMC2199726] [PubMed: 10402472]
- 11.
- Butz S, Larue L. Expression of catenins during mouse embryonic development and in adult tissues. Cell Adhes Commun. 1995;3(4):337–352. [PubMed: 8821035]
- 12.
- Yost C, Torres M, Miller JR. et al. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10(12):1443–1454. [PubMed: 8666229]
- 13.
- Aberle H, Bauer A, Stappert J. et al. Beta-catenin is a target for the ubiquitin-proteasome pathway. Embo J. 1997;16(13):3797–3804. [PMC free article: PMC1170003] [PubMed: 9233789]
- 14.
- Axelrod JD, Miller JR, Shulman JM. et al. Differential recruitment of Dishevelled provides signaling specificity in the planar cell polarity and Wingless signaling pathways. Genes Dev. 1998;12(16):2610–2622. [PMC free article: PMC317102] [PubMed: 9716412]
- 15.
- Yanagawa S, van LeeuwenF, Wodarz A. et al. The dishevelled protein is modified by wingless signaling in Drosophila. Genes Dev. 1995;9(9):1087–1097. [PubMed: 7744250]
- 16.
- Karasawa T, Yokokura H, Kitajewski J. et al. Frizzled-9 is activated by Wnt-2 and functions in Wnt/beta -catenin signaling. J Biol Chem. 2002;277(40):37479–37486. [PubMed: 12138115]
- 17.
- Novak A, Dedhar S. Signaling through beta-catenin and Lef/Tcf. Cell Mol Life Sci. 1999;56(5-6):523–537. [PubMed: 11212302]
- 18.
- Shtutman M, Zhurinsky J, Simcha I. et al. The cyclin D1 gene is a target of the beta-catenin/ LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96(10):5522–5527. [PMC free article: PMC21892] [PubMed: 10318916]
- 19.
- He TC, Sparks AB, Rago C. et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. [PubMed: 9727977]
- 20.
- Mann B, Gelos M, Siedow A. et al. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA. 1999;96(4):1603–1608. [PMC free article: PMC15532] [PubMed: 9990071]
- 21.
- Arnold SJ, Stappert J, Bauer A. et al. Brachyury is a target gene of the Wnt/beta-catenin signaling pathway. Mech Dev. 2000;91(1-2):249–258. [PubMed: 10704849]
- 22.
- Brabletz T, Jung A, Dag S. et al. Beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am J Pathol. 1999;155(4):1033–1038. [PMC free article: PMC1867011] [PubMed: 10514384]
- 23.
- Crawford HC, Fingleton BM, Rudolph-Owen LA. et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18(18):2883–2891. [PubMed: 10362259]
- 24.
- Gradl D, Kuhl M, Wedlich D. The Wnt/Wg signal transducer beta-catenin controls fibronectin expression. Mol Cell Biol. 1999;19(8):5576–5587. [PMC free article: PMC84410] [PubMed: 10409747]
- 25.
- Howe LR, Subbaramaiah K, Chung WJ. et al. Transcriptional activation of cyclooxygenase-2 in Wnt-1-transformed mouse mammary epithelial cells. Cancer Res. 1999;59(7):1572–1577. [PubMed: 10197631]
- 26.
- Takeda K, Yasumoto K, Takada R. et al. Induction of melanocyte-specific microphthalmia-associated transcription factor by Wnt-3a. J Biol Chem. 2000;275(19):14013–14016. [PubMed: 10747853]
- 27.
- Batlle E, Henderson JT, Beghtel H. et al. Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111(2):251–263. [PubMed: 12408869]
- 28.
- Fuse N, Yasumoto K, Takeda K. et al. Molecular cloning of cDNA encoding a novel microphthalmia-associated transcription factor isoform with a distinct amino-terminus. J Biochem (Tokyo). 1999;126(6):1043–1051. [PubMed: 10578055]
- 29.
- Amae S, Fuse N, Yasumoto K. et al. Identification of a novel isoform of microphthalmia-associated transcription factor that is enriched in retinal pigment epithelium. Biochem Biophys Res Commun. 1998;247(3):710–715. [PubMed: 9647758]
- 30.
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. [PubMed: 11057895]
- 31.
- Favelyukis S, Till JH, Hubbard SR. et al. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol. 2001;8(12):1058–1063. [PubMed: 11694888]
- 32.
- Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61(2):203–212. [PubMed: 2158859]
- 33.
- Dong LQ, Farris S, Christal J. et al. Site-directed mutagenesis and yeast two-hybrid studies of the insulin and insulin-like growth factor-1 receptors: The Src homology-2 domain-containing protein hGrb10 binds to the autophosphorylated tyrosine residues in the kinase domain of the insulin receptor. Mol Endocrinol. 1997;11(12):1757–1765. [PubMed: 9369444]
- 34.
- Dong LQ, Farris S, Christal J. et al. Site-directed mutagenesis and yeast two-hybrid studies of the insulin and insulin-like growth factor-1 receptors: The Src homology-2 domain-containing protein hGrb10 binds to the autophosphorylated tyrosine residues in the kinase domain of the insulin receptor. Mol Endocrinol. 1997;11(12):1757–1765. [PubMed: 9369444]
- 35.
- Morrione A, Valentinis B, Resnicoff M. et al. The role of mGrb10alpha in insulin-like growth factor I-mediated growth. J Biol Chem. 1997;272(42):26382–26387. [PubMed: 9334212]
- 36.
- Wang J, Dai H, Yousaf N. et al. Grb10, a positive, stimulatory signaling adapter in platelet-derived growth factor BB-, insulin-like growth factor I-, and insulin-mediated mitogenesis. Mol Cell Biol. 1999;19(9):6217–6228. [PMC free article: PMC84567] [PubMed: 10454568]
- 37.
- Stein EG, Gustafson TA, Hubbard SR. The BPS domain of Grb10 inhibits the catalytic activity of the insulin and IGF1 receptors. FEBS Lett. 2001;493(2-3):106–111. [PubMed: 11287005]
- 38.
- Dey BR, Frick K, Lopaczynski W. et al. Evidence for the direct interaction of the insulin-like growth factor I receptor with IRS-1, Shc, and Grb10. Mol Endocrinol. 1996;10(6):631–641. [PubMed: 8776723]
- 39.
- Sasaoka T, Rose DW, Jhun BH. et al. Evidence for a functional role of Shc proteins in mitogenic signaling induced by insulin, insulin-like growth factor-1, and epidermal growth factor. J Biol Chem. 1994;269(18):13689–13694. [PubMed: 7513704]
- 40.
- Rozakis-Adcock M, Fernley R, Wade J. et al. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature. 1993;363(6424):83–85. [PubMed: 8479540]
- 41.
- Lowenstein EJ, Daly RJ, Batzer AG. et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70(3):431–442. [PubMed: 1322798]
- 42.
- Matsuda M, Hashimoto Y, Muroya K. et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994;14(8):5495–5500. [PMC free article: PMC359069] [PubMed: 8035825]
- 43.
- Ohba Y, Mochizuki N, Yamashita S. et al. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/ R-Ras3. J Biol Chem. 2000;275(26):20020–20026. [PubMed: 10777492]
- 44.
- Gotoh T, Niino Y, Tokuda M. et al. Activation of R-Ras by Ras-guanine nucleotide-releasing factor. J Biol Chem. 1997;272(30):18602–18607. [PubMed: 9228027]
- 45.
- Craparo A, O'Neill TJ, Gustafson TA. NonSH2 domains within insulin receptor substrate-1 and SHC mediate their phosphotyrosine-dependent interaction with the NPEY motif of the insulin-like growth factor I receptor. J Biol Chem. 1995;270(26):15639–15643. [PubMed: 7541045]
- 46.
- Kuhne MR, Pawson T, Lienhard GE. et al. The insulin receptor substrate 1 associates with the SH2-containing phosphotyrosine phosphatase Syp. J Biol Chem. 1993;268(16):11479–11481. [PubMed: 8505282]
- 47.
- Qu CK. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000;10(4):279–288. [PubMed: 11191350]
- 48.
- Courtneidge SA, Fumagalli S, Koegl M. et al. The Src family of protein tyrosine kinases: Regulation and functions. Dev Suppl. 1993:57–64. [PubMed: 8049488]
- 49.
- Tobe K, Sabe H, Yamamoto T. et al. Csk enhances insulin-stimulated dephosphorylation of focal adhesion proteins. Mol Cell Biol. 1996;16(9):4765–4772. [PMC free article: PMC231477] [PubMed: 8756634]
- 50.
- Li W, Fan J, Woodley DT. Nck/Dock: An adapter between cell surface receptors and the actin cytoskeleton. Oncogene. 2001;20(44):6403–6417. [PubMed: 11607841]
- 51.
- Boney CM, Sekimoto H, Gruppuso PA. et al. Src family tyrosine kinases participate in insulin-like growth factor I mitogenic signaling in 3T3-L1 cells. Cell Growth Differ. 2001;12(7):379–386. [PubMed: 11457735]
- 52.
- Mason CS, Springer CJ, Cooper RG. et al. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. Embo J. 1999;18(8):2137–2148. [PMC free article: PMC1171298] [PubMed: 10205168]
- 53.
- Treisman R. Journey to the surface of the cell: Fos regulation and the SRE. Embo J. 1995;14(20):4905–4913. [PMC free article: PMC394592] [PubMed: 7588619]
- 54.
- Herrera R. Modulation of hepatocyte growth factor-induced scattering of HT29 colon carcinoma cells. Involvement of the MAPK pathway. J Cell Sci. 1998;111(Pt 8):1039–1049. [PubMed: 9512500]
- 55.
- Kodaki T, Woscholski R, Hallberg B. et al. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4(9):798–806. [PubMed: 7820549]
- 56.
- Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B. et al. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. Embo J. 1996;15(10):2442–2451. [PMC free article: PMC450176] [PubMed: 8665852]
- 57.
- Chan TO, Tsichlis PN. PDK2: A complex tail in one Akt. Sci STKE. 2001;2001(66):E1. [PubMed: 11752635]
- 58.
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273(49):32377–32379. [PubMed: 9829964]
- 59.
- Brunet A, Bonni A, Zigmond MJ. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–868. [PubMed: 10102273]
- 60.
- Tang ED, Nunez G, Barr FG. et al. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274(24):16741–16746. [PubMed: 10358014]
- 61.
- Smith JL, Schaffner AE, Hofmeister JK. et al. Ets-2 is a target for an akt (Protein kinase B)/jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol Cell Biol. 2000;20(21):8026–8034. [PMC free article: PMC86413] [PubMed: 11027273]
- 62.
- Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001;98(20):10983–10985. [PMC free article: PMC58668] [PubMed: 11572954]
- 63.
- Frame MC. Src in cancer: Deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602(2):114–130. [PubMed: 12020799]
- 64.
- Kaplan KB, Swedlow JR, Varmus HE. et al. Association of p60c-src with endosomal membranes in mammalian fibroblasts. J Cell Biol. 1992;118(2):321–333. [PMC free article: PMC2290043] [PubMed: 1378446]
- 65.
- Fincham VJ, Unlu M, Brunton VG. et al. Translocation of Src kinase to the cell periphery is mediated by the actin cytoskeleton under the control of the Rho family of small G proteins. J Cell Biol. 1996;135(6 Pt 1):1551–1564. [PMC free article: PMC2133963] [PubMed: 8978822]
- 66.
- Fincham VJ, Brunton VG, Frame MC. The SH3 domain directs acto-myosin-dependent targeting of v-Src to focal adhesions via phosphatidylinositol 3-kinase. Mol Cell Biol. 2000;20(17):6518–6536. [PMC free article: PMC86126] [PubMed: 10938128]
- 67.
- Bence-Hanulec KK, Marshall J, Blair LA. Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the alpha1 subunit on a specific tyrosine residue. Neuron. 2000;27(1):121–131. [PubMed: 10939336]
- 68.
- Arbet-Engels C, TartareDeckert S, Eckhart W. C-terminal Src kinase associates with ligand-stimulated insulin-like growth factor-I receptor. J Biol Chem. 1999;274(9):5422–5428. [PubMed: 10026153]
- 69.
- Azarnia R, Reddy S, Kmiecik TE. et al. The cellular src gene product regulates junctional cell-to-cell communication. Science. 1988;239(4838):398–401. [PubMed: 2447651]
- 70.
- Toyofuku T, Yabuki M, Otsu K. et al. Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ Res. 1999;85(8):672–681. [PubMed: 10521240]
- 71.
- Toyofuku T, Akamatsu Y, Zhang H. et al. C-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276(3):1780–1788. [PubMed: 11035005]
- 72.
- Rodier JM, Valles AM, Denoyelle M. et al. pp60c-src is a positive regulator of growth factor-induced cell scattering in a rat bladder carcinoma cell line. J Cell Biol. 1995;131(3):761–773. [PMC free article: PMC2120611] [PubMed: 7593195]
- 73.
- Boyer B, Roche S, Denoyelle M. et al. Src and Ras are involved in separate pathways in epithelial cell scattering. Embo J. 1997;16(19):5904–5913. [PMC free article: PMC1170221] [PubMed: 9312048]
- 74.
- Volberg T, Geiger B, Dror R. et al. Modulation of intercellular adherens-type junctions and tyrosine phosphorylation of their components in RSV-transformed cultured chick lens cells. Cell Regul. 1991;2(2):105–120. [PMC free article: PMC361726] [PubMed: 1650581]
- 75.
- Gomez S, del Mont Llosas M, Verdu J. et al. Independent regulation of adherens and tight junctions by tyrosine phosphorylation in Caco2 cells. Biochim Biophys Acta. 1999;1452(2):121–132. [PubMed: 10559465]
- 76.
- Hansson A, Thoren M. Activation of MAP kinase in Swiss 3T3 fibroblasts by insulin-like growth factor-I. Growth Regul. 1995;5(2):92–100. [PubMed: 7627095]
- 77.
- Casamassima A, Rozengurt E. Insulin-like growth factor I stimulates tyrosine phosphorylation of p130(Cas), focal adhesion kinase, and paxillin. Role of phosphatidylinositol 3'-kinase and formation of a p130(Cas).Crk complex. J Biol Chem. 1998;273(40):26149–26156. [PubMed: 9748296]
- 78.
- Lee CH, Li W, Nishimura R. et al. Nck associates with the SH2 domain-docking protein IRS-1 in insulin-stimulated cells. Proc Natl Acad Sci USA. 1993;90(24):11713–11717. [PMC free article: PMC48054] [PubMed: 8265614]
- 79.
- Myers JrMG, Backer JM, Sun XJ. et al. IRS-1 activates phosphatidylinositol 3'-kinase by associating with src homology 2 domains of p85. Proc Natl Acad Sci USA. 1992;89(21):10350–10354. [PMC free article: PMC50336] [PubMed: 1332046]
- 80.
- Alessi DR, Andjelkovic M, Caudwell B. et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996;15(23):6541–6551. [PMC free article: PMC452479] [PubMed: 8978681]
- 81.
- Parsons JT, Martin KH, Slack JK. et al. Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene. 2000;19(49):5606–5613. [PubMed: 11114741]
- 82.
- Fujita Y, Krause G, Scheffner M. et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4(3):222–231. [PubMed: 11836526]
- 83.
- Kirchhausen T. Clathrin adaptors really adapt. Cell. 2002;109(4):413–416. [PubMed: 12086597]
- 84.
- Pearse BM, Smith CJ, Owen DJ. Clathrin coat construction in endocytosis. Curr Opin Struct Biol. 2000;10(2):220–228. [PubMed: 10753805]
- 85.
- Barbieri MA, Kohn AD, Roth RA. et al. Protein kinase B/akt and rab5 mediate Ras activation of endocytosis. J Biol Chem. 1998;273(31):19367–19370. [PubMed: 9677351]
- 86.
- Grille SJ, Bellacosa A, Upson J. et al. The protein kinase akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63(9):2172–2178. [PubMed: 12727836]
- 87.
- Sun XJ, Pons S, Asano T. et al. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J Biol Chem. 1996;271(18):10583–10587. [PubMed: 8631859]
- 88.
- Warren SL, Nelson WJ. Nonmitogenic morphoregulatory action of pp60v-src on multicellular epithelial structures. Mol Cell Biol. 1987;7(4):1326–1337. [PMC free article: PMC365217] [PubMed: 3110593]
- 89.
- Tsukita S, Oishi K, Akiyama T. et al. Specific proto-oncogenic tyrosine kinases of src family are enriched in cell-to-cell adherens junctions where the level of tyrosine phosphorylation is elevated. J Cell Biol. 1991;113(4):867–879. [PMC free article: PMC2288988] [PubMed: 1709169]
- 90.
- Ukropec JA, Hollinger MK, Salva SM. et al. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem. 2000;275(8):5983–5986. [PubMed: 10681592]
- 91.
- Lee MM, Fink BD, Grunwald GB. Evidence that tyrosine phosphorylation regulates N-cadherin turnover during retinal development. Dev Genet. 1997;20(3):224–234. [PubMed: 9216062]
- 92.
- Lampugnani MG, Corada M, Andriopoulou P. et al. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110(Pt 17):2065–2077. [PubMed: 9378757]
- 93.
- Daniel JM, Reynolds AB. Tyrosine phosphorylation and cadherin/catenin function. Bioessays. 1997;19(10):883–891. [PubMed: 9363682]
- 94.
- Esser S, Lampugnani MG, Corada M. et al. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111(Pt 13):1853–1865. [PubMed: 9625748]
- 95.
- Nawroth R, Poell G, Ranft A. et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. Embo J. 2002;21(18):4885–4895. [PMC free article: PMC126293] [PubMed: 12234928]
- 96.
- Lickert H, Bauer A, Kemler R. et al. Casein kinase II phosphorylation of E-cadherin increases E-cadherin/beta-catenin interaction and strengthens cell-cell adhesion. J Biol Chem. 2000;275(7):5090–5095. [PubMed: 10671552]
- 97.
- Roura S, Miravet S, Piedra J. et al. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274(51):36734–36740. [PubMed: 10593980]
- 98.
- Playford MP, Bicknell D, Bodmer WF. et al. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci USA. 2000;97(22):12103–12108. [PMC free article: PMC17301] [PubMed: 11035789]
- 99.
- Hu P, O'Keefe EJ, Rubenstein DS. Tyrosine phosphorylation of human keratinocyte beta-catenin and plakoglobin reversibly regulates their binding to E-cadherin and alpha-catenin. J Invest Dermatol. 2001;117(5):1059–1067. [PubMed: 11710913]
- 100.
- Rubinfeld B, Albert I, Porfiri E. et al. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272(5264):1023–1026. [PubMed: 8638126]
- 101.
- Hecht A, Kemler R. Curbing the nuclear activities of beta-catenin. Control over Wnt target gene expression. EMBO Rep. 2000;1(1):24–28. [PMC free article: PMC1083688] [PubMed: 11256619]
- 102.
- Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90(5):871–882. [PubMed: 9298899]
- 103.
- Huber AH, Weis WI. The structure of the beta-catenin/E-cadherin complex and the molecular basis of diverse ligand recognition by beta-catenin. Cell. 2001;105(3):391–402. [PubMed: 11348595]
- 104.
- Graham TA, Weaver C, Mao F. et al. Crystal structure of a beta-catenin/Tcf complex. Cell. 2000;103(6):885–896. [PubMed: 11136974]
- 105.
- Chen JW, Pan W, D'Souza MP. et al. Lysosome-associated membrane proteins: Characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch Biochem Biophys. 1985;239(2):574–586. [PubMed: 3923938]
- 106.
- Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ. et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene. 2001;20(2):252–259. [PubMed: 11313952]
- 107.
- Cross DA, Alessi DR, Cohen P. et al. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. [PubMed: 8524413]
- 108.
- Grevengoed EE, Loureiro JJ, Jesse TL. et al. Abelson kinase regulates epithelial morphogenesis in Drosophila. J Cell Biol. 2001;155(7):1185–1198. [PMC free article: PMC2199330] [PubMed: 11756472]
- 109.
- Larue L, Antos C, Butz S. et al. A role for cadherins in tissue formation. Development. 1996;122(10):3185–3194. [PubMed: 8898231]
- 110.
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7(6):1321–1327. [PubMed: 11430833]
- 111.
- Dajani R, Fraser E, Roe SM. et al. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105(6):721–732. [PubMed: 11440715]
- 112.
- Fukumoto S, Hsieh CM, Maemura K. et al. Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem. 2001;276(20):17479–17483. [PubMed: 11278246]
- 113.
- Chen RH, Ding WV, McCormick F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J Biol Chem. 2000;275(23):17894–17899. [PubMed: 10749878]
- 114.
- Ding VW, Chen RH, McCormick F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem. 2000;275(42):32475–32481. [PubMed: 10913153]
- 115.
- Molenaar M, van de Wetering M, Oosterwegel M. et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86(3):391–399. [PubMed: 8756721]
- 116.
- Behrens J, von KriesJP, Kuhl M. et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–642. [PubMed: 8757136]
- 117.
- Huber O, Korn R, McLaughlin J. et al. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59(1):3–10. [PubMed: 8892228]
- 118.
- Horb ME, Thomsen GH. A vegetally localized T-box transcription factor in Xenopus eggs specifies mesoderm and endoderm and is essential for embryonic mesoderm formation. Development. 1997;124(9):1689–1698. [PubMed: 9165117]
- 119.
- Papaioannou VE, Silver LM. The T-box gene family. Bioessays. 1998;20(1):9–19. [PubMed: 9504043]
- 120.
- Jacobs-Cohen RJ, Spiegelman M, Bennett D. Abnormalities of cells and extracellular matrix of T/ T embryos. Differentiation. 1983;25(1):48–55. [PubMed: 6662286]
- 121.
- Wilson V, Manson L, Skarnes WC. et al. The T gene is necessary for normal mesodermal morphogenetic cell movements during gastrulation. Development. 1995;121(3):877–886. [PubMed: 7720590]
- 122.
- Rashbass P, Wilson V, Rosen B. et al. Alterations in gene expression during mesoderm formation and axial patterning in Brachyury (T) embryos. Int J Dev Biol. 1994;38(1):35–44. [PubMed: 7915533]
- 123.
- von Bredow DC, Nagle RB, Bowden GT. et al. Degradation of fibronectin fibrils by matrilysin and characterization of the degradation products. Exp Cell Res. 1995;221(1):83–91. [PubMed: 7589259]
- 124.
- Cuvelier A, Kuntz C, Sesboue R. et al. Metalloproteinases in the extracellular matrix: Structure and activity. Rev Mal Respir. 1997;14(1):1–10. [PubMed: 9082500]
- 125.
- Delmas V. unpublished.
- 126.
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–454. [PubMed: 12189386]
- 127.
- Cano A, Perez-Moreno MA, Rodrigo I. et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2(2):76–83. [PubMed: 10655586]
- 128.
- Batlle E, Sancho E, Franci C. et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2(2):84–89. [PubMed: 10655587]
- 129.
- Comijn J, Berx G, Vermassen P. et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. [PubMed: 11430829]
- 130.
- Perez-Moreno MA, Locascio A, Rodrigo I. et al. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276(29):27424–27431. [PubMed: 11309385]
- 131.
- Rodrigo I, Cato AC, Cano A. Regulation of E-cadherin gene expression during tumor progression: The role of a new Ets-binding site and the E-pal element. Exp Cell Res. 1999;248(2):358–371. [PubMed: 10222128]
- 132.
- Morali OG, Jouneau A, McLaughlin KJ. et al. IGF-II promotes mesoderm formation. Dev Biol. 2000;227(1):133–145. [PubMed: 11076682]
- 133.
- Sartor O, Cooper MR, Khleif SN. et al. Suramin decreases circulating levels of insulin-like growth factor-I. Am J Med. 1994;96(4):390. [PubMed: 8166161]
- 134.
- Sell C, Dumenil G, Deveaud C. et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14(6):3604–3612. [PMC free article: PMC358728] [PubMed: 8196606]
- 135.
- Xie SP, Pirianov G, Colston KW. Vitamin D analogues suppress IGF-I signalling and promote apoptosis in breast cancer cells. Eur J Cancer. 1999;35(12):1717–1723. [PubMed: 10674019]
- 136.
- Hogan B, Beddington R, Costantini F. et al. Manipulating the mouse embryo : A laboratory manualCold Spring Harbor Laboratory Press,1994 .
- 137.
- Lee JE, Pintar J, Efstratiadis A. Pattern of the insulin-like growth factor II gene expression during early mouse embryogenesis. Development. 1990;110(1):151–159. [PubMed: 1964408]
Publication Details
Author Information and Affiliations
Authors
Sylvia Julien-Grille, Robert Moore, Laurence Denat, Olivier G. Morali, Véronique Delmas, Alfonso Bellacosa, and Lionel Larue.Copyright
Publisher
Landes Bioscience, Austin (TX)
NLM Citation
Julien-Grille S, Moore R, Denat L, et al. The Role of Insulin-like Growth Factors in the Epithelial to Mesenchymal Transition. In: Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.




