

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
This chapter presents an analysis and a summarized synthesis of our present knowledge of the biology of the thyroid gland, phylogeny ,ontogeny ,anatomy ,structure ,general metabolism ,regulatory factors and hormones , signalling cascades and their regulations , ( eg TSH ), functions including iodine metabolism and thyroid hormones synthesis , control of gene expression ,differentiation and growth and cell proliferation .Emphasis is ,when possible , put on the human thyroid. The original primary literature,as well as reviews , over the last 50 years are comprehensively and critically analyzed with:600 references .
Controversies are presented . For complete coverage of this and related topics, please visit www.endotext.org.
PHYLOGENY
The primary event in the phylogeny of the thyroid was the development in living forms of the capability of collecting iodide ion and binding it to protein. These activities have been observed widely among plants and in the invertebrate members of the animal kingdom. Brown algal kelps are the most efficient accumulators of iodide identified with enrichment factors for iodine of up to 10 6 (1). In Laminariadigitata , for example, the iodine content can reach up to 5% of the dry weight. However, only a minor fraction of iodine is stored in the form of iodinated amino acid residues including monoiodotyrosine (MIT) and diiodotyrosine (2) (3) (4). The biochemical pathways involved in iodine uptake, accumulation and metabolism in these algae have still not been fully elucidated. Recent studies suggest that iodide is oxidized by a vanadium-dependent iodoperoxidase (5) yielding more lipophilic iodine species which diffuse across cell membranes and are subsequently sequestered as labile iodine species in the apoplastic compartment (6). Thus, the iodide uptake mechanism utilized by these algae appears very different from that of vertebrate thyroid follicular cells and the organification of iodine is still considered a by-product of the reactive environment.
In invertebrates, endogenous synthesis of iodothyronines including thyroxine (T4) and triiodothyronine (T3) has been clearly demonstrated for urochordates and cephalochordates (7), whereas evidence for endogenous iodothyronine synthesis outside the chordates is very limited (8;9) (10) (11). Nevertheless, invertebrates deserve attention when analyzing the evolution of the hormonal signalling function of iodothyronines. Already in 1896, Drechsel (12) recognized that sponges and corals contain large quantities of iodine as iodotyrosines. Iodohistidine and bromotyrosine have also been detected. Monoiodotyrosine (MIT) and diiodotyrosine (DIT) have been found in starfish, mollusks, annelids, crustaceae, and insects (13) (14) (15). In insects, several organs and tissues can concentrate radioiodide but there is no evidence that this results in thyroid hormone (TH) formation (16). One process that is likely to yield iodinated compounds is cuticle formation (17). It has been suggested that iodinated substances may be by-products of the process of "quinone tanning". The formation of benzoquinone cross-linkages in the molecular structure of scleroproteins is probably responsible for hardening of the cuticle, and it is known that, in the presence of inorganic iodide, benzoquinones can bring about the iodination of proteins invitro (18). Thus, the iodination of tyrosine may be mediated quite accidentally by quinones that are involved in the general tanning reaction of the exoskeleton. However, recent studies by Heyland et al. (19) (20) suggest that at least some echinoderms and mollusks might produce T4 and T3 which was detected by thin layer chromatography and confirmed by ELISA measurements. Interestingly, iodothyronine synthesis in these organisms was prevented by thiourea but not by perchlorate treatment.
Even though most invertebrates might not be able to endogenously synthesize TH, a wealth of data indicate that organic iodine species are taken up from the environment (e.g., via the food) and might function as signalling molecules with pleiotropic effects on various aspects of invertebrate physiology (21). In analogy to vitamins, Eales coined the term “ vitamones ” to describe this ancient function of iodinated compounds as external morphogenic signals governing larval development in some invertebrates. Recently, this model has been supported by several experimental studies (10;22;23). Moreover, insilico analyses of genome sequences available for several invertebrate species in conjunction with experimental studies in a few model species corroborate a deep ancestry of iodothyronine signalling, most likely at the origin of deuterostomes (24-26). Orthologs of vertebrate TH receptors (TRs) have been identified in several invertebrate species including deuterostomes and protostomes (26-30). However, functional data for invertebrate TRs are still limited to deuterostomes and, thus, the functional role of TR orthologs identified in platyhelminths, mollusks and crustaceans remains elusive.
Interestingly, the functional characterization of the single TR orthologs of the amphioxus Branchiostomafloridae and Branchiostomabelcheri and of the ascidian Cionaintestinalis consistently revealed a lack of effective TR binding by T3 (26;30;31) . Instead, TRIAC (triiodothyroacetic acid), an acetic T3 derivative, was found to bind strongly to amphioxus TRs, to stimulate coactivator recruitment to the TRs and to activate TR-dependent gene expression (26;30). Despite the lack of T3-TR interaction in amphioxus and ascidian species, exogenous T3 and TRIAC are both effective in stimulating chordate metamorphosis (26). One clue to interprete these findings came from the observation that TRIAC is a major metabolite of T4 and T3 in amphioxus suggesting that metabolism of T3 to TRIAC might be involved in the metamorphosis-stimulating activity of T3 in amphioxus (32). A key role for TRIAC in regulating amphioxus metamorphosis is further supported by the recent identification of a nonselenodeiodinase in the amphioxus B.floridae (33). This deiodinase has a cysteine instead of a selenocysteine in its catalytic center and effectively deiodinates TRIAC and TETRAC (tetraiodothyroacetic acid) but not T3 and T4. Together, these experimental data suggest that TRIAC, or a related derivative, but not T3 is the ancestral TH acting in chordates.
Gorbman (34) has hypothesized that during evolution, organisms became accustomed to a supply of iodotyrosines and iodothyronines derived from external sources, and eventually developed a requirement for the iodinated amino acids. The first evidence of an organ capable of providing iodothyronines and thus, related to the vertebrate thyroid, is found in the protochordates, comprising the subphyla Cephalochordata (amphioxus) and Urochordata (ascidians) (Fig. 1-1). In the origin and evolution of the thyroid gland, the protochordates occupy key positions in phylogeny, because cephalochordates are the most basal in the phylum Chordata and urochordates are the closest living relatives of vertebrates (7). In ascidians and amphioxus, an organ known as the endostyle lies on the floor of the pharynx and connects with the pharynx by a duct. Notably, an endostyle is still present in the basalmost vertebrates, the lamprey larvae (ammocoete).
From the present point of view, the significant evolutionary event was the development of iodination centers within the endostyle. The differentiated endostyles in protochordates and lamprey larvae are histologically divided in “ zones ” containing different cell types (35). In the ascidian C.intestinalis , these iodination centers are present in zones 7, 8 and 9 at the tip of the endostyle (36). The amphioxus endostyle contains seven zones and iodide organification has been observed in zones 5a, 5b, and 6 which are also located at the tip of the endostyle (37). In amphioxus, Barrington (38) has shown that an iodinated glycoprotein is formed in these iodination centers, probably on the surface of the cell. The endostyle secretes a mucus that passes down the duct into the pharynx and thence is moved along into the alimentary canal, presumably carrying iodinated protein along with it. Although early accounts were sometimes conflicting (39), accumulation of radioiodide, peroxidase activity as well as endogenous synthesis of T4 and T3 has been demonstrated in the endostyle of several protochordate species (40-42) (43;44) .
In the ascidian C.intestinales , the proposed organ homology between the endostyle and the vertebrate thyroid was strengthened in recent molecular studies. For Ciona orthologs of vertebrate thyroid-specific marker genes, including the transcription factors Pax2/5/8 , ciTTF 1 and Ci-FoxE , expression was demonstrated in several zones of the endostyle in adult Ciona (45-47) . In addition, insitu hybridization revealed expression of Ciona orthologs of thyroid peroxidase (ciTPO) and dual oxidase (ci-Duox) in the iodide-concentrating zone 7 of the endostyle (48). An interesting observation was that Pax2/5/8 and Ci-FoxE expression domains overlapped with those of ciTPO and ci-Duox whereas ciTTF 1 expression was not detectable in zone 7. In amphioxus, however, all three transcription factors, Pax2/5/8 , TTF 1 and FoxE4 , were expressed together with TPO in the iodide-concentrating zones (49).
The recent releases of genome sequences of C.intestinales and B.floridae greatly contributed to our understanding of the gene repertoire encoding for components of the thyroid system in protochordates (25;50) . Genome analyses identified orthologs encoding for sodium-iodide symporter-like proteins, thyroid peroxidase, dual oxidase, several deiodinases and a single TR. However, no sequence homologous to thyroglobulin (TG) were identified in protochordate genomes suggesting that other scaffold proteins might be used for iodotyrosine synthesis (51). In amphioxus, a TG-like protein has been described by Monaco et al. (52), but a molecular characterization of this protein is not yet available. Further, no clear homologs of thyrotropin-releasing hormone (TRH) or the two subunits of thyroid-stimulating hormone (TSH) have been detected in protochordate genomes, which is in accordance with the current view that protochordates do not have a pituitary gland (53). Based on a comparison of Ciona and vertebrate genomes, Campbell (51) concluded that some features of the vertebrate thyroid system appear well represented in urochordates but that critical genes involved in the neuroendocrine control of thyroid function are lacking.
Interestingly, a recent analysis of the spatial expression profile of TR mRNA in adult amphioxus demonstrated abundant expression in those endostyle zones associated with iodide organification and TH synthesis (30). This finding raises the possibility of a direct role of TH in the regulation of TH synthesis.
Despite the observation of developmental effects of T3 and T4 in echinoderms (e.g., sea urchin, sand dollar, sea star, sea biscuit) and the identification of several genes encoding for components of a functional TH signalling system (24;25), solid experimental data to corroborate TH synthesis, metabolism and TR-mediated biological activities are not yet available. Although data by Heyland et al. (54;55) suggest endogenous TH production in in sea urchin and sea biscits, the tissues and organs involved have not yet been identified (echinoderms do not have an endostyle).

Figure 1Phylogeny of the development of the thyroid gland.
The most primitive vertebrates in which a follicular thyroid gland can be definitely demonstrated are the jawless fishes (agnathans). Concerning the origin and the evolution of the thyroid gland, lampreys are of particular interest because they are the only known vertebrates that possess a larval endostyle that directly transforms into a follicular thyroid during metamorphosis (56) (57). Although the endostyle of the lamprey larvae (ammocoete) has a different structure and organization compared to the endostyle of protochordates, physiological and molecular characteristics are very similar. The lamprey endostyle shows iodide uptake and organification, the latter involving a protein that is apparently related to TG (58) (59;60) (61). The TG-like protein undergoes proteolytic digestion in the intestinal tract to liberate T4 and T3 which are probably taken up directly from the gut lumen to enter blood circulation (62;63). Given this pathway of TH synthesis and release, it is of particular interest that high 5 ’ -deiodinase activities were determined in the larval intestine (64). Comparative analyses of thyroid-related genes confirmed the expression of Pax2/5/8 (65) and TTF 1 orthologs (56) (66) in the lamprey endostyle. Interestingly, similar to ascidians, the expression domains of TTF 1 were not clearly overlapping with domains of iodide concentration, TG and T4 synthesis (56;67).
Very high plasma concentrations of TH have been determined in lamprey larvae (68). A unique feature of lamprey developmental endocrinology is a dramatic decrease of circulating TH levels concomitantly with the onset of metamorphosis. This developmental TH profile is in sharp contrast to metamorphosis in amphibians and various fishes where high TH titers are associated with metamorphosis. Moreover, metamorphosis in lamprey larvae can be induced by anti-thyroidal compounds such as perchlorate or methimazole suggesting that the abrupt decline of plasma TH levels might trigger the onset of metamorphosis (69).
During metamorphosis of the ammocoete into the adult lamprey, the endostyle loses its connection with the pharynx and becomes a thyroid composed of scattered follicles (70) These follicles are not encapsulated, but they have the typical biosynthetic functions associated with hormone formation in the adult vertebrates. In the lamprey, the biosynthesis of TG in larval forms has the same characteristics as that formed in thyroid follicles of the adult form, with a 12S as the precursor of the 18-19S protein. Total iodine content of TG is very low (0.002%) and about 5% is present in the form of T3 and T4 (71). Curiously, thyroid activity appears to play no role in the metamorphosis of the ammocoete, while the gland itself undergoes a remarkable morphological changes.
Some relationships of the thyroid to the gastrointestinal tract is apparent in phylogenetic studies. Dunn (72) has actually found ciliated thyroid cells in the mouse and shark, a reminder of the origin of the gland from endoderm. In mammals the gastric mucosa and the salivary glands retain a functional relationship to the thyroid in that they too can concentrate iodide (73), and the salivary gland contains a peroxidase.
Thus, a thyroid capable of forming iodotyrosines and iodothyronines is present in all vertebrates. Its level of function varies widely from species to species and season to season. With the exceptions noted below, thyroid activity in the poikilotherms is very low. Seasonal changes in thyroid activity have been found in both warm- and cold-blooded animals. Certain morphologic changes occur after the biochemical evolution of the thyroid has ceased. In the adult lamprey and in most bony fishes, the gland is not encapsulated. The follicles may be widely scattered, either singly or in small clusters, especially along the course of the ventral aorta and in the kidneys (74). In cartilaginous fish, the thyroid is encapsulated. In the higher vertebrate forms, the thyroid is a one- or two-lobed encapsulated structure.
Function of the Thyroid in Non-Mammalian Species
A functioning thyroid is evident in forms as primitive as lampreys and hagfishes. TH are critically important for the regulation of diverse biological processes associated with development, growth and metabolism in non-mammalian vertebrates (75) (76;77) (78). In particular, the vital importance of TH for the regulation of early developmental processes is not limited to human or mammalian species (79), but is well conserved throughout the vertebrate kingdom (80) (81) (82). In avian species, for example, TH are required for nervous system and skeleton development (83) and TH action has also been demonstrated to regulate both direct larval and metamorphic development in fish (84;85). In amphibians, TH are the primary morphogen regulating postembryonic development (metamorphosis) (81).
Many fish species undergo similar developmental phases as described for amphibians including a larval stage, juveniles and adults (82) with a larvae-to-juvenile transition often associated with metamorphic changes (86). Experimental manipulation of the thyroid status as well as the recent cloning of fish TRs and the characterization of TR developmental expression profiles clearly demonstrated the important role of THs for early fish development (87). At later life stages, TH have been shown to assist in the control of various physiological functions in fish including osmoregulation, metabolism, somatic growth, and behaviour (88) (82). In salmonids, for example, TH are important for migration from fresh water to salt water (smoltification), and the high T4 plasma levels during smoltification represent some of the highest circulating T4 levels in fish (89;90).
Initially, it was believed that TH had little or no stimulatory effect on the oxidative metabolism of cold-blooded species. Now it is known that the effect of the thyroid on metabolic activity in cold-blooded species is strongly dependent on environmental temperature. For example, T4 causes stimulation of metabolism in lizards at 32°C, but not when they are acclimated to low temperatures (91). The thyroid gland is also more active at higher temperatures (23°-32°C) than at 10°-15°C in snakes, fish, amphibians, turtles, and lizards (92). TH levels are also influenced by the nutritional status in both endothermic (birds, mammals) and ectothermic (fish) vertebrates with a decreased T3 and T4 to T3 conversion in caloric deficient states (93).
A most striking effect of TH is the induction of metamorphosis in anuran amphibians, first reported by Gundernatsch in 1912 (94). The physiological role of TH during anuran metamorphosis is best exemplified by the fact that surgical removal of the thyroid gland or chemical blockage of TH synthesis leads to complete cessation of metamorphic development (95). On the other hand, addition of minute amounts of T4 (1 nM) to the rearing water of tadpoles during premetamorphosis leads to a precocious induction of metamorphosis (81). Particularly the metamorphosis of the South African clawed frog Xenopuslaevis has been used for years as a highly successful animal model to understand TH function in a developmental context (81) (96) (97;98) (99). The relationship between the functional state of the thyroid system and the progress of postembryonic development is well documented in this species (81) (100). The premetamorphic period is characterized by rapid growth of tadpoles with only minor morphological changes (101) and TH levels are very low (100). Growth and differentiation of the hind limbs are the earliest TH induced modifications marking the onset of the prometamorphic period during which levels of circulating TH steadily increase (102) stimulating further morphogenesis and differentiation of the limbs. The emergence of fore limbs marks the beginning of metamorphic climax characterized by rapid and dramatic changes in morphology (e.g., intestinal remodelling, complete resorption of gill and tail tissue) under the influence of peaking TH levels (81) (103). Towards the end of metamorphosis, plasma TH decline and low levels are present in juvenile and adult frogs (100).
A fascinating aspect of anuran metamorphosis is that a single type of hormone, TH, induces different tissues and organs to undergo remodelling in a highly coordinated spatio-temporal fashion (81) (104) (105). Similar to mammals, TH synthesis is regulated by thyroid-stimulating hormone (TSH) (98). T4 is considered the major hormone secreted by the thyroid gland while the secretion of T3 is low in X.laevis tadpoles (106). Expression of glycoprotein TSH alpha-and beta subunits mRNAs in the pituitary of metamorphosing X.laevis tadpoles increase from low premetamorphic levels to maximum levels at early climax stages and decrease towards the end of metamorphosis (107) (108). Thus, there is a concurrent increase of TSH expression, thyroid activity and circulating T4 levels from premetamorphosis to early climax stages. Different hypotheses have been put forward to explain this condition. Some authors stressed the importance of climax stage induction of type II iodothyronine deiodinase (D2) expression in pituitary thyrotrophs as a molecular switch to establish a negative feedback control of TSH synthesis (109). In contrast, other studies could demonstrate negative feedback control of TSH expression by T4 at much earlier stages suggesting that the developmental TSH expression profile is the net result of negative feedback action of circulating TH and a concomitant increase in pituitary stimulation by hypothalamic factors (110) (111). Of relevance for the increased hypothalamic stimulation of pituitary thyrotrophs might be the TH-dependent maturation of the median eminence (112) as well as increased synthesis and release of hypothalamic peptide hormones (113).
The extent to which different plasma proteins such as thyroxine-binding globulin (TBG), transthyretin (TTR) and albumin account for TH binding in the blood varies among different groups of vertebrates (114). In humans and rodents, TBG and TTR are the main THBP, respectively, showing a higher affinity for T4 than T3 (115). TTR is assumed to be the main TH-binding plasma protein in metamorphosing tadpoles and many teleost fish. One characteristic property of amphibian and teleost TTRs is that they display a several-fold higher affinity for T3 than for T4 (116).
In their target cells, the biological action of TH is mediated by activation of nuclear TH receptors (TRs). Two TR subtypes (TR , TR ) which are encoded by separate genes have been described in X.laevis (82). Due to pseudo-tetraploidy, there are two TR (A and B) and two TR (A and B) in X.laevis (117). Similar to mammals, TRs can bind to TH response elements (TREs) weakly as homodimers whereas heterodimers of TR and 9 cis retinoic acid receptors (RXR) strongly bind with TREs (118). Extensive investigations on the gene expression profiles induced by T3 made X.laevis one of the leading resources for understanding TH action during vertebrate development (119) (120). Gene transcription in X. laevis tadpoles can be either up- or down-regulated by TH (121;122). For the category of genes that are up-regulated by TH, it has been shown that unliganded heterodimeric TR-RXR complexes can bind to TRE but repress transcription through recruitment of corepressor (123).
In X.laevis , low mRNA expression has been demonstrated for TR and TR in oocytes and embryos but the putative functions are still poorly defined (124) (125). During postembryonic development, the expression profiles of TR and TR show striking differences (81) (126) (127;128) . TR is expressed at high levels during early premetamorphic stages and expression is maintained at an elevated level throughout metamorphic development (129) (130). TR shows a more complex developmental expression profile, characterized by low expression during premetamorphosis and a dramatic up-regulation in parallel with the increasing TH plasma levels during prometamorphosis (131). When analyzed in individual tissues, TR was found to be up-regulated particularly during periods of active tissue remodelling (132) (133). Another important aspect of TH action in X.laevis tadpoles is that exogenous T3 regulates the expression of its cognate receptors (134). Among the most rapid changes in gene expression induced by T3, a dramatic up-regulation of TR gene expression has been observed in all organs and tissues analyzed (135) (136). In addition, recently developed transgenic models carrying dominant negative and constitutively activated TR mutants could clearly demonstrate the important role of TR in mediating the developmental effects of TH during X.laevis metamorphosis (137).
Three types of iodothyronine deiodinases (D1, D2, D3) have been identified in vertebrates which differ in tissue distribution, substrate specificity and sensitivity to inhibiting compounds (138). D1 and D2 catalyze primarily the removal of one iodide from the outer tyrosine ring of T4 to produce T3. D3 catalyzes the cleaving of one iodide from the inner tyrosine rings of T4 and T3 generating inactive iodothyronine derivatives (e.g. reverse T3 and diiodothyronines), respectively. In amphibian tadpoles, the coordinated progression of metamorphic development requires a high degree of local control of T3 production which apparently dominates over the general supply of T3, at least during metamorphosis (139). Only recently, a putative X.laevis homologue of mammalian D1 has been identified (140) but neither the expression profile nor the putative regulatory role of D1 during metamorphic development have been characterized so far. However, several studies have investigated the role of D2 and D3 in controlling TH action during metamorphosis of anuran tadpoles (141). The data derived from these studies support the view that both D2 and D3 play a central role in modulating the tissue responsiveness to TH by either increasing intracellular concentrations of biologically active T3 (e.g., D2 in hind limbs) or by preventing TH action via rapid inactivation of T4 and T3 (e.g., D3 in tadpole tail).
Central to the understanding of TH function in lower vertebrates are the manyfold interactions of TH with other hormones (e.g., corticosterone, prolactin, and growth hormone) contributing to a fine-tuning of developmental TH action. For example, several studies have shown that high concentrations of corticosterone can provoke both inhibitory or accelerating effects on amphibian metamorphic development, depending on whether treatment is initiated at early (inhibition) or at late developmental stages (acceleration) (142). Concerning the accelerating effects of corticosterone on the development of tadpoles at late stages, two molecular mechanisms have been proposed including corticosterone -induced increases in T3 receptor binding capacities and corticosterone-induced increases in peripheral deiodination of T4 to T3 (81). Similarly, corticosterone is known to affect peripheral deiodination of T4 to T3 in avian species, including inhibitory effects on D3 and D1 activities in chicken liver (143).
Another hormone that has received a great deal of attention with regard to modulation of TH dependent metamorphic development is prolactin. Early studies using mammalian prolactin preparations could demonstrate antagonistic effects of prolactin on TH action in various peripheral tissues (144). Inhibitory effects on TR autoinduction by TH have been suggested as a primary mechanism of prolactin action to antagonize TH action in peripheral tissues. It should be noted, however, that in a transgenic frog model of prolactin overexpression, no retardation of tadpole development was detectable, with the exception of blocked tail resorption in a limited number of transgene animals (145).
In chicken embryos, GH appears to be a potent inhibitor of hepatic D3 activity resulting in increased T3 availability (146). This GH action on TH metabolism probably represents a physiologically relevant hormonal response linking the nutritional state with the activity of the thyroid system. Given the great diversity and heterogeneity in fish physiology and ecology, it is not surprising that a multitude of hormonal interferences have been described in various model species. The reader is referred to several comprehensive reviews about this hormonal interplay in fish (147-149).
Hypothalamic and Pituitary Control
Hypothalamic-pituitary control of thyroid function in the most primitive vertebrates represented by hagfishes and lampreys is still poorly understood (150) (151). Although the pituitary of these primitive fishes might contain thyrotrophic factors (152), their nature and their hypothalamic-releasing factors are unknown. TRH and TSH were largely ineffective in stimulating thyroid activity as assessed by different experimental protocols (153). Instead, numerous studies support the concept that peripheral deiodination of TH might provide the major regulatory mechanism for the control of thyroid status (154). The intestinal tract seems to be a major site of this TH metabolism in larval lamprey (155).
Neuroendocrine control of thyroid activity has been established for teleost fishes, amphibians, birds and reptiles (156). TRH is regarded as the main regulator of TSH secretion in mammals, and TSH-releasing activity of this peptide hormone has also been observed in various non-mammalian vertebrates. In avian species, injection of TRH has been shown to preferentially increase circulating T3 instead of T4, an effect that was later related to a GH-dependent inhibition of hepatic D3 activity subsequent to a stimulation of somatotrophs by TRH (157). In fact, preferential binding of TRH was demonstrated for somatotrophs in chicken. In recent years, it became clear that another hypothalamic factor, corticotropin-releasing hormone (CRH), is a more potent stimulator of TSH release in non-mammalian vertebrates. In chicken, CRH was found to increase both T3 and T4 plasma levels via stimulation of TSH release (158). Studies by de Groef (159) could show a preferential expression of CRH receptors type 1 (CRH-R1) and type 2 (CRH-R2) in chicken corticotrophs and thyrotrophs, respectively. The role of CRH R2 in mediating the CRH effect on TSH release in chicken was corroborated in further studies using CRH R2-specific agonists and antagonists.
In amphibians, the regulation of TSH release by TRH and CRF appears to be dependent on the life stage. While TRH was able to increase TSH secretion in adult frogs but not in tadpoles (160;161), the finding that TRH stimulates TSH release at least in adult amphibians argues against the hypothesis that the TSH-stimulating activity of TRH has only recently been coopted in association with the development of endothermy. Similar to the studies with chicken, CRF proved to be the most potent releasing factor for TSH in the tadpole pituitary invitro (162;163). In vivo, injection of CRH increased plasma T4 levels in Xenopus tadpoles and accelerated metamorphic development in ranids. In addition, TSH synthesis and release by the amphibian pituitary is under the control of a negative TH feedback (164;165). Few studies have addressed CRH effects on thyrotrophs in fish but the available data indicate that CRH but not TRH stimulates TSH release in salmonids (166).
ONTOGENY
The main anlage of the thyroid gland develops as a median endodermal downgrowth from the tongue. It can be seen in the human embryo at the end of the third week (167). It is located near the primordium of the heart, and as the heart is pulled caudally, the thyroid anlage follows. At the 5 th week the thyroglossal duct starts to breakdown. At about the 30th day it has developed into a hollow bilobed structure, and by the 40th day, the original hollow stalk connecting it to the pharyngeal floor atrophies and then breaks. Shortly thereafter the lateral extensions of the median anlage make contact with the ultimobranchial bodies developing from the 4th pharyngeal pouches, the so-called lateral anlage of the thyroid. The ultimobranchial cells or neural cells accompanying them are the origin of calcitonin-secreting C-cells in the thyroid gland and may contribute to the formation of follicular cells as well (168). By the 8th week the cells have a tubular arrangement, and cell clusters are apparent. Two weeks later, when the embryo is approximately 80 mm long, follicles are present. Shortly after this time the follicles contain colloid, and the thyroid accumulates and binds iodide by the 11th-12th week (Fig. 2). Secondary follicles arise by budding from the primary follicles; they increase in number until the embryo reaches a length of about 160 mm. After this time the follicles increase in size, but the number remains the same. Under intense stimulation, the adult thyroid can form new follicles.

Fig. 1-2
Photograph of thyroid tissue from a fetus with a 50-mm crown-rump length, estimated gestational age 64 days. The arrows indicate two intracellular canaliculi. During incubation of the tissue in organ culture in vitro, there was no uptake or fixation of iodide. The figure shows the earliest stage of formation of colloid spaces. The tissue was fixed in ozmium, embedded in Epon epoxy resin, and sectioned at 1-mm thickness (x2,400). (From T.H. Shephard, J. Clin. Endocrinol. Metab., 27:945,1967, with permission).
Fujita and Machino (169) have studied the origins of the follicular lumen in the chick embryo. They found that colloid droplets, 1-5µm in diameter and enclosed by a limiting membrane, first appear within the cytoplasm of parenchymal cells. As the droplets enlarge, they approach the cell membrane and come in contact with the droplets of an adjoining cell. The limiting cell membrane disappears, and the droplets fuse. By an extension of this process to cells close to the original droplet, an acinar structure containing colloid and enclosed by a ring of parenchymal cells is formed. A similar process can be demonstrated in aggregates of isolated thyroid cells in vitro (170).
GROSS ANATOMY
Physical Appearance and Anatomic Location
The Germans call the thyroid the "shield gland" (Schilddrüse), and the English name, derived from the Greek, means the same thing. Such a term gives a most erroneous impression of its shape. It is interesting, however, that in the Minoan culture, a shield was used that had a shape somewhat like that of the mammalian thyroid gland. The gland as seen from the front is more nearly the shape of a butterfly. It wraps itself about and becomes firmly fixed by fibrous tissue to the anterior and lateral parts of the larynx and trachea. Anteriorly, its surface is convex; posteriorly, it is concave. The isthmus lies across the trachea anteriorly just below the level of the cricoid cartilage. The lateral lobes extend along either side of the larynx as roughly conical projections reaching the level of the middle of the thyroid cartilage. Their upper extremities are known as the upper poles of the gland. Similarly, the lower extremities of the lateral lobes are spoken of as the lower poles, although they make no such prominent projections as do the upper (Fig. 1-3).

Fig. 1-3
Gross Anatomy of the thyroid and surroundings (from: Netter FH, The Ciba Collection of Medical Illustrations, vol. 4, Endocrine system and selected metabolic diseases, Ciba, 1965, with permission).
The weight of the thyroid of the normal nongoitrous adult is 6-20 g depending on body size and iodine supply. The width and length of the isthmus average 20 mm, and its thickness is 2-6 mm. The lateral lobes from superior to inferior poles usually measure 4 cm. Their breadth is 15-20 mm, and their thickness is 20-39 mm.
The thyroid is enveloped by a thin, fibrous, nonstripping capsule that sends septa into the gland substance to produce an irregular, incomplete lobulation. No true lobulation or lobation exists. In fact, the gland is throughout a uniform agglomeration of follicles packed like berries into a bag. It has no true subdivisions. The lateral lobes lie in a bed between the trachea and the larynx medially and between the carotid sheath and the sternomastoid muscles laterally. The deep cervical fascia, dividing into an anterior and a posterior plane, lines this bed and makes a loosely applied false or surgical capsule for the lateral portions of the gland. In front are the thin, ribbon-like infrahyoid muscles. The thyroid is molded perfectly to fit the space available between the neighboring structures, and is superficially placed. It can usually be outlined by careful palpation in normal humans, but if the neck is thick and short or the sternomastoid muscles heavily developed, it may be impossible to feel the gland.
The shape and attachments of the organ are important in examination and diagnosis. The relation of the thyroid gland to the parathyroids, which are usually situated on the posterior surface of the lateral lobes of the gland within the surgical capsule, and to the recurrent laryngeal nerves, which run in the cleft between the trachea and esophagus just medial to the lateral lobes, are most important to the surgeon. The relationship to the trachea is important from the point of view of pressure symptoms.
The pyramidal lobe is a narrow projection of thyroid tissue extending upward from the isthmus and lying on the surface of the thyroid cartilage, to the right or left of the prominence of that structure. It is a vestige of the embryonic thyroglossal tract. The importance of the pyramidal lobe is in its relation to developmental anomalies and also in its propensity to undergo hypertrophy when the rest of the thyroid has been removed. Any pathologic process that is diffuse may involve the pyramidal lobe, for example, Graves' disease or Hashimoto's thyroiditis. It becomes thus an item of some importance diagnostically and in thyroid surgery. A pyramidal lobe is found by the surgeon in about 80% of patients.
Blood Supply
The thyroid gland has an abundant blood supply. It has been estimated that the normal flow rate is about 5 ml/g of thyroid tissue each minute. The blood volume of normal humans is about 5 liters and total blood flow 5 liters/min. This mass moves through the lungs about once a minute, through the kidneys once in five minutes, and through the thyroid approximately once an hour. Although the thyroid represents about 0.4‰ of body weight it accounts for 2% of total blood flow. In disease the flow through the gland may be increased up to 100-fold.
This abundant blood supply is provided from the four major thyroid arteries. The superior pair arise from the external carotid and descend several centimeters through the neck to reach the upper poles of the thyroid, where they break into a number of branches and enter the substance of the gland. The inferior pair spring from the thyrocervical trunk of the subclavian arteries and enter the lower poles from behind. Frequently, a fifth artery, the thyreoidea ima, from the arch of the aorta, enters the thyroid in the midline. There are free anastomoses between all of these vessels. In addition, a large number of smaller arteriolar vessels derived from collaterals of the esophagus and larynx supply the posterior aspect of the thyroid. The branching of the large arteries takes place on the surface of the gland, where they form a network. Only after much branching are small arteries sent deep into the gland. These penetrating vessels arborize among the follicles, finally sending a follicular artery to each follicle. This, in turn, breaks up into the rich capillary basket like network surrounding the follicle.
The veins emerge from the interior of the gland and form a plexus of vessels under the capsule. These drain into the internal jugular, the brachiocephalic, and occasionally the anterior jugular veins.
Lymphatics
A rich plexus of lymph vessels is in close approximation to the individual follicles, but no unique role in thyroid function has been assigned to this system. The major normal, if not only, secretory pathway for thyroid hormone is through the venous drainage of the thyroid rather than through the lymphatics, but thyroglobulin is mainly secreted in the lymph.
Innervation
The gland receives fibers from both sympathetic and parasympathetic divisions of the autonomic nervous system. The sympathetic fibers are derived from the cervical ganglia and enter the gland along the blood vessels. The parasympathetic fibers are derived from the vagus and reach the gland by branches of the laryngeal nerves. Both myelinated and nonmyelinated fibers are found in the thyroid, and occasionally in the ganglion cells as well. The nerve supply does not appear to be simply a secretory system. The major neurogenic modifications of thyroid physiology have to do with blood flow and are reviewed in Chapter 4. However neurotransmitters have direct effects on thyroid follicular cells, which vary from one species to another. The physiological relevance of these effects remains to be proved.
The Secretory Unit - The Follicle
The adult thyroid is composed of follicles, or acini. These have lost all luminal connection with other parts of the body and may be considered, from both the structural an functional points of view, as the primary, or secretory, units of the organ. The cells of the follicles are the makers of hormone; the lumina are the storage depots. In the normal adult gland the follicles are roughly spherical and vary considerably in size. The average diameter is 300 microns. The walls consist of a continuous epithelium one cell deep, the parenchyma of the thyroid. The epithelium of the normal gland is usually described as cuboidal, the cell height being of the order of 15 µm. In the resting gland the cells may become flatter. Under chronic TSH stimulation such as occurs with iodide deficiency, the height increases, and the term columnar is applied. Such stimulation, which increases colloid resorption, also leads to a reduction in size the follicular lumen. As a result, the height of the epithelium is often inversely proportional to the diameter of the lumen of the follicle.
Within the follicle and filling its lumen is the homogeneous colloid. This is a mixture of proteins, principally thyroglobulin, but there are other lightweight iodoproteins and serum proteins and albumin, originating from the serum, as well.
In addition to the acinar cells, there are individual cells or small groups of cells that are seen not to extend to the follicular lumen and which may appear as clusters between follicles (Fig. 1-4a,b). These light cells, or C-cells, are a distinct category probably derived from the neural crest via the ultimobranchial body, as shown by studies in quail chicks by Le Douarin and Le Lièvre (171). The C-cells secrete calcitonin (or "thyrocalcitonin") in response to an increase in serum calcium (172). This hormone is important in the regulation of bone resorption and to a lesser extent influences the concentration of serum calcium. Calcitonin acts primarily by suppressing resorption of calcium from bone and therefore lowers plasma free Ca ++ levels. C-cells also contain somatostatin, calcitonin gene related peptide, gastrin-releasing peptide, katacalcin and helodermin that could have either a stimulatory or inhibitory activity on thyroid hormone secretion. Their physiological relevance is doubtful (see "Other Regulatory Factors" in this chapter). The C-cells are also the origin of the "medullary" thyroid cancers. In adult human they represent 1% of the cell population.
Outside the follicles two other types of cells populate the thyroid the endothelial cells and fibroblasts. In normal dog thyroids the relative proportions of follicular, endothelial cells and fibroblasts are 70%, 20% and 10% (173).
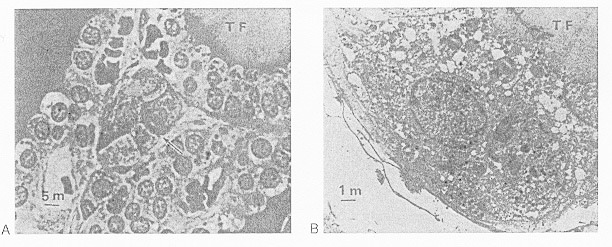
Fig. 1-4
(A) Light microscopy of a parafollicular cluster (arrow) in relationship to thyroid follicle (TF) (x900). (B) Parafollicular cell in characteristic position between follicular cells and follicular basement membrane, not abutting on colloid (TF) (x4,200). Tissue was obtained from normal thyroid tissue of a 26-year-old woman operated on because of a solitary thyroid adenomatous nodule. Specimens were fixed in glutaraldehyde and embedded in Araldite-502. (From Teitelbaum et al. Nature, 230,1971, with permission).
THE FINE STRUCTURE OF THE THYROID CELLS
The follicular organization and the polarity of the thyrocytes are essential to the specialized metabolism of the organ : with the vectorial transport of thyroglobulin and iodide at the apex, the synthesis of thyroid hormones at the apical membrane, the storage of iodine and thyroid hormone within thyroglobulin in the lumen and endocytosis of thyroglobulin also at the apex. The onset of thyroid function in embryo coincides with the appearance of this structure.
The acinar surface of thyroid parenchymal cells appears to be smooth in the light microscope, but the electron microscope shows that it is covered with tiny villi and some pseudopods. Each cell displays a cilium in the follicular lumen. The base of the cell abuts on a capillary and is separated from it by a two-layer basement membrane visible under the electron microscope. In the usual hematoxylin and eosin stain, the cell cytoplasm is neutrophilic, and colloid droplets may be present. The nucleus is at the base of the cell.
The colloid is variable in tinctorial response but tends to be strongly eosinophilic in resting follicles and pale-staining or even slightly basophilic when the gland is stimulated. In hyperactive follicles the margin of the colloid is scalloped by resorption vacuoles. These vacuoles may represent the "negatives" of the resorption process.
The villi are extensions of cytoplasm which increase cell secretory surface. In acutely stimulated thyroids, pseudopods extend out into the colloid and surround and ingest it by macropinocytosis. Over the course of several hours, the ingested droplets move toward the base of the cell (174). These droplets of resorbed colloid are processed for secretion as hormone by the gland (175).
The resolving power of the electron microscope has been turned upon the thyroid acinar cell by several investigators, among them Wissig (176), Dempsey and Peterson (177), Ekholm and Sjöstrand (178) and Herman (179). Wissig's and Ekholm's findings are presented here in detail and can be taken as typical of the cytologic picture of most species. The entire follicular cell is covered by an uninterrupted plasma membrane (Fig. 1-5 and Fig. 1-6). The apical surface of the cell is dome-shaped and is provided with numerous microvilli that are approximately 0.35 mm tall and 0.07 mm broad. This membrane is composed of two dark layers separated by a single pale layer and is 70 Å thick. Terminal bars join opposing cells at the apical margin, and desmosomes often occur on contacting cell surfaces. Vesicular structures, approximately 60 mm broad, appear in the microvilli and contain material that has the same density as colloid. Beneath the apical border there is a band of cytoplasm that is approximately 0.5 mm wide and devoid of organelles, although microtubular and microfilamentous structures are seen in this area. Beneath this band, a few apical vesicles of 400-15,000 Å are seen, and beneath this area and extending to the base of the cell are the channels of the endoplasmic reticulum, also known as ergoplasmic vesicles. These vesicles, or channels, are limited by a single membrane (the a cytomembrane) approximately 60-70 Å thick, and their outer surface is studded with ribosomes approximately 130-150 Å in diameter. In some areas the membrane covering the cytomembrane is devoid of ribonucleoprotein particles, and in between the vesicles the ribonucleoprotein granules may be seen to lie free. The endoplasmic vesicles are very pleiomorphic. Small vesicles are seen near the apical surface, 50 nm to several microns in diameter, and closed by a single-layer membrane 5 nm in thickness. These droplets appear especially in the apex of the cell and are thought to be secretion droplets. The material within them is frequently quite dense. Large vesicles of up to 1 micron appear especially in stimulated thyroids. These are called colloid droplets because the material within the vesicles is homogeneous and has the density of colloid and results from colloid endocytosis.

Fig. 1-5. A
thyroid follicular cell, including: (a) apical vessel of cell; (e) endoplasmic reticulum; (d) colloid droplets; (v) microvilli; (r) ribosomes on endoplasmic reticulum; (g) Golgi apparatus; (m) mitochondrion; (p) plasma membrane; (c) capillary cells; (n) nucleus; (b) basement membrane; (o) open "pore" endothelial cells ; (c) cilium. (Reproduced by permission of the Journal of Ultrastructural Research).

Fig. 1-6
Electron micrographs of rat thyroid. (a) Appearance after inactivation of the gland by two daily doses of T4. The micrograph shows two cell nuclei (N), well-developed rough-surfaced endoplasmic reticulum (RER), Golgi apparatus (G), mitochondria (M), lyosomes (L), and numerous dense, apical (exocytocic) vesicles (V). Because of the T4-induced TSH suppression, no colloid droplets are present (no hormone release); TG synthesis, however, is still going on, as indicated by the dense apical vesicle. (b) Appearance 20 minutes after intravenous administration of TSH (100 mU) to a rat treated with T4 for two days. The most characteristic features of these cells are the large number of colloid droplets (CD) and the almost complete disappearance of dense apical vesicles; TSH has induced an emptying of these vesicles into the follicle lumen. Other organelles are similar to those in Fig. 1-2. Note the close relation relation between colloid droplets and lysosomes (L). At the base of the follicle cells part of a blood capillary (C) is seen. (Micrographs kindly supplied by Professor Ragnar Ekholm, Goteborg, Sweden). The Golgi apparatus is located near the nucleus and consists of small vacuoles and vesicles 400-800 Å in diameter. No nucleoprotein granules are found on the surface of these vesicles. The content of the Golgi vesicles has a density similar to that of secretion droplets.
Numerous rod-shaped or irregular mitochondria are present. Their average diameter is 0.2 mm. They are bordered by a triple-layered membrane 160 Å in width consisting of two opaque layers and a less opaque interposed layer. The inner opaque layer is thrown up into folds, or cristae, which run irregularly, either in the long or the short axis of the mitochondrion.
The nucleus is enclosed within a double-walled envelope whose layers are separated by a less dense area approximately 200 Å thick. The outer nuclear membrane is continuous with the membranes forming the endoplasmic reticulum. The nuclear envelope has characteristic pores 400 Å in diameter.
The abutting plasma membranes of adjacent cells parallel one another and are about 70 Å thick. They are separated by a space 150 Å wide, which contains a material of the same density as the basement membrane. The membrane at the base of the cell is covered on the outer surface by a basement membrane that is approximately 400 Å in width. A thin layer of fibers about 400 Å in diameter may occur at the outer surface of the basement membrane. The basement membrane of the follicular cell is separated by a clear area from the basement membrane of the opposing capillary endothelium. At frequent intervals, the wall of the endothelial cell is interrupted by a pore approximately 450 Å in diameter. Here the lumen of the capillary appears to be in direct contact with the basement membrane of the endothelial cell. The thyroid follicle cells are separated by two layers of basement membrane from the capillaries, but the pores in the endothelial lining of the capillaries may allow, some plasma to come in direct contact with basement membrane. This arrangement should allow free diffusion of materials into and out of the acinar cell.
Ribosomes of the ergastoplasm synthesize thyroglobulin which is processed in the smooth reticulum and Golgi apparatus.
The colloid lumen is sealed by various cell-cell junctions: 1) the tight junctions of the zonula occludens, close to the apical border and which separate the basal from the apical membrane (main protein constituents : occludins); 2) further from the apex are the tight junctions (main protein components : cadherins) and 3) further the desmosome junctions (main protein components : desmogleins and desmocollins). All these junctions are linked to the cytoskeleton. Gap junctions (main protein components : connexins) provide joint channels allowing the passage of small molecules between the cells.
GENERAL METABOLISM OF THE FOLLICULAR THYROID CELL
The metabolism of the thyroid as related to hormone synthesis and secretion is discussed in Chapter 2. In this section, a review of some general aspects of metabolism of the thyroid acinar cell is provided. The metabolism of the thyroid has been studied by all the usual techniques - in vivo in men and mice, in situ, in in vitro perfusion, in slices, cells, homogenates, or subcellular fractions. Several species, including humans, have been investigated, often with obvious and consistent species-related differences. Conditions of tissue preparation and assays have varied widely.
Energy metabolism
Energy supply in the human thyroid cell is necessary for many activities like synthesis of nucleotides, proteins, nuclear acids, lipids, transport functions and other activities like phagocytosis, lysosome movement etc. It is mainly produced by mitochondrial oxidative phosphorylation (about 85%) and to a minor extent by cytosolic aerobic glycolysis (180). Energy metabolism in the human thyroid cell resembles in many aspects that in the dog thyroid. However, the absolute values of oxygen uptake, glucose uptake and lactate formation are significantly less in the human thyroid slices. Adenosine triphosphate (ATP) concentration in the thyroid cell is in the millimolar range and about 90% of ribonucleotides are in the form of triphosphates (181). Free fatty acids are probably the main source of energy in thyroid cells as respiration is maintained for long periods in vitro in the absence of exogenous substrate. As there is hardly any glycogen present in these conditions, most probably free fatty acids are the endogenous substrates. Compartmentation may make glycolytic ATP the main energy source for some membrane functions such as endocytosis of colloid. Indeed inhibition of glycolysis inhibits colloid endocytosis much more than total energy metabolism and addition of glucose counteracts this effect (181).
Mitochondrial inhibitors abolish the stimulatory effect of TSH on thyroid cell respiration (182). Cyclic AMP does not influence mitochondrial respiration in a direct manner. It is therefore assumed that the stimulatory effect of TSH on respiration is secondary to its enhancing effect on energy (i.e. ATP) consuming cellular processes.
Carbohydrate metabolism
The main source of energy delivery for metabolic processes in the thyroid cell are free fatty acids. However, glucose metabolism has an important function in the thyroid for several reasons. About 70% of the glucose taken up by dog or human thyroid slices is transformed to lactate, a further 5% is catabolized through the Embden-Meyerhof pathway and the Krebs cycle (183). Another 6% of glucose carbon is incorporated into protein and less than 1% into lipids and glycogen. The remaining part (about 10%) is oxidized through the hexose monophosphate pathway (HMP). Most of the enzymes participating in the Embden-Meyerhof pathway, HMP and Krebs cycle have been demonstrated in the human thyroid (184) (185) (186). As hexokinase instead of glucokinase is present in the thyroid, the rate of phosphorylation of glucose is probably independent of its concentration because of the low Km of hexokinase for glucose.
Glucose metabolism serves several purposes. The incorporation of glucose carbon into proteins is related to the function of the thyroid cell in protein synthesis i.e. synthesis of thyrogobulin which contains 10% carbohydrate. The metabolism of glucose along the HMP is related to the generation of NADPH and pentoses in this pathway. The production of pentoses is obviously necessary for generation of nucleotides. NADPH production is necessary in several respects (Fig. 7). It is needed for generation of H2O2 for oxidation, organification of iodide and thyroid hormone synthesis. NADPH is also an important cofactor in iodotyrosines deiodination. It is also needed to reduce oxidized glutathione after its generation by GSH peroxidase in the detoxification of the H2O2 leaking in the cell (187).
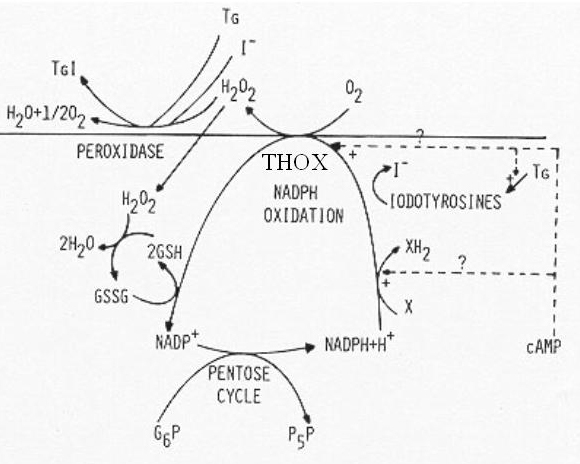
Fig. 1-7. Postulated NADP oxidation-reduction cycle in thyroid. Four mechanisms of NADPH oxidation are outlined: the reduction of any intermediate X by an NADPH-linked dehydrogenase, the deiodination of iodotyrosines released by thyroglobulinolysis, the generation of H2O2 by THOX (thyroid H2O2 generating enzyme), and the reduction of H2O2 through GSH peroxidase. TG and TGI: uniodinated and iodinated thyroglobulin. +, activation. (From: Dumont JE (187)).
H2O2 generation is stimulated by TSH through cAMP in dog thyroid and by Ca <sup>++</sup> and diacylglycerol in all investigated species, including humans and dogs.
TSH enhances carbohydrate metabolism in the dog thyroid (183). During the stimulation there is selective increase in the activity of the HMP whereas incorporation of glucose into proteins and lipids decreases (183). The activity of TSH in this metabolism is probably mediated by cAMP, since this nucleotide can reproduce the TSH effects on glucose uptake, catabolism, incorporation in protein and lipids and on the HMP pathway. TSH also causes an increase in NADPH and NADP+ concentration through increased NAD+ kinase activity (187).
The increased metabolic activity induced by TSH mainly reflects increased consumption by NADPH dependent processes stimulated via cAMP. For instance, the activity of the HMP pathway is predominantly dependent on the availability of the substrate NADP+ generated during oxidation of NADPH (187).
Mitochondrial Respiration
The mitochondrion has appropriately been termed the "powerhouse" of the cell. It provides about 85% of generated ATP in the thyroid cell, only 15% coming from glycolysis. The thyroids of different animal species contain mitochondria with their typical morphology, having electron transport chain, Krebs cycle enzymes, coupled oxidative phosphorylation and good respiratory control (182). The activity of mitochondria is controlled by adenosine diphosphate (ADP) levels. Also respiration linked Ca++ accumulation plays a general and fundamental role in vertebrate cell physiology (188). Free fatty acids are the preferred substrate of oxidation in the unstimulated thyroid, presumably through mitochondrial pathways (189). In thyroids of patients operated for hyperthyroid Graves' disease all enzyme activities studied were increased suggesting an increase in the mitochondrial population in chronically stimulated thyroid cells (181). TSH increases oxygen consumption in thyroid slices by 20 - 30% within a few minutes, independently of exogenous substrates. The increased respiration is oligomycin and antimycin sensitive. Thus, respiration is of largely mitochondrial origin and probably represents the effect of TSH in increasing metabolic activities and consequently ATP consumption (see section on Energy Metabolism) (187). TSH augments oxidation of pyruvate and acetate by thyroid slices. Compounds such as perchlorate, methimazole, iodide, thiocyanate and T4 have no significant direct action on thyroid mitochondria (182). In isolated thyroid mitochodria protein synthesis is dependent on intact electron transport and oxidative phosphorylation. It is inhibited by chloramphenicol but not by cycloheximide (190).
RNA and DNA Metabolism
Chronic TSH stimulation produces cell hypertrophy, and proliferation with a greater increase of RNA than of DNA. Since RNA and DNA synthesis are required for cell growth and division, it is not surprising that TSH stimulation causes rapid and continued increases in synthetic activities. When given in vivo, TSH stimulates uptake and incorporation of RNA precursors within one hour and net RNA increases in about 12 hours (191) (192). TSH stimulates cell uptake and synthesis of purine and pyrimidine precursors (193) (194) and purine and pyrimidine synthesis. Synthesis of both messenger RNA (mRNA) and ribosomal RNA (rRNA) is stimulated by TSH (195). The population of mRNA preferentially synthesized in response to TSH and cyclic AMP is important and includes specific thyroid gene expression such as thyroperoxidase (TPO), Na+/I- cotransporters (NIS) (196), thyroglobulin etc.. RNA degradation is not known to be influenced by TSH.
Formation of polyamines is closely linked to cell growth, although the mechanism is not known. TSH and cAMP enhance ornithine decarboxylase activity, the rate-limiting enzyme in polyamine synthesis (197).
Protein Metabolism
Thyroid tissue is composed of cells and a storage protein, and the kinetic behavior of each compartment varies enormously with the conditions. Thus, in the colloid especially, protein storage and degradation go on concurrently, and content at any time reflects a balance between these activities.
TSH enhances uptake of amino acids by isolated thyroid cells, and stimulates protein synthesis within 30 minutes to 4 hours in some preparations. Because of effects on thyroglobulin (TG) degradation, and dilution of amino acid precursor pools, stimulation of synthesis is more difficult to demonstrate in whole tissues (198) (199) (200). However, if thyroid slices are incubated in high concentration of leucine to obliterate any separate effect of TSH on cell uptake of amino acid, a clear stimulation of protein synthesis by TSH can be demonstrated in vitro in dog thyroid slices (200), and also in isolated thyroid cells but not in primary cultures of dog thyroid cells. Within 12-24 hours of chronic TSH stimulation in vivo, net protein content may be decreased by active TG hydrolysis, but after this, protein content is increased (201). This response remains nearly linear over four to five weeks as thyroid size in animals quintuples. The response is primarily due to production of new cells, since DNA and protein change in parallel.
Huge polysomes (40 to 80 ribosomal units) connected by mRNA have been demonstrated in the thyroid (202) (203) and were shown to incorporate precursors into TG-related peptides. (Fig.1-8). In dog thyroid slices, TSH also shifts thyroid monosomes to polysomes, and this is stimulated by cAMP. This action suggests a direct effect on translation (204).
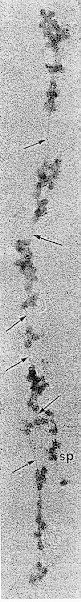
Fig. 1-8
Electron microscopic photograph of an enormous polysome containing 60 or 70 monosomes, the presumed source of TG synthesis. The arrows point to a thread possibly representing mRNA holding the polysome together. (From Keyhani et al., J. Microscop., 10:269,1971, with permission).
Lipid metabolism
Free fatty acids are the main fuel of the thyroid cell and they may be completely oxidized. Sufficient endogenous substrate is present to sustain respiration for several hours during in vitro incubation of thyroid slices (205) (206) (207). Studies on localization of lipids in human thyroids have shown that small amounts are only present in goitres from thyrotoxic patients, but that appreciable amounts are present in the normal human thyroid i.e. phospholipids, cholesterol and gangliosides: 5.2, 4.3 and 0.12 mmol/kg fresh tissue. C-cells contain most abundantly phospholipids. The human thyroid contains phospholipids in the proportion: phosphatidylcholine (41.8%), phosphathidylethanolamine (26.9%), phosphathidylserine (10.4%), phosphathidylinositol (4.4%), cardiolipin (3.4%), sphyngomyelin (12.4%) (208) (209) (210). TSH enhances the incorporation of precursors into most phospholipids. The effect is believed to reflect a direct stimulation of synthesis of phospholipids. However, as TSH also stimulates phospholipid degradation, increased phospholipids synthesis under the influence of TSH could correspond in part to this accelerated turnover rather than to an accumulation 123 . TSH also stimulates incorporation of inositol into phosphoinositides in a glucose free system. TSH specifically enhances synthesis of phosphatidic acid from glycerophosphate after in vivo administration (211) (212).
Electrolyte Transport and metabolism
The mean resting transmembrane potential as studied in rat, rabbit and guinea-pigs thyroid cells varies between -60 and -70 mV. The magnitude of the membrane potential was found te be dependent mainly upon the gradient for K+ across the membrane. A high intracellular K+ and low Na+ concentration is maintained by ouabain sensitive Na+ - K+ ATPase. The activity of this ATPase varies in direct relation to chronic TSH stimulation, probably corresponding to cell hypertrophy and hyperplasia. There is no evidence for direct action of TSH on this enzyme. Acute stimulation of thyroid cells induces a depolarization of the cell, which is accompanied by a decrease in membrane resistance. The depolarization may correspond to increased permeability to predominantly extracellular cations, such as Na+ or to a decreased permeability to predominantly intracellular cations, such as K+. Administration of TSH or veratridine, a sodium channel agonist, depolarized cultured thyroid cells and increased the secretion of radioiodine from the organically bound pool. Depolarization of the cells by increasing the potassium concentration in the medium failed to promote secretion of radioactive iodine indicating that the sodium influx rather than the depolarization itself, may mediate the secretory response (187).
THYROID REGULATORY FACTORS
In Physiology
Four major biologic variables are regulated in the thyrocyte as in any other cell type: function, cell size, cell number, and differentiation. The first three variables are quantitative and the latter is qualitative. In this chapter we consider the factors involved in these controls in physiology and in pathology, the main regulatory cascades through which these factors exert their effects, and the regulated processes, which are function, proliferation and cell death, gene expression, and differentiation. Whenever possible, we describe what is known in humans.
The two main factors that control the physiology of the thyroid after embryogenesis are the requirement for thyroid hormones and the supply of its main and specialized substrate iodide (Table 1-1). Thyroid hormone plasma levels and action are monitored by the hypothalamic supraoptic nuclei and by the thyrotrophs of the anterior lobe of the pituitary, where they exert a negative feedback. The corresponding homeostatic control is expressed by thyroid-stimulating hormone (TSH, thyrotropin). The hypophysis adjusts its secretion of TSH to the sensitivity of the thyroid, increasing TSH levels when thyrocyte sensitivity decreases (e.g. because of reduced TSH receptor expression) (213) . The TSH receptor is also stimulated by a new different natural hormone cloned by homology, thyrostimuline. The physiological role of this protein is unknown but its level is not controlled by a thyroid hormone feedback and it does not participate in the homeostatic control of the thyroid (214) . Iodide supply is monitored in part through its effects on the plasma level of thyroid hormones, but mainly in the thyroid itself, where it depresses various aspects of thyroid function and the response of the thyrocyte to TSH. These two major physiologic regulators control the function and size of the thyroid - TSH positively, iodide negatively (187;215-217) . These are the specific controls exerted at the level of the thyrocyte itself. The follicular cells themselves probably regulate the other thyroid cells, fibroblasts and endothelial cells through local extracellular signals such as NO, prostaglandins, growth factors etc.
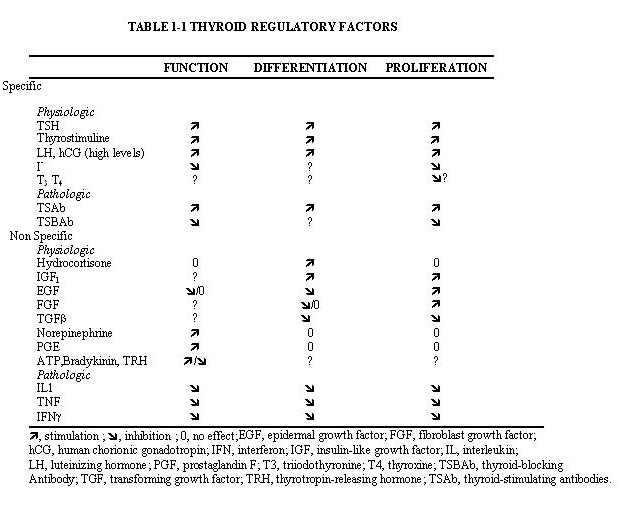
In mice embryo, other unknown factors control differentiation and organ growth which takes place normally in the absence of TSH receptor (218;219) . However, homozygous inactivating mutations of the TSH receptor in familial congenital hypothyroidism were found to be associated with a very hypoplastic thyroid gland (220). Although the thyroid contains receptors for thyroid hormones and a direct effect of these hormones on thyrocytes would make sense (221), as yet little evidence has indicated that such control plays a role in physiol ogy (222). However expression of dominant negative thyroid hormone receptors in mice represses PPARγ expression and induces thyroid tumors in thyroid (223) . Luteinizing hormone (LH) and human chorionic gonadotropin (hCG) at high levels directly stimulate the thyroid, and this effect accounts for the depression of TSH levels and sometimes elevated thyroid activity at the beginning of pregnancy (224-226) .
The thyroid gland is also influenced by various other nonspecific hormones (227). Hydrocortisone exerts a differentiating action in vitro (228) . Estrogens affect the thyroid by unknown mechanisms, directly or indirectly, as exemplified clinically in the menstrual cycle and in pregnancy and by higher prevalence of thyroid disease in females. Growth hormone induces thyroid growth, but its effects are thought to be mediated by locally produced somatomedins (IGF-I). Nevertheless the presence of basal TSH levels might be a prerequisite for the growth promoting action of IGF-I, because a GH replacement therapy did not increase thyroid size in patients deficient for both GH and TSH (229) . The anomalously low endemic goiter prevalence among pygmies living in iodine deficient areas (230), who are genetically resistant to IGF-I, is also compatible with an in vivo permissive effect of IGF-1 and IGF-1 receptor on TSH mitogenic action. Indeed thyroids of transgenic mice overexpressing IGF1 and IGF1 receptor develop hyperplasia and a degree of autonomy vs TSH: their serum TSH is lower and thyroid hormones level normal which shows that they require less TSH to maintain normal thyroid hormone levels (231;232) . In dog and human thyroid primary cultures, the presence of insulin receptors strictly depends on TSH, suggesting that thyroid might be a more specific target of insulin than generally considered (233;234) . It is permissive for TSH mitogenic action in vitro.
Effects of locally secreted neurotransmitters and growth factors on thyrocytes have been demonstrated in vitro and sometimes in vivo, and the presence of some of these agents in the thyroid has been ascertained. The set of neurotransmitters acting on the thyrocyte and their effects vary from species to species (215;235) . In human cells, well-defined direct, but short-lived responses to norepinephrine, ATP, adenosine, bradykinin, and thyrotropin-releasing hormone (TRH) have been observed (215;236;237) . In rat, as evidenced by superior cervical ganglion nervation, sympathetic activity positively modulates function and size of the thyroid (238).
Growth factor signaling cascades demonstrated in vitro can exert similar effects in vivo . In nude mice, the injection of EGF promotes DNA synthesis in thyroid and inhibits iodide uptake in xenotransplanted rat (239) and human thyroid tissues (240). By contrast, the injection of FGF induces a colloid goiter in mice with no inhibition of iodide metabolism or thyroglobulin and thyroperoxidase mRNA accumulation (241) . These effects are the exact replica of initial observations in the dog and other thyroid primary culture system (242),(243-246) . EGF and FGF have since been reported to be locally synthesized in the thyroid gland, as a possible response to thyroxine and TSH (247) respectively. Their exact role as autocrine and/or paracrine agents in the development, function and pathology of the thyroid gland of different species has yet to be clarified (248;249) . HGF does not activate mitogenesis in normal human thyrocyte. The Transforming Growth Factors (TGF)β constitute another category of cytokines that are produced locally by thyrocytes and influence their proliferation and the action of mitogenic factors (248;250) . TGFβ inhibits proliferation and prevents most of the effects of TSH and cAMP in human thyrocytes in vitro (251;252) . TGFβ is synthesized as an inactive precursor which can be activated by different proteases produced by thyrocytes. TGFβ expression is upregulated during TSH-induced thyroid hyperplasia in rats, suggesting an autocrine mechanism limiting goiter size (253). Activin A and the bone morphogenetic peptide (BMP), which are related to TGFβ, are also present in thyroid ( MP7 and MP8A, unpublished) and inhibit thyrocyte proliferation in vitro (254). Unlike TGFβ, they are directly synthesized as an active form. Elements of a Wnt/β catenin signaling pathway (Wnt factors, Frizzled receptors and disheveled isoforms) have been identified in human thyroid and thyroid cancer cell lines (255) . The eventual role in vivo in humans of most of these factors remains to be proved and clarified.
Thyroglobulin has been reported as a negative feedback inhibitor repressing the expression of specific thyroid transcription factors TTF1, TTF2, Pax8 and acting through a putative receptor at the apical membrane (256). However, as previous claims by the same group (the exophtalmic producing factor, ganglioside as the TSH receptor, etc) this one is neither substantiated nor supported by others.
Human thyroid cells contain androgen and estrogen receptors (257). Estrogens promote the growth of these cells (258) which may explain the higher prevalence of thyroid tumors and diseases in women, particularly between puberty and menopause. In mice, thyroid estrogen by downregulating CDKn1B (p27) facilitates the growth effects of the PI3K cascade (259).
In Pathology Mutated constitutively active TSH receptors and Gs proteins cause thyroid autonomous adenomas (260;261) . Mutations conferring higher sensitivity of the TSH receptor to LH/HCG cause hyperthyroidism in pregnancy (262) (263). Pathologic extracellular signals play an important role in autoimmune thyroid disease. Thyroid-stimulating antibodies (TSAbs), which bind to the TSH receptor and activate it, reproduce the stimulatory effects of TSH on the function and growth of the tissue. Their abnormal generation is responsible for the hyperthyroidism and goiter of Graves' disease. The kinetic characteristics of TSH and TSAbs differ: TSH effects on camp accumulation are rapid and disappear rapidly in the absence of the hormone (minutes) while TSAbs effects are slow and persistent (hours) (264) .
Thyroid-blocking antibodies (TBAbs) also bind to the TSH receptor but do not activate it and hence behave as competitive inhibitors of the hormone. Such antibodies are responsible for some cases of hypothyroidism in thyroiditis. Both stimulating and inhibitory antibodies induce transient hyperthyroidism or hypothyroidism in newborns of mothers with positive sera (216). The existence of thyroid growth immunoglobulins has been hypothesized to explain the existence of Graves' disease with weak hyperthyroidism and prominent goiter (265). The thyroid specificity of such immunoglobulins would imply that they recognize thyroid-specific targets. This hypothesis is now abandoned (266-268) . Discrepancies between growth and functional stimulation may instead reflect cell intrinsic factors. Local cytokines have been shown to influence, mostly negatively, the function, growth, and differentiation of thyrocytes in vitro and thyroid function in vivo. Because they are presumably secreted in loco in autoimmune thyroid diseases, these effects might play a role in the pathology of these diseases, but this notion has not yet been proved (215) (269). Moreover in selenium and iodine deficiency plus dietary supplementation of thiocyanate, secretion of TGFβ by macrophages has been implicated in the generation of thyroid fibrosis (270) (271) and the pathogenesis of thyroid failure in endemic cretinism. The overexpression of both FGF and FGF receptor 1 in thyrocytes from human multinodular goiter might explain their relative TSH-independence (272) . On the other hand, the subversion of tyrosine kinase pathways similar to those normally operated by local growth factors (i.e. the activation of Ret/PTC (273) and TRK (274), the overexpression of Met/HGF receptor sometimes in association with HGF (275), or erbB/EGF receptor in association with its ligand TGFα (276) have been causally associated with TSH-independent thyroid papillary carcinomas. An autocrine loop involving IGF-II and the insulin receptor isoform-A is also proposed to stimulate growth of some thyroid cancers (277) . Thyroid cancer cells often escape growth inhibition by TGFβ (278).
REGULATORY CASCADES
The great number of extracellular signals acting through specific receptors on cells in fact control a very limited number of regulatory cascades. We first outline these cascades, along with the signals that control them, and then describe in more detail the specific thyroid cell features: controls by iodide and the TSH receptor.
The Cyclic Adenosine Monophosphate Cascade
The cyclic adenosine monophosphate (cAMP) cascade in the thyroid corresponds, as far as it has been studied, to the canonic model of the β-adrenergic receptor cascade (216) (Fig. 1-9). It is activated in the human thyrocyte by the TSH and the β-adrenergic and prostaglandin E receptors. These receptors are classic seven-transmembrane receptors controlling transducing guanosine triphosphate (GTP)-binding proteins. Activated G proteins belong to the G s class and activate adenylyl cyclase; they are composed of a distinct α s subunit and nonspecific β and γ monomers. Activation of a G protein corresponds to its release of guanosine diphosphate (GDP) and binding of GTP and to its dissociation into α GTP and βγ dimers. α sGTP directly binds to and activates adenylyl cyclase. Inactivation of the G protein follows the spontaneous, more or less rapid hydrolysis of GTP to GDP by α s GTPase activity and the reassociation of α GDP with βγ. The effect of stimulation of the receptor by agonist binding is to increase the rate of GDP release and GTP binding, thus shifting the equilibrium of the cycle toward the α GTP active form. One receptor can consecutively activate several G proteins (hit-and-run model). The thyroid contains mainly three isoforms of adenylyl cyclase : III, VI and IX (279) . A similar system negatively controls adenylyl cyclase through G i . It is stimulated in the human thyroid by norepinephrine through α 2 -receptors. Adenosine at high concentrations directly inhibits adenylyl cyclase. The cAMP generated by adenylyl cyclase binds to the regulatory subunit of protein kinase A (PKA) that is blocking the catalytic subunit and releases this now-active unit. The activated, released catalytic unit of protein kinase phosphorylates serines and threonines in the set of proteins containing accessible specific peptides that it recognizes. These phosphorylations, through more or less complicated cascades, lead to the observed effects of the cascade. cAMP-dependent kinases have two isoenzymes (I, II), the first of which is more sensitive to cAMP, but as yet no clear specificity of action of these kinases has been demonstrated. In the case of the thyroid, this cascade is activated through specific receptors, by TSH in all species, and by norepinephrine receptors and prostaglandin E in humans, with widely different kinetics: prolonged for TSH and short lived (minutes) for norepinephrine and prostaglandins (280). Other neurotransmitters have been reported to activate the cascade in thyroid tissue, but not necessarily in the thyrocytes of the tissue (237). In the thyroid cAMP, besides PKA, activates EPAC (Exchange Proteins directly Activated by cAMP) or Rap guanosine nucleotide exchange factor-1 (GEF-1) and the less abundant GEF-2, which activate the small G protein Rap (281). However, despite high expression of EPAC1 in thyrocytes and its further increase in response to TSH, all the physiologically relevant cAMP-dependent functions of TSH studied in dog thyroid cells, including acute regulation of cell functions (including thyroid hormone secretion) and delayed stimulation of differentiation expression and mitogenesis, are mediated only by PKA activation (282) . The role of the cAMP/EPAC/Rap cascade in thyroid thus remains largely unknown. Activation of PKA inactivates small G proteins of the Rho family (RhoA, Rac1 and Cdc42), which reorganizes the actin cytoskeleton and could play an important role in stimulation of thyroid hormone secretion and induction of thyroid differentiation genes (283) . Of the other known possible effectors of cAMP, cyclic nucleotide-activated channels have not been looked for. For several effects of cyclic AMP (eg NIS and thyroglobulin induction, DNA synthesis) protein kinase A is required.
The cAMP cascade is also controlled by several negative feedbacks. The most important is the activation and induction by PKA of PDE4 D3 and other phosphodiesterases (284) (285)
The thyrocyte is very sensitive to internal c AMP : a mere doubling of its concentration is sufficient to elicit near maximal thyroglobulin phagocytosis (286).
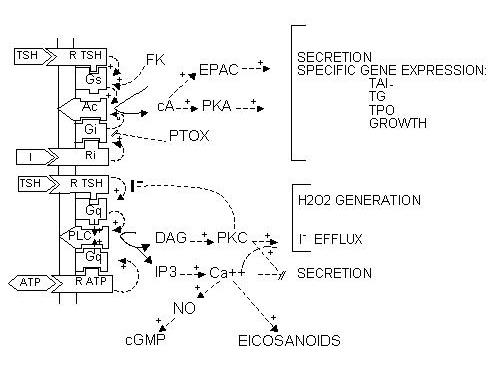
Fig .1.9
Regulatory cascades activated by thyroid-stimulating hormone (TSH) in human thyrocytes. In the human thyrocyte, H 2 O 2 (H2O2) generation is activated only by the phosphatidylinositol 4,5-bisphosphate (PIP 2 ) cascade, that is, by the Ca 2+ (Ca++) and diacylglycerol (DAG) internal signals. In dog thyrocytes, it is activated also by the cyclic adenosine monophosphate (cAMP) cascade. In dog thyrocytes and FRTL-5 cells, TSH does not activate the PIP 2 cascade at concentrations 100 times higher than those required to elicit its other effects. Ac, adenylate cyclase; cA, 3 ’ -5 ’ -cAMP, cGMP, 3 ’ -5 ’ -cyclic guanosine monophosphate; FK, forskolin; Gi, guanosine triphosphate (GTP) binding transducing protein inhibiting adenylate cyclase; Gq, GTP-binding transducing protein activating PIP 2 phospholipase C; Gs, GTP-binding transducing protein activating adenylate cyclase; I, putative extracellular signal inhibiting adenylate cyclase (e.g., adenosine through A 1 receptors); IP 3 , myoinositol 1,4,5-triphosphate; EPAC: cAMP dependent Rap guanyl nucleotide exchange factor; PKA, cAMP-dependent protein kinases; PKC, protein kinase C; PLC, phospholipase C; PTOX, pertussis toxin; R ATP, ATP purinergic P 2 receptor; R TSH, TSH receptor; Ri, receptor for extracellular inhibitory signal I; TAI, active transport of iodide; TG, thyroglobuline; TPO, thyroperoxidase.
The Ca2 + – Inositol 1,4,5-Triphosphate Cascade
The Ca 2+ – inositol 1,4,5-triphosphate (IP 3 ) cascade in the thyroid also corresponds, as far as has been studied, to the canonic model of the muscarinic or α 1 -adrenergic receptor – activated cascades. It is activated in the human thyrocyte by TSH, through the same receptors that stimulate adenylyl cyclase, and by ATP, bradykinin, and TRH — through specific receptors. In this cascade, as in the cAMP pathway, the activated receptor causes the release of GDP and the binding of GTP by the GTP-binding transducing protein (G q ) and its dissociation into α q and βγ. α GTP then stimulates phospholipase C. Gs and Gq compete for the same TSH receptor, with a higher affinity for Gs (287-289) . Phospholipase C hydrolyzes membrane phosphatidylinositol 4,5-bisphosphate (PIP 2 ) into diacylglycerol and IP 3 . IP 3 enhances calcium release from its intracellular stores, followed by an influx from the extracellular medium. The rise in free ionized intracellular Ca 2+ leads to the activation of several proteins, including calmodulin. The latter protein in turn binds to target proteins and thus stimulates them: cyclic nucleotide phosphodiesterase and, most importantly, calmodulin-dependent protein kinases. These kinases phosphorylate a whole set of proteins exhibiting serines and threonines on their specific peptides and thus modulate them and cause many observable effects of this arm of the cascade. Calmodulin also activates constitutive nitric oxide (NO) synthase in thyrocytes. The generated NO itself enhances soluble guanylyl cyclase activity in thyrocytes and perhaps in other thyroid cells and thus increases cGMP accumulation (290). Nothing is yet known about the role of cGMP in the thyroid cell but NO causes vasodilatation.
Diacylglycerol released from PIP 2 activates protein kinase C, or rather the family of protein kinases C, which by phosphorylating serines or threonines in specific accessible peptides in target proteins causes the effects of the second arm of the cascade (291). It inhibits phospholipase C or its G q , thus creating a negative feedback loop. In the human thyroid, the PIP 2 cascade is stimulated through specific receptors by ATP, bradykinin, TRH and by TSH (237) (292) (293). The effects of bradykinin and TRH are very short lived. Acetylcholine, which is the main activator of this cascade in the dog thyrocyte (294), is inactive on the human cell, although it activates nonfollicular (presumably endothelial) cells in this tissue (237).
Other Phospholipid-Linked Cascades
In dog thyroid cells and in a functional rat thyroid cell line (FRTL5), TSH activates PIP 2 hydrolysis weakly and at concentrations several orders of magnitude higher than those required to enhance cAMP accumulation. Of course, these effects have little biologic significance. However, in dog cells, at lower concentrations TSH increases the incorporation of labeled inositol and phosphate into phosphatidylinositol. Similar effects may exist in human cells, but they would be masked by stimulation of the PIP 2 cascade. They may reflect increased synthesis perhaps coupled to and necessary for cell growth (295).
Diacylglycerol can be generated by other cascades than the classic Ca 2+ -IP 3 pathway. Activation of phosphatidylcholine phospholipase D takes place in dog thyroid cells stimulated by carbamylcholine. Because it is reproduced by phorbol esters, that is, by stable analogues of diacylglycerol, it has been ascribed to phosphorylation of the enzyme by protein kinase C, which would represent a positive feedback loop (296). Although such mechanisms operate in many types of cells, their existence in human thyroid cells has not been demonstrated (297).
Release of arachidonate from phosphatidylinositol by phospholipase A 2 and the consequent generation of prostaglandins by a substrate-driven process are enhanced in various cell types through G protein – coupled receptors, by intracellular calcium, or by phosphorylation by protein kinase C. In dog thyroid cells all agents enhancing intracellular calcium concentration, including acetylcholine, also enhance the release of arachidonate and the generation of prostaglandins. In this species, stimulation of the cAMP cascade by TSH inhibits this pathway. In pig thyrocytes, TSH has been reported to enhance arachidonate release. In human thyroid, TSH, by stimulating PIP 2 hydrolysis and intracellular calcium accumulation, might be expected to enhance arachidonate release and prostaglandin generation, but such effects have not yet been proved.
Regulatory Cascades Controlled by Receptor Tyrosine Kinases
Many growth factors and hormones act on their target cells by receptors that contain one transmembrane segment. They interact with the extracellular domain and activate the intracellular domain, which phosphorylates proteins on their tyrosines. Receptor activation involves in some cases a dimerization and in others a conformational change. The first step in activation is interprotein tyrosine phosphorylation, followed by binding of various protein substrates on tyrosine phosphates containing segments of the receptor. Such binding through src homology domains (SH2) leads to direct activation or to phosphorylation of these proteins on their tyrosines and to membrane localization. In turn, these cause sequential activation of the ras and raf proto-oncogenes, mitogen-activated protein (MAP) kinase kinase, MAP kinase, and so on, on the one hand, and phosphatidylinositol-3-kinase (PI-3-kinase), protein kinase B (PKB), and TOR (target of rapamycin) on the other hand. The set of proteins phosphorylated by a receptor defines the pattern of action of this receptor. In thyroids of various species, insulin, IGF-I, EGF, FGF, HGF, but not platelet-derived growth factor activate such cascades (298;299) . In the human thyroid, effects of insulin, IGF-I, EGF, FGF, but not HGF have b een demonstrated (215;300-304) . Transforming growth factor-β, acting through the serine threonine kinase activity of its receptors and intermediate proteins (Smad), inhibits proliferation and specific gene expression in human thyroid cells (251;252;305) . TSH and cAMP do not activate either the MAPK-ERK nor the JUNK and p38 phosphorylation pathways in dog or human thyroids (306).
Cross-Signaling between the Cascades
Calcium, the intracellular signal generated by the PIP 2 cascade, activates calmodulin-dependent cyclic nucleotide phosphodiesterases and thus inhibits cAMP accumulation and its cascade (307). This activity represents a negative cross-control between the PIP 2 and the cAMP cascades. Activation of protein kinase C enhances the cAMP response to TSH and inhibits the prostaglandin E response, which suggests opposite effects on the TSH and prostaglandin receptors (308). No important effect of cAMP on the PIP 2 cascade has been detected. On the other hand, stimulation of protein kinase C by phorbol esters inhibits EGF action.
Cross-signaling between the cyclic AMP pathway and growth factor activated cascades have been observed in various cell types (309;310). In ovarian granulosa cells, FSH through cAMP activates MAP kinases and the PI3 kinase pathway (311). In FRTL5, but not in WRT cell lines, TSH through cAMP activates MAP kinases. In WRT cells but not in PCCl3 cells, TSH and cAMP activate PKB (312;313). Such cross signallings have not been observed in human or dog thyroid cells. Ras, MAPK, p38, Jun kinase and ERK5, as well as PI3 kinase and PKB, are not modulated by cAMP (314;315) (316) (317).
Other growth activating cascades have been little investigated in the thyroid. In dog and human cells TSH or cAMP have no effect on STAT phosphorylations i.e. on the JAK-STAT pathways. The NF β pathway has not yet been investigated in thyroid cells.
SPECIFIC CONTROL BY IODIDE
Iodide, the main substrate of the specific metabolism of the thyrocyte, is known to control the thyroid. Its main effects in vivo and in vitro are to decrease the response of the thyroid to TSH, to acutely inhibit its own oxidation (Wolff-Chaikoff effect), to reduce its trapping after a delay (adaptation to the Wolff-Chaikoff effect), and at high concentrations to inhibit thyroid hormone secretion (Fig. 1-10). The first effect is very sensitive in as much as small changes in iodine intake are sufficient to reset the thyroid system at different serum TSH levels without any other changes (e.g., thyroid hormone levels), which suggests that in physiologic conditions, modulation of the thyroid response to TSH by iodide plays a major role in the negative feedback loop (217;318). Iodide in vitro has also been reported to inhibit a number of metabolic steps in thyroid cells (319) (320). These actions might be direct or indirect as a result of an effect on an initial step of a regulatory cascade. Certainly, iodide inhibits the cAMP cascade at the level of G s or cyclase and the Ca 2+ -PIP 2 cascade at the level of G q or phospholipase C; such effects can account for the inhibition of many metabolic steps controlled by these cascades (321) (322). In one case in which this process has been studied in detail, the control of H 2 O 2 generation, that is, the limiting factor of iodide oxidation and thyroid hormone formation, iodide inhibited both the cAMP and the Ca 2+ -PIP 2 cascades at their first step but also the downstream effects of the generated intracellular signals cAMP, Ca 2+ , and diacylglycerol on H 2 O 2 generation (323). This effect account for the inhibition by iodide of its oxidation i.e. the Wolff-Chaikoff effects (324).
Until now, the mechanism of action of iodide on all the metabolic steps besides secretion fits the "XI" paradigm of Van Sande (325). These inhibitions are relieved by agents that block the trapping of iodide (e.g., perchlorate) or its oxidation (e.g., methimazole) — the Van Sande criteria. The effects are therefore ascribed to one or several postulated intracellular iodinated inhibitors called XI. The identity of such signals is still unproved. At various times several candidates have been proposed for this role, such as thyroxine, iodinated eicosanoids (iodolactone) (326) , and iodohexadecanal (327). The latter, the predominant iodinated lipid in the thyroid, can certainly account for the inhibition of adenylyl cyclase and of H 2 O 2 generation (328) (329) (330) . The stimulation of H 2 O 2 generation by iodide in follicles of human thyroid is inhibited by methimazole but not by perchlorate.
This suggests that the generation of Xi takes place at the membrane and that the intermediate Xi may diffuse in the membrane but not in the medium (unpublished). It should be emphasized that iodination of the various enzymes, as well as a catalytic role of iodide in the generation of O 2 radicals (shown to be involved in the toxic effects of iodide), could account for the Van Sande criteria with no need for the XI paradigm (325) (331) . Besides, an inhibition of thyroid secretion by iodide in antithyroid drugs treated hyperthyroid patients suggests a direct Xi independent effect.
Distinct from its inhibitory effects, iodide also activates H2O2 generation and therefore protein iodination in the thyroid of some species including humans. This effect is also inhibited by inhibitors of thyroperoxidase and NIS. It would link the generation of H2O2 to the availability of its cosubstrate iodide (332).
Iodide in vivo, at moderate doses in dog, decreases cell proliferation and the expression of TPO and NIS mRNA but not the synthesis or secretion of thyroid hormones. The downregulation of NIS explains the well known delayed decrease of iodide transport in response to iodide, i.e. the adaptation to the Wolff-Chaikoff effect (333).
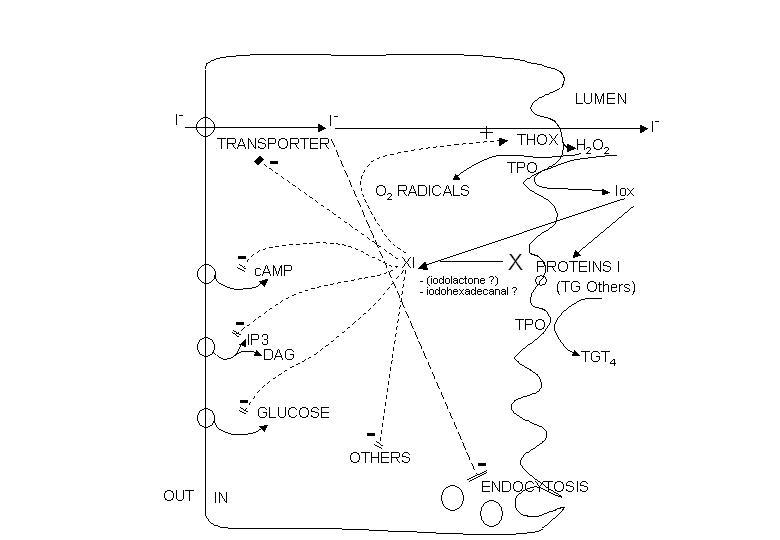
Fig 1-10
Effects of iodide on thyroid metabolism. All inhibitory effects of iodide, except in part the inhibition of secretion, are relieved by drugs that inhibit iodide trapping (e.g., perchlorate) or iodide oxidation (e.g., methimazole). All effects are direct inhibitions except the effect on iodide transport which bears on the transcription of Na/I- symporter gene. Three possible mechanisms corrresponding to this paradigm are outlined: generation of O 2 radicals, iodination of target proteins and synthesis of an XI compound. Any of these mechanisms could account for the various steps ascribed to XI inhibition by I - (indicated by slashes).
THE THYROTROPIN RECEPTOR
The Structure of the Thyrotropin Receptor
The receptors for TSH, FSH, and LH/CG are members of the rhodopsin-like G protein-coupled receptor family. As such, the TSH receptor has a “ serpentine ” domain containing seven transmembrane regions with many (but not all) of the features typical of this receptor family. In addition, and a hallmark of the subfamily of glycoprotein hormone receptors (334-336), it has a large (about 400 amino-acid residues) amino-terminal extracellular domain that contains sites that selectively bind TSH with high affinity (337). The higher sequence identity of the serpentine domains of the glycoprotein hormone receptors (about 70%), as compared with the extracellular domains (about 40%, Fig.11 ) suggests that the former are interchangeable modules capable of activating guanine-nucleotide-binding (G) proteins (mainly G s ) after specific binding of the individual hormones to the latter (338). Contrary to other rhodopsin-like G protein-coupled receptors, the glycoprotein hormones bind to their respective extracellular domains with high affinity in the absence of the serpentine domain (339-341). The intramolecular transduction of the signal between these two portions of the receptors involves a still incompletely defined mechanism specific to the glycoprotein hormone receptor family (see below). The relatively high sequence identity between the hormone-binding domains of the TSH and LH/CG receptors opens the possibility of spillover phenomena during normal or, even more so, molar or twin pregnancies, when serum CG concentrations are several orders of magnitude higher than are serum TSH concentrations. This provides an explanation to cases of gestational thyrotoxicosis (see below, Chapter 20 and Chapter 56).
Fig. 1-11
Both the the beta subunits of the glycoprotein hormones and the glycoprotein hormone receptors are encoded by paralogous genes. A: Similarity of the aminoacid sequences of the -subunits of human chrorionic gonadotropin (hCG), luteinizing hormone (LH), thyrotropin (TSH) and follicle stimulating hormone (FSH). Inset: Diagram of the general structure of the receptors for these hormones, showing the ectodomain (extracellular), serpentine (transmembrane) and endodomain (intracellular domain). B: Similarity of the aminoacid sequences of the LH/CG, FSH and TSH receptors (r). Sequence identities are indicated, separately for the extracellular domains of the three receptors. The pattern of shared similarities suggests co-evolution of the hormones and the extracellular domain of their receptors, resulting in generation of specificity barriers. The high similarity of the serpentine domains of the receptors is compatible with a conserved mechanism of intramolecular signal transduction. (Reproduced from Vassart G, Pardo L, Costagliola S. Molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci 2004;29:119, with permission of the publisher)
The TSH receptor contains six sites for N-glycosylation, of which four are effectively glycosylated (342). The functional role of the individual carbohydrate chains is still debated. It is likely that they contribute to the routing and stabilization of the receptor as it passes through the membrane system of the cell and is inserted into the cell membrane. Alone among the glycoprotein hormone receptors, the extracellular domain of the TSH receptor is cleaved, severing it from the serpentine domain (343). This phenomenon has been related to the presence in the extracellular domain of the receptor of a 50 amino-acid insertion for which there is no counterpart in the FSH receptor or LH/CG-receptor. The initial cleavage step, due to the action of a metalloprotease, takes place at around position 314 (within the 50-amino-acid insertion) from the amino terminus of the receptor, and is followed by removal of approximately 50 amino acids from the amino-terminal end of the serpentine-containing portion of the receptor (344;345). The amino-terminal end of the receptor remains bound to the extracellular end of the serpentine domain by disulfide bonds. The functional importance of this TSH receptor-specific postranslational modification remains unclear. Whereas all wild type TSH receptors on the surface of thyroid follicular cells seem to be in cleaved form, non-cleavable mutant constructs are functionally undistinguishable from cleaved receptors, when expressed in transfected cells (346). Residues 303-366 can be deleted from the hinge region with minimal effects o the function of the receptor (347). When transiently or permanently transfected in non-thyroid cells, wild type human TSH receptors are present at the cell surface as a mixture of monomers and cleaved dimers. There are indications from immunization experiments in mice that cleavage and possible shedding of the aminoterminal portion of the receptor would play a role in the generation of stimulating autoantibodies in patients with Graves disease (348;349) (see Chapter 17).
The TSH receptor is specifically inserted into the basolateral membrane of thyroid follicular cells. This phenomenon involves a signal (amino acids 731-746) unusually localized in the very C-terminal portion of the receptor, at a marked distance from the membrane (350).
The possibility that TSH receptors are present on the cell surface as dimers of cleaved dimers was raised after demonstration that most rhodopsin-like G-protein coupled receptors do dimerize (351). Functional complementation of TSH receptors with mutations in the extracellular and the serpentine domains has been observed after expression of receptor constructs in transfected cells (352). A definitive demonstration that glycoprotein hormone receptors do dimerize in vivo and that dimers interact functionally has been provided by similar complementation experiments performed in LH/CG receptor knockout mice. Mice co-expressing two inactive mutant receptors are fertile (353). Direct demonstration of dimerization of the TSH receptor has been provided by Bioluminescence Resonance Energy Transfer (BRET) and the dimers have been shown to display negative cooperativity for binding of TSH (352). Whether this allosteric behavior of the receptor has (patho)physiological significance remains to be determined..
The Thyrotropin Receptor Gene
The gene coding for the human TSH receptor is located on the long arm of chromosome 14 (14q31)(354;355). It is is organized into 10 exons. The extracellular domain is encoded by a series of 9 exons, each of which corresponds to one or an integer number of leucine-rich repeat segments (see below). The carboxyl-terminal half of the receptor containing the carboxyl-terminal part of the extracellular domain and the serpentine domain is encoded by a single large exon (356), in keeping with the fact that the genes for many G protein-coupled receptor have no introns. A likely evolutionary scenario derives from this gene organization: the glycoprotein hormone receptor genes would have evolved from the condensation of an intron-less classic G protein-coupled receptor with a mosaic gene encoding a protein with leucine-rich repeat segments (357). Triplication of this ancestral gene and subsequent divergence led to the receptors for TSH, FSH, and LH/CG. The existence of 10 exons in both the TSH and FSH receptor genes (as opposed to the 11-exon LH/CG-receptor gene), suggests the following evolutionary steps: first, duplication of an ancestral glycoprotein hormone receptor gene, yielding the LH/CG-receptor gene and the ancestors of the TSH- and FSH-receptor genes. After losing one intron, the latter duplicated subsequently into the TSH- and FSH- receptor genes. The family of the glycoprotein hormone receptors comprises five additional members presenting a similar pattern made of leucine-rich repeats in the ectodomain, upstream of a rhodopsin-like serpentine domain: two of them (LGR7, LGR8) encode insulin-like 3 and relaxin receptors, respectively (358), the remaining three (LGR4, LGR5, LGR6) are orphan receptors involved in regulation of epithelial stem cell biology (359).
The promoter of the TSH receptor gene has the characteristics of a housekeeping gene promoter, being GC-rich and devoid a TATA box. In rats it stimulates transcription from multiple start sites (360) and it contains a functional thyroid transcription factor (TTF)-1 recognition site (361). Expression of the TSH-receptor gene is largely thyroid-specific. Constructs made of a chloramphenicol acetyltransferase reporter gene under control of the 5 ’ -flanking region of the rat TSH-receptor gene are expressed when transfected into FRTL5 cells and FRT cells but not into non-thyroid HeLa or rat liver (BRL) cells (360). However, TSH receptor mRNA has been clearly demonstrated in fat tissue of guinea pigs (362), in adipocytes (363;364) and in ependymal cells of the mediobasal hypothalamus in the mouse, where it plays a role in adjustment of reproductive physiology to the length of the days (365;366) . TSH receptors may also be present in lymphocytes, extraocular tissue, cartilage, and bone, but their functional importance in these tissues is uncertain (367;368). Expression of the TSH receptor in thyroid cells is extremely robust. It is moderately upregulated by TSH in vitro and downregulated by iodide in vivo (369).
Recognition of the Receptor by Thyrotropin
The three-dimensional structures are available for hCG and FSH FSH (370-372) which allows accurate modelization of TSH on these templates. The crystal structure of the human FSHr-FSH complex (373) has confirmed that the ectodomain of glycoprotein hormone receptors belongs to the family of proteins with leucine-rich repeats (LRRs) (374). The concave inner surface of the receptor (Fig.12) is an untwisted, non-inclined b-sheet formed by ten LRRs. Whereas the N-terminal portion of the beta-sheet (LRR1 – 7) is nearly flat, the C-terminal portion (LRR7 – 10) has the horseshoe-like curvature of LRR proteins. The crystal structure of part of the TSHr ectodomain in complex with thyroid-stimulating or – blocking autoantibodies has recently been obtained (375;376). Notably, both the structure of the ectodomain of TSHr and the receptor binding arrangements of the autoantibodies are very similar to those reported for the FSHr-FSH complex. The ectodomain of glycoprotein hormone receptors also contains, downstream of the LRR region, a cysteine cluster domain of unknown structure (the hinge region), involved in receptor inhibition/activation and containing sites for tyrosine sulfation important for hormone binding (see below).

Fig. 1-12
Schematic representation of the structure of the TSH receptor. (A) Two-dimensional representation with indication of the various domains. The blue boxes correspond to amino-terminal and carboxyl-terminal cysteine-rich portions of the extracellular domain, flanking leucine-rich repeats (LRR, yellow box). (B) General view of the follicle-stimulating hormone receptor (FSHr)-FSH crystal structure as a template to model the interaction between TSH and the TSH receptor (692).The concave inner surface of the receptor, formed by ten leucine rich repeats (LRR2 – 9, shown in blue), contact the middle section of the hormone molecule, both the C-terminal segment of the subunit and the “ seat-belt ” segment of the -subunit (shown in red). (C) EachLRR is composed of theX1-X2-L-X3-L-X4-X5 residues (where X is any amino acid, and L usually is Leu, Ile, or Val), forming the central X2-L-X3-L-X4 a typical beta-strand, while X1 and X5 are parts of the adjacent loops. (D) Molecular model of the transmembrane domain of the TSH receptor, constructed from the crystal structure of bovine rhodopsin. The color code of the -carbon ribbons is: transmembrane helix 1 (crimson), 2 (goldenred), 3 (dark red), 4 (gray), 5 (red), 6 (orange), and 7 (blue), and Helix8 (blue). The structures of available class A rhodopsin-like GPCRs are similar at the transmembrane domain.
Replacement by site-directed mutagenesis of the residues facing the hormone in the leucine-rich repeat portion of the TSH receptor (see figure 12 ) with their counterparts in the LH/CG receptor has been performed to assess the structural bases of binding selectivity (377). Exchanging eight amino acids of the TSH receptor for the corresponding amino acids in the LH/CG receptor resulted in a mutant receptor which bound human CG as well as the wild-type LH/CG receptor. While gaining sensitivity to human CG, the mutant receptor retained normal sensitivity to TSH, making it a dual-specificity receptor. It is necessary to exchange 12 additional amino-acid residues to fully transform this mutant receptor into a bonafide LH/CG receptor (378). From an evolutionary point of view, these observations indicate that the specificity of hormone receptors is based on both attractive and repulsive residues, and that residues at different homologous positions have been selected to this result in the different receptors.
Inspection of electrostatic surface maps of models of the wild-type TSH and LH/CG receptors and some of the mutants is revealing in this respect (379;380). The LH/CG receptor has an acidic groove in the middle of its horseshoe, extending to the lower part of it (corresponding to the carboxyl-terminal ends of the strands). Generation of a similar distribution of charges in the dual-specificity and reverse-specificity TSH receptor mutants suggests that this is important for recognition of human CG. A detailed modelization of the interactions between TSH and the ectodomain of its receptor has been realized (381).
In addition to the hormone-specific interactions genetically encoded in the primary structure of glycoprotein hormone receptors and their ligands, there are important non-hormone-specific ionic interactions involving sulfated tyrosine residues present in the extracellular domains of all three receptors (382). In the TSH receptor, both tyrosine residues of a conserved Tyr-Asp-Tyr motif located close to the border between the extracellular domain and the first transmembrane helix are sulfated ( Fig.13 ), although only sulfation of the first tyrosine of the motif seems to be functionally important (383), contributing importantly to the affinity of the receptor for TSH, without interfering with specificity. The functional role of this postranslational modification of the TSH receptor has been confirmed by demonstration of profound hypothyroidism due to resistance to TSH in mice with inactivation of Tpst2, one of the enzymes responsible for tyrosine sulfation (384;385).

Fig. 1-13
Linear representation of the TSH receptor. Sequences common to all rhodopsin-like G protein-coupled receptors and sequences specific to the glycoprotein hormone receptor gene family are both implicated in activation of the TSH receptor. Key residues are indicated (red dots) as well as conserved motifs: SO3 -- stands for postranslational sulfation of the indicated tyrosine residues (693). The black boxes stand for transmembrane helices and I1-I3, E1-E3, for intracellular and extracellular loops, respectively; LRR, leucine-rich repeats.
Activation of the Serpentine Portion of the Thyrotropin Receptor
Being a member of the G-protein coupled receptor family, the serpentine domain of the TSH receptor is likely to share with rhodopsin common mechanisms of activation (386;387). However, sequence variations in this domain of the glycoprotein hormone receptors suggest the existence of idiosyncrasies associated with hormone-specific mechanisms of activation (Fig. 13). The crystallographic structure of several GPCRs belonging to family A has now been determined (388-396) giving templates for realistic modeling of the serpentine portion of class A GPCRs, including theTSH receptor. A host of artificial and natural mutants of GPCRs have been studied over the past 20 years. This led to scenarios of GPCR activation which have only very recently been confronted with direct structural data (394;397;398). The first activating mutation identified in the α2adrenergic receptor suggested that activation resulted from the release of a structural lock between transmembrane helices 3 and 6 keeping the wild type receptor inactive (399;400). Structural data obtained from opsin at low pH (expected to mimic the active state) (401), a constitutively active rhodopsin mutant (394), and an active state of the α2adrenergic receptor stabilized by a nanobody (398) point to a mechanism of activation involving subtle changes in the conformations of the ligand-binding pocket associated with a more dramatic movement of transmembrane helix 6 (TM6) secondary to rupture of the same TM3-TM6 “ ionic lock ” . The result is the “ opening ” of a cavity between the cytoplasmic ends of TM3, 5 and 6 allowing interaction with- and activation of the G protein (401;402).
The many gain-of-function somatic and germline mutations that have been found in the serpentine domain of the TSH receptor in autonomous toxic adenomas and hereditary nonautoimmune hyperthyroidism (see Chapters 19 and Chapter 25) are expected to trigger a similar conformational change in the TSH receptor (for a complete list of mutations, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/). Of note, a mutation affecting Asp619, at the basis of TM6, which would rupture the TM3-TM6 ionic lock, was amongst the very first activating mutations identified in toxic adenomas (403;404). As many activating mutations affecting a given residue have been found repeatedly over the past 20 years, it is likely that we are getting close to a saturation map for spontaneous gain of function mutations. Combined analyses of these natural mutants with extensive site-directed mutagenesis have identified key interactions implicated in activation of the serpentine portion of the TSH receptor (352;405).
Interaction between the Extracellular and Serpentine Domains of the Thyrotropin Receptor
The dichotomy between hormone binding (to the ectodomain) and activation of the G protein (by the serpentine domain) poses the question of how the activation signal travels intra-molecularly between the two domains after binding of TSH. Two important clues cast light on this issue. Firstly, experiments with aminoterminally truncated receptors demonstrated that the ectodomain exerts a negative effect on the serpentine domain (406;407). Indeed, constructs devoid of the ectodomain display increase signaling via Gαs leading to the notion that in pharmacological terms, the ectodomain behaves as an inverse agonist of the serpentine domain. When compared to wild type receptors fully stimulated by TSH, the activation state of aminoterminally truncated constructs is however far from maximal. Secondly, and more important, mutations of a single residue (Ser 281) present in a highly conserved motif of the ectodomain of glycoprotein hormone receptors (YP S HCCAF) ( Fig. 14 ), result in strong activation of the receptor (408). The segment containing this motif, sometimes referred to as the “ hinge ” region, plays an important role in activation of all three glycoprotein hormone receptors (409). The functional effect of substitutions of S281 in the TSH receptor likely results in a local “ loss-of-structure ” , because the more de-structuring the substitutions, the stronger the activation (410-412). The most active mutants cause increase in cellular cAMP similar to that achieved by saturating concentration of TSH on the wild type receptor (413).
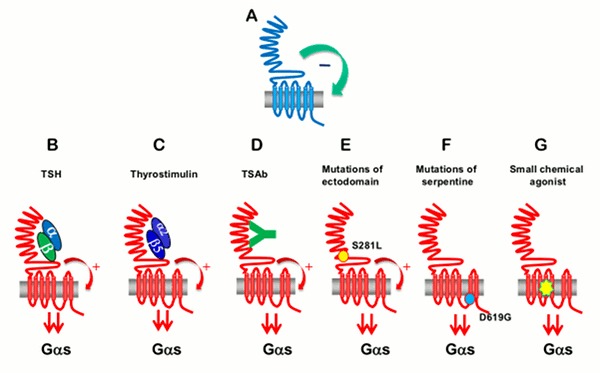
Fig. 1-14
Model for activation of the thyrotropin (TSH) receptor by various agonists. Interactions between the extracellular domain and the serpentine domain are implicated in the activation mechanism. The TSH receptor is represented with its extracellular domain containing a concave, hormone-binding structure facing rightwards, and a transmembrane serpentine domain. The basal state of the receptor is characterized by an inhibitory interaction between the extracellular domain and the serpentine domain (indicated by the (-) green sign). In the absence of agonist, the extracellular domain would function as a tethered inverse agonist of the serpentine domain. Binding of physiological agonists (TSH , B; thyrostimulin 2 5, C), or stimulating autoantibodies (D) switches the ectodomain from inverse agonist to full agonist of the serpentine domain (indicated by the (+) red sign). Mutation of the Ser in position 281of the ectodomain has the same effect (yellow dot, E). Other mutations may activate directly the serpentine domain by breaking silencing locks between transmembrane helices (the example of Asp619Gly is illustrated; blue dot, F). The serpentine domain may also be activated, by binding of low molecular chemical agonists directly to transmembrane segments (yellow star, G). The serpentine domain in the basal state is shown as a compact blue structure. The fully activated serpentine domain is shown as relaxed red structures with arrows indicating activation of G s.
These results led to the following model for activation of the TSH receptor (Fig.14) (414;415). In the resting state, the extracellular domain would inhibit the activity of an inherently noisy rhodopsin-like serpentine domain. Upon activation, by binding of TSH, or secondary to mutation of S281, a structural module including the hinge region would switch from inverse agonist to full agonist of the serpentine domain. The ability of the strongest S281 mutants to activate the receptor fully in the absence of TSH suggests that the ultimate agonist of the serpentine domain would be the “ activated ” extracellular domain, with no need for a direct interaction between TSH and the serpentine domain. Several arguments support such a model: (i) mutations in the extracellular loops which strongly activate the intact receptor are unable to activate constructs devoid of ectodomain, while mutations affecting the transmembrane helices or the intracellular loops of the same construct are effective. This suggests that, in the intact receptor, the extracellular loops would be part of a module containing the “ hinge region ” involved in activation. (ii) a monoclonal antibody displaying inverse agonistic activity has been generated and its epitope has been localized within the hinge region (416). (iii) the TSH receptor can be activated by molecules, like autoantibodies (see Chapter 17) and thyrostimulin (417), sharing little if any structural homology with TSH. A parsimonious explanation is that these diverse agonists would bind to the ectodomain, switching it into an agonist of the serpentine domainwith no need for additional specific interaction with the serpentine.
Activation by Chorionic Gonadotropin
The sequence similarity between TSH and hCG, and between their receptors, allows for some degree of promiscuous activation of the TSH receptor by CG during the first trimester of pregnancy, when serum human CG concentrations are highest. The inverse relation between serum TSH and CG concentrations in most pregnant women is clear indication that their thyroid gland is stimulated by CG (418) (see Chapter 20 and Chapter 56). While most pregnant women are euthyroid, thyrotoxicosis may occur if CG production is excessive (as it occurs in twin pregnancies or chorionic tumors (see Chapter 20), or in rare women who have a mutant TSH receptor with increased sensitivity to CG (419).
Activation by Thyrotropin-Receptor Antibodies
The serum antibodies found in most patients with Graves' thyrotoxicosis and some patients with hypothyroidism caused by chronic autoimmune thyroiditis can stimulate or block the TSH receptor, respectively (see Chapter 17, Chapter 34). Epitopes recognized by TSAbs have been identified from precise mapping of binding site of murine or human monoclonal antibodies endowed with TSAb activity (420-422). However, the precise mechanisms implicated in activation of the receptor by TSAbs (and by TSH) are still unknown. Although most TSAbs do compete with TSH for binding to the receptor and despite similarity in interaction surfaces the precise targets of the hormone and autoantibodies are likely to be different, at least in part.. It has indeed be shown that the sulfated tyrosine residues, which are important for TSH binding (see above), are not implicated in recognition of TSH receptor by TSH receptor-stimulating antibodies (423). Also, most TSH receptor-stimulating antibodies stimulate cyclic AMP accumulation in cells transfected with TSH receptors more slowly than does TSH (424).
Activation by low molecular weight drug-like chemicals
High-throughput screening of low molecular weight chemical libraries identified specific TSH receptor agonists which were found to bind to the serpentine portion of the receptor (425). Oral administration of the agonist to mice stimulated thyroid, resulting in increased serum thyroxine and thyroidal radioiodide uptake (426). Apart from their interest to unravel the mechanism of activation of the receptor, these molecules constitute leads for development of drugs to use in place of recombinant human TSH, e.g. in patients with thyroid cancer.
Downregulation of the Thyrotropin Receptor
Desensitization of some G protein-coupled receptors involves phosphorylation of specific residues by G-protein receptor kinases (homologous desensitization) or protein kinase A (heterologous desensitization) (427). Acute desensitization of the receptor in the presence of TSH, presumably by phosphorylation, is weak and delayed(428). When compared with other G protein-coupled receptors, the TSH receptor contains few serine or threonine residues in its intracellular loops and intracellular carboxyl-terminal domain that can be phosphorylated, which probably accounts for the limited but definite desensitization and internalization observed in heterologous transfected cells after stimulation by TSH (429). Weak down-regulation, confounded by the long life of both TSHR mRNA and protein, does occur, but has little functional role (430). Chronic administration to mice invivo of stimulating monoclonal antibodies causes sustained hyperthyroidism, with no sign of desensitization (431). Using transgenic mice expressing a cAMP sensor in thyrocytes (432) showed recently that TSH receptors internalized after stimulation by TSH continue to signal to Gαs. This may provide an explanation to the persistence of thyrotoxicosis in patients with TSH-secreting pituitary adenomas and in patients with Graves ’ disease.
CONTROL OF THYROID FUNCTION
Thyroid Hormone Synthesis
Thyroid hormone synthesis requires the uptake of iodide by active transport, thyroglobulin biosynthesis, oxidation and binding of iodide to thyroglobulin, and within the matrix of this protein, oxidative coupling of two iodotyrosines into iodothyronines. All these steps are regulated by the cascades just described.
Iodide Transport
Iodide is actively transported by the iodide Na + /I – symporter (NIS) against an electrical gradient at the basal membrane of the thyrocyte and diffuses, following the electrical gradient, by a specialized channel (pendrin or another channel) (433;434) from the cell to the lumen at the apical membrane. The opposite fluxes of iodide, from the lumen to the cell and from the cell to the outside, are generally considered to be passive and nonspecific. At least five types of control have been demonstrated (319;320;433) .
1. Rapid and transient stimulation of iodide efflux by TSH in vivo, which might reflect a general increase in membrane permeability. The cascade involved is not known.
2. Rapid activation of iodide apical efflux from the cell to the lumen by TSH. This effect, which contributes to the concentration of iodide at the site of its oxidation, is mediated, depending on the species, by Ca 2+ and/or cAMP (294;433) . In human cells it is mainly controlled by Ca 2+ and therefore by the TSH effect on phospholipase C.
3. Delayed increase in the capacity (Vmax) of the active iodide transport NIS in response to TSH. This effect is inhibited by inhibitors of RNA and protein synthesis and is due to activation of iodide transporter gene expression. This effect of TSH is reproduced by cAMP analogues in vitro and is therefore mediated by the cAMP cascade (187). mRNA expression is enhanced by TSH and cAMP and decreased by iodide (333;435). TSH enhancement of thyroid blood flow, more or less delayed depending on the species, also contributes to increase the uptake of iodide (187). Iodine levels in the thyroid are also inversely related to blood flow (436).
4. Rapid inhibition by iodide of its own transport in vivo and in vitro. This inhibitory effect requires an intact transport and oxidation function, that is, it fulfills the criteria of an XI effect. After several hours the capacity of the active transport mechanism is greatly impaired (adaptation to the Wolff-Chaikoff effect) (320). The mechanism of the first effect is unknown but probably initially involves direct inhibition of the transport system itself (akin to the desensitization of a receptor), followed later by inhibition of NIS gene expression and NIS synthesis (akin to the downregulation of a receptor) (333).
5. Inhibition by iodide of thyroid blood flow. This effect may be direct as it takes place in patients treated with thyroperoxidase inhibitors and therefore does not fit the XI paradigm. By decreasing the iodide input it decreases the uptake.
Iodide Binding to Protein and Iodotyrosine Coupling
Iodide oxidation and binding to thyroglobulin and iodotyrosine coupling in iodothyronines are catalyzed by the same enzyme, thyroperoxidase, with H 2 O 2 used as a substrate (437). The same regulations apply to the two steps. H 2 O 2 is generated by a NADPH oxidase system of which two proteins DUOX (dual oxidases) or THOX have been identified by cloning (438;439) . The system is very efficient in the basal state inasmuch as little of the iodide trapped can be chased by perchlorate in vivo. Also, in vitro and in vivo the amount of iodine bound to proteins mainly depends on the iodide supply. Nevertheless, in human thyroid in vitro, stimulation of the iodination process takes place even at low concentrations of the anion, thus indicating that iodination is a secondary limiting step. Such stimulation is caused in all species by intracellular Ca 2+ and is therefore a consequence of activation of the Ca 2+ -PIP 2 cascade. In many species, phorbol esters and diacylglycerol, presumably through protein kinase C, also enhance iodination (440). It is striking that in a species such as the human, in which TSH activates the PIP 2 cascade, cAMP inhibits iodination, whereas in a species (dog) in which TSH activates only the cAMP cascade, cAMP enhances iodination. Obviously in the latter species a supplementary cAMP control was necessary (440-442).
Thyroperoxidase does not contain any obvious phosphorylation site in its intracellular tail. On the other hand, all the agents that activate iodination also activate H 2 O 2 generation, and inhibition of H 2 O 2 generation decreases iodination, which therefore suggests that iodination is an H 2 O 2 substrate – driven process and that it is mainly controlled by H 2 O 2 generation and iodide supply (440;443). Congruent with the relatively high K m of thyroperoxidase for H 2 O 2 , H 2 O 2 is generated in disproportionate amounts with regard to the quantity of iodide oxidized. Negative control of iodination by iodide (the Wolff-Chaikoff effect) is accompanied and mostly explained by the inhibition of H 2 O 2 generation. This effect of I – is relieved by perchlorate and methimazole and thus pertains to the XI paradigm (325;440).
Iodotyrosine coupling to iodotyrosines is catalyzed by the same system and is therefore subject to the same regulations as iodination. However, coupling requires that suitable tyrosyl groups in thyroglobulin be iodinated, that is, that the level of iodination of the protein be sufficient. In the case of severe iodine deficiency or when thyroglobulin exceeds the iodine available, insufficient iodination of each thyroglobulin molecule will preclude iodothyronine formation whatever the activity of the H 2 O 2 generating system and thyroperoxidase. On the other hand, when the iodotyrosines involved in the coupling are present, coupling is controlled by the H 2 O 2 concentration but independent of iodide (437). In this case, H 2 O 2 control has a significance even at very low iodide concentrations.
H 2 O 2 generation requires the reduced form of nicotinamide-adenine dinucleotide phosphate (NADPH) as a coenzyme and is thus accompanied by NADPH oxidation. Limitation of the activity of the pentose phosphate pathway by NADP + insures that NADPH oxidation for H 2 O 2 generation causes stimulation of this pathway. Also, excess H 2 O 2 leaking back into the thyrocyte is reduced by glutathione (GSH) peroxidase, and the oxidized GSH (GSSG) produced is reduced by NADPH-linked GSH reductase. Thus both the generation of H 2 O 2 and the disposal of excess H 2 O 2 by pulling NADP oxidation and the pentose pathway lead to activation of this pathway — historically one of the earliest and unexplained effects of TSH (187;440).
On the long-term, in vivo or in vitro, the activity of the whole iodination system obviously also depends on the level of its constitutive enzymes. In human thyrocytes H2O2 generation but not TPO is stimulated by direct activation of DUOX by the Gq-PLC-Ca ++ cascade while TPO expression, but not DUOX is upregulated by the TSH-cAMP cascade (444). It is therefore not surprising that activation of thyrocytes by the cAMP cascade increases the corresponding gene expression whereas dedifferentiating treatments with EGF and phorbol esters inhibit this expression and thus reduce the capacity and activity of the system. Apparent discrepancies in the literature about the effects of phorbol esters on iodination are mostly explained by the kinetics of these effects (acute stimulation of the system, delayed inhibition of expression of the involved genes).
Thyroid Hormone Secretion
Secretion of thyroid hormone requires endocytosis of human thyroglobulin, its hydrolysis, and the release of thyroid hormones from the cell. Thyroglobulin can be ingested by the thyrocyte by three mechanisms (187;445-447) .
In macropinocytosis , pseudopods engulf clumps of thyroglobulin. In all species this process is triggered by acute activation of the cAMP/PKA cascade and therefore by TSH. Stimulation of macropinocytosis is preceded and accompanied by an enhancement of thyroglobulin exocytosis and thus of the membrane surface (443;448;449). In dog thyroid slices (450) and even (325) primary cultures, TSH and PKA activation acutely induces phagocytosis (451), which appears as the invitro manifestation of the macropinocytosis of thyroglobulin involved in stimulated thyroid hormone secretion. This process might be mediated by inactivation of the Rho family small G proteins), resulting in microfilament depolymerization and stress fiber disruption accompanied by dephosphorylation of cofilin (452) and myosin light chains (453) .
By micropinocytosis , the second process, small amounts of colloid fluid are ingested. This process does not appear to be greatly influenced by acute modulation of the regulatory cascades. It is enhanced in chronically stimulated thyroids and thyroid cells with induction of vesicle transport proteins Rab 5 and 7 (454) (455;456). It probably accounts for most of basal secretion.
A third (hypothesized) process is receptor-mediatedendocytosis ; it is enhanced in chronically stimulated thyroid cells (457-459). The protein involved could be megalin (460) or and asyaloglycoprotein. This process probably accounts for the transcytosis of low hormonegenic thyroglobulin (461) which is found in the serum .
Contrary to the last named, the first two processes are not specific for the protein. They can be distinguished by the fact that macropinocytosis is inhibited by microfilament and microtubule poisons and by lowering of the temperature (below 23°C) (187;462). Whatever its mechanism, endocytosis is followed by lysosomal digestion with complete hydrolysis of thyroglobulin. The main iodothyronine in thyroglobulin is thyroxine. However, during its secretion a small fraction is deiodinated by type I 5 and in man type II 5 -deiodinase to triiodothyronine (T 3 ), thus increasing relative T 3 (the active hormone) secretion (463).
The free thyroid hormones are released by the thyroid hormone transporter MC7 an unknown mechanism, which may be diffusion or transport. The iodotyrosines are deiodinated by specific deiodinases and their iodide recirculated in the thyroid iodide compartments. Under acute stimulation, a release (spillover) of amino acids and iodide from the thyroid is observed. A mechanism for lysosome retention of poorly iodinated thyroglobulin on N -acetylglucosamine receptors and recirculation to the lumen has been proposed. Under normal physiologic conditions, endocytosis is the limiting step of secretion, but after acute stimulation, hydrolysis might become limiting with the accumulation of colloid droplets. Secretion by macropinocytosis is triggered by activation of the cAMP cascade and inhibited by Ca 2+ at two levels: cAMP accumulation and cAMP action. It is also inhibited in some thyroids by protein kinase C downstream from cAMP. Thus the PIP 2 cascade negatively controls macropinocytosis (308).
The thyroid also releases thyroglobulin. Inasmuch as this thyroglobulin was first demonstrated by its iodine, at least part of this thyroglobulin is iodinated; thus it must originate from the colloid lumen. Release is inhibited in vitro by various metabolic inhibitors and therefore corresponds to active secretion (448;464). The most plausible mechanism is transcytosis from the lumen to the thyrocyte lateral membranes (449). As for thyroid hormone, this secretion is enhanced by activation of the cAMP cascade and TSH and inhibited by Ca 2+ and protein kinase C activation. Because thyroglobulin secretion does not require its iodination, it reflects the activation state of the gland regardless of the efficiency of thyroid hormone synthesis. Thyroglobulin serum levels and their increase after TSH stimulation constitute a very useful index of the functional state of the gland when this synthesis is impaired, as in iodine deficiency, congenital defects in iodine metabolism, treatment with antithyroid drugs, and the like (465). Regulated thyroglobulin secretion should not be confused with the release of this protein from thyroid tumors, which corresponds in large part to exocytosis of newly synthesized thyroglobulin in the extracellular space rather than in the nonexistent or disrupted follicular lumen. In inflammation or after even mild trauma, opening of the follicles can cause unregulated leakage of lumen thyroglobulin.
Transcytosis or leakage from the lumen yields iodinated thyroglobulin while exocytotic thyroglobulin is not iodinated.
Functional Heterogeneity
It has long been known that at any given time the function of the thyroid follicles is not homogeneous. For instance, after injection of radioiodide, some follicles will incorporate important amounts of radioiodine while others will not incorporate at all. Similarly, after stimulation with TSH in in vivo thyroids or in in vitro incubated slices, some cells will develop pseudopods for macropinocytosis whithin 15 min while others submitted to the same stimulus will only respond after one to two hours (175;466).
In a more recent study, Gérard et al (467) (468) showed in human thyroids that while some follicles exhibit marked expression of pendrin, TPO and THOX, others did not. The expressing follicles were those containing iodinated thyroglobulin. They correspond to larger capillary networks and to the expression in the follicular cells of vascular regulators nitric oxide synthase and endothelin. This shows the existence of active and inactive angiofollicular units. It suggests that over time angiofollicular units cycle from active to inactive states and that this is controlled by the follicular cells. It would be interesting to know if the inactive state corresponds to a lower sensitivity to TSH.
CONTROL OF THYROID-SPECIFIC GENE EXPRESSION
The study of specific gene expression and proliferation at the biochemical and mechanistic level requires long term in vitro incubations, i.e. cell culture. It is therefore easy and tempting to rely on cell lines such as the rat thyroid FRTL-5, WRT and PCCl3 cells. However these cells, sometimes different from one lab to another, are different from each other and are very different from the cells in vivo, especially the human cells. Primary cultures are closer to the in vivo situation but they are difficult to obtain and not as reproducible. However, in monolayers, but not in reorganized follicles, follicular structure is fully and cell polarity and structure are partially lost. As most authors generalize the results of work done on their pet systems to "The Thyroid" the literature is very confusing and full of contradictions (469;470). Among the various possibilities demonstrated in such systems only those validated in vivo in transgenic animals and in human cells interest us.
A positive in vivo effect of TSH on general protein synthesis has been well documented. This effect is mimicked by cAMP agonists and is part of the trophic effect of TSH on the thyrocyte. It involves stimulation of transcription and translation; however, the detailed mechanisms implicated are not known. In cells in culture there is no such effect and the TSH cyclic AMP cascade has rather an inhibitory effect. Stimulation of protein synthesis and hypertrophy of the cell in culture results from IGF1 action on PI3 kinase (471). The in vivo effect of TSH might be mediated by IGF-1.
The so-called thyroid-specific genes encode proteins that are either found in the thyroid exclusively, like thyroglobulin and thyroperoxidase, or that, although being also found in a few additional tissues, are primarily involved in thyroid function, like TSH receptor and sodium/iodide symporter. The transcription of these genes in the thyroid appears to rely on the coordinated action of a master set of transcription factors that includes at least the homeodomain protein TTF-1 (also known as Nkx 2.1 or T/ebp or Titf1), the paired-domain protein Pax 8, and perhaps also the forkhead-domain protein TTF-2 (also known as FoxE1) (472;473). Loss of function mutant mice for TTF-1, Pax 8 or TTF-2 have been generated and allowed to identify a crucial role for these transcription factors in the development of the thyroid also. However, as none of these animals develop a normal mature thyroid, they could not be used to investigate the exact role of these key factors in the control of gene expression in the mature thyroid. A conditional loss of function mutant mouse for TTF-1 has also been generated (368). Only partial inactivation of TTF-1 could be achieved in this animal which precluded the analysis of TTF-1 role in the developed thyroid at the molecular level. Most of the work concerning this last aspect has been conducted either in primary cultures of thyrocytes (474) or in immortalized thyroid cell lines like FRTL-5 and PCCl3 (475). Although the data gathered to date agree on most basic aspects, significant differences have sometimes been observed between primary versus immortalized cell models (476). Part of these discrepancies may result from the existence of occasional species-specific differences (477).
The main regulator of thyroid function, the TSH signal, which is predominantly conveyed inside the cell by cAMP and PKA, upregulates the expression of transcription factor Pax 8, both in primary cells (478) and established cell lines (479). However, mice genetically deprived of TSH or of functional TSH receptor do not show reduced amounts of Pax 8 in their thyroids as compared to wild type animals (480) suggesting that compensatory mechanisms may ensure an adequate production of this factor when thyroid development takes place in the absence of the normal physiological stimulus. Besides this control on the amount of Pax 8 protein, there is no firm evidence that TSH, or cAMP, exerts any other control at the level of the master thyroid transcription factors identified presently (476;481;482). The expression of several other transcription factors was shown to be upregulated, often at least transiently, in response to TSH/cAMP in the thyroid, namely c-myc (483) , c-fos (483), fos B, jun B, jun D (484), CREM (485) , NGFI-B (486) and CHOP(487), for example. An hypothetical role in the control exerted by TSH/cAMP on the expression of the thyroid-specific genes has been proposed for some of these factors (488;489), but no final link has been established yet (490). A recent report proposes that the dopamine and cAMP-regulated neuronal phosphoprotein DARPP-32 could play an essential role in this control (382).
It is noteworthy that in addition to its control on the transcription of the individual thyroid-specific genes, which is detailed below, TSH also regulates gene expression by acting at some post-transcriptional steps, as shown in the case of thyroglobulin (491). Finally, many effects of TSH and cAMP on gene expression (including on thyroid-specific genes such as thyroglobulin) might be rather indirect and depend in part on the profound modifications of cell morphology and cytoskeleton that result from PKA activation (245;492).
TGF-β has been shown to downregulate the expression of thyroid-specific genes (493;494). It seems to involve a reduction in the level of Pax 8 activity that is mediated by Smad proteins (495;496). In human thyroid primary cultures, TGF-β inhibits most effects of cAMP on gene expression (252). As above, this might be related in part to an inhibition of morphological effects of TSH/cAMP. In all the species tested so far, EGF strongly represses thyroglobulin and thyroperoxidase gene expression as well as iodide transport (245;430;497-499). FGF has a similar action in some species including bovine (500). The mechanisms have not been explored. The apparent dedifferentiation induced by EGF in dog thyrocytes is associated with an enhanced vimentin expression and a progressive induction of a fusiform fibroblast-like morphology, which is suggestive of an epithelial-mesenchymal transition (501) (502). This process is reversible after elimination of EGF and re-addition of TSH. As recently quantified by SAGE analysis in the thyroid cell line PCCl3, exposure to a high dose of iodide also decreases the expression of most of the thyroid-specific genes within the thyrocyte (389).
Thyroglobulin
The regulatory DNA elements of the thyroglobulin gene have been characterized in several species (472;477;503). The proximal promoter, as defined in transfection experiments, extends over 200 base-pairs and contains binding sites for transcription factors TTF-1, TTF-2 and Pax 8 (see Fig. 1-15). An upstream enhancer element containing binding sites for TTF-1 has been identified in beef and man (504). In the latter, the enhancer region is longer and harbors additional binding sites for TTF-1 and cAMP responsive element binding (CREB) protein (505). Both TTF-1 and Pax 8 proteins were individually shown to exert a major control on thyroglobulin gene transcription (506;507). By contrast, TTF-2 activity appears to be dispensable as the thyroglobulin gene is expressed in cells devoid of TTF-2 protein (508). Synergism in the transcriptional activation of the gene by TTF-1 and Pax 8 appears to rely on a direct interaction between these two factors (509), and on their coordinated action involving both the enhancer and proximal promoter sequences (510). Transactivation of the thyroglobulin promoter by TTF-1 and Pax 8 has been reported to be enhanced by the coactivator TAZ in vitro (396). But the overexpresion of TAZ observed in papillary thyroid carcinoma is not associated with an increased expression of the thyroglobulin gene (397). The coactivator p300 has also been independently reported to be involved in this transactivation mechanism (398). On the other side, poly(ADP-ribose) polymerase-1 was reported to counteract the transactivation of the thyroglobulin promoter by Pax 8 through a direct interaction with this factor impairing its DNA-binding activity (399). Recently, the osteoblast-specific transcription factor Runx2 (also known as Cbfa1 or AML3) was shown to be expressed in the thyroid and to control thyroglobulin gene expression by direct binding to the thyroglobulin proximal promoter region (see Fig. 1-15). Runx2 deficiency in mice causes a marked reduction in thyroglobulin gene expression leading to hypothyroidism (400). Again however, Runx2 overexpression in papillary thyroid cancers is not associated with an increased expression of the thyroglobulin gene (401).

Fig. 1-15
Schematic of the known regulatory elements of thyroglobulin, thyroperoxidase, sodium/iodide symporter and TSH receptor genes. The organization of the proximal promoter and upstream enhancer elements of the different genes is depicted as determined in the species studied so far. Coordinates of the proximal promoters are in base pairs and refer to the transcription start site as +1. The positions of the upstream enhancer elements relative to the transcription start site are not indicated as they vary extensively among the different species.
The known thyroglobulin gene regulatory elements were shown to be sufficient to drive the thyroid-restricted expression of a linked gene in living mice (511). This thyroid-restricted expression likely results from the requirement for the simultaneous presence of both TTF-1 and Pax 8, which occurs in thyroid only. It is associated with the tissue-specific demethylation of thyroglobulin gene sequences (512). Demethylation of the DNA is supposed to relieve the constitutive silencing of the gene (513).
Thyroglobulin gene transcription has been shown to require the presence of circulating TSH in the adult rat (514) and to be highly dependent on an elevated cAMP level in dog thyroid tissue slices, in primary cultured cells (515), and, to a much lower extent, in immortalized thyroid cell lines like FRTL-5 (516). Although they are devoid of classical cAMP-responsive element (CRE), the proximal promoter sequences are essentially involved in this control, as indicated by the observation of TSH/cAMP-induced changes in their chromatin structure (517) and their TSH/cAMP-dependent activity in transfection experiments (518). It has however been demonstrated recently that the onset of thyroglobulin gene expression during thyroid development takes place normally in mouse strains deprived of either circulating TSH or functional TSH receptors (519;520). This may be consistent with the observation that the thyroglobulin gene displays a low level of cAMP-independent transcription in primary cultured thyrocytes (515), which might depend on insulin, as observed in different culture models (521-523). In primary cultures of dog thyrocytes, the transcriptional activation of the thyroglobulin gene by cAMP after transient TSH withdrawal is also delayed as compared to that of the thyroperoxidase gene (515), and, unlike thyroperoxidase gene expression, it requires an active protein synthesis (515). The increase in Pax 8 concentration consecutive to TSH/cAMP stimulation of the thyrocyte is not sufficient to account for the observed control on thyroglobulin gene transcription, as TSH is still required for transcriptional activation even in cells expressing high levels of Pax 8 protein (524). Thus, besides TTF-1 and Pax 8, at least one additional, still unidentified, factor is likely to play a key role in the control of thyroglobulin gene expression, as also suggested by the observation that, in the course of thyroid development, both TTF-1 and Pax 8 are present well before thyroglobulin gene is expressed.
In addition to the full length thyroglobulin mRNA, a shorter transcript accumulates in the rat thyroid in response to TSH stimulation (525). This transcript results from differential splicing and polyadenylation of the primary transcript, and encodes a protein limited to the very N-terminal part of thyroglobulin. As this truncated protein still contains a major hormonogenic site(526), it could suggest that, in conditions in which the balance of thyroid metabolism would favor hormone synthesis over iodine storage (e.g., shortage of iodine), the rat thyrocyte would manufacture a shorter thyroglobulin with a preserved hormonogenic ability but lacking many of the nonhormonogenic tyrosines.
Thyroperoxidase
In the species studied so far, the architecture of the proximal promoter region of the thyroperoxidase gene strikingly resembles that of the corresponding region of the thyroglobulin gene (527;528) (see Fig. 1-15 ). The upstream enhancer element also encompasses a pair of TTF-1 binding sites and contains an additional binding site for Pax 8, as compared to its counterpart in the thyroglobulin gene (529;530). Here again, the combination of the upstream enhancer and proximal promoter supports the synergistic action of TTF-1 and Pax 8 on gene transcription (531). The transcriptional co-activator p300 has also been reported to be involved in the activation of this promoter (413).
Despite the existence of this high similarity, thyroperoxidase gene transcription is more tightly and more rapidly controled by TSH and cAMP than that of the thyroglobulin gene in primary cultured thyrocytes, and does not require a new protein synthesis (515;532). Contrary to the thyroglobulin gene also, the thyroperoxidase gene is not expressed in the absence of circulating TSH or functional TSH receptors in intact animals (533). On the other hand, the constitutive hyperactivation of the cAMP cascade leads to an increased expression of the gene as compared to the normal situation (534). In spite of their lack of a classical CRE, the proximal promoter sequences have been shown to mediate this TSH/cAMP control on transcription in transfection experiments (535). Exposure to a high dose of iodide reduces thyroperoxidase gene expression as well as that of thyroglobulin, sodium/iodide symporter and thyrotropin receptor genes in PCCl3 thyroid cells (389). Low doses of iodide also decrease thyroperoxidase gene expression in vivo, while the expression of thyroglobulin remains unaffected (333). Thus, apart from their basic dependence on the presence of the transcription factors TTF-1 and Pax 8, which insures their shared thyroid-restricted expression, the thyroperoxidase and thyroglobulin genes distinguish themselves significantly regarding the control of their transcription. It is worth mentioning in this context that a synergistic action of Pax8 and pRb, the retinoblastoma protein, appears to be required for thyroperoxidase promoter activation, whereas this is not the case for thyroglobulin promoter activation (415). It has been postulated recently that the hormone-induced developmental activation of the thyroperoxidase gene involves the concerted action of TTF-2 and NF-1, both of which bind neighbouring sequences in the gene promoter (see figure 1-15) resulting in the initial opening of the chromatin structure of the promoter (416).
The existence of a major thyroperoxidase mRNA isoform has been detected in man (536). It appears to encode a protein devoid of its normal enzymatic activity.
Sodium/iodide Symporter
Although the sodium/iodide symporter plays a key role in thyroid hormonogenesis, the expression of the corresponding gene is not restricted to the thyroid. Accordingly, the proximal promoter sequences identified so far do not exhibit a thyroid-specific activity in vitro (537;538), even if this activity may be marginally increased in the presence of TTF-1 (539). The robust and appropriately controlled expression of this gene in the thyroid seems to be mediated essentially by the upstream enhancer element which contains binding sites for both TTF-1 and Pax 8, and a cAMP responsive element (CRE)-like DNA motif which is involved in the control by TSH/cAMP (540;541) (see Fig. 1-15). The cAMP-response element modulator (CREM) has recently been proposed to be involved in this control (422). The Ras oncoprotein was also shown to reduce sodium/iodide symporter gene expression by targeting Protein Kinase A-dependent Pax 8 transcriptional activity (423). As for the thyroperoxidase gene, TSH signaling is indispensable for sodium/iodide symporter gene transcriptional activation in vivo (542;543), and iodide downregulates the expression of the gene (333). In addition, synergy between Pax8 and pRb appears to be required for the activation of both thyroperoxidase and sodium/iodide symporter promoters (415). A very similar control is thus exerted on the expression of both of these genes in the thyroid in spite of the fact that, overal, the known regulatory regions of the thyroperoxidase gene bear more resemblances to those of the thyroglobulin gene than to those of the sodium/iodide symporter gene. It has been reported recently that the bacterial endotoxin lipopolysaccharide enhances sodium/iodide symporter gene transcription through the direct binding of Nuclear Factor-κB (NF-κB) to the upstream enhancer element and its interaction with adjacently bound Pax 8 factor (423) (see Fig. 1-15). The basic Helix-Loop-Helix transcription factor Hairy/enhancer of split 1 (Hes 1), which is part of the Notch signalling pathway, has also been shown to control sodium/iodide symporter gene expression, as well as that of the thyroperoxidase gene, although no binding sites for this factor have been identified within these promoters as yet. Hes 1 appears to be required in mice for developing a functional thyroid and its decreased expression in thyroid tumors parallels the reduction of thyroid differentiation markers expression observed in these cells (424;425).
Thyropin Receptor
Like the gene described above, the TSH receptor gene is also expressed in tissues other than the thyroid. Again, the promoter elements identified presently, which include binding sites for thyroid hormone receptor (TR)-α1/retinoid-X receptor (RXR) heterodimer (544), GA-binding protein (GABP) (545), cAMP responsive element binding (CREB) protein(546)and TTF-1 (547) (see Fig. 1-15), do not display a clear thyroid-specific activity in transfection experiments, as could be expected. Contrary to the promoters described so far, the promoter of the TSH receptor gene does not contain a TATA-box motif, but encompasses a GC-rich region preceding the multiple neighbouring transcription start sites. Consistent with the presence of TTF-1 binding sites in the promoter region, the TSH receptor genes exhibits a decreased activity in animals expressing reduced level of TTF-1 (548). No other regulatory DNA element specifically involved in the thyroid-specific expression of this gene has been identified as yet. On the other hand, DNA demethylation events in the promoter region have been observed in thyroid cells expressing the TSH receptor gene, as compared to non-expressing cells (549).
The control exerted on the expression of the TSH receptor gene in the thyroid seems to be more complex than the ones described previously. Discordant effect of TSH/cAMP on the expression of this gene have been reported depending on the nature of the experimental system used (550). The presence in the promoter region of a CRE-like DNA motif which appears to be able to bind the CREB protein(551), a transcriptional activator directly activated by cAMP, as well as the CREM isoform ICER(552), a transcriptional repressor induced by cAMP, could explain both reported increase and decrease in gene expression following TSH stimulation, depending on the relative amounts of these factors (and likely of other CRE-binding proteins also) preexisting in the studied cells and the kinetics of the individual observations. Moreover, the binding site of the TRα1/RXR heterodimer identified in this promoter encompasses the CRE-like motif (see figure 1-15), which may add a further level of complexity depending on the availability of thyroid hormone in the experimental system.
On the other hand, the possible existence of species-specific differences could also account for the occurrence of seemingly discrepant reports (430). The accumulation of TSH receptor mRNA requires an active protein synthesis in primary cultured dog thyrocytes (430), which is reminiscent of what was observed for the thyroglobulin gene (see above). Considering the facts that, after the TSH receptor gene, the thyroglobulin gene is the most affected in its expression by a reduced TTF-1 availability as compared to the other known thyroid-specific genes(553), and that, alike the TSH receptor gene itself, the thyroglobulin gene is activated independently of the TSH/TSHR signaling during thyroid development(554;555), it suggests that these two genes may share at least partially similar control mechanisms. Recently, thyrotropin receptor mRNA was also shown to be decreased in PCCl3 cells exposed to a high iodide concentration (556).
Control of TSH receptor gene expression has been studied in the FRTL5 cell line (550;557;558), the canine thyrocyte in primary culture (430), cultured human thyrocytes (559;560), and human thyroid cancer (471;561) . The general conclusion emerging from these studies is the extreme robustness of TSH receptor gene expression as compared with the other markers of thyroid cell differentiation (thyroglobulin and thyroperoxidase). In the dog, levels of TSH receptor mRNA remain virtually unchanged in animals subjected for 28 days to hyperstimulation by TSH secondary to treatment with methimazole or to TSH withdrawal achieved by administration of thyroxine (430). In the same study, the effect of TSH or forskolin has been investigated in dog thyrocytes in primary culture. This experimental system has the advantage that the differentiation state of the cells can be manipulated at will: cAMP agonists maintain expression of the differentiated phenotype, whereas agents such as EGF, tetradecanoyl phorbol acetate (TPA), and serum lead to "dedifferentiation" (498). The results demonstrate that the dedifferentiating agents reduce accumulation of the receptor mRNA. However, contrary to what is observed with thyroglobulin and thyroperoxidase mRNA, the inhibition is never complete. TSH or forskolin is capable of promoting reaccumulation of the receptor mRNA, a maximum being reached after 20 hours. As with thyroglobulin but at variance with the thyroperoxidase gene, this stimulation requires ongoing protein synthesis (430). Chronic stimulation of cultured dog thyrocytes by TSH for several days does not lead to any important downregulation in mRNA. Similar data have been obtained with human thyrocytes in primary culture (430;560) . By contrast, n egative regulation of receptor mRNA accumulation has been observed in immortal FRTL5 cells after treatment with TSH or TSAB (550;558). This difference versus human and canine cells must probably be interpreted in the general framework of the other known differences in phenotype and regulatory behavior of this cell line as compared with primary cultured thyrocytes (see below) (562) .
The effect of malignant transformation on the amounts of TSH receptor mRNA has been studied in spontaneous tumors in humans (471;561), in a murine transgenic model of thyroid tumor promoted by expression of the simian virus-40 large T oncogene(563), and in FRTL5 cells transformed with v- ras (557). In the two last models, expression of the TSH receptor gene was suppressed: the tumor or cell growth became TSH independent. In the transgenic animal model, loss of TSH receptor mRNA seemed to take place gradually, with early tumors still displaying some TSH dependence for growth. In the human tumors a spectrum of phenotypes was observed. As expected, anaplastic tumors had completely lost the receptor mRNA, as well as other markers of thyrocyte differentiation (thyroglobulin and thyroperoxidase). In papillary carcinoma, variable amounts of TSH receptor mRNA were invariably found (561), even in the tumors that had lost the capacity to express the thyroglobulin or thyroperoxidase genes (561). These data agree well with the observations of thyrocytes in primary culture: expression of the TSH receptor gene is robust and it persists in the presence of agents (or after several steps in tumor progression) that promote extinction of the other markers of thyroid cell differentiation. This evidence leads to the conclusion that the basic marker of the thyroid phenotype is probably the TSH receptor itself, which makes sense: the gene encoding the sensor of TSH — the major regulator of thyroid function, growth, and differentiated phenotype — is virtually constitutive in thyrocytes. From a pragmatic viewpoint, these data provide a rationale for the common therapeutic practice of suppressing TSH secretion in patients with a differentiated thyroid tumor (564).
Thyroid oxidases
Two distinct genes, ThOX1 and ThOX2 (also known as DUOX-1 and -2), both significantly related to the gene encoding the phagocyte NADPH oxidase gp91 Phox , are expressed in the thyroid essentially but not only (565;566). In the dog, ThOX mRNAs accumulate in response to TSH/cAMP stimulation (567). This effect is much less apparent in man (568), and in the rat conflicting results were obtained in vivo and in the established FRTL-5 cell line respectively (569). Two other genes encoding proteins required for the maturation and function of the thyroid oxidases were identified in the close vicinity of both ThOX/DUOX genes, and named DUOXA-1 and -2 respectively (570). Proper hydrogen peroxide production requires that each DUOXA protein associates with the corresponding DUOX protein (i.e. DUOXA1 with DUOX1 and DUOXA2 with DUOX2) at the cell membrane (571). In both ThOX-DUOXA pairs, the genes encoding the ThOX and DUOXA partners are arranged in a head to head configuration, the tiny DNA region separating the two transcription starts acting as a bidirectional promoter (439). Noteworthy, ThOX1+DUOXA-1 and ThOX2+DUOXA2 promoters exhibit totally unrelated architectures, the former displaying all features of a CpG-rich island, the second containing canonical TATA-box and Initiator elements (see Fig. 1-16 ). This fundamental difference in promoter organization suggests that both genes pairs are subjected to distinct transcriptional controls. In transient transfection experiment, both cloned ThOX-1 and -2 promoters do not show thyroid-cell restricted activity, do not respond to TSH or cAMP (440), and do not appear to depend on transactivation by either TTF-1 or Pax 8 (441), at least in the transcriptional direction investigated in these studies. However, an independent study identified the endogenous ThOX-2 promoter as a target for either Pax8 or TTF-1 (442). A likely explanation for this discrepancy is that this transactivation involves regulatory sequences other than the ones identified and cloned so far (440;441).

Fig. 1-16
Organization of the bidirectional promoters of ThOX and DUOXA genes as determined in the rat (439). The transcription starts are symbolized by arrows and the distances in base pairs separating them are indicated in both cases.
Other thyroid-specific genes
Recently, a few other genes have been found to be highly expressed in the thyroid and/or to play an important role in this tissue, and have been added to the list of the previously known thyroid-specific genes. Notably, the gene encoding Pendrin, a protein at least partly involved in the apical export of iodide ions, has been shown to be under the control of TTF-1 (443). The Tensin 3 gene has been reported to present a particularly high expression level in the thyroid as compared to other tissues, and to exhibit decreased levels of expression in thyroid tumors, but no data are available as yet regarding the molecular mechanisms involved (444). Serial analysis of gene expression (SAGE) aiming at the identification of genes preferentially expressed in the thyroid led to the isolation of C16orf89 encoding a protein of presently unknown function. The expression of C16orf89 is stimulated by TSH and parallels that of the sodium-iodide symporter during thyroid development (445). Genetic analysis of the predisposition to hypothyroidism in mice containing only one intact allele encoding TTF-1 and Pax 8 identified Dnajc17 as a gene highly expressed in the thyroid and playing an essential role in thyroid development. This gene encodes a protein belonging to the type III heat-shock protein-40 family (446).
CONTROL OF GROWTH AND DIFFERENTIATION
Thyroid Cell Turnover The thyroid is composed of thyrocytes (70%), endothelial cells (20%), and fibroblasts (10%) (proportions measured in dog thyroid) (173). Human thyroids: 80% follicular cells for 20% stromal endothelial cells and fibroblasts 80% (572;572). In a normal adult the weight and composition of the tissue remain relatively constant. Because a low but significant proliferation is demonstrated in all types of cells, it must be assumed that the generation of new cells is balanced by a corresponding rate of cell death (215;572;573) . The resulting turnover is on the order of one per 5 to 10 years for human thyrocytes, that is, six to eight renewals in adult life, as in other species. In one child the turnover was 2 per year (573) . Normal cell population can therefore be modulated mainly at the level of proliferation but also secondarily of cell death. In growth situations, that is, either in normal development or after stimulation, the different cell types grow more or less in parallel, which implies coordination between them (248;574-576). Because TSH receptors and iodine metabolism and signaling coexist only in the thyrocyte in thyroid, this cell, sole receiver of the physiologic information, must presumably control the other types of cells by paracrine factors such as FGF, IGF-I, NO, and the like (577). The successful isolation of human thyroid endothelial cells will allow a more detailed study of these interactions (578) . TSH has been demonstrated to upregulate the production of vascular endothelial cell growth factor (VEGF) by human thyrocytes (579) . It is interesting in this regard that the vascular support of the follicles reflects their activity suggesting the concept of angiofollicular units (580;581) .
The Mitogenic Cascades
The study of the control of thyroid cell proliferation has been much confused by the unwarranted extrapolation of data obtained in different model systems, including different rat thyroid cell lines at different stages in their evolution, to the human thyroid. Sentences like “ Agent X stimulates pathway Y in PCCl3 cells, thus human thyroids could ne treated by agent Z which inhibits agent X action ” are not acceptable even if only implied (582).
In the thyroid at least three families of distinct mitogenic pathways have been well defined (Fig. 1-17): (1) the hormone receptor – Gs-adenylyl cyclase – cAMP-dependent protein kinase system, (2) the hormone receptor – tyrosine protein kinase pathways, and (3) the hormone receptor – Gq-phospholipase C cascade (215;583) . The thyroid also autoregulates its size by an unknown mechanism. Thyroxin treated dogs, and humans, compensate the loss of one thyroid lobe independently of TSH (333).
The receptor – tyrosine kinase pathway may be subdivided into two branches; some growth factors, such as EGF, induce proliferation and repress differentiation expression, whereas others, such as FGF in dog cells or IGF-I and insulin, are either mitogenic or are necessary for the proliferation effect of other factors without being mitogenic by themselves, but they do not inhibit differentiation expression (521;584) . In human thyroid cells, IGF-I is required for the mitogenic action of TSH or EGF but by itself it only weakly stimulates proliferation (304). In dog and human thyrocytes in primary cultures, after induction of insulin receptors by TSH, physiological concentrations of insulin permit the proliferative action of TSH (233;585) . In PCCl3 and rat cells, and in mouse thyroid in vivo (586), IGF-I is weakly mitogenic per se (587), whereas in pig thyroid cells it produces a strong effect (588).

Fig 1-17a
Mitogenic pathways in the thyroid. Data from the thyroid cell systems are integrated into the present general scheme of cell proliferation cascades. In the first line, known activators of various cascades in dog and human thyroid cells are shown. Various levels indicate a time sequence and postulated causal relationships from initial interaction of extracellular signal with its receptor to endpoints: proliferation and differentiation expression. In dog but not in human thyroid cells, acetylcholine through muscarinic receptors activates the phospholipase C cascade. PCNA, proliferating cell nuclear antigen; cAPK, cyclic adenosine monophosphate-dependent kinase; CDK, cyclin-dependent kinase; DAG, diacylglycerol, + , stimulation; + , inhibition; ; ♦ + , induction; GFR, growth factor receptor; ODC, ornithine decarboxylase; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PLC, phospholipase C; RSK, ribosomal S6 kinase.
It should be noted that TSH directly stimulates proliferation while maintaining the expression of differentiation. Differentiation expression, as evaluated by NIS or by thyroperoxidase and thyroglobulin mRNA content or nuclear transcription, is induced by TSH, forskolin, cholera toxin, and cAMP analogues (215). These effects are obtained in all the cells of a culture, as shown by in situ hybridization experiments (521) . They are reversible; they can be obtained either after the arrest of proliferation or during the cell division cycle (499;521) . Moreover, the expression of differentiation, as measured by iodide transport, is stimulated by concentrations of TSH lower than those required for proliferation (304).
All the proliferation effects of TSH are mimicked by nonspecific modulators of the cAMP cascade, that is, cholera toxin and forskolin (which stimulate adenylate cyclase), cAMP analogues (which activate the cAMP-dependent protein kinases), and even synergistic pairs of cAMP analogues acting on the different sites of these two kinases (288;304;589) . They are reproduced in vitro and in vivo by expression of the adenosine A 2 receptor, which is constitutively activated by endogenous adenosine (534), and by constitutively active Gsα (590) and cholera toxin (591) . They are inhibited by antibodies blocking G s (592) . Inhibition of cAMP-dependent protein kinases (PKA) inhibits the proliferation and differentiation effects of cAMP (593;594). Moreover, stimulation of PKA by selective cAMP analogs that do not activate EPAC proteins is sufficient to fully mimic mitogenic effects of TSH and forskolin in dog thyrocytes (593). There is, therefore, no doubt that the mitogenic and differentiating effects of TSH are mainly and probably entirely mediated by cAMP-dependent protein kinases. A complementary role of the Rap guanyl nucleotide exchange factor EPAC and of Rap has been proposed in rat thyroid cell lines (595;596) but not observed in canine thyroid primary cultures (593).
EGF also induces proliferation of thyroid cells from various species (215;304;597) . However, the action of EGF is accompanied by a general and reversible loss of differentiation expression assessed as described above (498). The effects of EGF on differentiation can be dissociated from its proliferative action. Indeed, they are obtained in cells that do not proliferate in the absence of insulin and in human cells, in which the proliferative effects are weaker, or in pig cells at concentrations lower than the mitogenic concentrations (215).
Finally, the tumor-promoting phorbol esters, the pharmacologic probes of the protein kinase C system, and analogues of diacylglycerol also enhance the proliferation and inhibit the differentiation of thyroid cells. These effects are transient because of desensitization of the system by protein kinase C inactivation.
Activation of the Gq/phospholipase C (PLC)/PKC cascade by a more physiological agent such as carbamylcholine in dog thyroid cells does not reproduce all the effects of phorbol esters. In particular, prolonged stimulation of this cascade by carbamyl choline permits the cAMP-dependent mitogenesis of dog thyrocytes (598), but unlike phorbol esters it does not induce proliferation in the presence of insulin (599) . The Ras protooncogene is strongly activated by phorbol esters but more weakly by carbachol (317). Thus we cannot necessarily equate the effects of phorbol esters and prolonged stimulation of the PLC cascade. The dedifferentiating effects of phorbol esters do not require their mitogenic action either. Thus the effects of TSH, EGF, and phorbol esters on differentiation expression are largely independent of their mitogenic action (215).
In several thyroid cell models, very high insulin concentrations are necessary for growth even in the presence of EGF. We now know that this prerequisite mainly reflects a requirement for IGF-I receptor (215;302;587;600) . It is interesting that in FRTL5 cells, as in cells from thyroid nodules, this requirement may disappear, probably because the cells secrete their own somatomedins and thus become autonomous with regard to these growth factors (302;601) . By contrast, in primary cultures of normal dog and human thyrocytes, very low concentrations of insulin, acting on insulin receptors, are sufficient to support the mitogenic effects of TSH and cAMP when insulin receptors have been induced to high levels by TSH (233;602) . This puzzling regulation, which is reminiscent of the induction of insulin receptors during the differentiation of adipocytes, suggests that thyroid might well be revealed as a more specific target of circulating insulin than hitherto recognized.
In the action of growth factors on receptor protein tyrosine kinase pathways, the effects on differentiation expression vary with the species and the factor involved: from stimulation (e.g., insulin, as well as IGF-II in dog and FRTL5 cells) (603) to an absence of effect (604), to transitory inhibition of differentiation during growth (FGF and HGF in dog cells (605;606) to full but reversible dedifferentiation effects (EGF in dog and human cells) (498;607) . Ret/PTC rearrangements, activating mutations of Ras, as well as oncogenic mutation of B-Raf, which are responsible of most differentiated carcinoma, constitutively activate the signaling cascades of growth factors (608-610) .
The kinetics of the induction of thymidine incorporation into nuclear DNA of dog thyroid cells is similar for TSH, forskolin, EGF, and TPA. Whatever the stimulant, a minimal delay of about 16 to 20 hours takes place before the beginning of labeling, that is, the beginning of DNA synthesis (611). This time is the minimal amount required to prepare the necessary machinery. For the cAMP and EGF pathways, the stimulatory agent has to be present during this whole prereplicative period; any interruption in activation (e.g., by washing out the stimulatory forskolin) greatly delays the start of DNA synthesis (612). This limitation explains why norepinephrine and prostaglandin E, which also activate the cAMP cascade, do not induce growth and differentiation: the rapid desensitization of their receptors does not allow a sustained rise in cAMP levels.
The three main types of mitogenic cascade, specifically, the growth factor – protein tyrosine kinase, phorbol ester – protein kinase C, and TSH-cAMP cascades, are fully distinct at the level of their primary intracellular signal and/or the first signal-activated protein kinase (215).
Iodide actually inhibits the cAMP and the Ca 2+ -phosphatidylinositol cascades and in a more delayed and chronic effect decreases the sensitivity of the thyroid to the TSH growth response. These effects are relieved, according to the general paradigm of Van Sande, by perchlorate and methimazole (325;613).
Steps in the Mitogenic Cascades (Fig. 1-17)
The phenomenology of EGF, TPA, and TSH proliferative action cells has been partially elucidated using dog thyroid primary cultures (215;474;614). The mechanisms of TSH/cAMP mitogenic effects have also been investigated using immortal rat thyroid cell lines (FRTL-5, WRT and PC Cl3 cells) (313). Whereas the signaling cascades involved in the action of growth factors and IGF-I are likely to be well conserved in the different thyroid systems, as generally observed in the other cell types, the mechanistic logics of cell cycle regulation by cAMP has disappointingly turned out to strongly diverge in the various thyroid in vitro models (615). These divergences do not only reflect species differences (215). Among the apparently similar rat thyroid cell lines, or even among different subclones of FRTL-5 cells, major differences have been observed (616). For instance, the PI3 kinase/PKB cascade is activated by cAMP in WRT cells (617), but inhibited by cAMP in PC Cl3 cells (618) . The induction of c-jun by TSH/cAMP in FRTL-5 cells and its repression by cAMP in WRT cells (619) as in dog (620) and human thyrocytes likely reflect major differences in upstream signaling cascades, and should result in divergent expression of downstream target genes, such as cyclin D1. Cyclin D1 synthesis, an accepted endpoint of mitogenic cascades, is indeed induced by cAMP in FRTL-5 and PC Cl3 cells, but rather repressed by cAMP in dog and human thyroid primary cultures (621;622) . The reasons for such discrepancies are unclear. Some signaling features, when they lead to selective proliferative advantages, might have been acquired during the establishment and continuous cultures of the cell lines and stabilized by subcloning. Many mechanisms demonstrated in the dog thyroid primary culture system so far apply to normal human thyrocytes (623), but much remains to be defined (624) . In the following lines, we thus rely mostly on these systems.
Three biochemical aspects of the proliferative response occurring at different times of the prereplicative phase have been considered. The pattern of protein phosphorylation induced within minutes by TSH is reproduced by forskolin and cAMP analogues. It totally diverges from the phosphorylations induced by EGF and phorbol esters (625). EGF, HGF and phorbol ester actions rapidly converge on the activation of Ras (317) and the resulting activation of p42/p44 MAP kinases and p90 RSK (316;626;627) . PI-3-kinase and its effector enzyme PKB are activated for several hours only by insulin and IGF-I, the effect of EGF being short lived (628) . This activity is therefore the one specific feature of insulin action and presumably the mechanism of the facilitating effect on mitogenesis. In dog thyrocytes, only HGF can trigger cell proliferation in the absence of insulin/IGF-I; this is explained by the fact that only this factor strongly activates both PI3 kinase and MAP kinase cascades (629) . Only insulin, IGF-I and HGF also markedly enhance general protein synthesis and induce cell hypertrophy (630). By contrast, TSH and cAMP are very unique as mitogens, as they do not activate Ras, the PI3kinase/PKB pathway, or any of different classes of MAP kinases in dog thyrocytes (316;317;631;632). TSH and cAMP also do not activate MAPkinases in human thyrocytes (633). The phosphorylation and activation of p70 S6K and thus likely of mTOR cascade constitutes the only early convergence point of growth factor and cAMP-dependent mitogenic cascades (634;635). A recent study has demonstrated the crucial role of this cascade for TSH-elicited thyroid follicular hyperplasia invivo in mice (636). Indeed, as found in dog thyroid primary cultures (637) and PCCl3 cells (Blancquaert and Roger, unpublished), TSH stimulates in mice the mTOR/ p70 S6K axis without activating PKB, and a rapamycin derivative abrogates the hyperplastic (but, interestingly, not the hypertrophic) responses to TSH (638). The cAMP-dependent mitogenesis and gene expression also appears to require the phosphorylation by PKA and activity of CREB/CREM transcription factors (639;640).
As in other types of cells, EGF and TPA first enhance c-fos and c-myc mRNA and protein concentrations in dog thyrocytes. On the other hand, TSH and forskolin strongly, but for a short period, enhance the c-myc mRNA concentration and with the same kinetics as the enhancement of the c-fos mRNA concentration by EGF/TPA. In fact, cAMP first enhances and then decreases c-myc expression. This second phenomenon is akin to what has been observed in the fibroblast, in which cAMP negatively regulates growth. As in fibroblasts, EGF and TPA enhance c-jun, junB, junD, and egr1 expression. However, as in fibroblasts, activators of the cAMP cascade decrease c-jun and egr1 expression. c-Jun is therefore not, as has been claimed, a gene whose expression is universally necessary for growth (484;620) .
The investigation of the pattern of proteins synthesized in response to the various proliferation stimuli has suggested very early that the proliferation of dog thyroid cells is controlled during G1 phase by at least two largely distinct, cAMP-dependent or cAMP-independent, pathways (641;642). Recent microarray analyses have confirmed and extended this concept in human thyrocytes (643;644). Nevertheless, the different mitogenic cascades are expected to finally modulate the level and activity of proteins that are the primary regulators of the cell cycle machinery.
As generally considered, mitogenic signals regulate mammalian cell cycle by stimulating the accumulation of D-type cyclins and their assembly through a ill-defined mechanism with their partner the cyclin-dependent kinases (cdk) 4 and 6. These complexes operate in mid-to-late G1 phase to promote progression through the restriction point, and thus commit cells to replicate their genome (645). In the current model, this key decision depends on the initiation by cyclin D-cdk complexes of the phosphorylation of the growth/tumor suppressor protein pRb, which triggers the activation of transcription factors, including those of the E2F family, the synthesis of cyclin E and then cyclin A, and cdk2 activation by these cyclins. Activated cdk2 in turn further phosphorylates pRb and other substrates and initiates and organizes the progression through the DNA synthesis phase (646). The down regulation of cdk inhibitors of the CIP/KIP family, including p27 kip1 , by mitogenic factors and/or their sequestration by cyclin D-cdk complexes participate to cdk2 activation, but their proposed role of adaptor and/or nuclear anchor for cyclin D-cdk complexes suggests positive influences on cell cycle progression as well (647). These mechanisms have been well studied in dog thyroid cells (Fig. 1-17). As expected, the different mitogenic stimulations (TSH, cAMP, growth factors) require the activity of cdk4 (648), and converge on the inactivating phosphorylation of pRb and related proteins p107 and p130 (649), on the phosphorylation and nuclear translocation of cdk2, and on the induction of cyclin A and cdc2 (650). These effects are dependent on insulin action (651;652). What is strikingly different between the cascades is the mechanism of D-type cyclin-cdk4 activation. TSH, unlike all the other known mitogenic factors, does not induce the accumulation of cyclins D (653), but it paradoxically stimulates the expression of the cdk “ inhibitor ” p27 kip1 (654). However the predominant cyclin D3 is required for the proliferation stimulated by TSH, but not in the proliferation of dog thyrocytes stimulated by EGF or HGF that induce cyclins D1 and D2 in addition to increasing cyclin D3 levels (653). The formation and the nuclear translocation of essential cyclin D3-cdk4 complexes depend on the synergistic interaction of TSH and insulin (653;655). These complexes are absent from cells stimulated by TSH or insulin alone. Paradoxically, in the absence of insulin TSH inhibits the basal accumulation of cyclin D3 (656). On the opposite insulin alone stimulates the required cyclin D3 accumulation and it overcomes in large part the inhibition by TSH (657), but it is unable to assemble cyclin D3-cdk4 complexes in the absence of TSH. In the presence of insulin, TSH (cAMP) unmasks some epitopes of cyclin D3 and induces the assembly of cyclin D3-cdk4 complexes and their import into nuclei (653;658) where these complexes are anchored by their association with p27 kip1 (659;660). This also sequesters p27 away of cdk2 complexes (661), thus contributing to cdk2 activation. Moreover, cAMP exerts an additional crucial function in very late G1 phase to stimulate the enzymatic activity of cyclin D3-cdk4-p27 complexes, which involves the stimulation of the activating Thr172-phosphorylation of cdk4 (662). TGF selectively inhibits the cAMP-dependent proliferation of dog thyrocytes by preventing the association of the cyclin D3-cdk4 complex with nuclear p27 kip1 and the Thr172-phosphorylation of cdk4 (663;664) (665)
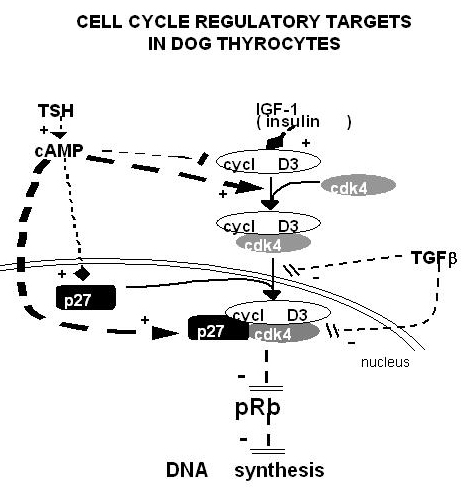
Fig. 1-17b
Targets of cell cycle regulatory effects of TSH, insulin/IGF-1 and TGFβ, as demonstrated in the dog thyroid primary culture system. Diamond/rectangle arrowheads represent inductions/repressions: the other dashed arrows are activations (+) and inhibitions (-). TSH (cAMP) does not induce cyclins D but assembles and then activates the cyclin D3-cdk4-p27 holoenzyme. IGF-1 and insulin allow the accumulation of the required cyclin D3. TGFβ inhibits the nuclear translocation of the cyclin D3-cdk4 complex, its association to p27 and its activation by TSH(cAMP). See text for full explanation.
The investigation of cell cycle regulatory proteins has thus clearly established that both cdk4 activation and pRb phosphorylation result from distinct but complementary actions of TSH and insulin, rather than from their interaction at an earlier step of the signaling cascades (666;667) (Fig. 1-17). Together with the fact that the necessary increase of cell mass before division depends on insulin/IGF-I but not TSH (630), these observations provide a molecular basis for the well established physiological concept that in the regulation of normal thyroid cell proliferation, TSH is the “ decisional ” mitotic trigger, while locally produced IGF-I and/or circulating insulin are supporting “ permissive ” factors (215). Of note, in all these experiments, the facilitative action of insulin can be replaced by activation of the Gq/PLC cascade by carbamylcholine (668).
Studies of protein phosphorylation, proto-oncogene expression, and cell cycle regulatory proteins in dog thyrocytes allow discrimination between two models of cAMP action on proliferation in this system: a direct effect on the thyrocyte or an indirect effect through the secretion and autocrine action of another growth factor. If the effect of TSH through cAMP involved such an autocrine loop, one would expect to find faster kinetics of action of the growth factor and at least some common parts in the patterns of protein phosphorylation and protein synthesis induced by cAMP and the growth factor. The results do not support such an hypothesis, at least for the growth factors tested (215) (Fig. 1-16). Moreover, the data on cAMP action in the dog and human thyrocyte systems do not support a major role for various mechanisms involving cross-signaling of cAMP with growth factor pathways, as claimed in rat thyroid cell line studies (reviewed and discussed in (669) ). Indeed, in primary cultures of normal human thyrocytes, EGF+serum increases cyclin D1 and p21 accumulation, and it stimulates the assembly and activity of cyclin D1-cdk4-p21. By contrast, TSH (cAMP) represses cyclin D1 and p21, but it stimulates the activating phosphorylation of cdk4 and the pRb-kinase activity of preexisting cyclin D3-cdk4 complexes (670). Cyclin D1 or cyclin D3 are thus differentially used in the distinct mitogenic stimulations by growth factors and TSH, and potentially in hyperproliferative diseases generated by the overactivation of their respective signaling pathways.
The validity of these concepts in vivo has been established by using transgenic mice models. The expression in thyroid of oncogene E7 of HPV-16, which sequestrates pRb protein, leads to thyroid growth and euthyroid goiter. Expression in the thyroid of the adenosine A 2 receptor, which behaves as a constitutive activator of adenylyl cyclase, induces thyroid growth, goitrogenesis, and hyperthyroidism (534). Similar, albeit weaker phenotypes are obtained in mice expressing constitutive Gs (the G protein activating adenylyl cyclase) (671) or cholera toxin (672) . A contrario, the expression in thyroid of a dominant negative CREB provokes a marked thyroid hypotrophy, suggesting the crucial role of CREB and its activating phosphorylation by PKA (673) . By contrast, transgenic mice overexpressing both human IGF-I and IGF-I receptor in their thyroid (TgIGF-I – TgIGF-IR) and the downregulation of PTEN the PIP3 3 ’ phosphatase develop only a mild thyroid hyperplasia and respond to some extent to a goitrogenic effect of antithyroid drugs while maintaining a comparatively low serum TSH level. This indicates some autonomy of these thyroids, as in acromegalic patients, and a much greater sensitivity to endogenous TSH (674) . Very recently, thyrocyte-specific deficiency of Gq/G 11 (the G proteins activating PLC ) in mice was shown to impair not only the TSH-stimulated iodine-organification and thyroid hormone synthesis, but also TSH-dependent development of goiter (675). It remains to be defined whether this impaired follicular cell hyperplasia could result in part from the lack of induction of VEGF and angiogenesis (676) which normally accompany goitrogenesis. Nevertheless, the phenotype of these mice underscores the role in TSH-dependent goitrogenesis of PLC, which is activated by TSH but even more strongly by neurotransmitters. Noteworthy, section of inferior laryngeal nerve in rats was similarly found to impair both thyroid function and growth stimulated by TSH (677) . Moreover, activation of Gq /PLC by carbamycholine can facilitate cAMP-dependent mitogenesis in dog thyrocytes cultured without insulin or IGF-I (678) . On the other hand, expression of Ret, which is a rearranged constitutive growth factor receptor, in papillary thyroid carcinoma (PTC), leads to growth, cancer, and hypothyroidism (679;680).
Proliferation and Differentiation (Fig 1-18)

Fig 1-18
Main controls of the principal biologic variables of the human thyrocyte. EGF, epidermal growth factor; FGF, fibroblast growth factor; GH, growth hormone; HGF, hepatocyte growth factor; I – , iodide; IGF-I, insulin-like growth factor; IFN, interferon; IL-1, interleukin-1; TGF , tumor growth factor- ; TNF, tumor necrosis factor; TSAb, thyroid-stimulating immunoglobulins; positive control (stimulation); + : negative control (inhibition) .
Fig 1-18. Main controls of the principal biologic variables of the human thyrocyte. EGF, epidermal growth factor; FGF, fibroblast growth factor; GH, growth hormone; HGF, hepatocyte growth factor; I - , iodide; IGF-I, insulin-like growth factor; IFN, interferon; IL-1, interleukin-1; TGF , tumor growth factor- ; TNF, tumor necrosis factor; TSAb, thyroid-stimulating immunoglobulins; positive control (stimulation); + : negative control (inhibition) .
The incompatibility at the cell level of a proliferation and differentiation program is commonly accepted in biology. In general, cells with a high proliferative capacity are poorly differentiated, and during development such cells lose this capacity as they progressively differentiate. Some cells even lose all potential to divide when reaching their full differentiation, a phenomenon called terminal differentiation. Conversely, in tumor cells, proliferation and differentiation expression are inversely related. Activation of Ras and p42/p44 MAPkinases, induction of c-jun, sustained expression of c-myc, induction of cyclin D1 and down regulation of p27 kip1 , all have been shown to be causatively associated not only with proliferation, but also with loss of differentiation in a large variety of systems, sometimes independently of proliferation effects. It is therefore not surprising that in thyroid cells the general mitogenic agents and pathways, phorbol esters and the protein kinase C pathway, EGF, and in calf and porcine cells, FGF and the protein tyrosine kinase pathway, induce both proliferation and the loss of differentiation expression. The effects of the cAMP cascade are in striking contrast with this general concept. Indeed, TSH and cAMP induce proliferation of dog thyrocytes while maintaining differentiation expression; both proliferation and differentiation programs can be triggered by TSH in the same cells at the same time (521). This situation is by no means unique because neuroblasts in the cell cycle may also simultaneously differentiate. It is tempting to relate this apparent paradox to the unique characteristics of the cAMP-dependent mitogenic pathway, such as the lack of activation (or even the inhibition) of the Ras/MAPkinase/c-jun/cyclin D1 cascade, as demonstrated in dog and human thyrocytes. For instance, if one generalization could be made about proto-oncogenes, it is the dedifferentiating role of c-myc. A rapid and dramatic decrease in c-myc mRNA by antisense myc sequences induces differentiation of a variety of cell types. It is therefore striking that in the case of the thyrocyte, in which activation of the cAMP cascade leads to both proliferation and differentiation, the kinetics of the c-myc gene expression appears to be tightly controlled. After a first phase of 1 hour of higher level of c-myc mRNA, c-myc expression is decreased below control levels. In this second phase, cAMP decreases c-myc mRNA levels, as it does in proliferation-inhibited fibroblasts. It even depresses EGF-induced expression. The first phase could be necessary for proliferation, whereas the second phase could reflect stimulation of differentiation by TSH (483;681). The specific involvement of cyclin D3 in the cAMP-dependent mitogenic stimulation of dog and human thyrocytes, but not for their response to growth factors, is also interesting in this context (653;682) . Indeed, unlike cyclins D1 and D2, cyclin D3 is highly expressed in several quiescent tissues in vivo, and its expression is not only stimulated by mitogenic factors but also induced during several differentiation processes associated with a repression of cyclin D1 (683). We have recently shown that the differential utilization of cyclin D1 or cyclin D3 affects the site specificity of the pRb-kinase of cdk4, including in dog and human thyrocytes (684;685). In addition to inhibiting E2F-dependent gene transcription related to cell cycle progression, pRb plays positive roles in the induction of tissue-specific gene expression by directly interacting with a variety of transcription factors, including Pax8 in thyroid cells (686). Whether, the selective utilization of cyclin D3 in the TSH cascade, associated with a more restricted pRb-kinase activity, could allow the preservation of some differentiation-related functions of pRb thus remains to be examined.
We now consider the distinct cAMP-dependent mitogenic pathway, which appears to be adjuncted to the more general mechanisms used by growth factors, as pertaining to the specialized differentiation program of thyroid cells (309). In dog thyrocytes, the proliferation in response to serum or growth factors specifically extincts their capacity to respond to TSH/cAMP as a mitogenic stimulus (687). Similarly, in less differentiated thyroid cancers generated by the subversion of growth factor mechanisms, the TSH-dependence of growth is generally found to be lost.
Because the cell renewal rate is very low in the thyroid (once every 8 years in adults), the role of apoptosis is unimportant. However, under different circumstances the apoptotic role can greatly increase, such as after the arrest of an important stimulation in vitro (688) and in vivo (689) (690;691).
References
1. Verhaeghe EF, Fraysse A, Guerquin-Kern JL et al. Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanism for its accumulation. J Biol Inorg Chem 2008; 13(2):257-269.
2. Verhaeghe EF, Fraysse A, Guerquin-Kern JL et al. Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanism for its accumulation. J Biol Inorg Chem 2008; 13(2):257-269.
3. Tong W, Chaikoff IL. Metabolism of 131I by the marine alga, Nereocystis leutkana. J Biol Chem 1955; 215:473.
4. Shah M, uilloud RG, Kannamkumaratha SS, Caruso JA. Iodide speciation studies in commercialy available seaweed by coupling different chromatographic techniques with UV and ICP-MS detection.e. J Anat At Spectrom 2005; 20:176.
5. Colin C, Leblanc C, Michel G et al. Vanadium-dependent iodoperoxidases in Laminaria digitata, a novel biochemical function diverging from brown algal bromoperoxidases. J Biol Inorg Chem 2005; 10(2):156-166.
6. Verhaeghe EF, Fraysse A, Guerquin-Kern JL et al. Microchemical imaging of iodine distribution in the brown alga Laminaria digitata suggests a new mechanism for its accumulation. J Biol Inorg Chem 2008; 13(2):257-269.
7. Sherwood NM, Adams BA, Tello JA. Endocrinology of protochordates. Can J Zool 2005; 83:225.
8. Heyland A, Price DA, Bodnarova-Buganova M, Moroz LL. Thyroid hormone metabolism and peroxidase function in two non-chordate animals. J Exp Zoolog B Mol Dev Evol 2006; 306(6):551-566.
9. Heyland A, Reitzel AM, Price DA, Moroz LL. Endogenous thyroid hormone synthesis in facultative planktotrophic larvae of the sand dollar Clypeaster rosaceus: implications for the evolutionary loss of larval feeding. Evol Dev 2006; 8(6):568-579.
10. Flatt F, Moroz LL, Tatar M, Heyland A. Comparing thyroid and insect hormone signaling. Integr Comp Biol 2006; 46:777.
11. Eales JG. Iodine metabolism and thyroid-related functions in organisms lacking thyroid follicles: are thyroid hormones also vitamins? Proc Soc Exp Biol Med 1997; 214(4):302-317.
12. Drechsel HFE. Beiträge zur Chemie einiger Seethiere. II. Über das Achsenskelett der Gorgonia cavolini. Z Biol 1896; 33:85.
13. Eales JG. Iodine metabolism and thyroid-related functions in organisms lacking thyroid follicles: are thyroid hormones also vitamins? Proc Soc Exp Biol Med 1997; 214(4):302-317.
14. Davey K. From insect ovaries to sheep red blood cells: a tale of two hormones. J Insect Physiol 2007; 53(1):1-10.
15. Heyland A, Moroz LL. Cross-kingdom hormonal signaling: an insight from thyroid hormone functions in marine larvae. J Exp Biol 2005; 208(Pt 23):4355-4361.
16. Davey K. From insect ovaries to sheep red blood cells: a tale of two hormones. J Insect Physiol 2007; 53(1):1-10.
17. Eales JG. Iodine metabolism and thyroid-related functions in organisms lacking thyroid follicles: are thyroid hormones also vitamins? Proc Soc Exp Biol Med 1997; 214(4):302-317.
18. Tong W, Chaikoff IL. Activation of iodine utilization in thyroid-gland homogenates by cytochrome C and quinones. Biochim Biophys Acta 1960; 37:189.
19. Heyland A, Price DA, Bodnarova-Buganova M, Moroz LL. Thyroid hormone metabolism and peroxidase function in two non-chordate animals. J Exp Zoolog B Mol Dev Evol 2006; 306(6):551-566.
20. Heyland A, Reitzel AM, Price DA, Moroz LL. Endogenous thyroid hormone synthesis in facultative planktotrophic larvae of the sand dollar Clypeaster rosaceus: implications for the evolutionary loss of larval feeding. Evol Dev 2006; 8(6):568-579.
21. Eales JG. Iodine metabolism and thyroid-related functions in organisms lacking thyroid follicles: are thyroid hormones also vitamins? Proc Soc Exp Biol Med 1997; 214(4):302-317.
22. Davey K. From insect ovaries to sheep red blood cells: a tale of two hormones. J Insect Physiol 2007; 53(1):1-10.
23. Heyland A, Moroz LL. Cross-kingdom hormonal signaling: an insight from thyroid hormone functions in marine larvae. J Exp Biol 2005; 208(Pt 23):4355-4361.
24. Hodin J. Expanding networks: Signaling components in and a hypothesis for the evolution of metamorphosis. Integr Comp Biol 2006; 46(6):719-742.
25. Paris M, Escriva H, Schubert M et al. Amphioxus postembryonic development reveals the homology of chordate metamorphosis. Curr Biol 2008; 18(11):825-830.
26. Paris M, Laudet V. The history of a developmental stage: metamorphosis in chordates. Genesis 2008; 46(11):657-672.
27. Ollikainen N, Chandsawangbhuwana C, Baker ME. Evolution of the thyroid hormone, retinoic acid, ecdysone and liver X receptors. Integr Comp Biol 2006; 46:815.
28. Wu W, Niles EG, LoVerde PT. Thyroid hormone receptor orthologues from invertebrate species with emphasis on Schistosoma mansoni. BMC Evol Biol 2007; 7:150.
29. Carosa E, Fanelli A, Ulisse S, Di Lauro R, Rall JE, Jannini EA. Ciona intestinalis nuclear receptor 1: a member of steroid/thyroid hormone receptor family. Proc Natl Acad Sci U S A 1998; 95(19):11152-11157.
30. Wang S, Zhang S, Zhao B, Lun L. Up-regulation of C/EBP by thyroid hormones: a case demonstrating the vertebrate-like thyroid hormone signaling pathway in amphioxus. Mol Cell Endocrinol 2009; 313(1-2):57-63.
31. Davey K. From insect ovaries to sheep red blood cells: a tale of two hormones. J Insect Physiol 2007; 53(1):1-10.
32. Paris M, Hillenweck A, Bertrand S et al. Active metabolism of thyroid hormone during metamorphosis of amphioxus. Integr Comp Biol 2010; 50(1):63-74.
33. Klootwijk W, Friesema EC, Visser TJ. A Nonselenoprotein from Amphioxus Deiodinates Triac But Not T3. Is Triac the Primordial Bioactive Thyroid Hormone? Endocrinology 2011.
34. Gorbman A. Some aspects of the comparative biochemistry of iodine utilization and the evolution of thyroidal function. Physiol Rev 1955; 35:336.
35. Hiruta J, Mazet F, Yasui K, Zhang P, Ogasawara M. Comparative expression analysis of transcription factor genes in the endostyle of invertebrate chordates. Dev Dyn 2005; 233(3):1031-1037.
36. Barrington EJW. The distribution and significance of organically bound iodine in the ascidian, Ciona intestinalis L. J Marine Biol Assoc UK 1957; 36:1.
37. Fredriksson G, Ericson LE, Olsson R. Iodine binding in the endostyle of larval Branchiostoma lanceolatum (Cephalochordata). Gen Comp Endocrinol 1984; 56(2):177-184.
38. Barrington EJW. Some endocrinological aspects of the protochordata. In: Gorbman A, editor. Comparative Endocrinology. New York: John Wiley & Sons Inc, 1959: 250.
39. Salvatore G. Thyroid hormone biosynthesis in Agnatha and Protochordata. Gen Comp Endocrinol 1969; 2:535.
40. Covelli I, Salvatore G, Sena L, Roche J. Sur la formation d'hormones thyroidiennes et de leurs precurseurs par Branchiostoma lanceolatum Pallas (Amphioxus). Compt Rend Soc Biol 1960; 154:1165.
41. Tong W, Kerkof P, Chaikoff IL. Identification of labeled thyroxine and triiodothyronine in Amphioxus treated with 131I. Biochim Biophys Acta 1962; 56:326.
42. Dunn AD. Studies on iodoproteins and thyroid hormones in ascidians. Gen Comp Endocrinol 1980; 40:473.
43. Fredriksson G, Ericson LE, Olsson R. Iodine binding in the endostyle of larval Branchiostoma lanceolatum (Cephalochordata). Gen Comp Endocrinol 1984; 56(2):177-184.
44. Fredriksson G, Lebel JM, Leloup J. Thyroid hormones and putative nuclear T3 receptors in tissues of the ascidian, Phallusia mammillata cuvier. Gen Comp Endocrinol 1993; 92(3):379-387.
45. Hiruta J, Mazet F, Yasui K, Zhang P, Ogasawara M. Comparative expression analysis of transcription factor genes in the endostyle of invertebrate chordates. Dev Dyn 2005; 233(3):1031-1037.
46. Ogasawara M, Shigetani Y, Suzuki S, Kuratani S, Satoh N. Expression of thyroid transcription factor-1 (TTF-1) gene in the ventral forebrain and endostyle of the agnathan vertebrate, Lampetra japonica. Genesis 2001; 30(2):51-58.
47. Ogasawara M, Satou Y. Expression of FoxE and FoxQ genes in the endostyle of Ciona intestinalis. Dev Genes Evol 2003; 213(8):416-419.
48. Hiruta J, Mazet F, Ogasawara M. Restricted expression of NADPH oxidase/peroxidase gene (Duox) in zone VII of the ascidian endostyle. Cell Tissue Res 2006; 326(3):835-841.
49. Hiruta J, Mazet F, Yasui K, Zhang P, Ogasawara M. Comparative expression analysis of transcription factor genes in the endostyle of invertebrate chordates. Dev Dyn 2005; 233(3):1031-1037.
50. Dehal P, Satou Y, Campbell RK et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science 2002; 298(5601):2157-2167.
51. Campbell RK, Satoh N, Degnan BM. Piecing together evolution of the vertebrate endocrine system. Trends genet 2004; 20:359.
52. Monaco F, Dominici R, Andreoli M, De Pirro R, Roche J. Thyroid hormone formation in thyroglobulin synthesized in the Amphioxus (Branchiostoma lanceolatum pallas). Comp Biochem Physiol B 1981; 70:341.
53. Sherwood NM, Tello JA, Roch GJ. Neuroendocrinology of protochordates: insights from Ciona genomics. Comp Biochem Physiol A Mol Integr Physiol 2006; 144(3):254-271.
54. Sherwood NM, Tello JA, Roch GJ. Neuroendocrinology of protochordates: insights from Ciona genomics. Comp Biochem Physiol A Mol Integr Physiol 2006; 144(3):254-271.
55. Heyland A, Reitzel AM, Price DA, Moroz LL. Endogenous thyroid hormone synthesis in facultative planktotrophic larvae of the sand dollar Clypeaster rosaceus: implications for the evolutionary loss of larval feeding. Evol Dev 2006; 8(6):568-579.
56. Kluge B, Renault N, Rohr KB. Anatomical and molecular reinvestigation of lamprey endostyle development provides new insight into thyroid gland evolution. Dev Genes Evol 2005; 215:32.
57. Wright GM, Youson JH. Transformation of the endostyle of the anadromous sea lamprey, Petromyzon marinus L., during metamorphosis. I. Light microscopy and autoradiography with 125I1. Gen Comp Endocrinol 1976; 30(3):243-257.
58. Wright GM, Youson JH. Transformation of the endostyle of the anadromous sea lamprey, Petromyzon marinus L., during metamorphosis. I. Light microscopy and autoradiography with 125I1. Gen Comp Endocrinol 1976; 30(3):243-257.
59. Manzon RG, Youson JH. KClO(4) inhibits thyroidal activity in the larval lamprey endostyle in vitro. Gen Comp Endocrinol 2002; 128(3):214-223.
60. Wright GM, Filosa MF, Youson JH. Light and electron microscopic immunocytochemical localization of thyroglobulin in the thyroid gland of the anadromous sea lamprey, Petromyzon marinus L., during its upstream migration. Cell Tissue Res 1978; 187(3):473-478.
61. Suzuki S, Kondo Y. Thyroidal morphogenesis and biosynthesis of thyroglobulin before and after metamorphosis in the lamprey, Lampetra reissneri. Gen Comp Endocrinol 1973; 21:451.
62. Clements M, Gorbman A. Protease in ammocoetes endostyle. Biol Bull 1955; 108:258.
63. Gorbman A. Problems in the comparative morphology and physiology of the vertebrate thyroid gland. In: Gorbman A, editor. Comparative Endocrinology. New York: John Wiley & Sons Inc, 1959: 266.
64. Eales JG, Holmes JA, McLeese JM, Youson JH. Thyroid hormone deiodination in various tissues of larval and upstream-migrant sea lampreys, Petromyzon marinus. Gen Comp Endocrinol 1997; 106(2):202-210.
65. McCauley DW, Bronner-Fraser M. Conservation of Pax gene expression in ectodermal placodes of the lamprey. Gene 2002; 287(1-2):129-139.
66. Ogasawara M, Shigetani Y, Suzuki S, Kuratani S, Satoh N. Expression of thyroid transcription factor-1 (TTF-1) gene in the ventral forebrain and endostyle of the agnathan vertebrate, Lampetra japonica. Genesis 2001; 30(2):51-58.
67. Hiruta J, Mazet F, Yasui K, Zhang P, Ogasawara M. Comparative expression analysis of transcription factor genes in the endostyle of invertebrate chordates. Dev Dyn 2005; 233(3):1031-1037.
68. Lintlop SP, Youson JH. Concentration of triiodothyronine in the sera of the sea lamprey, Petromyzon marinus, and the brook lamprey, Lampetra lamottenii, at various phases of the life cycle. Gen Comp Endocrinol 1983; 49(2):187-194.
69. Manzon RG, Holmes JA, Youson JH. Variable effects of goitrogens in inducing precocious metamorphosis in sea lampreys (Petromyzon marinus). J Exp Zool 2001; 289(5):290-303.
70. Wright GM, Youson JH. Transformation of the endostyle of the anadromous sea lamprey, Petromyzon marinus L., during metamorphosis. I. Light microscopy and autoradiography with 125I1. Gen Comp Endocrinol 1976; 30(3):243-257.
71. Monaco F, Andreoli M, La Posta A, Cataudella S, Roch J. Biosynthesis of thyroglobulin in the endostyle of larva (ammocoetes) of a fresh water lamprey, Lampetra planeri B1. C R Soc Biol (Paris) 1977; 171:308.
72. Dunn TB. Ciliated cells of the thyroid of the mouse. JNCI 1944; 4:555.
73. Brown-Grant K. Extrathyroidal iodide concentrating mechanisms. Physiol Rev 1961; 41:189.
74. Baker-Cohen KF. Renal and other heterotopic thyroid tissue in fishes. In: Gorbman A, editor. Comparative Endocrinology. New York: John Wiley & Sons Inc, 1959: 283.
75. Boelaert K, Franklyn JA. Thyroid hormone in health and disease. J Endocrinol 2005; 187(1):1-15.
76. McNabb FM. Avian thyroid development and adaptive plasticity. Gen Comp Endocrinol 2006; 147(2):93-101.
77. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
78. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
79. Zoeller RT, Rovet J. Timing of thyroid hormone action in the developing brain: clinical observations and experimental findings. J Neuroendocrinol 2004; 16(10):809-818.
80. McNabb FM. Avian thyroid development and adaptive plasticity. Gen Comp Endocrinol 2006; 147(2):93-101.
81. Shi YB. Amphibian metamorphosis. From Morphol to Mol Biol 1999; John Wiley & Sons.
82. Leatherland JF. Environmental physiology of the teleostean thyroid gland: a review. Env Biol Fish 1982; 7:83.
83. McNabb FM. Avian thyroid development and adaptive plasticity. Gen Comp Endocrinol 2006; 147(2):93-101.
84. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
85. Brown DD. The role of thyroid hormone in zebrafish and axolotl development. Proc Natl Acad Sci U S A 1997; 94(24):13011-13016.
86. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
87. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
88. Power DM, Llewellyn L, Faustino M et al. Thyroid hormones in growth and development of fish. Comp Biochem Physiol C Toxicol Pharmacol 2001; 130(4):447-459.
89. Dickhoff WW, Folmar LC, Gorbman A. Changes in plasma thyroxine during smoltification of Coho Salmon, Oncorhynchus kisutch. Gen Comp Endocriol 1978; 36:229.
90. Nishikawa K, Hirashima T, Suzuki S, Suzuki M. Changes in circulating L-thyroxine and L-triiodothyronine of the masu salmon, Oncorhynchus masou accompanying the smoltification, measured by radioimmunoassay. Endocrinol Jpn 1979; 26:731.
91. Maker MJ. Metabolic responses of isolated tissues to thyroxine administered in vivo. Endocrinology 1964; 74:994.
92. Turner JE, Tipton SR. Environmental temperature and thyroid function in the green water snake. Natrix cyclopion. Gen Comp Endocrinol 1972; 18:195.
93. Eales JG. The influence of nutritional state on thyroid function in various vertebrates. Amer Zool 1988; 28:351-362.
94. Gudernatsch JF. Feeding experiments on tadpoles. I. The influence of specific organs given as food on growth and differentiation. A contribution to the knowledge of organs with internal secretion. Arch Enwicklungsmech Organ 1912; 35:457.
95. Dodd MHI, Dodd JM. The biology of metamorphosis. Physiology of the Amphibia 1976; Academic Press:467.
96. Brown DD, Cai L. Amphibian metamorphosis. Dev Biol 2007; 306(1):20-33.
97. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
98. Denver RJ, Glennemeier KA, Boorse GC. Endocrinology of complex life cycles. Amphibian Horm , Brain and behavior 2002; Academic Press:469.
99. Boorse GC, Denver RJ. Expression and hypophysiotropic actions of corticotropin-releasing factor in Xenopus laevis. Gen Comp Endocrinol 2004; 137(3):272-282.
100. Leloup J, Buscaglia M. La triiodothyronine, hormone de la métamorphose des amphibiens. C R Acad Sci Paris Ser D 1977; 284:2261.
101. Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin). Garland Publ Inc 1994.
102. Fredriksson G, Lebel JM, Leloup J. Thyroid hormones and putative nuclear T3 receptors in tissues of the ascidian, Phallusia mammillata cuvier. Gen Comp Endocrinol 1993; 92(3):379-387.
103. Brown DD, Cai L. Amphibian metamorphosis. Dev Biol 2007; 306(1):20-33.
104. Brown DD, Cai L. Amphibian metamorphosis. Dev Biol 2007; 306(1):20-33.
105. Furlow JD, Neff ES. A developmental switch induced by thyroid hormone: Xenopus laevis metamorphosis. Trends Endocrinol Metab 2006; 17(2):40-47.
106. Rosenkilde P, Ussing AP. Regulation of metamorphosis. Biol & Physiol of Amphibians 1990; 38:125.
107. Buckbinder L, Brown DD. Expression of the Xenopus laevis prolactin and thyrotropin genes during metamorphosis. Proc Natl Acad Sci U S A 1993; 90(9):3820-3824.
108. Opitz R, Trubiroha A, Lorenz C et al. Expression of sodium-iodide symporter mRNA in the thyroid gland of Xenopus laevis tadpoles: developmental expression, effects of antithyroidal compounds, and regulation by TSH. J Endocrinol 2006; 190(1):157-170.
109. Cai L, Brown DD. Expression of type II iodothyronine deiodinase marks the time that a tissue responds to thyroid hormone-induced metamorphosis in Xenopus laevis. Dev Biol 2004; 266(1):87-95.
110. Manzon RG, Denver RJ. Regulation of pituitary thyrotropin gene expression during Xenopus metamorphosis: negative feedback is functional throughout metamorphosis. J Endocrinol 2004; 182(2):273-285.
111. Boorse GC, Denver RJ. Expression and hypophysiotropic actions of corticotropin-releasing factor in Xenopus laevis. Gen Comp Endocrinol 2004; 137(3):272-282.
112. Etkin W. Hypothalamic sensitivity of thyroid feedback in the tadpole. Neuroendocrinology 1965; 1:293.
113. Manzon RG, Denver RJ. Regulation of pituitary thyrotropin gene expression during Xenopus metamorphosis: negative feedback is functional throughout metamorphosis. J Endocrinol 2004; 182(2):273-285.
114. Larsson M, Pettersson T, Carlstrom A. Thyroid hormone binding in serum of 15 vertebrate species: isolation of thyroxine-binding globulin and prealbumin analogs. Gen Comp Endocrinol 1985; 58(3):360-375.
115. Schreiber G. The evolutionary and integrative roles of transthyretin in thyroid hormone homeostasis. J Endocrinol 2002; 175(1):61-73.
116. Schreiber G. The evolutionary and integrative roles of transthyretin in thyroid hormone homeostasis. J Endocrinol 2002; 175(1):61-73.
117. Yaoita Y, Shi YB, Brown DD. Xenopus laevis alpha and beta thyroid hormone receptors. Proc Natl Acad Sci U S A 1990; 87(18):7090-7094.
118. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
119. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
120. Shi YB, Ritchie JW, Taylor PM. Complex regulation of thyroid hormone action: multiple opportunities for pharmacological intervention. Pharmacol Ther 2002; 94(3):235-251.
121. Helbing CC, Werry K, Crump D, Domanski D, Veldhoen N, Bailey CM. Expression profiles of novel thyroid hormone-responsive genes and proteins in the tail of Xenopus laevis tadpoles undergoing precocious metamorphosis. Mol Endocrinol 2003; 17(7):1395-1409.
122. Das B, Cai L, Carter MG et al. Gene expression changes at metamorphosis induced by thyroid hormone in Xenopus laevis tadpoles. Dev Biol 2006; 291(2):342-355.
123. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
124. Cossette SM, Drysdale TA. Early expression of thyroid hormone receptor beta and retinoid X receptor gamma in the Xenopus embryo. Differentiation 2004; 72(5):239-249.
125. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
126. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
127. Furlow JD, Neff ES. A developmental switch induced by thyroid hormone: Xenopus laevis metamorphosis. Trends Endocrinol Metab 2006; 17(2):40-47.
128. Yaoita Y, Shi YB, Brown DD. Xenopus laevis alpha and beta thyroid hormone receptors. Proc Natl Acad Sci U S A 1990; 87(18):7090-7094.
129. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
130. Yaoita Y, Shi YB, Brown DD. TI. Xenopus levis ? and ß thyroid hormone receptor. Proc Natl Acad Sci USA 1990; 87:7090.
131. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
132. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
133. Yaoita Y, Shi YB, Brown DD. Xenopus laevis alpha and beta thyroid hormone receptors. Proc Natl Acad Sci U S A 1990; 87(18):7090-7094.
134. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
135. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
136. Yaoita Y, Shi YB, Brown DD. Xenopus laevis alpha and beta thyroid hormone receptors. Proc Natl Acad Sci U S A 1990; 87(18):7090-7094.
137. Buchholz DR, Paul BD, Fu L, Shi YB. Molecular and developmental analyses of thyroid hormone receptor function in Xenopus laevis, the African clawed frog. Gen Comp Endocrinol 2006; 145(1):1-19.
138. Kohrle J, Jakob F, Contempre B, Dumont JE. Selenium, the thyroid, and the endocrine system. Endocr Rev 2005; 26(7):944-984.
139. Brown DD. The role of deiodinases in amphibian metamorphosis. Thyroid 2005; 15(8):815-821.
140. Kuiper GG, Klootwijk W, Morvan DG et al. Characterization of recombinant Xenopus laevis type I iodothyronine deiodinase: substitution of a proline residue in the catalytic center by serine (Pro132Ser) restores sensitivity to 6-propyl-2-thiouracil. Endocrinology 2006; 147(7):3519-3529.
141. Brown DD. The role of deiodinases in amphibian metamorphosis. Thyroid 2005; 15(8):815-821.
142. Kikuyama S, Kawamura K, Tanaka S, Yamamoto K. Aspects of amphibian metamorphosis: hormonal control. Int Rev Cytol 1993; 145:105-148.
143. Decuypere E, Van As P, Van der GS, Darras VM. Thyroid hormone availability and activity in avian species: a review. Domest Anim Endocrinol 2005; 29(1):63-77.
144. Baker BS, Tata JR. Prolactin prevents the autoinduction of thyroid hormone receptor mRNAs during amphibian metamorphosis. Dev Biol 1992; 149(2):463-467.
145. Huang H, Brown DD. Prolactin is not a juvenile hormone in Xenopus laevis metamorphosis. Proc Natl Acad Sci U S A 2000; 97(1):195-199.
146. Decuypere E, Van As P, Van der GS, Darras VM. Thyroid hormone availability and activity in avian species: a review. Domest Anim Endocrinol 2005; 29(1):63-77.
147. Leatherland JF. Endocrine factors affecting thyroid economy of teleost fish. Am Zool 1988; 28:319.
148. Eales JG, Nrown SB. Measurement and regulation of thyroidal status in telest fos. Rev Fish Biol Fish 1993; 3:299.
149. Orozco A, Valverde R. Thyroid hormone deiodination in fish. Thyroid 2005; 15(8):799-813.
150. Dickhoff WW, Crim JW, Gorbman A. Lack of effect of synthetic thyrotropin releasing hormone on Pacific hagfish (Eptatretus stouti) pituitary-thyroid tissues in vitro. Gen Comp Endocrinol 1978; 35(1):96-98.
151. Pickering AD. Effects of hypophysectomy on the activity of the endostyle and thyroid gland in the larval and adult river lamprey, Lampetra fluviatilis L. Gen Comp Endocrinol 1972; 18(2):335-343.
152. Dickhoff WW, Crim JW, Gorbman A. Lack of effect of synthetic thyrotropin releasing hormone on Pacific hagfish (Eptatretus stouti) pituitary-thyroid tissues in vitro. Gen Comp Endocrinol 1978; 35(1):96-98.
153. Dickhoff WW, Crim JW, Gorbman A. Lack of effect of synthetic thyrotropin releasing hormone on Pacific hagfish (Eptatretus stouti) pituitary-thyroid tissues in vitro. Gen Comp Endocrinol 1978; 35(1):96-98.
154. Orozco A, Valverde R. Thyroid hormone deiodination in fish. Thyroid 2005; 15(8):799-813.
155. Eales JG, Holmes JA, McLeese JM, Youson JH. Thyroid hormone deiodination in various tissues of larval and upstream-migrant sea lampreys, Petromyzon marinus. Gen Comp Endocrinol 1997; 106(2):202-210.
156. De Groef B, Van der GS, Darras VM, Kuhn ER. Role of corticotropin-releasing hormone as a thyrotropin-releasing factor in non-mammalian vertebrates. Gen Comp Endocrinol 2006; 146(1):62-68.
157. Darras VM, Berghman LR, Vanderpooten A, Kuhn ER. Growth hormone acutely decreases type III iodothyronine deiodinase in chicken liver. FEBS Lett 1992; 310(1):5-8.
158. Geris KL, Kotanen SP, Berghman LR, Kuhn ER, Darras VM. Evidence of a thyrotropin-releasing activity of ovine corticotropin-releasing factor in the domestic fowl (Gallus domesticus). Gen Comp Endocrinol 1996; 104(2):139-146.
159. De Groef B, Goris N, Arckens L, Kuhn ER, Darras VM. Corticotropin-releasing hormone (CRH)-induced thyrotropin release is directly mediated through CRH receptor type 2 on thyrotropes. Endocrinology 2003; 144(12):5537-5544.
160. Darras VM, Kuhn ER. Increased plasma levels of thyroid hormones in a frog Rana ridibunda following intravenous administration of TRH. Gen Comp Endocrinol 1982; 48(4):469-475.
161. Denver RJ. Several hypothalamic peptides stimulate in vitro thyrotropin secretion by pituitaries of anuran amphibians. Gen Comp Endocrinol 1988; 72(3):383-393.
162. Denver RJ. Several hypothalamic peptides stimulate in vitro thyrotropin secretion by pituitaries of anuran amphibians. Gen Comp Endocrinol 1988; 72(3):383-393.
163. Okada R, Yamamoto K, Koda A et al. Development of radioimmunoassay for bullfrog thyroid-stimulating hormone (TSH): effects of hypothalamic releasing hormones on the release of TSH from the pituitary in vitro. Gen Comp Endocrinol 2004; 135(1):42-50.
164. Manzon RG, Denver RJ. Regulation of pituitary thyrotropin gene expression during Xenopus metamorphosis: negative feedback is functional throughout metamorphosis. J Endocrinol 2004; 182(2):273-285.
165. Kaneko M, Fujisawa H, Okada R, Yamamoto K, Nakamura M, Kikuyama S. Thyroid hormones inhibit frog corticotropin-releasing factor-induced thyrotropin release from the bullfrog pituitary in vitro. Gen Comp Endocrinol 2005; 144(2):122-127.
166. Larsen DA, Swanson P, Dickey JT, Rivier J, Dickhoff WW. In vitro thyrotropin-releasing activity of corticotropin-releasing hormone-family peptides in coho salmon, Oncorhynchus kisutch. Gen Comp Endocrinol 1998; 109(2):276-285.
167. Trueba SS, Auge J, Mattei G et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis-associated malformations. J Clin Endocrinol Metab 2005; 90(1):455-462.
168. Soyama F. Development and differentiation of lateral thyroid. Endocrinol Jpn 1973; 20:565.
169. Fujita H, Machino M. On the follicle formation of the thyroid gland in the chick embryo. Exp Cell Res 1961; 25:204.
170. Rousset B, Poncet C, Dumont JE, Mornex R. Intracellular and extracellular sites of iodination in dispersed dog thyroid cells. Biochem J 1980; 192:801.
171. LeDouarin N, LeLièvre C. Démonstration de l'origine neurale des cellules à calcitonine du corps ultimobranchial chez l'embryon de poulet. CR Acad Sci 1970; D270:2857.
172. Wolfe HJ, Voelkel EF, Tashjian JA. Distribution of calcitonin-containing cells in the normal adult human thyroid gland: a correlation of morphology with peptide content. J Clin Endocrinol Metab 1974; 38:688.
173. Dow CJDJEKP. Etudes du pourcentage de cellules epitheliales, fibroblastes et cellules endothéliales dans les thyroïdes de chien. C R Soc Biol (Paris) 1986; 19:449-453.
174. Nadler NJ, Sarkar SK, Leblond CP. Origin of intracellular colloid droplets in the rat thyroid. Endocrinology 1962; 71:120.
175. Dumont JE, Rocmans P. In vivo effects of thyrotropin on the metabolism of the thyroid gland. J Physiol 1964; 174:26-45.
176. Wissig SL. The anatomy of secretion in the follicular cells of the thyroid gland. I. The fine structure of the gland in the normal rat. J Biophys Biochem Cytol 1960; 7:419.
177. Dempsey EW, Peterson RR. Electron microscopic observations on the thyroid glands of normal, hypophysectomized cold-exposed and thiouracil-treated rats. Endocrinology 1955; 56:46.
178. Ekholm R. Thyroid gland. In: Kurtz Stanley M, editor. Electron Microscopic Anatomy. New York: Academic Press, 1964: 221-237.
179. Herman L. An electron microscope study of the salamander thyroid during hormonal stimulation. J Biophys Biochem Cytol 1960; 7:143.
180. Otten J, Dumont JE. Glucose metabolism in normal human thyroid tissue in vitro. Eur J Clin Invest 1972; 2:213.
181. Dumont JE, Willems C, Van Sande J, Nève P. Regulation of the release of thyroid hormones: Role of cyclic AMP. Ann NY Acad Sci 1971; 185:291.
182. Lamy FM, Rodesch FR, Dumont JE. Action of thyrotropin on thyroid energetic metabolism. Exp Cell Res 1967; 46:518.
183. Dumont JE, Tondeur-Montenez T. Action de l'hormone thyreotrope sur le métabolisme énergetique du tissue thyroidien. III Evalution au moyen du 14C glucose des voies du métabolisme du glucose, dans le tissue thyrodien de chien. Biochim Biophys Acta 1965; 3:258.
184. Dumont JE. Carbohydrate metabolism in the thyroid gland. J.Clin.Endocrinol.Metab. 20, 1246-1258. 1960.
Ref Type: Journal (Full)
185. Rheinwein D, Engelhardt A. Enzymmuster der menschlichen Schilddrüse II. Euthyreote und hyperthyreote Strumen. Klin Wochenschr 1964; 42:736.
186. Rheinwein D, Engelhardt A. Enzymmuster der Menschlichen Schilddrüse I. Normale Schilddrüse. Klin Wochenschr 1964; 42:731.
187. Dumont JE. The action of thyrotropin on thyroid metabolism. Vitam Horm 1971; 29:287-412.
188. Carafoli EN, Lehninger AL. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem J 1971; 122:681.
189. Freinkel N. Action of pituitary thyrotropin on the inorganic phosphorus of thyroid tissue in vitro. Nature 1963; 198:889.
190. Mockel J, Dumont JE. Protein synthesis in isolated thyroid mitochondria. Endocrinology 1972; 91:817.
191. Ochi Y, DeGroot LJ. Stimulation of RNA and phospholipid synthesis by long-acting thyroid stimulator and by thyroid stimulating hormone. Biochim Biophys Acta 1968; 170:198.
192. Kleiman D, Pisarev MA, Spaulding SW. Early effect of thyrotropin on ribonucleic acid transcription in the thyroid. Endocrinology 1979; 104:693.
193. Lamy F, Willems C, Lecocq R, Delcroix C, Dumont JE. Stimulation by thyrotropin in vitro of uridine incorporation into the RNA of thyroid slices. Horm Metab Res 1971; 3:414.
194. Lindsay RH, Cash AG, Hill JB. TSH stimulation of orotic acid conversion to pyrimidine nucleotides and RNA in bovine thyroid. Endocrinology 1969; 84:534.
195. Cartouzou G, Attali JC, Lissitzky S. Acides ribonucleiques messagers de la glande thyroide. 1. RNA a marquage rapide des noyaux et des polysomes. Eur J Biochem 1968; 4:41.
196. van Staveren WC, Solis DW, Delys L et al. Gene expression in human thyrocytes and autonomous adenomas reveals suppression of negative feedbacks in tumorigenesis. Proc Natl Acad Sci U S A 2006; 103(2):413-418.
197. Sheinman SJ, Burrow GN. In vitro stimulation of thyroid ornithine decarboxylase activity and polyamines by thyrotropin. Endocrinology 1977; 101:1088.
198. Tong W. TSH stimulation of 14C-amino acid incorporation into protein by isolated bovine thyroid cells. Endocrinology 1967; 80:1101.
199. Wagar G. Action of cyclic adenosine 3',5' monophosphate on 1-14C-leucine incorporation in a system of rough microsomes from bovine thyroind gland. Acta Endocrinol 1976; 81:96.
200. Lecocq RE, Dumont JE. Stimulation by thyrotropin of amino acid incorporation into proteins in dog thyroid slices in vitro. Biochim Biophys Acta 1972; 281:434.
201. Creek RO. Effect of thyrotropin on the weight, protein, ribonucleic acid, and the radioactive phosphorus of chick thyroids. Endocrinology 1965; 76:1124.
202. Keyhani E, Claude A, Lecocq RE, Dumont JE. An electron microscopic study of ribosomes and polysomes isolated from sheep thyroid gland. J Microsc 1971; 10:269.
203. Kondo Y, DeNayer P, Salabe G, Robbins J, Rall JE. Function of isolated bovine thyroid polyribosomes. Endocrinology 1968; 83:1123.
204. Lecocq RE, Dumont JE. In vivo and in vitro effects of thyrotropin on ribosomal pattern of dog thyroid. Biochim Biophys Acta 1973; 299:304.
205. Freinkel N. Aspects of the endocrine regulation of lipid metabolism. In: Dawson MC, Rhodes DN, editors. Metabolism and physiological significance of lipids. New York: John Wiley & Sons Inc, 1965: 455.
206. Shah SN, Lossow WJ, Trujillo JL, Chaikoff IC. Metabolic characteristics of preparations of isolated sheep thyroid gland cells. II-Fatty acid oxidation. Endocrinology 1965; 77:103.
207. Freinkel N. Further observations concerning the action of pituitary thyrotropin on the intermediary metabolism of sheep thyroid tissue in vitro. Endocrinology 1960; 66:851.
208. Kasabian SS, Pisarev VB. Histochemical characteristics of lipids in different forms of goitre. Arch Pathol 1976; 38:27.
209. Svennerholm L. Gangliosides of human thyroid gland. Biochim Biophys Acta 1985; 835:231.
210. Levis GM, Carli JN, Malamos B. The phospholipids of the thyroid gland. Clin Chim Acta 1972; 41:335.
211. Schneider PS. Thyroidal synthesis of phosphatidic acid. Endocrinology 1968; 82:969.
212. Scott TW, Good BF, Ferguson KA. Comparative effects of LATS and pituitary thyrotropin on the intermediate metabolism of thyroid tissue in vitro. Endocrinology 1966; 79:949.
213. Moeller LC, Alonso M, Liao X et al. Pituitary-thyroid setpoint and thyrotropin receptor expression in consomic rats. Endocrinology 2007; 148(10):4727-4733.
214. Nakabayashi K, Kudo M, Kobilka B, Hsueh AWJ. Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. Journal of Biological Chemistry 2000; 275(39):30264-30271.
215. Dumont JE, Lamy F, Roger PP, Maenhaut C. Physiological and pathological regulation of thyroid cell proliferation and differentiation by thyrotropin and other factors. Physiological Rev 1992; 72:667-697.
216. Vassart G, Dumont JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocrine Rev 1992; 13:596-611.
217. Brabant G, Bergmann P, Kirsch CM, Köhrie J, Hesch RD, von zur Mühlen A. Early adaptation of thyrotropin and thyroglobulin secretion to experimentally decreased iodine supply in man. Metabolism 1992; 41:1093-1096.
218. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15776-15781.
219. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
220. Abramowicz MJ, Duprez L, Parma J, Vassart G, Heinrichs C. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. Journal of Clinical Investigation 1997; 99(12):3018-3024.
221. Toyoda N, Nishikawa M, Horimoto M. Synergistic effect of thyroid hormone and thyrotropin on iodothyronine 5'-adenosinase in FRTL-5 rat thyroid cells. Endocrinology 1990; 127:1199-1205.
222. Paire A, Bernier-Valentin F, Rabilloud R, Watrin C, Selmi-Ruby S, Rousset B. Expression of alpha- and beta-subunits and activity of Na+ K+ ATPase in pig thyroid cells in primary culture: modulation by thyrotropin and thyroid hormones. Molecular and Cellular Endocrinology 1998; 146:93-101.
223. Ying H, Suzuki H, Zhao L, Willingham MC, Meltzer P, Cheng SY. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor gamma during thyroid carcinogenesis. Cancer Research 2003; 63(17):5274-5280.
224. Glinoer D, de Nayer P, Bourdoux P et al. Regulation of maternal thyroid during pregnancy. Journal of Clinical Endocrinology and Metabolism 1990; 71:276-287.
225. Hershman JM, Lee HY, Sugawara M et al. Human chorionic gonadotropin stimulates iodide uptake, adenylate cyclase, and deoxyribonucleic acid synthesis in cultured rat thyroid cells. J Clin Endocrinol Metab 1988; 67:74-79.
226. Glinoer D. The regulation of thyroid function in pregnancy: Pathways of endocrine adaptation from physiology to pathology. Endocrine Rev 1997; 18(3):404-433.
227. Dumont JE, Maenhaut C, Pirson I, Baptist M, Roger PP. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab 1991; 5(4):727-754.
228. Gerard CM, Roger PP, Dumont JE. Thyroglobulin gene expression as a differentiation marker in primary cultures of calf thyroid cells. Molecular and Cellular Endocrinology 1989; 61(1):23-35.
229. Cheung NW, Lou JC, Boyages SC. Growth hormone does not increase thyroid size in the absence of thyrotropin: a study in adults with hypopituitarism. Journal of Clinical Endocrinology and Metabolism 1996; 81(3):1179-1183.
230. Dormitzer PR, Ellison PT, Bode HH. Anomalously low endemic goiter prevalence among Efe pygmies. American Journal of Physiology-Anthropology 1989; 78(4):527-531.
231. Clement S, Refetoff S, Robaye B, Dumont JE, Schurmans S. Low TSH requirement and goiter in transgenic mice overexpressing IGF-I and IGF-I receptor in the thyroid gland. Endocrinology 2001; 142(12):5131-5139.
232. Govaerts C, Lefort A, Costagliola S et al. A conserved Asn in transmembrane helix 7 is an on/off switch in the activation of the thyrotropin receptor. Journal of Biological Chemistry 2001; 276(25):22991-22999.
233. Burikhanov R, Coulonval K, Pirson I, Lamy F, Dumont JE, Roger PP. Thyrotropin via cyclic AMP induces insulin receptor expression and insulin co-stimulation of growth and amplifies insulin and insulin-like growth factor signaling pathways in dog thyroid epithelial cells. Journal of Biological Chemistry 1996; 271:29400-29406.
234. Van Keymeulen A, Dumont JE, Roger PP. TSH induces insulin receptors that mediate insulin costimulation of growth in normal human thyroid cells. Biochemical and Biophysical Research Communications 2000; 279(1):202-207.
235. Ahrén B. Regulatory peptides in the thyroid gland - a review on their localization and function. Acta Endocrinologica 1991; 124:225-232.
236. Raspé E, Laurent E, Andry G, Dumont JE. ATP, bradykinine, TRH and TSH activate the Ca2+-phophatidyl inositol cascade of human thyrocytes in primary culture. Molecular and Cellular Endocrinology 1991; 81:175-183.
237. Raspé E, Andry G, Dumont JE. Adenosine triphosphate, bradykinin, and thyrotropin-releasing hormone regulate the intracellular Ca2+ concentration and the 45Ca2+ efflux of human thyrocytes in primary culture. J Cell Physiol 1989; 140:608-614.
238. Young JB, Burgi-Saville ME, Burgi U, Landsberg L. Sympathetic nervous system activity in rat thyroid: potential role in goitrogenesis. Am J Physiol Endocrinol Metab 2005; 288(5):E861-E867.
239. Osawa S, Spaulding SW. Epidermal growth factor inhibits radioiodine uptake but stimulates deoxyribonucleic acid synthesis in new born rat thyroids, grown in nude mice. Endocrinology 1990; 127:604.
240. Paschke R, Eck T, Herfurth J, Usadel KH. Stimulation of proliferation and inhibition of function of xenotransplanted human thyroid tissue by epidermal growth factor. Journal of Endocrinology and Investigation 1995; 18(5):359-363.
241. De Vito WJ, Chanoine JP, Alex S et al. Effect of in vivo administration of recombinant acidic fibroblast growth factor on thyroid function in the rat: induction of colloid goiter. Endocrinology 1992; 131(2):729-735.
242. Roger PP, Dumont JE. Factors controlling proliferation and differentiation of canine thyroid cells cultured in reduced serum conditions: effects of thyrotropin, cyclic AMP and growth factors. Molecular and Cellular Endocrinology 1984; 36(1-2):79-93.
243. Westermark K, Karlsson FA, Westermark B. Epidermal growth factor modulates thyroid growth and function in culture. Endocrinology 1983; 112(5):1680-1686.
244. Eggo MC, Bachrach LK, Fayet G et al. The effects of growth factors and serum on DNA synthesis and differentiation in thyroid cells in culture. Molecular and Cellular Endocrinology 1984; 38(2-3):141-150.
245. Lamy F, Taton M, Dumont JE, Roger PP. Control of protein synthesis by thyrotropin and epidermal growth factor in human thyrocytes: role of morphological changes. Mol Cell Endocrinol 1990; 73:195.
246. Kraiem Z, Sadeh O, Yosef M, Aharon A. Mutual antagonistic interactions between the thyrotropin (adenosine 3',5'-monophosphate) and protein kinase C/epidermal growth factor (tyrosine kinase) pathways in cell proliferation and differentiation of cultured human thyroid follicles. Endocrinology 1995; 136(2):585-590.
247. Becks GP, Logan A, Phillips ID et al. Increase of basic fibroblast growth factor (FGF) and FGF receptor messenger RNA during rat thyroid hyperplasia: temporal changes and cellular distribution. Journal of Endocrinology 1994; 142(2):325-338.
248. Bidey SP, Hill DJ, Eggo MC. Growth factors and goitrogenesis. J Endocrinol 1999; 160:321-332.
249. Derwahl M, Broecker M, Kraiem Z. Clinical review 101: Thyrotropin may not be the dominant growth factor in benign and malignant thyroid tumors. Journal of Clinical Endocrinology and Metabolism 1999; 84(3):829-834.
250. Roger PP, Dumont JE. Thyrotropin-dependent insulin-like growth factor I mRNA expression in thyroid cells. European Journal of Endocrinology 1995; 132:601-602.
251. Grubeck-Loebenstein B, Buchan G, Sadeghi R et al. Transforming growth factor beta regulates thyroid growth. J Clin Invest 1989; 83:764-770.
252. Taton M, Lamy F, Roger PP, Dumont JE. General inhibition by transforming growth factor beta1 of thyrotropin and cAMP responses in human thyroid cells in primary culture. Molecular and Cellular Endocrinology 1993; 95:13-21.
253. Logan A, Smith C, Becks GP, Gonzalez AM, Phillips ID, Hill DJ. Enhanced expression of transforming growth factor-beta 1 during thyroid hyperplasia in rats. Journal of Endocrinology 1994; 141:45-57.
254. Franzen A, Piek E, Westermark B, ten Dijke P, Heldin NE. Expression of transforming growth factor-beta1, activin A, and their receptors in thyroid follicle cells: negative regulation of thyrocyte growth and function. Endocrinology 1999; 140:4300-4310.
255. Helmbrecht K, Kispert A, von Wasielewski R, Brabant G. Identification of a Wnt/beta-catenin signaling pathway in human thyroid cells. Endocrinology 2001; 142(12):5261-5266.
256. Suzuki K, Mori A, Lavaroni S et al. Thyroglobulin regulates follicular function and heterogeneity by suppressing thyroid-specific gene expression. Biochimie 1999; 81(4):329-340.
257. Sheridan PJ, McGill HC, Jr., Lissitzky JC, Martin PM. The primate thyroid gland contains receptors for androgens. Endocrinology 1984; 115(6):2154-2159.
258. Manole D, Schildknecht B, Gosnell B, Adams E, Derwahl M. Estrogen promotes growth of human thyroid tumor cells by different molecular mechanisms. J Clin Endocrinol Metab 2001; 86(3):1072-1077.
259. Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A. Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene 2010; 29(42):5678-5686.
260. Lyons J, Landis CA, Harsh G et al. Two G protein oncogenes in human endocrine tumors. Science 1990; 249(4969):655-659.
261. Parma J, Van Sande J, Swillens S, Tonacchera M, Dumont JE, Vassart G. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3',5'-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol 1995; 9:725-733.
262. Vasseur C, Rodien P, Beau I et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. New England Journal of Medicine 2003; 349(8):753-759.
263. Smits G, Olatunbosun O, Delbaere A, Pierson R, Vassart G, Costagliola S. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. New England Journal of Medicine 2003; 349(8):760-766.
264. Van Sande J, Costa MJ, Massart C et al. Kinetics of thyrotropin-stimulating hormone (TSH) and thyroid-stimulating antibody binding and action on the TSH receptor in intact TSH receptor-expressing CHO cells. Journal of Clinical Endocrinology and Metabolism 2003; 88(11):5366-5374.
265. Drexhage H, Mooij P, Wilders-Truschnig MM. Thyroid growth stimulating immunoglobulins in sporadic and endemic colloid goitre. Thyroidology 1990; 2:99-105.
266. Dumont JE, Roger PP, Ludgate M. Assays for thyroid growth immunoglobulins and their clinical implications: methods, concepts and misconceptions. Endocrine Rev 1987; 8:448-452.
267. Zakarija M, Jin S, McKenzie JM. Evidence supporting the identity in Graves's disease of thyroid-stimulating antibody and thyroid growth-promoting immunoglobulin G as assayed in FRTL5 cells. J Clin Invest 1988; 81:879-884.
268. Zakarija M, McKenzie JM. Do thyroid growth-promoting immunoglobulins exist ? Journal of Clinical Endocrinology and Metabolism 1990; 70:308-310.
269. Dumont JE, Maenhaut C, Pirson I, Baptist M, Roger PP. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab 1991; 5(4):727-754.
270. Contempré B, Le Moine O, Dumont JE, Denef JF, Many MC. Selenium deficiency and thyroid fibrosis. A key role for macrophages and transforming growth factor beta (TGF-beta). Molecular and Cellular Endocrinology 1996; 124:7-15.
271. Contempre B, de Escobar GM, Denef JF, Dumont JE, Many MC. Thiocyanate induces cell necrosis and fibrosis in selenium- and iodine-deficient rat thyroids: A potential experimental model for myxedematous endemic cretinism in central Africa. Endocrinology 2004; 145(2):994-1002.
272. Thompson SD, Franklyn JA, Watkinson JC, Verhaeg JM, Sheppard MC, Eggo MC. Fibroblast growth factors 1 and 2 and fibroblast growth factor receptor 1 are elevated in thyroid hyperplasia. Journal of Clinical Endocrinology and Metabolism 1998; 83(4):1336-1341.
273. Fusco A, Santoro M, Grieco M et al. RET/PTC activation in human thyroid carcinomas. Journal of Endocrinology and Investigation 1995; 18(2):127-129.
274. Pierotti MA, Bongarzone I, Borrello MG et al. Rearrangements of TRK proto-oncogene in papillary thyroid carcinomas. Journal of Endocrinology and Investigation 1995; 18(2):130-133.
275. Trovato M, Villari D, Bartolone L et al. Expression of the hepatocyte growth factor and c-met in normal thyroid, non-neoplastic, and neoplastic nodules. Thyroid 1998; 8(2):125-131.
276. Aasland R, Akslen LA, Varhaug JE, Lillehaug JR. Co-expression of the genes encoding transforming growth factor-alpha and its receptor in papillary carcinomas of the thyroid. International Journal Cancer 1990; 46(3):382-387.
277. Vella V, Pandini G, Sciacca L et al. A novel autocrine loop involving IGF-II and the insulin receptor isoform-A stimulates growth of thyroid cancer. Journal of Clinical Endocrinology and Metabolism 2002; 87(1):245-254.
278. Blaydes JP, Schlumberger M, Wynford-Thomas D, Wyllie FS. Interaction between p53 and TGF beta 1 in control of epithelial cell proliferation. Oncogene 1995; 10:307-317.
279. Vanvooren V, Allgeier A, Cosson E et al. Expression of multiple adenylyl cyclase isoforms in human and dog thyroid. Mol Cell Endocrinol 2000; 170(1-2):185-196.
280. Van Sande J, Mockel J, Boeynaems JM, Dor P, Andry G, Dumont JE. Regulation of cyclic nucleotide and prostaglandin formation in human thyroid tissues and in autonomous nodules. J Clin Endocrinol Metab 1980; 50:776-785.
281. de Rooij J, Zwartkruis FJT, Verheijen MHG et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998; 396:474-477.
282. Dremier S, Milenkovic M, Blancquaert S et al. Cyclic adenosine 3',5'-monophosphate (cAMP)-dependent protein kinases, but not exchange proteins directly activated by cAMP (Epac), mediate thyrotropin/cAMP-dependent regulation of thyroid cells. Endocrinology 2007; 148(10):4612-4622.
283. Fortemaison N, Blancquaert S, Dumont JE et al. Differential involvement of the actin cytoskeleton in differentiation and mitogenesis of thyroid cells: inactivation of Rho proteins contributes to cyclic adenosine monophosphate-dependent gene expression but prevents mitogenesis. Endocrinology 2005; 146(12):5485-5495.
284. Raymond JR, Hnatowich M, Lefkowitz RJ, Caron MG. Adrenergic receptors. Models for regulation of signal transduction processes. Hypertension 1990; 15:119-131.
285. van Staveren WC, Solis DW, Delys L et al. Gene expression in human thyrocytes and autonomous adenomas reveals suppression of negative feedbacks in tumorigenesis. Proc Natl Acad Sci U S A 2006; 103(2):413-418.
286. Ketelbant-Balasse P, Van Sande J, Neve P, Dumont JE. Time sequence of 3',5'-cyclic AMP accumulation and ultrastructural changes in dog thyroid slices after acute stimulation by TSH. Horm Metab Res 1976; 8(3):212-215.
287. Cleator JH, Ravenell R, Kurtz DT, Hildebrandt JD. A dominant negative Galphas mutant that prevents thyroid-stimulating hormone receptor activation of cAMP production and inositol 1,4,5-trisphosphate turnover: competition by different G proteins for activation by a common receptor. J Biol Chem 2004; 279(35):36601-36607.
288. Van Sande J, Lefort A, Beebe S et al. Pairs of cyclic AMP analogs, that are specifically synergistic for type I and type II cAMP-dependent protein kinases, mimic thyrotropin effects on the function, differentiation expression and mitogenesis of dog thyroid cells. Eur J Biochem 1989; 183:699-708.
289. Van Sande J, Dequanter D, Lothaire P, Massart C, Dumont JE, Erneux C. Thyrotropin stimulates the generation of inositol 1,4,5-trisphosphate in human thyroid cells. J Clin Endocrinol Metab 2006; 91(3):1099-1107.
290. Esteves R, Van Sande J, Dumont JE. Nitric oxide as a signal in thyroid. Molecular and Cellular Endocrinology 1992; 90:R1-R3.
291. Munari-Silem Y, Audebet C, Rousset B. Protein kinase C in pig thyroid cells: activation , translocation and endogenous substrate phosphorylating activity in response to phorbol esters. Molecular and Cellular Endocrinology 1987; 54:81-90.
292. Van Sande J, Raspe E, Perret J et al. Thyrotropin activates both the cyclic AMP and the PIP2 cascades in CHO cells expressing the human cDNA of TSH receptor. Mol Cell Endocrinol 1990; 74:R1-R6.
293. Laurent E, Mockel J, Van Sande J, Graff I, Dumont JE. Dual activation by thyrotropin of the phospholipase C and cAMP cascades in human thyroid. Mol Cell Endocrinol 1987; 52:273.
294. Raspé E, Dumont JE. Control of the dog thyrocyte plasma membrane iodide permeability by the Ca2+-phosphatidylinositol and adenosine 3',5'-monophosphate cascades. Endocrinology 1994; 135:986-995.
295. Mockel J, Laurent E, Lejeune C, Dumont JE. Thyrotropin does not activate the phosphatidylinositol bisphosphate hydrolyzing phospholipase C in the dog thyroid. Molecular and Cellular Biology 1991; 82(2-3):221-227.
296. Mockel J, Lejeune C, Dumont JE. Relative contribution of phosphoinositides and phosphatidylcholine hydrolysis to the actions of carbamylcholine, thyrotropin, and phorbol esters on dog thyroid slices: regulation of cytidine monophosphate-phosphatidic acid accumulation and phospholipase-D activity. II. Actions of phorbol esters. Endocrinology 1994; 135:2497-2503.
297. Lejeune C, Mockel J, Dumont JE. Relative contribution of phosphoinositides and phosphatidylcholine hydrolysis to the actions of carbamylcholine, thyrotropin (TSH), and phorbol esters on dog thyroid slices: regulation of cytidine monophosphate-phosphatidic acid accumulation and phospholipase-D activity. I. Actions of carbamylcholine, calcium ionophores, and TSH. Endocrinology 1994; 135:2488-2496.
298. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
299. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
300. Van Keymeulen A, Dumont JE, Roger PP. TSH induces insulin receptors that mediate insulin costimulation of growth in normal human thyroid cells. Biochemical and Biophysical Research Communications 2000; 279(1):202-207.
301. Thompson SD, Franklyn JA, Watkinson JC, Verhaeg JM, Sheppard MC, Eggo MC. Fibroblast growth factors 1 and 2 and fibroblast growth factor receptor 1 are elevated in thyroid hyperplasia. Journal of Clinical Endocrinology and Metabolism 1998; 83(4):1336-1341.
302. Williams DW, Williams ED, Wynford-Thomas D. Evidence for autocrine production of IGF-1 in human thyroid adenomas. Mol Cell Endocrinol 1989; 61:139-147.
303. Errick JE, Ing KW, Eggo MC, Burrow GN. Growth and differentiation in cultured human thyroid cells: effects of epidermal growth factor and thyrotropin. In Vitro Cell Developmental Biology 1986; 22(1):28-36.
304. Roger PP, Taton M, Van Sande J, Dumont JE. Mitogenic effects of thyrotropin and adenosine 3',5'-monophosphate in differentiated normal human thyroid cells in vitro. J Clin Endocrinol Metab 1988; 66:1158-1165.
305. Heldin NE, Bergström D, Hermansson A et al. Lack of responsiveness to TGF-b1 in a thyroid carcinoma cell line with functional type I and type II TGF-b receptors and Smad proteins, suggests a novel mechanism for TGF-b insensitivity in carcinoma cells. Molecular and Cellular Endocrinology 1999; 153:79-90.
306. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
307. Dumont JE, Miot F, Erneux C et al. Negative regulation of cyclic AMP levels by activation of cyclic nucleotide phosphodiesterases: the example of the dog thyroid. Adv Cyclic Nucl Res 1984; 16:325-336.
308. Mockel J, Van Sande J, Decoster C, Dumont JE. Tumor promoters as probes of protein kinase C in dog thyroid cell: inhibition of the primary effects of carbamylcholine and reproduction of some distal effects. Metabolism 1987; 36:137-143.
309. Roger PP, Reuse S, Maenhaut C, Dumont JE. Multiple facets of the modulation of growth by cAMP. Vitamine and Hormones 1995; 51:59-191.
310. Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends in Cell Biology 2002; 12(6):258-266.
311. Richards JS. New signaling pathways for hormones and cyclic adenosine 3',5'-monophosphate action in endocrine cells. Mol Endocrinol 2001; 15(2):209-218.
312. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
313. Rivas M, Santisteban P. TSH-activated signaling pathways in thyroid tumorigenesis. Mol Cell Endocrinol 2003; 213:31-45.
314. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
315. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
316. Lamy F, Wilkin F, Baptist M, Posada J, Roger PP, Dumont JE. Phosphorylation of mitogen-activated protein kinases is involved in the epidermal growth factor and phorbol ester, but not in the thyrotropin/cAMP, thyroid mitogenic pathways. Journal of Biological Chemistry 1993; 268:8398-8401.
317. Van Keymeulen A, Roger PP, Dumont JE, Dremier S. TSH and cAMP do not signal mitogenesis through Ras activation. Biochemical and Biophysical Research Communications 2000; 273:154-158.
318. Bray GA. Increased sensitivity of the thyroid in iodine-depleted rats to the goitrogenic effects of thyrotropin. J Clin Invest 1968; 47:1640-1647.
319. Wolff J. Congenital goiter with defective iodide transport. Endocrine Rev 1983; 4:240.
320. Wolff J. Iodide goiter and the pharmacologic effects of excess iodide. Am J Med 1969; 47:101-124.
321. Cochaux P, Van Sande J, Swillens S, Dumont JE. Iodide-induced inhibition of adenylate cyclase activity in horse and dog thyroid. European Journal of Biochemistry 1987; 170:435-442.
322. Laurent E, Mockel J, Takazawa K, Erneux C, Dumont JE. Stimulation of generation of inositol phosphates by carbamylcholine and its inhibition by phorbol esters and iodide in dog thyroid cells. Biochemical Journal 1989; 263:795-801.
323. Corvilain B, Laurent E, Lecomte M, Van Sande J, Dumont JE. Role of the cyclic adenosine 3',5'-monophosphate and the phosphatidylinositol-Ca2+ cascades in mediating the effects of thyrotropin and iodide on hormone synthesis and secretion in human thyroid slices. Journal of Clinical Endocrinology and Metabolism 1994; 79:152.
324. Wolff J. Excess iodide inhibits the thyroid by multiple mechanisms. Adv Exp Med Biol 1989; 261:211-244.
325. Van Sande J, Grenier G, Willems C, Dumont JE. Inhibition by iodide of the activation of the thyroid cyclic 3',5'-AMP system. Endocrinology 1975; 96:781-786.
326. Dugrillon A, Bechtner G, Uedelhoven WM, Weber PC, Gärtner R. Evidence that an iodolactone mediates the inhibitory effect of iodine on thyroid cell proliferation but not on adenosine 3',5'-monophosphate formation. Endocrinology 1990; 127:337-343.
327. Panneels V, Macours P, Van den BH, Braekman JC, Van Sande J, Boeynaems JM. Biosynthesis and metabolism of 2-iodohexadecanal in cultured dog thyroid cells. Journal of Biological Chemistry 1996; 271(38):23006-23014.
328. Panneels V, Macours P, Van den BH, Braekman JC, Van Sande J, Boeynaems JM. Biosynthesis and metabolism of 2-iodohexadecanal in cultured dog thyroid cells. Journal of Biological Chemistry 1996; 271(38):23006-23014.
329. Panneels V, Van Sande J, Van den BH et al. Inhibition of human thyroid adenylyl cyclase by 2-iodoaldehydes. Molecular and Cellular Endocrinology 1994; 106(1-2):41-50.
330. Panneels V, Van den BH, Jacoby C et al. Inhibition of H2O2 production by iodoaldehydes in cultured dog thyroid cells. Molecular and Cellular Endocrinology 1994; 102(1-2):167-176.
331. Many MC, Mestdagh C, Van Den Hove MF, Denef JF. In vitro study of acute toxic effects of high iodide doses in human thyroid follicles. Endocrinology 1992; 131:621-630.
332. Corvilain B, Collyn L, Van Sande J, Dumont JE. Stimulation by iodide of H(2)O(2) generation in thyroid slices from several species. Am J Physiol Endocrinol Metab 2000; 278(4):E692-E699.
333. Uyttersprot N, Pelgrims N, Carrasco N et al. Moderate doses of iodide in vivo inhibit cell proliferation and the expression of thyroperoxidase and Na+/I- symporter mRNAs in dog thyroid. Mol.Cell.Endocrinol. 131, 195-203. 1997.
Ref Type: Journal (Full)
334. Dias JA, Van Roey P. Structural biology of human follitropin and its receptor. Arch Med Res 2001; 32(6):510-519.
335. Szkudlinski MW, Fremont V, Ronin C, Weintraub BD. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiological Reviews 2002; 82(2):473-502.
336. Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 2002; 23(2):141-174.
337. Smits G, Campillo M, Govaerts C et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. EMBO J 2003; 22(11):2692-2703.
338. Vassart G. Le récepteur de la TSH. In: Leclere J, Orgiazzi J, Rousset B, Schlienger JL, Wemeau JL, editors. La Thyroïde. Paris: la thyroïde, 1992: 64-74.
339. Ieiri T, Cochaux P, Targovnik H et al. A 3' splice site mutation in the thyroglobulin gene responsible for congenital goitre with hypothyroidism. J Clin Invest 1991; 88:1901-1905.
340. Remy JJ, Nespoulous C, Grosclaude J et al. Purification and structural analysis of a soluble human chorionogonadotropin hormone-receptor complex. J Biol Chem 2001; 276(3):1681-1687.
341. Cornelis S, Uttenweiler-Joseph S, Panneels V, Vassart G, Costagliola S. Purification and characterization of a soluble bioactive amino-terminal extracellular domain of the human thyrotropin receptor. Biochemistry 2001; 40(33):9860-9869.
342. Cornelis S, Uttenweiler-Joseph S, Panneels V, Vassart G, Costagliola S. Purification and characterization of a soluble bioactive amino-terminal extracellular domain of the human thyrotropin receptor. Biochemistry 2001; 40(33):9860-9869.
343. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies. Endocrine Rev 1998; 19:673-716.
344. Couet J, de Bernard S, Loosfelt H, Saunier B, Milgrom E, Misrahi M. Cell surface protein disulfide-isomerase is involved in the shedding of human thyrotropin receptor ectodomain. Biochemistry 1996; 35(47):14800-14805.
345. de Bernard S, Misrahi M, Huet JC et al. Sequential cleavage and excision of a segment of the thyrotropin receptor ectodomain. Journal of Biological Chemistry 1999; 274(1):101-107.
346. Rapoport B, Chazenbalk GD, Jaume JC, McLachlan SM. The thyrotropin (TSH) receptor: interaction with TSH and autoantibodies [In Process Citation]. Endocr Rev 1998; 19(6):673-716.
347. Hamidi S, Chen CR, Mizutori-Sasai Y, McLachlan SM, Rapoport B. Relationship between thyrotropin receptor hinge region proteolytic posttranslational modification and receptor physiological function. Mol Endocrinol 2011; 25(1):184-194.
348. McLachlan SM, Taverne J, Atherton MC et al. Cytokines, thyroid autoantibody synthesis and thyroid cell survival in culture. Clin Exp Immunol 1990; 79:175-181.
349. Rapoport B, McLachlan SM. The thyrotropin receptor in Graves' disease. Thyroid 2007; 17(10):911-922.
350. Beau I, Groyer-Picard MT, Desroches A et al. The basolateral sorting signals of the thyrotropin and luteinizing hormone receptors: an unusual family of signals sharing an unusual distal intracellular localization, but unrelated in their structures. Mol Endocrinol 2004; 18(3):733-746.
351. Angers S, Salahpour A, Bouvier M. Dimerization: an emerging concept for G protein-coupled receptor ontogeny and function. Annu Rev Pharmacol Toxicol 2002; 42:409-35.:409-435.
352. Urizar E, Claeysen S, Deupi X et al. An activation switch in the rhodopsin family of G protein-coupled receptors: the thyrotropin receptor. J Biol Chem 2005; 280(17):17135-17141.
353. Rivero-Müller A, Chou Y, Ji I et al. Proc Natl Acad Sci U S A 2009; 00(00):00.
354. Libert F, Parmentier M, Lefort A, Dumont JE, Vassart G. Complete nucleotide sequence of a putative G protein coupled receptor: RDC4. Nucleic Acids Res 1990; 18:1916.
355. Rousseau-Merck MF, Misrahi M, Loosfelt H, Atger M, Milgrom E, Berger R. Assignment of the human thyroid stimulating hormone receptor (TSHR) gene to chromosome 14q31. Genomics 1990; 8(2):233-236.
356. Gross B, Misrahi M, Sar S, Milgrom E. Composite structure of the human thyrotropin receptor gene. Biochem Biophys Res Commun 1991; 177:679-687.
357. Gross B, Misrahi M, Sar S, Milgrom E. Composite structure of the human thyrotropin receptor gene. Biochem Biophys Res Commun 1991; 177:679-687.
358. Kong RC, Shilling PJ, Lobb DK, Gooley PR, Bathgate RA. Membrane receptors: structure and function of the relaxin family peptide receptors. Mol Cell Endocrinol 2010; 320(1-2):1-15.
359. Barker N, Clevers H. Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology 2010; 138(5):1681-1696.
360. Ikuyama S, Niller HH, Shimura H, Akamizu T, Kohn LD. Characterization of the 5'-flanking region of the rat thyrotropin receptor gene. Molecular Endocrinology 1992; 6:793-804.
361. Civitareale D, Ghibelli L, Di Lauro R. Partial purification of a thyroid specific nuclear protein recognizing the thyroglobulin promoter. Horm Metab Res Suppl 1987; 17:73-77.
362. Roselli-Rehfuss L, Robbins LS, Cone RD. Thyrotropin receptor messenger ribonucleic acid is expressed in most brown and white adipose tissues in the guinea pig. Endocrinology 1992; 130:1857-1861.
363. Bell A, Gagnon A, Grunder L, Parikh SJ, Smith TJ, Sorisky A. Functional TSH receptor in human abdominal preadipocytes and orbital fibroblasts. American Journal of Physiology-Cell Physiology 2000; 279(2):C335-C340.
364. Crisp MS, Lane C, Halliwell M, WynfordThomas D, Ludgate M. Thyrotropin receptor transcripts in human adipose tissue. Journal of Clinical Endocrinology and Metabolism 1997; 82(6):2003-2005.
365. Nakao N, Ono H, Yamamura T et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature 2008; 452(7185):317-322.
366. Ono H, Hoshino Y, Yasuo S et al. Involvement of thyrotropin in photoperiodic signal transduction in mice. Proc Natl Acad Sci U S A 2008; 105(47):18238-18242.
367. Abe Y. [Studies on the thyrotropin receptor and adenylate cyclase activity in various thyroid diseases: II. The properties of TSH receptor and adenylate cyclase in human thyroid tumors (author's transl)]. Nippon Naibunpi Gakkai Zasshi 1980; 56:754-764.
368. Bassett JH, Williams AJ, Murphy E et al. A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol Endocrinol 2008; 22(2):501-512.
369. Uyttersprot N, Allgeier A, Baptist M et al. The cAMP in thyroid. From the TSH receptor to mitogenesis and tumorigenesis. In: Corbin J, Francis S, editors. Signal Transduction in Health and Disease, Advances in Second Messenger and Phosphorylation Research. Philadelphia: Lippincott-Raven Publishers, 1997: 125-140.
370. Lapthorn AJ, Harris DC, Littlejohn A et al. Crystal-Structure of Human Chorionic-Gonadotropin. Nature 1994; 369(6480):455-461.
371. Wu H, Lustbader JW, Liu Y, Canfield RE, Hendrickson WA. Structure of human chorionic gonadotropin at 2.6 A resolution from MAD analysis of the selenomethionyl protein. Structure 1994; 2(6):545-558.
372. Fox KM, Dias JA, Van Roey P. Three-dimensional structure of human follicle-stimulating hormone. Molecular Endocrinology 2001; 15(3):378-389.
373. Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor
1. Nature 2005; 433(7023):269-277.
374. Kajava AV, Vassart G, Wodak SJ. Modeling of the 3-Dimensional Structure of Proteins with the Typical Leucine-Rich Repeats. Structure 1995; 3(9):867-877.
375. Sanders J, Chirgadze DY, Sanders P et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid 2007; 17(5):395-410.
376. Sanders P, Young S, Sanders J et al. Crystal structure of the TSH receptor bound to a blocking type TSHR autoantibody. J Mol Endocrinol 2011.
377. Smits G, Campillo M, Govaerts C et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. Embo Journal 2003; 22(11):2692-2703.
378. Smits G, Campillo M, Govaerts C et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. Embo Journal 2003; 22(11):2692-2703.
379. Smits G, Campillo M, Govaerts C et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. Embo Journal 2003; 22(11):2692-2703.
380. Caltabiano G, Campillo M, De Leener A et al. The specificity of binding of glycoprotein hormones to their receptors. Cell Mol Life Sci 2008; 65(16):2484-2492.
381. Caltabiano G, Campillo M, De Leener A et al. The specificity of binding of glycoprotein hormones to their receptors. Cell Mol Life Sci 2008; 65(16):2484-2492.
382. Costagliola S, Franssen JD, Bonomi M et al. Generation of a mouse monoclonal TSH receptor antibody with stimulating activity. Biochem Biophys Res Commun 2002; 299(5):891-896.
383. Costagliola S, Franssen JDF, Bonomi M et al. Generation of a mouse monoclonal TSH receptor antibody with stimulating activity. Biochemical and Biophysical Research Communications 2002; 299(5):891-896.
384. Westmuckett AD, Hoffhines AJ, Borghei A, Moore KL. Early postnatal pulmonary failure and primary hypothyroidism in mice with combined TPST-1 and TPST-2 deficiency. Gen Comp Endocrinol 2008; 156(1):145-153.
385. Sasaki N, Hosoda Y, Nagata A et al. A mutation in Tpst2 encoding tyrosylprotein sulfotransferase causes dwarfism associated with hypothyroidism. Mol Endocrinol 2007; 21(7):1713-1721.
386. Palczewski K, Kumasaka T, Hori T et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000; 289(5480):739-745.
387. Ridge KD, Abdulaev NG, Sousa M, Palczewski K. Phototransduction: crystal clear. Trends in Biochemical Sciences 2003; 28(9):479-487.
388. Palczewski K, Kumasaka T, Hori T et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science 2000; 289(5480):739-745.
389. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature 2009; 459(7245):356-363.
390. Jaakola VP, Griffith MT, Hanson MA et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008; 322(5905):1211-1217.
391. Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struct Biol 2009; 19(4):386-395.
392. Wu B, Chien EY, Mol CD et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010; 330(6007):1066-1071.
393. Choe HW, Kim YJ, Park JH et al. Crystal structure of metarhodopsin II. Nature 2011; 471(7340):651-655.
394. Standfuss J, Edwards PC, D'Antona A et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 2011; 471(7340):656-660.
395. Rasmussen SG, Choi HJ, Rosenbaum DM et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007; 450(7168):383-387.
396. Rasmussen SG, Choi HJ, Fung JJ et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011; 469(7329):175-180.
397. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature 2009; 459(7245):356-363.
398. Rasmussen SG, Choi HJ, Fung JJ et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011; 469(7329):175-180.
399. Cotecchia S, Kobilka BK, Daniel KW et al. Multiple second messenger pathways of alpha-adrenergic receptor subtypes expressed in eukaryotic cells. J Biol Chem 1990; 265(1):63-69.
400. Kjelsberg MA, Cotecchia S, Ostrowski J, Caron MG, Lefkowitz RJ. Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site. Evidence for a region which constrains receptor activation. J Biol Chem 1992; 267(3):1430-1433.
401. Scheerer P, Park JH, Hildebrand PW et al. Crystal structure of opsin in its G-protein-interacting conformation. Nature 2008; 455(7212):497-502.
402. Hofmann KP, Scheerer P, Hildebrand PW et al. A G protein-coupled receptor at work: the rhodopsin model. Trends Biochem Sci 2009; 34(11):540-552.
403. Parma J, Duprez L, Van Sande J et al. Diversity and prevalence of somatic mutations in the thyrotropin receptor and Gs alpha genes as a cause of toxic thyroid adenomas. Journal of Clinical Endocrinology and Metabolism 1997; 82(8):2695-2701.
404. Parma J, Duprez L, Van Sande J et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature 1993; 365:649-651.
405. Kleinau G, Hoyer I, Kreuchwig A et al. From molecular details of the interplay between transmembrane helices of the thyrotropin receptor to general aspects of signal transduction in family A GPCRs. J Biol Chem 2011.
406. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain from a Tethered Inverse Agonist to an Agonist. Mol Endocrinol 2002; 16(4):736-746.
407. Zhang M, Tong KP, Fremont V et al. The extracellular domain suppresses constitutive activity of the transmembrane domain of the human TSH receptor: implications for hormone-receptor interaction and antagonist design. Endocrinology 2000; 141(9):3514-3517.
408. Duprez L, Parma J, Costagliola S et al. Constitutive activation of the TSH receptor by spontaneous mutations affecting the N-terminal extracellular domain. FEBS Letters 1997; 409:469-474.
409. Nakabayashi K, Kudo M, Kobilka B, Hsueh AWJ. Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. Journal of Biological Chemistry 2000; 275(39):30264-30271.
410. Nakabayashi K, Kudo M, Kobilka B, Hsueh AWJ. Activation of the luteinizing hormone receptor following substitution of Ser-277 with selective hydrophobic residues in the ectodomain hinge region. Journal of Biological Chemistry 2000; 275(39):30264-30271.
411. Ho SC, Goh SS, Khoo DH. Association of Graves' disease with intragenic polymorphism of the thyrotropin receptor gene in a cohort of Singapore patients of multi-ethnic origins. Thyroid 2003; 13(6):523-528.
412. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain from a Tethered Inverse Agonist to an Agonist. Mol Endocrinol 2002; 16(4):736-746.
413. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain from a Tethered Inverse Agonist to an Agonist. Mol Endocrinol 2002; 16(4):736-746.
414. Vlaeminck-Guillem V, Ho SC, Rodien P, Vassart G, Costagliola S. Activation of the cAMP Pathway by the TSH Receptor Involves Switching of the Ectodomain from a Tethered Inverse Agonist to an Agonist. Mol Endocrinol 2002; 16(4):736-746.
415. Vassart G, Pardo L, Costagliola S. A molecular dissection of the glycoprotein hormone receptors. Trends in Biochemical Sciences 2004; 29:in press.
416. Chen CR, McLachlan SM, Rapoport B. Identification of key amino acid residues in a thyrotropin receptor monoclonal antibody epitope provides insight into its inverse agonist and antagonist properties. Endocrinology 2008; 149(7):3427-3434.
417. Nakabayashi K, Matsumi H, Bhalla A et al. Thyrostimulin, a heterodimer of two new human glycoprotein hormone subunits, activates the thyroid-stimulating hormone receptor. J Clin Invest 2002; 109(11):1445-1452.
418. Glinoer D. The regulation of thyroid function in pregnancy: Pathways of endocrine adaptation from physiology to pathology. Endocrine Reviews 1997; 18(3):404-433.
419. Rodien P, Bremont C, Samson ML et al. Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. New England Journal of Medicine 1998; 339:1823-1826.
420. Costagliola S, Bonomi M, Morgenthaler NG et al. Delineation of the discontinuous-conformational epitope of a monoclonal antibody displaying full in vitro and in vivo thyrotropin activity. Mol Endocrinol 2004; 18(12):3020-3034.
421. Furmaniak J, Sanders J, Smith BR. Thyrotropin receptor structure--in the crystal new horizons shine. Endocr Pract 2009; 15(1):56-60.
422. Sanders J, Chirgadze DY, Sanders P et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid 2007; 17(5):395-410.
423. Costagliola S, Franssen JDF, Bonomi M et al. Generation of a mouse monoclonal TSH receptor antibody with stimulating activity. Biochemical and Biophysical Research Communications 2002; 299(5):891-896.
424. Van Sande J, Costa MJ, Massart C et al. Kinetics of thyrotropin-stimulating hormone (TSH) and thyroid-stimulating antibody binding and action on the TSH receptor in intact TSH receptor-expressing CHO cells. Journal of Clinical Endocrinology and Metabolism 2003; 88(11):5366-5374.
425. Neumann S, Huang W, Titus S et al. Small-molecule agonists for the thyrotropin receptor stimulate thyroid function in human thyrocytes and mice. Proc Natl Acad Sci U S A 2009; 106(30):12471-12476.
426. Neumann S, Huang W, Titus S et al. Small-molecule agonists for the thyrotropin receptor stimulate thyroid function in human thyrocytes and mice. Proc Natl Acad Sci U S A 2009; 106(30):12471-12476.
427. Lefkowitz RJ, Cotecchia S, Kjelsberg MA et al. Adrenergic receptors: recent insights into their mechanism of activation and desensitization. Adv Second Messenger Phosphoprotein Res 1993; 28:1-9.
428. Delbeke D, Van Sande J, Swillens S, Erneux C, Dumont JE. Cooling enhances adenosine 3':5' monophosphate accumulation in thyrotropin stimulated dog thyroid slices. Metabolism 1982; 31(8):797-804.
429. Singh SP, McDonald D, Hope TJ, Prabhakar BS. Upon thyrotropin binding the thyrotropin receptor is internalized and localized to endosome. Endocrinology 2004; 145(2):1003-1010.
430. Maenhaut C, Brabant G, Vassart G, Dumont JE. In vitro and in vivo regulation of thyrotropin receptor mRNA levels in dog and human thyroid cells. J Biol Chem 1992; 267:3000-3007.
431. Flynn JC, Gilbert JA, Meroueh C et al. Chronic exposure in vivo to thyrotropin receptor stimulating monoclonal antibodies sustains high thyroxine levels and thyroid hyperplasia in thyroid autoimmunity-prone HLA-DRB1*0301 transgenic mice. Immunology 2007; 122(2):261-267.
432. Calebiro D, Nikolaev VO, Gagliani MC et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 2009; 7(8):e1000172.
433. Nilsson M, Björkman U, Ekholm R, Ericson LE. Polarized efflux of iodide in porcine thyrocytes occurs via a cAMP-regulated iodide channel in the apical plasma membrane. Acta Endocrinologica 1992; 126:67-74.
434. Rodriguez AM, Perron B, Lacroix L et al. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. Journal of Clinical Endocrinology and Metabolism 2002; 87(7):3500-3503.
435. Saito T, Endo T, Kawaguchi A et al. Increased expression of the Na + /I - symporter in cultured human thyroid cells exposed to thyrotropin and in Graves' thyroid tissue. Journal of Clinical Endocrinology and Metabolism 1997; 82:3331-3336.
436. Arntzenius AB, Smit LJ, Schipper J. Inverse relation between iodine intake and thyroid blood flow: color doppler flow imaging in euthyroid humans. Journal of Clinical Endocrinology and Metabolism 1991; 73:1051-1055.
437. Nunez J, Pommier J. Formation of thyroid hormones. Vitam Horm 1982; 39:175-229.
438. Dupuy C, Ohayon R, Valent A, Noel-Hudson MS, Deme D, Virion A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J Biol Chem 1999; 274(52):37265-37269.
439. De Deken X, Wang D, Dumont JE, Miot F. Characterization of ThOX proteins as components of the thyroid H(2)O(2)-generating system. Exp Cell Res 2002; 273(2):187-196.
440. Corvilain B, Van Sande J, Laurent E, Dumont JE. The H2O2-generating system modulates protein iodination and the activity of the pentose phosphate pathway in dog thyroid. Endocrinology 1991; 128:779-785.
441. Björkman U, Ekholm R. Hydrogen peroxide generation and its regulation in FRTL-5 and porcine thyroid cells. Endocrinology 1992; 130:393-399.
442. Song Y, Massart C, Chico-Galdo V et al. Species specific thyroid signal transduction: conserved physiology, divergent mechanisms. Mol Cell Endocrinol 2010; 319(1-2):56-62.
443. Björkman U, Ekholm R. Accelerated exocytosis and H2O2 generation in isolated thyroid follicles enhance protein iodination. Endocrinology 1988; 122:488-494.
444. Song Y, Driessens N, Costa M et al. Roles of hydrogen peroxide in thyroid physiology and disease. J Clin Endocrinol Metab 2007; 92(10):3764-3773.
445. Dumont JE, Boeynaems JM, Decoster C et al. Biochemical mechanisms in the control of thyroid function and growth. Adv Cyclic Nucl Res 1978; 9:723-734.
446. Bernier-Valentin F, Kostrouch Z, Rabilloud R, Rousset B. Analysis of the thyroglobulin internationalization process using in vitro reconstituted thyroid follicles: evidence for a coated vesicle-dependent endocytic pathway. Endocrinology 1991; 129:2194-2201.
447. Deshpande V, Venkatesh SG. Thyroglobulin, the prothyroid hormone: chemistry, synthesis and degradation. Biochimica et Biophysica Acta 1999; 1430:157-178.
448. Chambard M, Depetris D, Gruffat D, Gonzalez S, Mauchamp J, Chabaud O. Thyrotropin regulation of apical and basal exocytosis of thyroglobulin by porcine thyroid monolayers. J Mol Endocrinol 1990; 4:193-199.
449. Herzog V. Pathways of endocytosis in thyroid follicle cells. Internat Rev Cytol 1984; 91:107-139.
450. Ketelbant-Balasse P, Van Sande J, Neve P, Dumont JE. Time sequence of 3',5'-cyclic AMP accumulation and ultrastructural changes in dog thyroid slices after acute stimulation by TSH. Horm Metab Res 1976; 8(3):212-215.
451. Deery WJ, Heath JP. Phagocytosis induced by thyrotropin in cultured thyroid cells is associated with myosin light chain dephosphorylation and stress fiber disruption. J Cell Biol 1993; 122(1):21-37.
452. Saito T, Lamy F, Roger PP, Lecocq R, Dumont JE. Characterization and identification as cofilin and destrin of two thyrotropin- and phorbol ester-regulated phosphoproteins in thyroid cells. Exp Cell Res 1994; 212(1):49-61.
453. Deery WJ, Heath JP. Phagocytosis induced by thyrotropin in cultured thyroid cells is associated with myosin light chain dephosphorylation and stress fiber disruption. J Cell Biol 1993; 122(1):21-37.
454. Van Den Hove MF, Croizet-Berger K, Tyteca D, Selvais C, de Diesbach P, Courtoy PJ. Thyrotropin activates guanosine 5'-diphosphate/guanosine 5'-triphosphate exchange on the rate-limiting endocytic catalyst, Rab5a, in human thyrocytes in vivo and in vitro. J Clin Endocrinol Metab 2007; 92(7):2803-2810.
455. Croizet-Berger K, Daumerie C, Couvreur M, Courtoy PJ, Van Den Hove MF. The endocytic catalysts, Rab5a and Rab7, are tandem regulators of thyroid hormone production. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(12):8277-8282.
456. Rocmans PA, Ketelbant-Balasse P, Dumont JE, Neve P. Hormonal secretion by hyperactive thyroid cells is not secondary to apical phagocytosis. Endocrinology 1978; 103:1834-1848.
457. Van Den Hove MF, Couvreur M, De Visscher M. A new mechanism for the reabsorption of thyroid iodoproteins: selective fluid pinocytosis. European Journal of Biochemistry 1982; 122:415-422.
458. Lemansky P, Herzog V. Endocytosis of thyroglobulin is not mediated by mannose-6-phosphate receptors in thyrocytes. Evidence for low-affinity-binding sites operating in the uptake of thyroglobulin. European Journal of Biochemistry 1992; 209:111-119.
459. Marino M, McCluskey RT. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. American Journal of Physiology-Cell Physiology 2000; 279(5):C1295-C1306.
460. Marino M, Zheng G, McCluskey RT. Megalin (gp330) is an endocytic receptor for thyroglobulin on cultured fisher rat thyroid cells. Journal of Biological Chemistry 1999; 274:12898-12904.
461. Lisi S, Pinchera A, McCluskey RT et al. Preferential megalin-mediated transcytosis of low-hormonogenic thyroglobulin: a control mechanism for thyroid hormone release. Proc Natl Acad Sci U S A 2003; 100(25):14858-14863.
462. Delbeke D, Van Sande J, Swillens S, Erneux C, Dumont JE. Cooling enhances adenosine 3':5' monophosphate accumulation in thyrotropin stimulated dog thyroid slices. Metabolism 1982; 31(8):797-804.
463. Laurberg P. Mechanisms governing the relative proportions of thyroxine and 3,5,3'-triiodothyronine in thyroid secretion. Metabolism 1984; 33(4):379-392.
464. Unger J, Boeynaems JM, Van Herle A, Van Sande J, Rocmans P, Mockel J. In vitro nonbutanol-extractable iodine release in dog thyroid. Endocrinology 1979; 105(1):225-231.
465. Van Herle AJ, Vassart G, Dumont JE. Control of thyroglobulin synthesis and secretion. (First of two parts). New England Journal of Medicine 1979; 301(5):239-249.
466. Neve P, Dumont JE. Time sequence of ultrastructural changes in the stimulated dog thyroid. Z Zellforsch Mikrosk Anat 1970; 103(1):61-74.
467. Gerard AC, Xhenseval V, Colin IM, Many MC, Denef JF. Evidence for co-ordinated changes between vascular endothelial growth factor and nitric oxide synthase III immunoreactivity, the functional status of the thyroid follicles, and the microvascular bed during chronic stimulation by low iodine and propylthiouracyl in old mice. Eur J Endocrinol 2000; 142(6):651-660.
468. Gerard CM, Many MC, Daumerie C et al. Structural changes in the angiofollicular units between active and hypofunctioning follicles align with differences in the epithelial expression of newly discovered proteins involved in iodine transport and organification. Journal of Clinical Endocrinology and Metabolism 2002; 87(3):1291-1299.
469. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
470. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Reviews 2001; 22(5):631-656.
471. Ohta K, Endo T, Onaya T. The mRNA levels of thyrotropin receptor, thyroglobulin and thyroid peroxidase in neoplastic human thyroid tissues. Biochem Biophys Res Commun 1991; 174:1148-1153.
472. Damante G, Tell G, Di Lauro R. A unique combination of transcription factors controls differentiation of thyroid cells. Progress in Nucleic Acid Research Molecular Biology 2001; 66:307-56.:307-356.
473. Damante G, DiLauro R. Thyroid-Specific Gene-Expression. Biochimica et Biophysica Acta-Gene Structure and Expression 1994; 1218(3):255-266.
474. Roger PP, Christophe D, Dumont JE, Pirson I. The dog thyroid primary culture system: a model of the regulation of function, growth and differentiation expression by cAMP and other well-defined signaling cascades. European Journal of Endocrinology 1997; 137:579-598.
475. Medina DL, Suzuki K, Pietrarelli M, Okajima F, Kohn LD, Santisteban P. Role of insulin and serum on thyrotropin regulation of thyroid transcription factor-1 and Pax-8 genes expression in FRTL-5 thyroid cells. Thyroid 2000; 10(4):295-303.
476. Pouillon V, Pichon B, Donda A, Christophe D. TTF-2 does not appear to be a key mediator of the effect of cyclic AMP on thyroglobulin gene transcription in primary cultured dog thyrocytes. Biochemical and Biophysical Research Communications 1998; 242:327-331.
477. Donda A, Javaux F, Van Renterghem P, Gervy-Decoster C, Vassart G, Christophe D. Human, bovine, canine and rat thyroglobulin promoter sequences display species-specific differences in an in vitro study. Mol Cell Endocrinol 1993; 90:R23-R26.
478. Van Renterghem P, Vassart G, Christophe D. Pax 8 expression in primary cultured dog thyrocyte is increased by cyclic AMP. Biochimica et Biophysica Acta 1996; 1307:97-103.
479. Mascia A, Nitsch L, Di Lauro R, Zannini M. Hormonal control of the transcription factor Pax8 and its role in the regulation of thyroglobulin gene expression in thyroid cells. Journal of Endocrinology 2002; 172(1):163-176.
480. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
481. Van Renterghem P, Dremier S, Vassart G, Christophe D. Study of TTF-1 gene expression in dog thyrocytes in primary culture. Molecular and Cellular Endocrinology 1995; 112:83-93.
482. Zannini M, Acebron A, DeFelice M et al. Mapping and functional role of phosphorylation sites in the thyroid transcription factor-1 (TTF-1). Journal of Biological Chemistry 1996; 271(4):2249-2254.
483. Reuse S, Maenhaut C, Dumont JE. Regulation of protooncogenes c-fos and c-myc expressions by protein tyrosine kinase, protein kinase C, and cyclic AMP mitogenic pathways in dog primary thyrocytes: a positive and negative control by cyclic AMP on c-myc expression. Experimental Cell Research 1990; 189:33-40.
484. Deleu S, Pirson I, Clermont F, Nakamura T, Dumont JE, Maenhaut C. Immediate early gene expression in dog thyrocytes in response to growth, proliferation, and differentiation stimuli. J Cell Physiol 1999; 181:342-354.
485. Lalli E, Sassonecorsi P. Thyroid-Stimulating Hormone (Tsh)-Directed Induction of the Crem Gene in the Thyroid-Gland Participates in the Long-Term Desensitization of the Tsh Receptor. Proceedings of the National Academy of Sciences of the United States of America 1995; 92(21):9633-9637.
486. Pichon B, Jimenez-Cervantes C, Pirson I, Maenhaut C, Christophe D. Induction of nerve growth factor-induced gene-B (NGFI-B) as an early event in the cyclic adenosine monophosphate response of dog thyrocytes in primary culture. Endocrinology 1996; 137:4691-4698.
487. Pomerance M, Carapau D, Chantoux F et al. CCAAT/enhancer-binding protein-homologous protein expression and transcriptional activity are regulated by 3 ',5 '-cyclic adenosine monophosphate in thyroid cells. Mol Endocrinol 2003; 17(11):2283-2294.
488. Lalli E, Sassonecorsi P. Thyroid-Stimulating Hormone (Tsh)-Directed Induction of the Crem Gene in the Thyroid-Gland Participates in the Long-Term Desensitization of the Tsh Receptor. Proceedings of the National Academy of Sciences of the United States of America 1995; 92(21):9633-9637.
489. Pomerance M, Carapau D, Chantoux F et al. CCAAT/enhancer-binding protein-homologous protein expression and transcriptional activity are regulated by 3 ',5 '-cyclic adenosine monophosphate in thyroid cells. Mol Endocrinol 2003; 17(11):2283-2294.
490. Pichon B, Vassart G, Christophe D. A canonical nerve growth factor-induced gene-B response element appears not to be involved in the cyclic adenosine monophosphate-dependent expression of differentiation in thyrocytes. Molecular and Cellular Endocrinology 1999; 154:21-27.
491. Davies E, Dumont JE, Vassart G. Thyrotropin-stimulated recruitment of free monoribosomes on to membrane-bound thyroglobulinsythesizing polyribosomes. Biochemical Journal 1978; 172:227-231.
492. Fortemaison N, Blancquaert S, Dumont JE et al. Differential involvement of the actin cytoskeleton in differentiation and mitogenesis of thyroid cells: inactivation of Rho proteins contributes to cyclic adenosine monophosphate-dependent gene expression but prevents mitogenesis. Endocrinology 2005; 146(12):5485-5495.
493. Colletta G, Cirafici AM, Dicarlo A. Dual Effect of Transforming Growth Factor-Beta on Rat-Thyroid Cells - Inhibition of Thyrotropin-Induced Proliferation and Reduction of Thyroid-Specific Differentiation Markers. Cancer Research 1989; 49(13):3457-3462.
494. Nicolussi A, D'Inzeo S, Santulli M, Colletta G, Coppa A. TGF-beta control of rat thyroid follicular cells differentiation. Molecular and Cellular Endocrinology 2003; 207(1-2):1-11.
495. Nicolussi A, D'Inzeo S, Santulli M, Colletta G, Coppa A. TGF-beta control of rat thyroid follicular cells differentiation. Molecular and Cellular Endocrinology 2003; 207(1-2):1-11.
496. Costamagna E, Garcia B, Santisteban P. The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. Journal of Biological Chemistry 2004; 279(5):3439-3446.
497. Gerard CM, Roger PP, Dumont JE. Thyroglobulin gene expression as a differentiation marker in primary cultures of calf thyroid cells. Molecular and Cellular Endocrinology 1989; 61(1):23-35.
498. Roger PP, Van Heuverswyn B, Lambert C, Reuse S, Vassart G, Dumont JE. Antagonistic effects of thyrotropin and epidermal growth factor on thyroglobulin mRNA level in cultured thyroid cells. Eur J Biochem 1985; 152:239-245.
499. Pohl V, Abramowicz M, Vassart G, Dumont JE, Roger PP. Thyroperoxidase mRNA in quiescent and proliferating thyroid epithelial cells: expression and subcellular localization studied by in situ hydridization. European Journal of Cell Biology 1993; 62:94-104.
500. Gerard CM, Roger PP, Dumont JE. Thyroglobulin gene expression as a differentiation marker in primary cultures of calf thyroid cells. Molecular and Cellular Endocrinology 1989; 61(1):23-35.
501. Roger PP, Dumont JE. Factors controlling proliferation and differentiation of canine thyroid cells cultured in reduced serum conditions: effects of thyrotropin, cyclic AMP and growth factors. Molecular and Cellular Endocrinology 1984; 36(1-2):79-93.
502. Coclet J, Lamy F, Rickaert F, Dumont JE, Roger PP. Intermediate filaments in normal thyrocytes: modulation of vimentin expression in primary cultures. Mol Cell Endocrinol 1991; 76(1-3):135-148.
503. Blackwood L, Onions DE, Argyle DJ. Characterization of the feline thyroglobulin promoter. Domestic Animal Endocrinology 2001; 20(3):185-201.
504. Christophe-Hobertus C, Christophe D. Two binding sites for thyroid transcription factor 1 (TTF-1) determine the activity of the bovine thyroglobulin gene upstream enhancer element. Molecular and Cellular Endocrinology 1999; 149(1-2):79-84.
505. Berg V, Vassart G, Christophe D. A zinc-dependent DNA-binding activity co-operates with cAMP-responsive-element-binding protein to activate the human thyroglobulin enhancer. Biochemical Journal 1997; 323:349-357.
506. Mascia A, DeFelice M, Lipardi C et al. Transfection of TTF-1 gene induces thyroglobulin gene expression in undifferentiated FRT cells. Biochimica et Biophysica Acta-Gene Structure and Expression 1997; 1354(2):171-181.
507. di Magliano MP, Di Lauro R, Zannini M. Pax8 has a key role in thyroid cell differentiation. Proceedings of the National Academy of Sciences of the United States of America 2000; 97(24):13144-13149.
508. Mascia A, DeFelice M, Lipardi C et al. Transfection of TTF-1 gene induces thyroglobulin gene expression in undifferentiated FRT cells. Biochimica et Biophysica Acta-Gene Structure and Expression 1997; 1354(2):171-181.
509. Di Palma T, Nitsch R, Mascia A, Nitsch L, Di Lauro R, Zannini M. The paired domain-containing factor Pax8 and the homeodomain-containing factor TTF-1 directly interact and synergistically activate transcription. Journal of Biological Chemistry 2003; 278(5):3395-3402.
510. Miccadei S, De Leo R, Zammarchi E, Natali PG, Civitareale D. The synergistic activity of thyroid transcription factor 1 and Pax 8 relies on the promoter/enhancer interplay. Mol Endocrinol 2002; 16(4):837-846.
511. Ledent C, Parmentier M, Vassart G. Tissue-specific expression and methylation of a thyroglobulin- chloramphenicol acetyltransferase fusion gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 1990; 87:6176-6180.
512. Libert F, Vassart G, Christophe D. Methylation and expression of the human thyroglobulin gene. Biochimica et Biophysica Acta 1986; 134:1109-1113.
513. Pichon B, Christophe-Hobertus C, Vassart G, Christophe D. Unmethylated thyroglobulin promoter may be repressed by methylation of flanking DNA sequences. Biochemical Journal 1994; 298:537-541.
514. Van Heuverswyn B, Streydio C, Brocas H, Refetoff S, Dumont JE, Vassart G. Thyrotropin controls transcription of the thyroglobulin gene. Proc Natl Acad Sci USA 1984; 81:5941-5945.
515. Gerard CM, Lefort A, Christophe D et al. Control of thyroperoxidase and thyroglobulin transcription by cAMP: evidence for distinct regulatory mechanisms. Mol Endocrinol 1989; 3:2110-2118.
516. Avvedimento VE, Tramontano D, Ursini MV. The level of thyroglobulin mRNA is regulated by TSH both in vitro and in vivo. Biochemical and Biophysical Research Communications 1984; 122:472-477.
517. Hansen C, Gerard C, Vassart G, Stordeur P, Christophe D. Thyroid-specific and cAMP-dependent hypersensitive regions in thyroglobulin gene chromatin. European Journal of Biochemistry 1988; 178:387-393.
518. Christophe D, Gérard C, Juvenal G et al. Identification of a cAMP-responsive region in thyroglobulin gene promoter. Mol Cell Endocrinol 1989; 64:5-18.
519. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15776-15781.
520. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
521. Pohl V, Roger PP, Christophe D, Pattyn G, Vassart G, Dumont JE. Differentiation expression during proliferative activity induced through different pathways: in situ hybridization study of thyroglobulin gene expression in thyroid epithelial cells. J Cell Biol 1990; 111:663-672.
522. Ortiz L, Zannini M, Di Lauro R, Santisteban P. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin, and insulin-like growth factor I. Journal of Biological Chemistry 1997; 272(37):23334-23339.
523. Fayet G, Hovsepian S. Isolation of a normal human thyroid cell line: Hormonal requirement for thyroglobulin regulation. Thyroid 2002; 12(7):539-546.
524. Mascia A, Nitsch L, Di Lauro R, Zannini M. Hormonal control of the transcription factor Pax8 and its role in the regulation of thyroglobulin gene expression in thyroid cells. Journal of Endocrinology 2002; 172(1):163-176.
525. Graves PN, Davies TF. A second thyroglobulin messenger RNA species (rTg-2) in rat thyrocytes. Mol Endocrinol 1990; 4:155-161.
526. Mercken L, Simons MJ, Vassart G. The 5'-end of bovine thyroglobulin mRNA encodes a hormonogenic peptides. FEBS Letters 1982; 149:285-287.
527. Abramowicz MJ, Vassart G, Christophe D. Thyroid peroxidase gene promoter confers TSH responsiveness to heterologous reporter genes in transfection experiments. Biochem Biophys Res Commun 1990; 166(3):1257-1264.
528. Francis-Lang H, Price M, Polycarpou-Schwarz M, Di Lauro R. Cell-type-specific expression of the rat thyroperoxidase promoter indicates common mechanism for thyroid-specific gene expression. Molecular and Cellular Biology 1992; 12:576-588.
529. Mizuno K, Gonzalez FJ, Kimura S. Thyroid-specific enhancer-binding protein (T/EBP): cDNA cloning, functional characterization, and structural identity with thyroid transcription factor TTF-1. Molecular and Cellular Biology 1991; 11:4927-4933.
530. Esposito C, Miccadei S, Saiardi A, Civitareale D. PAX 8 activates the enhancer of the human thyroperoxidase gene. Biochemical Journal 1998; 331:37-40.
531. Miccadei S, De Leo R, Zammarchi E, Natali PG, Civitareale D. The synergistic activity of thyroid transcription factor 1 and Pax 8 relies on the promoter/enhancer interplay. Mol Endocrinol 2002; 16(4):837-846.
532. Gérard C, Lefort A, Libert F, Christophe D, Dumont JE, Vassart G. Transcriptional regulation of the thyroperoxydase gene by thyrotropin and forskolin. Molecular and Cellular Endocrinology 1988; 60:239-242.
533. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
534. Ledent C, Dumont JE, Vassart G, Parmentier M. Thyroid expression of an A2 adenosine receptor transgene induces thyroid hyperplasia and hyperthyroidism. EMBO J 1991; 11:537-542.
535. Abramowicz MJ, Vassart G, Christophe D. Thyroid peroxidase gene promoter confers TSH responsiveness to heterologous reporter genes in transfection experiments. Biochem Biophys Res Commun 1990; 166(3):1257-1264.
536. Niccoli P, Fayadat L, Panneels V, Lanet J, Franc JL. Human thyroperoxidase in its alternatively spliced form (TPO2) is enzymatically inactive and exhibits changes in intracellular processing and trafficking. Journal of Biological Chemistry 1997; 272(47):29487-29492.
537. Tong Q, Ryu KY, Jhiang SM. Promoter characterization of the rat Na+/I- symporter gene. Biochemical and Biophysical Research Communications 1997; 239(1):34-41.
538. Behr M, Schmitt TL, Espinoza CR, Loos U. Cloning of a functional promoter of the human sodium/iodide-symporter gene. Biochemical Journal 1998; 331:359-363.
539. Endo T, Kaneshige M, Nakazato M, Ohmori M, Harii N, Onaya T. Thyroid transcription factor-1 activates the promoter activity of rat thyroid Na+/I- symporter gene. Mol Endocrinol 1997; 11(11):1747-1755.
540. Rodriguez AM, Perron B, Lacroix L et al. Identification and characterization of a putative human iodide transporter located at the apical membrane of thyrocytes. Journal of Clinical Endocrinology and Metabolism 2002; 87(7):3500-3503.
541. Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA. A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3 ',5 '-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol 2002; 16(10):2266-2282.
542. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15776-15781.
543. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
544. Saiardi A, Falasca P, Civitareale D. Synergistic Transcriptional Activation of the Thyrotropin Receptor Promoter by Cyclic Amp-Responsive-Element-Binding Protein and Thyroid Transcription Factor-1. Biochemical Journal 1995; 310:491-496.
545. Yokomori N, Tawata M, Saito T, Shimura H, Onaya T. Regulation of the rat thyrotropin receptor gene by the methylation-sensitive transcription factor GA binding protein. Mol Endocrinol 1998; 12(8):1241-1249.
546. Saiardi A, Falasca P, Civitareale D. Synergistic Transcriptional Activation of the Thyrotropin Receptor Promoter by Cyclic Amp-Responsive-Element-Binding Protein and Thyroid Transcription Factor-1. Biochemical Journal 1995; 310:491-496.
547. Civitareale D, Castelli MP, Falasca P, Saiardi A. Thyroid Transcription Factor-1 Activates the Promoter of the Thyrotropin Receptor Gene. Mol Endocrinol 1993; 7(12):1589-1595.
548. Moeller LC, Kimura S, Kusakabe T, Liao XH, Van Sande J, Refetoff S. Hypothyroidism in thyroid transcription factor 1 haploinsufficiency is caused by reduced expression of the thyroid-stimulating hormone receptor. Mol Endocrinol 2003; 17(11):2295-2302.
549. Yokomori N, Tawata M, Saito T, Shimura H, Onaya T. Regulation of the rat thyrotropin receptor gene by the methylation-sensitive transcription factor GA binding protein. Mol Endocrinol 1998; 12(8):1241-1249.
550. Saji M, Akamizu T, Sanchez M. Regulation of thyrotropin receptor gene expression in rat FRTL-5 thyroid cells. Endocrinology 1992; 130:520-533.
551. Saiardi A, Falasca P, Civitareale D. Synergistic Transcriptional Activation of the Thyrotropin Receptor Promoter by Cyclic Amp-Responsive-Element-Binding Protein and Thyroid Transcription Factor-1. Biochemical Journal 1995; 310:491-496.
552. Lalli E, Sassonecorsi P. Thyroid-Stimulating Hormone (Tsh)-Directed Induction of the Crem Gene in the Thyroid-Gland Participates in the Long-Term Desensitization of the Tsh Receptor. Proceedings of the National Academy of Sciences of the United States of America 1995; 92(21):9633-9637.
553. Moeller LC, Kimura S, Kusakabe T, Liao XH, Van Sande J, Refetoff S. Hypothyroidism in thyroid transcription factor 1 haploinsufficiency is caused by reduced expression of the thyroid-stimulating hormone receptor. Mol Endocrinol 2003; 17(11):2295-2302.
554. Postiglione MP, Parlato R, Rodriguez-Mallon A et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15462-15467.
555. Marians RC, Ng L, Blair HC, Unger P, Graves PN, Davies TF. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(24):15776-15781.
556. Leoni SG, Galante PA, Ricarte-Filho JC, Kimura ET. Differential gene expression analysis of iodide-treated rat thyroid follicular cell line PCCl3. Genomics 2008; 91(4):356-366.
557. Berlingieri MT, Akamizu T, Fusco A. Thyrotropin receptor gene expression in oncogene-transfected rat thyroid cells: correlation between transformation, loss of thyrotropin-dependent growth, and loss of thyrotropin receptor gene expression. Biochemical and Biophysical Research Communications 1990; 173:172-178.
558. Akamizu T, Ikuyama S, Saji M et al. Cloning, chromosomal assignment, and regulation of the rat thyrotropin receptor: expression of the gene is regulated by thyrotropin, agents that increase cAMP levels, and thyroid autoantibodies. Proceedings of the National Academy of Sciences of the United States of America 1990; 87:5677-5681.
559. Kung AW, Collison K, Banga JP, McGregor AM. Effect of Graves' IgG on gene transcription in human thyroid cell cultures. Thyroglobulin gene activation. FEBS Letters 1988; 232:12-16.
560. Huber GK, Weinstein SP, Graves PN, Davies TF. The positive regulation of human thyrotropin (TSH) receptor messenger ribonucleic acid by recombinant human TSH is at the intranuclear level. Endocrinology 1992; 130:2858-2864.
561. Brabant G, Maenhaut C, Kohrle J et al. Human thyrotropin receptor gene: expression in thyroid tumors and correlation to markers of thyroid differentiation and dedifferentiation. Molecular and Cellular Endocrinology 1991; 82:R7-12.
562. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
563. Ledent C, Dumont JE, Vassart G, Parmentier M. Thyroid adenocarcinomas secondary to tissue-specific expression of Simian virus-40 large T-antigen in transgenic mice. Endocrinology 1991; 129:1391-1401.
564. Mazzaferri EL. Papillary and follicular thyroid cancer: a selective approach to diagnosis and treatment. Annual Review of Medicine 1981; 32:73-91.
565. De Deken X, Wang D, Many MC et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. Journal of Biological Chemistry 2000; 275(30):23227-23233.
566. Dupuy C, Pomerance M, Ohayon R et al. Thyroid oxidase (THOX2) gene expression in the rat thyroid cell line FRTL-5. Biochemical and Biophysical Research Communications 2000; 277(2):287-292.
567. De Deken X, Wang D, Many MC et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. Journal of Biological Chemistry 2000; 275(30):23227-23233.
568. De Deken X, Wang D, Many MC et al. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. Journal of Biological Chemistry 2000; 275(30):23227-23233.
569. Dupuy C, Pomerance M, Ohayon R et al. Thyroid oxidase (THOX2) gene expression in the rat thyroid cell line FRTL-5. Biochemical and Biophysical Research Communications 2000; 277(2):287-292.
570. Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 2006; 281(27):18269-18272.
571. Grasberger H, Refetoff S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J Biol Chem 2006; 281(27):18269-18272.
572. Christov K. Cell population kinetics and DNA content during thyroid carcinogenesis. Cell Tissue Kinetics 1985; 18:119-131.
573. Coclet J, Foureau F, Ketelbant P, Galand P, Dumont JE. Cell population kinetics in dog and human adult thyroid. Clinical Endocrinology 1989; 31:655-665.
574. Dumont JE, Maenhaut C, Pirson I, Baptist M, Roger PP. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab 1991; 5(4):727-754.
575. Smeds S, Wollman SH. 3H-thymidine labeling of endothelial cells in thyroid arteries, veins, and lymphatics during thyroid stimulation. Laboratory Investigation 1983; 48:285-291.
576. Many MC, Denef JF, Haumont S. Precocity of the endothelial proliferation during a course of rapid goitrogenesis. Acta Endocrinologica 1984; 105:487-491.
577. Dumont JE, Maenhaut C, Pirson I, Baptist M, Roger PP. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab 1991; 5(4):727-754.
578. Patel VA, Logan A, Watkinson JC et al. Isolation and characterization of human thyroid endothelial cells. American Journal of Physiology-Endocrinology and Metabolism 2003; 284(1):E168-E176.
579. Sato K, Yamazaki K, Shizume K et al. Stimulation by thyroid-stimulating hormone and Grave's immunoglobulin G of vascular endothelial growth factor mRNA expression in human thyroid follicles in vitro and flt mRNA expression in the rat thyroid in vivo. Journal of Clinical Investigation 1995; 96:1295-1302.
580. Gerard CM, Many MC, Daumerie C et al. Structural changes in the angiofollicular units between active and hypofunctioning follicles align with differences in the epithelial expression of newly discovered proteins involved in iodine transport and organification. Journal of Clinical Endocrinology and Metabolism 2002; 87(3):1291-1299.
581. Gerard AC, Xhenseval V, Colin IM, Many MC, Denef JF. Evidence for co-ordinated changes between vascular endothelial growth factor and nitric oxide synthase III immunoreactivity, the functional status of the thyroid follicles, and the microvascular bed during chronic stimulation by low iodine and propylthiouracyl in old mice. Eur J Endocrinol 2000; 142(6):651-660.
582. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
583. Takasu N, Komiya I, Nagasawa Y et al. Stimulation of porcine thyroid cell alkalinization and growth by EGF, phorbol ester, and diacylglycerol. Am J Physiol 1990; 258:E445-E450.
584. Roger PP, Dumont JE. Factors controlling proliferation and differentiation of canine thyroid cells cultured in reduced serum conditions: effects of thyrotropin, cyclic AMP and growth factors. Molecular and Cellular Endocrinology 1984; 36(1-2):79-93.
585. Van Keymeulen A, Dumont JE, Roger PP. TSH induces insulin receptors that mediate insulin costimulation of growth in normal human thyroid cells. Biochemical and Biophysical Research Communications 2000; 279(1):202-207.
586. Clement S, Refetoff S, Robaye B, Dumont JE, Schurmans S. Low TSH requirement and goiter in transgenic mice overexpressing IGF-I and IGF-I receptor in the thyroid gland. Endocrinology 2001; 142(12):5131-5139.
587. Tramontano D, Cushing GW, Moses AC, Ingbar SH. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergyzes the stimulation of DNA synthesis induced by TSH and Graves' IgG. Endocrinology 1986; 119:940-942.
588. Saji M, Tsushima T, Isozaki O et al. Interaction of insulin-like growth factor I with porcine thyroid cells cultured in monolayer. Endocrinology 1987; 121:749-756.
589. Roger PP, Servais P, Dumont JE. Stimulation by thyrotropin and cyclic AMP of the proliferation of quiescent canine thyroid cells cultured in a defined medium containing insulin. FEBS Letters 1983; 157(2):323-329.
590. Michiels FM, Caillou B, Talbot M et al. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 1994; 91(22):10488-10492.
591. Zeiger MA, Saji M, Gusev Y et al. Thyroid-specific expression of cholera toxin A1 subunit causes thyroid hyperplasia and hyperthyroidism in transgenic mice. Endocrinology 1997; 138(8):3133-3140.
592. Meinkoth JL, Goldsmith PK, Spiegel AM et al. Inhibition of thyrotropin-induced DNA synthesis in thyroid ollicular cells by microinjection of an antibody to the stimulatory G protein of adenylate cyclase, Gs. Journal of Biological Chemistry 1992; 267:13239-13245.
593. Dremier S, Pohl V, Poteet-Smith C et al. Activation of cyclic AMP-dependent kinase is required but may not be sufficient to mimic cyclic AMP-dependent DNA synthesis and thyroglobulin expression in dog thyroid cells. Mol Cell Biol 1997; 17:6717-6726.
594. Kupperman E, Wen W, Meinkoth JL. Inhibition of thyrotropin-stimulated DNA synthesis by microinjection of inhibitors of cellular Ras and cyclic AMP-dependent protein kinase. Molecular and Cellular Biology 1993; 13(8):4477-4484.
595. Saavedra AP, Tsygankova OM, Prendergast GV, Dworet JH, Cheng G, Meinkoth JL. Role of cAMP, PKA and Rap1A in thyroid follicular cell survival. Oncogene 2002; 21(5):778-788.
596. Ribeiro-Neto F, Urbani J, Lemee N, Lou LG, Altschuler DL. On the mitogenic properties of Rap1b: cAMP-induced G(1)/S entry requires activated and phosphorylated Rap1b. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(8):5418-5423.
597. Dumont JE, Maenhaut C, Pirson I, Baptist M, Roger PP. Growth factors controlling the thyroid gland. Baillieres Clin Endocrinol Metab 1991; 5(4):727-754.
598. Van Keymeulen A, Deleu S, Bartek J, Dumont JE, Roger PP. Respective roles of carbamylcholine and cyclic adenosine monophosphate in their synergistic regulation of cell cycle in thyroid primary cultures. Endocrinology 2001; 142(3):1251-1259.
599. Raspe E, Reuse S, Roger PP, Dumont JE. Lack of Correlation Between the Activation of the Ca2+-Phosphatidylinositol Cascade and the Regulation of Dna-Synthesis in the Dog Throcyte. Experimental Cell Research 1992; 198(1):17-26.
600. Ollis CA, Hill DJ, Munro DS. A role for insulin-like growth factor-I in the regulation of human thyroid cell growth by thyrotropin. J Clin Endocrinol 1989; 123:495-500.
601. Maciel RMB, Mores AC, Villone G, Tramontano D, Ingbar SH. Demonstration of the production and physiological role of insulin-like growth factor II in rat thyroid follicular cells in culture. J Clin Invest 1988; 82:1546-1553.
602. Van Keymeulen A, Dumont JE, Roger PP. TSH induces insulin receptors that mediate insulin costimulation of growth in normal human thyroid cells. Biochemical and Biophysical Research Communications 2000; 279(1):202-207.
603. Ortiz L, Zannini M, Di Lauro R, Santisteban P. Transcriptional control of the forkhead thyroid transcription factor TTF-2 by thyrotropin, insulin, and insulin-like growth factor I. Journal of Biological Chemistry 1997; 272(37):23334-23339.
604. De Vita G, Berlingieri MT, Visconti R et al. Akt/protein kinase B promotes survival and hormone-independent proliferation of thyroid cells in the absence of dedifferentiating and transforming effects. Cancer Research 2000; 60(14):3916-3920.
605. Roger PP, Dumont JE. Factors controlling proliferation and differentiation of canine thyroid cells cultured in reduced serum conditions: effects of thyrotropin, cyclic AMP and growth factors. Molecular and Cellular Endocrinology 1984; 36(1-2):79-93.
606. Dremier S, Taton M, Coulonval K, Nakamura T, Matsumoto K, Dumont JE. Mitogenic, dedifferentiating, and scattering effects of hepatocyte growth factor on dog thyroid cells. Endocrinology 1994; 135:135-140.
607. Errick JE, Ing KW, Eggo MC, Burrow GN. Growth and differentiation in cultured human thyroid cells: effects of epidermal growth factor and thyrotropin. In Vitro Cell Developmental Biology 1986; 22(1):28-36.
608. Gire V, Marshall CJ, Wynford-Thomas D. Activation of mitogen-activated protein kinase is necessary but not sufficient for proliferation of human thyroid epithelial cells induced by mutant Ras. Oncogene 1999; 18(34):4819-4832.
609. Melillo RM, Santoro M, Ong SH et al. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Molecular and Cellular Biology 2001; 21(13):4177-4187.
610. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Research 2003; 63(7):1454-1457.
611. Roger PP, Servais P, Dumont JE. Induction of DNA synthesis in dog thyrocytes in primary culture: synergistic effects of thyrotropin and cyclic AMP with epidermal growth factor and insulin. J Cell Physiol 1987; 130(1):58-67.
612. Roger PP, Servais P, Dumont JE. Regulation of dog thyroid epithelial cell cycle by forskolin, an adenylate cyclase activator. Experimental Cell Research 1987; 172:282-292.
613. Becks GP, Eggo MC, Burrow GN. Organic iodide inhibits deoxyribonucleic acid synthesis and growth in FRTL5 cells. Endocrinology 1988; 123:545-550.
614. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
615. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
616. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
617. Tsygankova OM, Saavedra A, Rebhun JF, Quilliam LA, Meinkoth JL. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Molecular and Cellular Biology 2001; 21(6):1921-1929.
618. Lou LG, Urbani J, Ribeiro-Neto F, Altschuler DL. cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. Journal of Biological Chemistry 2002; 277(36):32799-32806.
619. Tominaga T, Dela Cruz J, Burrow GN, Meinkoth JL. Divergent patterns of immediate early gene expression in response to thyroid-stimulating hormone and insulin-like growth factor I in Wistar rat thyrocytes. Endocrinology 1994; 135(3):1212-1219.
620. Reuse S, Pirson I, Dumont JE. Differential regulation of protooncogenes c-jun and jun D expressions by protein tyrosine kinase, protein kinase C, and cyclic-AMP mitogenic pathways in dog primary thyrocytes: TSH and cyclic-AMP induce proliferation but downregulate C-jun expression. Experimental Cell Research 1991; 196:210-215.
621. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
622. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
623. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
624. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
625. Contor L, Lamy F, Lecocq R, Roger PP, Dumont JE. Differential protein phosphorylation in induction of thyroid cell proliferation by thyrotropin, epidermal growth factor, or phorbol ester. Mol Cell Biol 1988; 8:2494-2503.
626. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
627. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
628. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
629. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
630. Deleu S, Pirson I, Coulonval K et al. IGF-1 or insulin, and the TSH cyclic AMP cascade separately control dog and human thyroid cell growth and DNA synthesis, and complement each other in inducing mitogenesis. Mol Cell Endocrinol 1999; 149:41-51.
631. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
632. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
633. Vandeput F, Perpete S, Coulonval K, Lamy F, Dumont JE. Role of the different mitogen-activated protein kinase subfamilies in the stimulation of dog and human thyroid epithelial cell proliferation by cyclic adenosine 5 '-monophosphate and growth factors. Endocrinology 2003; 144(4):1341-1349.
634. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
635. Cass LA, Meinkoth JL. Differential effects of cyclic adenosine 3',5'-monophosphate on p70 ribosomal S6 kinase. Endocrinology 1998; 139:1991-1998.
636. Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res 2007; 67(17):8002-8006.
637. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
638. Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res 2007; 67(17):8002-8006.
639. Uyttersprot N, Costagliola S, Dumont JE, Miot F. Requirement for cAMP-response element (CRE) binding protein/CRE modulator transcription factors in thyrotropin-induced proliferation of dog thyroid cells in primary culture. European Journal of Biochemistry 1999; 259(1-2):370-378.
640. Nguyen LQ, Kopp P, Martinson F, Stanfield K, Roth SI, Jameson JL. A dominant negative CREB (cAMP response element-binding protein) isoform inhibits thyrocyte growth, thyroid-specific gene expression, differentiation, and function. Mol Endocrinol 2000; 14(9):1448-1461.
641. Coulonval K, Vandeput F, Stein RC, Kozma SC, Lamy F, Dumont JE. Phosphatidylinositol 3-kinase, protein kinase B and ribosomal S6 kinases in the stimulation of thyroid epithelial cell proliferation by cAMP and growth factors in the presence of insulin. Biochemical Journal 2000; 348:351-358.
642. Lamy F, Roger PP, Lecocq R, Dumont JE. Differential protein synthesis in the induction of thyroid cell proliferation by thyrotropin, epidermal growth factor or serum. Eur J Biochem 1986; 155:265-272.
643. van Staveren WC, Solis DW, Delys L et al. Gene expression in human thyrocytes and autonomous adenomas reveals suppression of negative feedbacks in tumorigenesis. Proc Natl Acad Sci U S A 2006; 103(2):413-418.
644. Hebrant A, van Staveren WC, Delys L et al. Long-term EGF/serum-treated human thyrocytes mimic papillary thyroid carcinomas with regard to gene expression. Exp Cell Res 2007; 313(15):3276-3284.
645. Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol 1996; 8(6):805-814.
646. Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway and the restriction point. Curr Opin Cell Biol 1996; 8(6):805-814.
647. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Development 1999; 13(12):1501-1512.
648. Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G(1) checkpoint. Molecular and Cellular Biology 1996; 16(12):6917-6925.
649. Coulonval K, Maenhaut C, Dumont JE, Lamy F. Phosphorylation of the three Rb protein family members is a common step of the cAMP-, the growth factor, and the phorbol ester-mitogenic cascades but is not necessary for the hypertrophy induced by insulin. Experimental Cell Research 1997; 233(2):395-398.
650. Baptist M, Lamy F, Gannon J, Hunt T, Dumont JE, Roger PP. Expression and subcellular localization of CDK2 and cdc2 kinases and their common partner cyclin A in thyroid epithelial cells: comparison of cyclic AMP-dependent and -independent cell cycles. J Cell Physiol 1996; 166:256-273.
651. Coulonval K, Maenhaut C, Dumont JE, Lamy F. Phosphorylation of the three Rb protein family members is a common step of the cAMP-, the growth factor, and the phorbol ester-mitogenic cascades but is not necessary for the hypertrophy induced by insulin. Experimental Cell Research 1997; 233(2):395-398.
652. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
653. Depoortere F, Van Keymeulen A, Lukas J et al. A requirement for cyclin D3-cyclin-dependent kinase (cdk)-4 assembly in the cyclic adenosine monophosphate-dependent proliferation of thyrocytes. J Cell Biol 1998; 140:1427-1439.
654. Depoortere F, Dumont JE, Roger PP. Paradoxical accumulation of the cyclin-dependent kinase inhibitor p27kip1 during the cAMP-dependent mitogenic stimulation of thyroid epithelial cells. Journal of Cell Science 1996; 109(Pt 7):1759-1764.
655. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
656. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
657. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
658. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
659. Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1). Molecular Biology of the Cell 2000; 11(3):1061-1076.
660. Coulonval K, Bockstaele L, Paternot S, Dumont JE, Roger PP. The cyclin D3-CDK4-p27(kip1) holoenzyme in thyroid epithelial cells: activation by TSH, inhibition by TGFbeta, and phosphorylations of its subunits demonstrated by two-dimensional gel electrophoresis. Experimental Cell Research 2003; 291(1):135-149.
661. Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1). Molecular Biology of the Cell 2000; 11(3):1061-1076.
662. Paternot S, Coulonval K, Dumont JE, Roger PP. Cyclic AMP-dependent phosphorylation of cyclin D3-bound CDK4 determines the passage through the cell cycle restriction point in thyroid epithelial cells. Journal of Biological Chemistry 2003; 278(29):26533-26540.
663. Depoortere F, Pirson I, Bartek J, Dumont JE, Roger PP. Transforming growth factor beta(1) selectively inhibits the cyclic AMP-dependent proliferation of primary thyroid epithelial cells by preventing the association of cyclin D3-cdk4 with nuclear p27(kip1). Molecular Biology of the Cell 2000; 11(3):1061-1076.
664. Coulonval K, Bockstaele L, Paternot S, Dumont JE, Roger PP. The cyclin D3-CDK4-p27(kip1) holoenzyme in thyroid epithelial cells: activation by TSH, inhibition by TGFbeta, and phosphorylations of its subunits demonstrated by two-dimensional gel electrophoresis. Experimental Cell Research 2003; 291(1):135-149.
665. Bockstaele L, Kooken H, Libert F et al. Regulated activating Thr172 phosphorylation of cyclin-dependent kinase 4(CDK4): its relationship with cyclins and CDK "inhibitors". Mol Cell Biol 2006; 26(13):5070-5085.
666. Van Keymeulen A, Deleu S, Bartek J, Dumont JE, Roger PP. Respective roles of carbamylcholine and cyclic adenosine monophosphate in their synergistic regulation of cell cycle in thyroid primary cultures. Endocrinology 2001; 142(3):1251-1259.
667. Van Keymeulen A, Bartek J, Dumont JE, Roger PP. Cyclin D3 accumulation and activity integrate and rank the comitogenic pathways of thyrotropin and insulin in thyrocytes in primary culture. Oncogene 1999; 18(51):7351-7359.
668. Van Keymeulen A, Deleu S, Bartek J, Dumont JE, Roger PP. Respective roles of carbamylcholine and cyclic adenosine monophosphate in their synergistic regulation of cell cycle in thyroid primary cultures. Endocrinology 2001; 142(3):1251-1259.
669. Kimura T, Van Keymeulen A, Golstein J, Fusco A, Dumont JE, Roger PP. Regulation of thyroid cell proliferation by TSH and other factors: a critical evaluation of in vitro models. Endocrine Rev 2001; 22(5):631-656.
670. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
671. Michiels FM, Caillou B, Talbot M et al. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 1994; 91(22):10488-10492.
672. Zeiger MA, Saji M, Gusev Y et al. Thyroid-specific expression of cholera toxin A1 subunit causes thyroid hyperplasia and hyperthyroidism in transgenic mice. Endocrinology 1997; 138(8):3133-3140.
673. Nguyen LQ, Kopp P, Martinson F, Stanfield K, Roth SI, Jameson JL. A dominant negative CREB (cAMP response element-binding protein) isoform inhibits thyrocyte growth, thyroid-specific gene expression, differentiation, and function. Mol Endocrinol 2000; 14(9):1448-1461.
674. Clement S, Refetoff S, Robaye B, Dumont JE, Schurmans S. Low TSH requirement and goiter in transgenic mice overexpressing IGF-I and IGF-I receptor in the thyroid gland. Endocrinology 2001; 142(12):5131-5139.
675. Kero J, Ahmed K, Wettschureck N et al. Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J Clin Invest 2007; 117(9):2399-2407.
676. Kero J, Ahmed K, Wettschureck N et al. Thyrocyte-specific Gq/G11 deficiency impairs thyroid function and prevents goiter development. J Clin Invest 2007; 117(9):2399-2407.
677. Romeo HE, Diaz MC, Ceppi J, Zaninovich AA, Cardinali DP. Effect of inferior laryngeal nerve section on thyroid function in rats. Endocrinology 1988; 122(6):2527-2532.
678. Van Keymeulen A, Deleu S, Bartek J, Dumont JE, Roger PP. Respective roles of carbamylcholine and cyclic adenosine monophosphate in their synergistic regulation of cell cycle in thyroid primary cultures. Endocrinology 2001; 142(3):1251-1259.
679. Jhiang SM, Sagartz JE, Tong Q et al. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 1996; 137(1):375-378.
680. Powell DJ, Jr., Russell J, Nibu K et al. The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Research 1998; 58(23):5523-5528.
681. Pirson I, Coulonval K, Lamy F, Dumont JE. c-myc expression is controlled by the mitogenic cAMP-cascade in thyrocytes. J Cell Physiol 1996; 168:59-70.
682. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
683. Bartkova J, Lukas J, Strauss M, Bartek J. Cyclin D3: requirement for G1/S transition and high abundance in quiescent tissues suggest a dual role in proliferation and differentiation. Oncogene 1998; 17(8):1027-1037.
684. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
685. Paternot S, Dumont JE, Roger PP. Differential utilization of cyclin D1 and cyclin D3 in the distinct mitogenic stimulations by growth factors and TSH of human thyrocytes in primary culture. Mol Endocrinol 2006; 20(12):3279-3292.
686. Miccadei S, Provenzano C, Mojzisek M, Natali PG, Civitareale D. Retinoblastoma protein acts as Pax 8 transcriptional coactivator. Oncogene 2005; 24(47):6993-7001.
687. Roger PP, Baptist M, Dumont JE. A mechanism generating heterogeneity in thyroid epithelial cells: suppression of the thyrotropin/cAMP-dependent mitogenic pathway after cell division induced by cAMP-independent factors. Journal of Cell Biology 1992; 117:383-393.
688. Dremier S, Golstein J, Mosselmans R, Dumont JE, Galand P, Robaye B. Apoptosis in dog thyroid cells. Biochem Biophys Res Commun 1994; 200:52-58.
689. Rognoni JB, Penel C, Golstein J, Galand P, Dumont JE. Cell-Kinetics of Thyroid Epithelial-Cells During Hyperplastic Goiter Involution. Journal of Endocrinology 1987; 114(3):483-&.
690. Riesco JM, Juanes JA, Carretero J et al. Cell proliferation and apoptosis of thyroid follicular cells are involved in the involution of experimental non-tumoral hyperplastic goiter. Anatomy and Embryology 1998; 198:439-450.
691. Tamura M, Kimura H, Koji T et al. Role of apoptosis of thyrocytes in a rat model of goiter. A possible involvement of Fas system. Endocrinology 1998; 139:3643-3646.
692. Caltabiano G, Campillo M, De Leener A et al. The specificity of binding of glycoprotein hormones to their receptors. Cell Mol Life Sci 2008.
693. Costagliola S, Panneels V, Bonomi M et al. Tyrosine sulfation is required for agonist recognition by glycoprotein hormone receptors. EMBO J 2002; 21(4):504-513.
- ABSTRACT
- PHYLOGENY
- ONTOGENY
- GROSS ANATOMY
- THE FINE STRUCTURE OF THE THYROID CELLS
- GENERAL METABOLISM OF THE FOLLICULAR THYROID CELL
- THYROID REGULATORY FACTORS
- REGULATORY CASCADES
- SPECIFIC CONTROL BY IODIDE
- THE THYROTROPIN RECEPTOR
- CONTROL OF THYROID FUNCTION
- CONTROL OF THYROID-SPECIFIC GENE EXPRESSION
- CONTROL OF GROWTH AND DIFFERENTIATION
- References
- Review Physiology of the Hypothalamic-Pituitary-Thyroid Axis.[Endotext. 2000]Review Physiology of the Hypothalamic-Pituitary-Thyroid Axis.Mariotti S, Beck-Peccoz P. Endotext. 2000
- Review Chapter 2 Thyroid Hormone Synthesis And Secretion.[Endotext. 2000]Review Chapter 2 Thyroid Hormone Synthesis And Secretion.Rousset B, Dupuy C, Miot F, Dumont J. Endotext. 2000
- A Prospective Study to Evaluate the Possible Role of Cholecalciferol Supplementation on Autoimmunity in Hashimoto's Thyroiditis.[J Assoc Physicians India. 2023]A Prospective Study to Evaluate the Possible Role of Cholecalciferol Supplementation on Autoimmunity in Hashimoto's Thyroiditis.Bhakat B, Pal J, Das S, Charaborty SK, SircarMedical NR, Kolkata, RGKar, NorthBengal, Siliguri. J Assoc Physicians India. 2023 Jan; 71(1):1.
- Review TSH Receptor Mutations and Diseases.[Endotext. 2000]Review TSH Receptor Mutations and Diseases.Kleinau G, Vassart G. Endotext. 2000
- Review Ultrasonography of the Thyroid.[Endotext. 2000]Review Ultrasonography of the Thyroid.Blum M. Endotext. 2000
- Ontogeny, Anatomy, Metabolism and Physiology of the Thyroid - EndotextOntogeny, Anatomy, Metabolism and Physiology of the Thyroid - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...