

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Weight gain or body fat redistribution are common side effects of many widely used drugs. Weight gain amounts varying between a few kg to an increase of 10% or more of initial body weight have been described. Often accompanying this weight gain are worsened health risks, including an increased incidence of the metabolic syndrome, type 2 diabetes, and other cardiovascular risk factors. With many drug classes, such as β-receptor antagonists, anti-psychotic drugs, corticosteroids, neurotropic drugs, and those used in the therapy of HIV, both significant weight gain and metabolic disturbances occur in susceptible patients. In this review, we provide an overview of drugs that affect body weight, fat distribution, and metabolism. Attention is given to the possible pathogenic mechanisms underlying these effects and their metabolic consequences. Potential preventive, alternative, or therapeutic measures are suggested where applicable. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Many widely used drugs cause weight gain that—especially in susceptible individuals—may lead to patients becoming overweight or obese. Other drugs predominately influence body fat redistribution through increases in central adiposity, including visceral fat accumulation, and/or subcutaneous fat atrophy (lipodystrophy). Accompanying these changes are increases in insulin resistance, dyslipidemia, metabolic syndrome, and risk for type 2 diabetes (T2DM), non-alcoholic steatohepatitis (NASH), cardiovascular disease, cancer, and even increased mortality. These body weight and metabolic side-effects warrant close monitoring and potentially additional therapies to minimize their health impact, thereby increasing medical costs and contributing to non-compliance, which risks worsening of the underlying condition.
Weight gain is consistently associated with many older agents for the treatment of diabetes and with neuropsychotropic medications, including atypical antipsychotics, antidepressants, and antiepileptic drugs (1). For other drug classes, e.g. β-blocking agents, data are less consistent or well-studied. Glucocorticoids are associated with weight gain and lipodystrophy, as are retroviral agents used in the therapy of human immunodeficiency virus (HIV). Also, drugs used to manage lipid disorders, such as MTTP inhibitors and anti-sense apo-B oligonucleotides, are associated with changes in body fat distribution, especially liver lipid accumulation. Unfortunately, the mechanisms behind these effects on body weight and fat distribution are often poorly understood, which hampers identification of high-risk patients for prevention, development of lower risk-drugs, and possible treatments (2).
In this chapter, drugs affecting body weight, fat distribution, and glucometabolic outcomes will be reviewed, as well as the possible mechanisms contributing to these side effects. Recent studies will be highlighted that have been undertaken to identify predictors of weight gain and metabolic complications, and where possible, options for prevention and therapy will be discussed.
DRUGS ASSOCIATED WITH WEIGHT GAIN
Medications for Diabetes
Insulin, sulfonylurea (SU), and thiazolidinediones (TZD) are medications used in the management of diabetes that may cause substantial weight gain when compared to placebo (1). Metformin and Dipeptidyl Peptidase-4 (DPP-4) inhibitors are considered to be weight neutral, whereas sodium glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide 1 (GLP-1) receptor analogues (GLP1RA) are associated with weight loss on average (3).
INSULIN AND SULFONYLUREAS
Insulin causes weight gain by multiple mechanisms (4). Sulfonylureas cause weight gain by increasing endogenous insulin levels. Appetite stimulation, sometimes triggered by hypoglycemia and fluctuating glycemia, is probably the most important factor in body fat increase. Defensive snacking in order to prevent hypoglycemia or compensate for it, can be observed in some patients. Poor glycemic control increases metabolic rate and consequently, improving glycemic control decreases metabolism. Improving metabolic control also reduces glycosuria and retention of otherwise lost calories. Finally, the anabolic effects of insulin can increase protein synthesis and inhibit lipolysis and proteolysis, resulting in a gain of lean body mass (3,4).
The weight promoting properties of insulin are dose dependent and are more pronounced in injection regiments that include rapid-acting insulin compared to basal insulin only (5). The addition of metformin to insulin therapy, reduces the effects of insulin on body weight by decreasing energy intake (6).
The weight gain by sulfonylurea is most pronounced in the first months of therapy and then reaches a plateau. In the UKPDS and ADOPT studies a mean weight gain of this therapeutic class is approximately 4 kg during the first year of treatment (7,8).
THIAZOLIDINEDIONES
Thiazolidinediones also cause a substantial time and dose-dependent weight gain ranging from 1.5 to 4 kg in the first year of treatment (1,9). The mechanisms by which TZD’s cause weight gain include fluid retention, promotion of lipid storage, and adipogenesis through activation of peroxisome proliferator-activator receptor gamma (PPARg) (10,11). The fat accumulation is almost uniquely subcutaneous, with stable or even decreasing amounts of visceral fat (12). Thiazolidinediones also improve hepatic steatosis and inflammation in patients with non-alcoholic steatohepatitis (NASH), although safety concerns including osteoporosis and fluid retention with pioglitazone hamper their use (11,13). Newer PPAR drugs for the treatment of NASH are currently being studied in phase 3 trials (14).
Antihypertensive Drugs
Hypertension frequently accompanies obesity and T2DM. Therefore, drugs that promote weight gain and other metabolic side effects are of obvious concern in patients with obesity and hypertension (15).
BETA-BLOCKERS
The propensity of β-blockers to cause weight gain has been known for years (16). Their use is associated with a mean weight gain of 1.2 kg compared to controls, although among β-blockers, variable effects on weight, ranging from no significant change to an increase of 4 kg or more after one year of treatment, have been described. Most weight gain occurs in the first few months, after which no further weight gain is apparent (16).
Mechanisms whereby β-receptor antagonists are thought to affect body weight include reductions in total energy expenditure through lowering of basal metabolic rate and thermogenic response to meals, and by inhibition of lipolysis in response to adrenergic stimulation (17). In addition, β-receptor antagonists can promote fatigue and reductions in patient activity (18-20). Polymorphisms in human genes involved in catecholamine signal transduction affecting fat cell lipolysis might partly explain individual susceptibility to β-receptor antagonist-induced weight gain (21). β-blockers may also selectively promote the accumulation of abdominal fat, which is more sensitive to catecholamines than peripheral fat (22). This preponderance of abdominal fat accumulation may be, in part, responsible for the abnormalities related to carbohydrate and lipid metabolism associated with β-adrenergic blockade (23).
Several large trials have linked β-receptor antagonists to dysglycemia and new onset diabetes, even without significant weight gain (24). In particular, non-vasodilating beta-blockers (atenolol, metoprolol and propranolol) are associated with a worsening of glycemic and lipid parameters. In contrast, vasodilating beta-blockers (nebivolol, labetolol and carvedilol) have more favorable effects on glucose and lipid profiles (25). Nebivolol has been shown to induce lipolysis, reduce adipocyte lipid droplet size, and promote thermogenic and mitochondrial genes through a β3 adrenergic receptor affect (26). Therefore, selective agents with a vasodilating component such as nebivolol and carvedilol should be prioritized when β-blockers are needed in a population with high risk for metabolic side effects (15).
CALCIUM CHANNEL BLOCKERS
Calcium channel blockers are considered weight neutral and do not show adverse effects on glucose and/or lipid metabolism. However, flunarizine, a calcium channel blocker used in the prophylaxis of migraine, is associated with increased appetite and weight gain up to 4 kg. These properties have been linked to its blocking effects on both the calcium channel receptor and the dopamine receptor (27,28).
Psychotropic Medications
Obesity is two to three times more common among patients with psychiatric disorders than the general population, and individuals who are obese suffer more frequently from psychiatric illnesses than those who are normal weight. Underlying causes of this interaction between obesity and psychiatric disease likely include a clustering of adverse metabolic risk behaviors, such as unhealthy eating and insufficient physical activity, as well as substance abuse that accompany many psychiatric conditions (29). But the pathophysiological neural processes that lead to psychiatric diseases also seem to share common brain pathways with those that lead to unwanted weight gain, obesity, metabolic syndrome, and cardiovascular disease risk factors, each of which can influence the risk for the others (30). Mounting evidence points to a critical role for two major pathways: inflammatory processes including related alterations of brain functions and chronic stimulation of the hypothalamic-pituitary-adrenal (HPA) axis (30,31).
Psychiatric disorders are often characterized by a chronic stimulation of the HPA axis and sustained cortisol elevation, which have been linked with abdominal obesity, hepatic steatosis, insulin resistance, and cardiovascular disease (31). Chronic psychosocial stress has also been linked with inflammation and metabolic alterations, including weight gain with a predominance of visceral fat accumulation and insulin resistance (30). On the other hand, increased adiposity leads to chronic low-grade activation of inflammatory processes, which have been shown to have a potent role in the pathophysiological brain alterations associated with psychiatric disease (31). It is therefore possible that adiposity-driven inflammation contributes to the development of mood disorders and their growing prevalence worldwide.
Medical therapies for depression, mood disorders, and other psychiatric illnesses have been associated with sometimes very large weight gain (Table 1). Epidemiologic data show a positive correlation between weight gain and the time exposed to psychotropic medication or the number of different psychotropic drugs used (32). However, the variation in mean weight gain is large between the different drug classes and even within the same class. For most psychiatric treatments, no correlation is found with weight gain and original diagnosis or severity of the underlying psychiatric condition, treatment outcome, weight at the onset of the disease or treatment, age, or sex, which impedes prediction of those patients who will or will not have metabolic side effects (32). What has been consistently shown is that weight gain in the first month after the start of treatment is a strong predictor of long-term weight gain (33). Therefore, weight should be monitored before and shortly after starting a psychotropic drug therapy and a 5% increase above baseline weight after the first month should prompt physicians to reconsider therapeutic options or to initiate weight-controlling strategies (33,34).
Table 1.
Overview of the Psychotropic Drugs and Their Mean Effect on Weight
| Drug class | Weight loss | Weight neutral (< 1 kg/y) | Minor weight gain (1-5 kg/y) | Major weight gain (> 5 kg/y) | |
|---|---|---|---|---|---|
| Antidepressants | Tricyclic agents | amitriptylline nortriptylline imipramine desipramine dosulepine doxepine clomipramine | |||
| SSRI | escitalopram | paroxetine citalopram fluoxetine sertraline | |||
| SNRI | duloxetin venlafaxin | ||||
| MAO-inhibitors | moclobemide | phenelzine | |||
| Other | bupropion reboxetine | trazodone nefazodone | mirtazapine maprotiline mianserine | ||
| Antipsychotics | Typical | molindone | haloperidol perphenazine | ||
| Atypical | aripiprazol ziprasidone lurasidone paliperidone iloperidone asenapine | amisulpiride quietiapine risperidone sertindole | clozapine olanzapine | ||
| Anticonvulsants | topiramate zonisamide | lamotrigine levtiracetam Tiagabine oxcarbazepine | gabapentine pregabalin | valproate carbamazepine | |
| Mood stabilizers | lithium |
ANTIDEPRESSANT MEDICATIONS
The magnitude of weight gain during antidepressant therapy differs significantly by class.
Tricyclic Agents
The greatest potential to induce weight gain has been shown with the tricyclic agents’ amitriptyline and nortriptyline. Antidepressant–induced weight gain has been clearly established in the acute and maintenance period of depression therapy and is not related to disease severity. Medications from this drug class are also associated with weight gain when used for other indications, such as neuropathic pain or anxiety. To date, no predisposing factors to weight gain resulting from these drugs has been clearly identified (35).
Serotonin Agents
During initial treatment, several selective serotonin reuptake inhibitors (SSRI’s) (citalopram, fluoxetine, sertraline) and serotonin and norepinephrine reuptake inhibitors (SNRI’s) (venlafaxine and duloxetine) are associated with a slight weight loss. However, with chronic therapy many have shown weight gain. Paroxetine is considered to be the SSRI with the greatest long-term weight gain, possibly due to its affinity for the cholinergic receptor (35,36).
LITHIUM
In randomized controlled trials, the incidence of significant weight gain (more than 5% of initial body weight) has been described to be as high as 60% of the patients on lithium therapy for bipolar disorder. Risk factors for weight gain are a high baseline weight, younger age, co-administration of antidepressants, and female sex (39).
The exact mechanism by which lithium exerts these adverse effects on weight is still unknown. Possible mechanisms include a direct effect on hypothalamic centers controlling appetite, increased thirst and increased intake of high caloric drinks, changes in food preference, and its influence on thyroid function with increased incidence of hypothyroidism (39,40).
ANTIPSYCHOTICS
The number of individuals in the population receiving antipsychotic drugs is surprisingly high, most commonly for psychosis, although antipsychotic drugs are also widely used to treat other psychiatric conditions like bipolar disorders, attention deficit disorder, and dementia in the elderly (41,42).
Typical and Atypical Antipsychotics
Several major chemical classes of antipsychotic drugs have been developed, mainly the phenothiazines (e.g., chlorpromazine), the butyrophenones (e.g., haloperidol), and the thioxanthines (e.g., flupenthixol). All these “conventional,” or typical, neuroleptics are effective because they are dopamine D2 receptor antagonists, but they all have major neurological side effects (43). Therefore, newer drugs, the atypical antipsychotics or second-generation antipsychotics (SGAP), are increasingly replacing the conventional neuroleptics. These atypical antipsychotics are characterized by a combined activity on both the D2 and 5-HT2a receptors. Besides their antagonistic effects on these receptors, they possess diverse pharmacologic interactions with a number of neurotransmitter receptors (44).
Up to 80% of patients taking antipsychotic medication experience weight gain that exceeds their ideal body weight by 20% or more (45). Weight gain variability is high in between these drugs, which has been ascribed to both a high affinity for the H1-histaminic receptor as well as, to a lesser extent, the α1-adrenergic and 5-HT2c-receptors (44). The largest weight gain is consistently associated with olanzapine and clozapine. Weight gain promoting effects of the antipsychotics seem to be more pronounced in people with a normal body weight at baseline and more in women than in men. Weight gain associated with long-term treatment is time- and dose-dependent and can be predicted by weight increases in the first weeks of treatment. Drug-naïve patients gain significantly more weight than patients exposed to antipsychotics in the past and studies in pediatric patients demonstrate greater absolute weight gain in this group than in adults. Patients who have greater treatment-emergent weight gain are more likely to benefit from treatment with antipsychotics (33,46-48).
Although SGAP’s influence food intake by altering neurotransmitter function in the hypothalamus, thus leading to excess caloric consumption, obesity, and insulin resistance (49), weight gain is not the only concern for patients taking antipsychotic medication (Figure 2). SGAP’s can also promote lipogenesis and enhance antilipolytic effects of insulin, thereby favoring lipid accumulation and adipocyte enlargement and inducing insulin resistance (50). Metabolic sequela includes glucose dysregulation and an increased risk for developing metabolic syndrome and type 2 diabetes (51,52). Although an increased prevalence of metabolic syndrome has been reported in drug naïve patients with diverse psychoses, there is a significant association with longer disease duration and with the intake of clozapine in particular (51,53). In one 5-year study of clozapine-treated patients, 52% experienced one or more episodes of hyperglycemia and 30% were diagnosed as having type 2 diabetes (54). Newer SGAP appear to have fewer metabolic side effects (1,2,55,56).
The development of diabetes in patients taking antipsychotic has also been reported in patients without significant weight changes. SGAP’s are associated with a marked increase in insulin resistance in muscle, adipose tissue, and liver (49), possibly mediated by impaired GLUT-4 and GLUT-5 glucose transporter function (57). In addition, a direct impairment of pancreatic β-cell function and decreased insulin secretion has been linked to the affinity of these drugs for the 5HT-1a and 5HT-2 serotonin receptors of the β-cells (49).
Manifestations of insulin resistance, impaired glucose tolerance, metabolic syndrome (including elevations in triglyceride levels and reductions in HDL cholesterol), and type 2 diabetes contribute to the higher incidence of cardiovascular disease in patients taking these drugs. People with psychosis have a 20% shorter life expectancy than the general population, mainly driven by an increase in cardiovascular disease (53,58). In view of the high cardiometabolic risk associated with antipsychotic drug use, the American Diabetes Association and American Psychiatric Association (ADA/APA) Consensus Development Conference recommends close monitoring of weight and metabolic and cardiovascular risk factors in all patients taking SGAP’s (34).
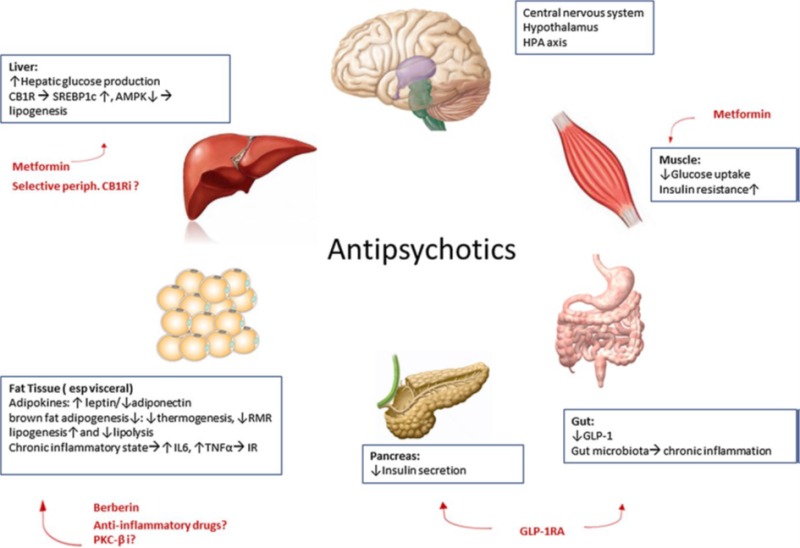
Figure 1.
Schematic representation of the central and peripheral mechanisms of antipsychotic-induced weight gain and metabolic side effects as well as current and future preventive and therapeutic options. HPA axis: hypothalamic–pituitary–adrenal axis, GLP-1: glucagon like peptide-1, GLP1RA: GLP-1 receptor agonist, RMR: resting metabolic rate, IL6: interleukin-6, TNF-α: tumor necrosis factor-α, IR: insulin resistance, PKC-βi: protein kinase C-β inhibitor, CB1R: cannabinoid receptor type 1, periph CB1Ri: peripheral cannabinoid receptor type 1 inhibitor, SREBP1c: sterol regulatory element-binding proteins type 1c, AMPK: AMP-activated protein kinase (Reprinted by permission from Springer Nature Customer Service Centr GmbH: J Endocrinol Invest. 2017;40(11):1165-1174) (2).
Therapy and Prevention of Antipsychotic Weight Gain
Many studies have evaluated pharmacological and non-pharmacological approaches to prevent or treat weight gain that accompanies SGAP treatment. Of non-pharmacological interventions, no significant difference was found between individual and group interventions, or cognitive-behavioral versus nutritional counselling. Adherence to the weight management program appears the best prognostic factor for achieved weight loss. To promote therapeutic alliance, weight management programs should be flexible and individualized to the patient’s needs, age and stage of their disease and incorporate daily recreational-based activities. Benefits are thought to be the greatest when delivered as early as possible, before weight gain has occurred (59).
Switching to another antipsychotic drug with less potential for weight and cardiometabolic side effects has been endorsed by the ADA/APA consensus guidelines in those with more than 5% weight gain or worsening of their lipid or glycemia parameters, following studies that show benefits of this strategy to limit further weight gain or reduce weight and reverse components of the metabolic syndrome (34,60) (Table 2). For example, use of the SNRI reboxetine may reduce olanzapine-induced weight gain in schizophrenia patients. Weight and metabolic benefits have also been reported by switching to, or the addition of, topiramate, amantadine, fluvoxamine, and orlistat (61-63). With metformin, attenuation or reduction of weight gain and amelioration of the metabolic side effects of SGAP therapy has been demonstrated, with greater benefits the earlier metformin was started (61,64). In a rodent study, the addition of metformin and berberine prevented the loss of brown fat induced by olanzapine and was associated with favorable changes in expression of several genes controlling energy expenditure (65).
Table 2.
Mean Weight Reducing Effects of Switching to a Less Metabolically Active SGAP or Addition of Weight Reducing Drugs (60,63).
| Action | Mean weight reduction (kg) | 95% CI |
|---|---|---|
| Switch to: | ||
| aripiprazole or quietiapine from olanzapine | -1.94 | -3.90 to 0.08 |
| Addition of: | ||
| Metformin | -2.94 | -4.89 to -0.99 |
| Topiramate | -2.52 | -4.87 to -0.16 |
| Reboxetine | -1.90 | -3.07 to -0.72 |
In patients who are obese or with diabetes, glucagon-like protein-1 receptor agonists (GLP-1 RA) demonstrate long-lasting weight loss and benefits on glucose metabolism (1,3). Growing evidence suggests that patients who are overweight and those with psychosis exhibit similar structural brain changes, cognitive deficits, and central neuropeptide alterations, suggesting an overlap between the pathophysiological pathways of these disorders (66). GLP-1 RA’s have been shown to provide neuroprotective effects in cerebral degenerative diseases such as Parkinson’s disease, Huntington’s chorea, and Alzheimer’s dementia (66). Liraglutide, a once daily injected GLP-1 RA, reverses SGAP-induced weight gain, impaired glucose tolerance, metabolic side effects and behavioral depression (67-69).
Mifepristone, a glucocorticoid and progestin receptor antagonist, attenuated increases in weight and reduced the metabolic changes induced by risperidone and olanzapine, suggesting mechanistic involvement of the hypothalamic-pituitary-adrenal axis in the weight and cardiometabolic side effects of antipsychotic medications (70). The orally effective selective protein kinase C-β (PKC- β) inhibitor ruboxistaurin, which is used in treatment of diabetes-associated retinopathy and macular edema, attenuates the effects on adipose tissue differentiation by clozapine in rodents. If this is shown to be relevant for humans, it could offer a new target for the prevention of antipsychotic-induced weight gain (71).
Because of the associations between inflammation, adiposity and psychiatric disease, other therapeutic options being explored to improve psychiatric symptoms without adverse metabolic sequelae include COX-2 selective non- steroidal anti-inflammatory drugs, and monoclonal antibodies against anti TNF-α and Interleukin-6 (72,73).
Anti-Seizure Drugs
Many of the anti-epileptic treatments are associated with weight change. Most prominent are valproate and carbamazepine, inducing weight gain in 71% and 43% of the patients, respectively. Pregabalin and gabapentin can also induce weight gain and are of particular importance since they are used more and more in the treatment of neuropathic pain, including in patients with diabetes. Weight neutral anti-epileptic drugs include lamotrigine, levetiracetam and phenytoin. Some others are associated with weight loss, including felbamate, topiramate, and zonisamide (74).
Weight-inducing effects of valproate are thought to result from interactions with appetite-regulating neuropeptides and cytokines within the hypothalamus as well as effects on energy expenditure (75). Greater weight gain is associated with longer duration of treatment with valproate, although most weight gain is observed within the first year. Specific categories of patients shown to be more susceptible to weight gain with this medication include women (vs. men), post-pubertal adolescents (vs. younger children), and those who are overweight before treatment begins. On the other hand, weight gain is not related to valproate dosage or serum levels (75).
Peripheral actions are thought to mediate valproates adverse effects on glucose and lipid metabolism that contribute to weight-independent worsening of insulin resistance and risk for type 2 diabetes. Directs effects in adipose tissue have been shown to increase leptin resistance, decrease adiponectin levels, as well as increase free fatty acids, all resulting in insulin resistance (76). On the other hand, valproic acid is associated with β-cell dysfunction and impaired insulin secretion by increasing oxidative stress, and direct inhibition of the GLUT-1 transporter, thereby hampering insulin secretion in an insulin resistant state, promoting hyperglycemia and type 2 diabetes (76-78).
Besides the increased risk for hyperglycemia and type 2 diabetes, an increased risk of other features of the metabolic syndrome (e.g., dyslipidemia) as well as endothelial dysfunction has been demonstrated. It has additionally been reported that up to 60% of the patients taking valproate also develop non-alcoholic steatohepatitis, further contributing to insulin resistance, chronic inflammation, and increased risk for cardiovascular disease (79).
With the other anticonvulsants, weight gain can be considerable in susceptible patients (74). However, in contrast to valproate, metabolic side effects accompanying use of carbamazepine, pregabalin and gabapentin are thought to be secondary to the induced weight gain rather than weight-independent mechanisms (80). On the other hand, topiramate and zonisamide have been shown to decrease body weight, even when studied in populations with obesity and overweight without any seizure history. Adding topiramate to another antiepileptic, antipsychotic or antidepressant drug, or changing anticonvulsant therapy for topiramate or zonisamide can help prevent or treat weight gain that accompanies psychiatric or anticonvulsant therapy (63,81). In the USA, the combination therapy topiramate/phentermine is approved as an antiobesity drug (82).
Drugs associated with weight gain and lipodystrophy
Corticosteroids
Although weight gain is considered a common side-effect of long-term treatment with glucocorticoids, prospective studies examining weight gain are scarce. Self-reported data in patients using chronic corticoid therapy show substantial weight gain in up to 70% of all patients (83). The weight gain associated with glucocorticoid therapy can be massive with over 10 kg increases in approximately 20% of patients in their first treatment year of treatment. The risk of weight gain with glucocorticoids is dose dependent and significantly increases with intakes above the equivalent of oral or parenteral 5 mg prednisone per day. Inhaled corticosteroids and single epidural steroid injections have no effect on body weight.
Glucocorticoids may induce an increase in food intake and dietary preference for high-caloric, high-fat ‘comfort foods’ through changes in the activity of AMP-activated protein kinase in the hypothalamus (84-86). Glucocorticoids decrease thermogenesis and uncoupling protein 1 (UCP-1) expression in brown adipose tissue, thereby influencing metabolic rate (87). Chronic glucocorticoid therapy or a state of chronic hyperactivation of the hypothalamic–pituitary–adrenal (HPA) axis is associated with activation of the endocannabinoid (eCB) system, which is a potent regulator of food intake and decreases energy expenditure (88).
Glucocorticoids also affect body fat distribution by increasing visceral fat mass, thereby increasing insulin resistance and the risk for impaired glucose tolerance, diabetes and cardiovascular disease (89). Although the mechanisms for this are not completely understood, glucocorticoids acutely stimulate lipolysis through the activation of hormone-sensitive lipase and an increased catecholamine responsiveness. When this disproportionately affects fat stores in the extremities, it can lead to loss of these depots, or lipodystrophy, the risk of which has been reported to be higher among females and younger patients and increases with a higher baseline body mass index (90). On the other hand, during chronic administration or exposure to high endogenous plasma corticoid concentration such as in Cushing’s syndrome, they promote both adipocyte hypertrophy by increasing synthesis and storage of lipids and adipose tissue hyperplasia by increasing differentiation of preadipocytes to mature adipocytes. Visceral adipose tissue has a higher glucocorticoid receptor density as compared with other fat depots, which might favor enhanced expansion of visceral adipose tissue (89). These differential effects on visceral and subcutaneous fat may be mediated by differential regulation of key metabolic genes including lipoprotein lipase, 11-beta-hydroxysteroid-dehydrogenase-1 (11β-HSD-1) and UCP-1 (89).
Glucocorticoid induced overexpression of 11β-HSD-1 in adipose tissue leads to an increase of plasma triglycerides and cholesterol levels, while 11β-HSD-1 overexpression in liver promotes insulin resistance, hepatic steatosis, and increased lipid synthesis (91,92).
In the liver, glucocorticoids act through peripheral stimulation of the cannabinoid-1 receptor (CB1R), inducing hepatic lipogenesis, steatosis and dyslipidemia. By enhancing CB1R in adipose tissue, glucocorticoids induce insulin resistance (IR) and obesity. Blocking the peripheral CB1R attenuates all aspects of metabolic dysregulation by glucocorticoids, leading the path to potential therapeutic option by selective peripheral CB1R blockers (88). These properties make specific 11β-HSD-1 inhibitors or peripheral CB1R blockers promising candidate drugs to reverse or prevent glucocorticoid-induced side effects.
Hypolipidemic Drugs
Protein convertase subtilisin kexin type 9 (PCSK9) regulates plasma low-density lipoprotein levels and low-density lipoprotein receptor expression in several tissues. Fully human monoclonal PCSK-9 inhibitors (alirocumab and evolocumab) and small interfering RNA molecules designed to target PCSK9 messenger RNA (inclisiran) have demonstrated substantial and sustained reductions in LDL-cholesterol levels, as well as significant reductions in major cardiovascular events (93).
Although no obvious side effects on body weight and body fat composition or on glucose tolerance were reported in these trials, patients with genetically low PCSK9 (R46L polymorphism) have a two-fold increased prevalence of hepatic steatosis and greater epicardial fat thickness (94). PCSK9 variants associated with lower LDL cholesterol have also been associated with higher fasting glucose levels, bodyweight, and waist-to-hip ratio, and an increased risk of type 2 diabetes (95). PCSK9 KO mice display higher visceral adipose tissue (but not subcutaneous adipose tissue), compared with wild type mice, suggesting that genetically determined PCSK9 deficiency might be associated with ectopic fat accumulation (94). It has been shown that besides the effects of LDL-R regulation in liver tissue, PCSK9 regulates VLDL-R, ApoE2 R and the CD36 receptor. PCSK9 deficiency results in reduced post-prandial lipemia and triglyceride rich lipoprotein production, while its overexpression promotes hepatic lipogenesis (96). Therefore, in genetic studies in animals and humans, PCSK9 is plays a pivotal role in fat metabolism: it regulates circulating cholesterol levels via hepatic LDL-R, but it also influences visceral adipogenesis via adipose VLDL-R regulation (97).
Therapies that reduce the rate of VLDL secretion represent an attractive alternative for reducing plasma concentrations of pro-atherogenic lipoproteins. Mipomersen is a second-generation anti-sense oligonucleotide that inhibits the translation of the apolipoprotein B by binding to the mRNA sequence of apolipoprotein B. Mipomersen reduces apolipoprotein B synthesis in the liver and the production of VLDL (and hence to IDL and LDL as well) and increases catabolism of VLDL. On the other hand, the hepatic production rate of triglycerides is unaffected (98). In clinical trials, mipomersen is associated with an approximately four times higher risk for hepatic steatosis. In contrast to NAFLD associated with common obesity, however, mipomersen-induced liver steatosis is not associated with an increase in inflammation or fibrosis (99).
Microsomal triglyceride transfer protein (MTP) is the key protein that delivers the lipid droplet to nascent VLDL and chylomicron particles during the assembly and secretion of lipoproteins in liver and intestine. MTP inhibition (e.g. lomitapide) decreases the secretion of chylomicrons and VLDL, thereby reducing the production of triglyceride rich lipoproteins and ultimately the production of LDL. Treatment with this drug also induces intra-hepatic fat accumulation, but in contrast to mipomersen, MTP inhibition is associated with greater (up to six-fold) increases in hepatic fat content and more severe increases in transaminase levels (100).
Acyl-CoA: cholesterol O-acyl transferase 2 (ACAT2) plays an important role in maintaining cellular cholesterol homeostasis. When absorbed cholesterol enters the body, it is esterified by ACAT2 and directed to the liver, where it is stored within hepatocytes in lipid droplets as cholesteryl esters. Recently it was shown in high-fat fed mice that mechanisms linking accumulation of hepatic cholesteryl esters with hepatic triglyceride accumulation depend not just on de-novo triglyceride synthesis and lipogenesis secondary to hyperinsulinemia and hyperglycemia, but also on the presence of cholesteryl ester in hepatocytes limiting mobilization of triglycerides from the liver (101). Studies have shown that ACAT inhibitors are effective for the treatment of hypercholesterolemia and atherosclerosis in rodents, however data in humans have been disappointing (102). The recent data coupling lower cholesteryl esters content to higher TG mobilization, as is realized by selective ACAT2 inhibitors, needs further study not only for treating hypercholesterolemia and atherosclerosis but also for the therapy of NAFLD.
Antiretroviral Therapy
Shortly after the introduction of effective highly-active antiretroviral therapy (HAART) for the treatment of human immunodeficiency virus (HIV) disease, it became clear that patients on these medications often had disorders of fat storage and/or wasting (lipodystrophy). The term “HIV-associated lipodystrophy syndrome” was introduced to describe a typical loss of subcutaneous fat in the limbs and face (lipoatrophy) and central or truncal fat accumulation (lipohypertrophy) (Table 3). Lipohypertrophy is characterized by intra-abdominal visceral fat accumulation and localized fat accumulation in breasts, the dorsocervical region and under the skin as lipomas. While in some patients mixed patterns are observed, some patients exhibit pure lipoatrophy and others have only fat accumulation (103). The prevalence of lipodystrophy in HIV infected patients ranges from 10 to 80%. This wide range is due to differences in study population, race, age and duration of HIV infection or antiretroviral therapy, but also due to different definitions of lipodystrophy and methods of diagnosis (104).
Table 3:
Classification of Antiretroviral Drugs and the Risk for Metabolic Consequences
| Drug | Lipohypertrophy / weight gain | Lipoatrophy | Insulin resistance | Dyslipidemia |
|---|---|---|---|---|
| Nucleoside and nucleotide reverse transcriptase inhibitors (NRTI’s) | ||||
| Abacavir (ABC) | 0 | 0 | 0 | + |
| Didanosine (ddI) | +/- | +/- | + | + |
| Emtricitabine (FTC) | 0 | 0 | 0 | 0 |
| Lamivudine (3TC) | 0 | 0 | 0 | + |
| Stavudine (d4T) | ++ | +++ | ++ | ++ |
| Tenofovir (TDF) | 0 | 0 | 0 | 0 |
| Zidovudine (AZT or ZDV) | + | ++ | ++ | + |
| Non- nucleoside reverse transcriptase inhibitors (NNRTI’s) | ||||
| Delavirdine(DLV) | +/- | +/- | 0 | + |
| Efavirenz (EFV) | +/- | + | + | ++ |
| Etrivirine (ETR) | +/- | 0 | 0 | 0 |
| Nevirapine (NVP) | 0 | 0 | 0 | ++ |
| Rilpivirine (RPV) | + | 0 | 0 | 0 |
| Protease inhibitors (PI’s) | ||||
| Amprenavir (APV) | + | + | + | + |
| Atazanavir (ATV) | ++ | 0 | 0 | + |
| Darunavir (DRV) | + | 0 | +/- | +/- |
| Indinavir (IDV) | + | +/- | +++ | + |
| Lopinavir (LPV) | + | +/- | +++ | ++ |
| Nelfinavir (NFV) | + | +/- | + | ++ |
| Ritonavir (RTV) | + | +/- | +++ | +++ |
| Saquinavir (SQV) | + | +/- | +/- | +/- |
| Tipranavir (TPV) | + | +/- | +++ | ++ |
| Fosamprenavir (FPV) | ++ | 0 | 0 | + |
| Fusion inhibitor | ||||
| Enfuvirtide (T20) | 0 | 0 | 0 | 0 |
| Integrase inhibitor | ||||
| Raltegravir (RAL) | + | 0 | 0 | 0 |
| Dolutegravir (DTG) | + | 0 | ||
| CCR5 antagonist = entry inhibitors | ||||
| Maraviroc (MVC) | No or positive effects (animal data) | 0 | 0 | 0 |
| Post attachment inhibitors | ||||
| Ibalizumab | No data | No data | 0 | 0 |
(+ = increase; 0 = neutral: +/- = discrepant)
Risk factors for lipoatrophy are male gender, older age, lower weight before therapy, lower CD4 cell counts, a higher baseline viral load, and co-infection with hepatitis C. Certain mitochondrial haplotypes and nuclear genetic polymorphisms are associated with an increased risk for lipoatrophy (104,105). In addition, there is a clear association of lipoatrophy with stavudine and zidovudine use, while switching to other retroviral drugs or using an NRTI-sparing regimen reverses lipoatrophy (106).
While the distribution of lipoatrophy is specific for HIV infected patients and anti-HIV therapy (Table 3), abdominal fat accumulation seems not to be associated with specific antiretroviral drugs and carries the same risk for metabolic syndrome as non-medication-associated visceral fat accumulation. Risk factors associated with fat accumulation during HAART are increasing age, female sex, weight before start of antiretroviral therapy, dietary factors, and longer duration of HIV treatment (104). In the AIDS Clinical Trial Group study (A5175), the prevalence of overweight or obesity increased from 25% to 40% after 144 weeks of HAART. In another trial a 30% increase in visceral fat mass was seen after 96 weeks of therapy (107). Whether lipohypertrophy can be directly attributed to HAART or represents the effects of treating HIV itself, is still a matter of debate. Indeed, central fat gain generally occurs at similar rates in patients randomized to different HAART regimens, is not associated with any specific antiretroviral drug or drug class, and does not reverse on switching antiretrovirals (106,108). In addition to the promotion of generalized weight gain, increases of as little as 5% of visceral adipose tissue are associated with increased metabolic risk, cardiovascular side effects, and even 5-year mortality. Stated differently, the cardiometabolic risk of weight gain in HIV patients is much higher than comparable weight gain in non-HIV infected controls (109).
Therefore, the European AIDS Clinical Society recommends monitoring for changes in body composition of HIV patients by using body mass index, waist circumference, waist-to-hip ratio, and to screen regularly for clinical lipodystrophy in all patients at HIV diagnosis, before starting HAART, and annually thereafter. Fat atrophy should be distinguished from general wasting associated with advanced AIDS, where besides wasting of fat mass, lean body mass is also lost. To distinguish visceral fat accumulation from simple obesity, skin fold measures can help since in HIV-induced abdominal fat accumulation, subcutaneous fat is normal or decreased, while it mostly increases in patients with simple obesity (110).
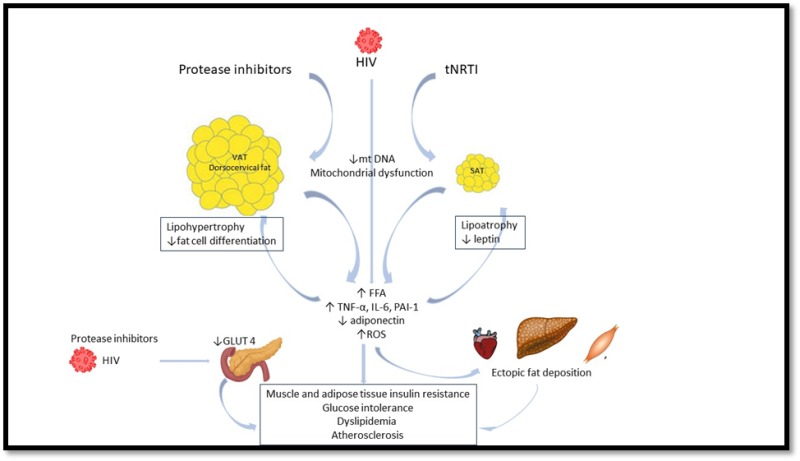
Figure 2.
Major pathogenetic pathways in HIV-induced lipodystrophy and its metabolic consequences (adapted from Debarle MC et al (111))
Pathogenetic Mechanisms of HIV Induced Lipodystrophy
Lipodystrophy is considered to be multifactorial, resulting from the complex interaction of host factors, HIV-related factors, and antiretroviral drug specific factors (Figure 3). While lipodystrophy is clearly linked to antiretroviral therapy, disturbances in adipose tissue gene expression are present in treatment-naïve patients with HIV, indicating that HIV-1 infection itself likely creates alterations in adipose tissue that are worsened by antiretroviral therapy (108,112).
When used in monotherapy, lipoatrophy is not noticed in patients using protease-inhibitors (PI). However, the co-administration of PI’s with nucleoside and nucleotide reverse transcriptase inhibitors (NRTI’s), such as stavudine and zidovudine, play an additive role in the NRTI-induced lipodystrophy (106). The older nucleoside analogues inhibit mitochondrial DNA polymerase-γ within adipocytes causing mtDNA depletion and mitochondrial dysfunction and oxidative stress in subcutaneous adipose tissue (SAT). Together with a genetic predisposition and mitochondrial dysfunction secondary to HIV itself, NRTI’s contribute in mitochondrial toxicity and increased oxidative stress, inhibiting adipogenesis and adipocyte differentiation, and promoting apoptosis, lipolysis, and dyslipidemia (105,113). Lipoatrophy is associated with inflammation, as shown by an increased macrophage number and expression of TNF-α, IL-6 and IL-8 together with increased fibrosis (114).
Adipose tissue serves as a reservoir for HIV virus, altering adipose tissue environment and causing adipose tissue inflammation. The HIV accessory viral protein R (Vpr) inhibits PPAR-γ, impairs the expression of genes related to adipocyte metabolism including adiponectin and activates glucocorticoid target gene expression, inducing macrophage infiltration and adipose tissue hypertrophy (108). Therefore, it appears that HIV-1 infection initiates a first wave of alterations in adipose tissue that is amplified by HAART and ultimately results in lipoatrophy.
PI’s are more closely associated with lipo-accumulation (115). They interfere with adipocyte maturation and differentiation by alterations in gene expression of several transcriptase factors (SREBP-1, PPAR-γ, C/EBPα and β genes) and genes encoding for acyl coenzyme-A synthase, lipoprotein lipase, GLUT-4, leptin and adiponectin, resulting in impaired fatty acid and glucose uptake, increased lipolysis and peripheral fat loss, increased triglyceride esterification and central fat accumulation. The imbalance in the production of adipokines (adiponectin and leptin) and infiltration of immune cells into adipose tissue exacerbate the pro-inflammatory environment (114).
PPAR-γ expression is also reduced by NRTI’s. The non-nucleoside reverse transcriptase inhibitor (NNRTI) efavirenz decreases expression of SREBP-1c, thus decreasing intracellular stores of triglycerides, and exerts anti-adipogenic effects in cultured adipose cells (116).
Mitochondrial DNA depletion is common to both subcutaneous and visceral or dorsocervical depots in HIV lipodystrophy and mitochondrial dysfunction in visceral adipose tissue (VAT) was found to be similar to that in SAT. In SAT, mitochondrial dysfunction is linked with lipoatrophy, whereas in VAT lipohypertrophy results. These observations indicate that different responses occurring in subcutaneous and visceral fat depots during HAART treatment are likely related to intrinsic differences in physiology between these depots (114).
Increases in proinflammatory cytokines, such as TNF-α and interleukin-6 further contribute to the development of lipodystrophy and their metabolic consequences. TNF-α inhibits adipocyte differentiation, increases apoptosis and mitochondrial toxicity and activates 11β-HSD-1 resulting in increased lipid accumulation in adipocytes, lipolysis and insulin resistance. In addition, increased fat tissue fibrosis and lipo-hypertrophy, are associated with ectopic lipid accumulation in liver, muscle and heart further increasing cardiometabolic complications (111).
Older PI‘s (indinavir, lopinavir and ritonavir) are associated with abnormalities in glucose tolerance and their use is associated with a threefold increase in the risk of diabetes compared to other treatment options. PI‘s inhibit the uptake of glucose into cells by interfering with the GLUT-4 glucose transporter and decrease insulin secretion through effects on β-cell function. Following chronic treatment, this insulin resistance leads to an inadequate suppression of lipolysis and endogenous glucose production as well as a decreased peripheral glucose uptake. Newer PI’s like darinavir and atazanavir and Integrase inhibitors (INSTI’s) have only limited effects on glucose metabolism. Of the nucleoside analogues, stavudine and zidovudine have been associated with the greatest increase in insulin resistance (114,117).
Finally, the hypothalamic-pituitary-growth hormone axis may also be involved in the metabolic changes associated with lipodystrophy. Mean growth hormone levels and growth hormone pulse amplitude are reduced in HIV-infected men with body-fat changes receiving HAART, compared with men without body-fat changes and healthy control subjects (118).
Abnormalities of lipid metabolism in HIV-infected patients were described before the advent of HAART and are mainly characterized by increases in triglycerides by a decreased triglyceride clearance and increased hepatic VLDL synthesis and apolipoprotein-E levels. In advanced AIDS disease, reduced HDL-cholesterol and a predominance of small, dense LDL particles have been reported (119). The chronic inflammation of the HIV infection itself and the associated increase in circulating cytokines can induce dyslipidemia. With the use of HAART, these lipid abnormalities tend to increase in severity and in their prevalence, with sometimes dramatic increases in lipid concentrations, particularly triglycerides (119,120). The prevalence and severity of lipid abnormalities varies widely depending on the type of HAART, nutritional status, and HIV disease stage. Risk factors seem to be a higher viral load, a family history of lipid abnormalities, less physical activity, increasing weight, greater BMI and greater trunk-to-limb fat ratio (104,119). Lipid changes and therapy of dyslipidemia in patients on retroviral therapy are described elsewhere in Endotext (121).
Therapy of HIV Associated Lipodystrophy
In patients with visceral fat accumulation, combined aerobic and strength training is generally recognized to reduce visceral fat and biomarkers for inflammation (108). Switching from a thymidine analogue NRTI (e.g. stavudine or zidovudine) to an alternative agent is considered to be a reasonable strategy to reverse or slow progression of lipoatrophy. Switching from stavudine to other NRTIs has been shown to improve mitochondrial indices, reduce fat apoptosis, and decrease some adipose tissue markers of inflammation (114,122). Switching to newer antiretroviral drugs has no effect on VAT accumulation. A switch from NRTI and NNRTI to protease inhibitors showed no weight changes whereas a switch to newer integrase inhibitors may cause even greater weight gain (123).
As mentioned above, pituitary growth hormone (GH) secretion is altered in HIV patients, and about one-third of patients meet criteria for GH deficiency. Growth hormone therapy in patients with ART induced lipodystrophy, reduces visceral adiposity, but is associated with supraphysiologic levels of IGF-I and symptoms of growth hormone excess. The FDA approved tesamorelin, a recombinant human GH releasing hormone, for the treatment of excess abdominal fat in HIV-infected patients. Tesamorelin decreases VAT by 15 to 18%, with a significant improvement of triglyceride and cholesterol levels. The effects of tesamorelin appear to be highly specific for the visceral-fat compartment, with relatively little effect on subcutaneous fat. The preferential reduction in VAT is important, given the peripheral lipoatrophy. The reduction in VAT is associated with greater baseline visceral fat mass, suggesting that larger effects might be seen among patients with more accumulation of visceral fat. Tesamorelin also improves lipid profiles, triglyceride levels and the ratio of total to HDL cholesterol. Despite this reduction in VAT, a small but statistically significant increase in HbA1c is seen in subjects receiving tesamorelin. Therefore, monitoring of IGF-1 and glycemic parameters is warranted (124).
In patients with glucose intolerance and central obesity, metformin treatment was associated with small reductions of VAT. However, in patients with lipoatrophy, metformin should be used with caution, since a further decrease of subcutaneous fat can be induced. Doleglutavir (an integrase inhibitor) increases metformin concentration. The total daily dose of metformin should therefore not exceed 1000 mg in patients co-administrated metformin with doleglutavir (104).
While the therapeutic goals and the management of diabetes in patients with HIV with or without HAART is similar to the guidelines in the general population, evidence suggests that insulin sensitizers may be preferable to insulin secretagogues. A meta-analysis of all studies with thiazolidinedione therapy (pioglitazone) showed a significantly higher limb fat mass gain in patients treated with pioglitazone. However, some studies, demonstrated a lack of effect in patients on thymidine–NRTI therapy, explained by the decreased expression of PPARγ in those patients. Any benefit appears to be small, therefore pioglitazone should be reserved to patients with severe insulin resistance and /or diabetes (125,126).
For patients with morbid obesity and/or major obesity-related diseases, bariatric surgery can be considered. An average of 20% reduction of initial BMI, improved body composition and metabolic status was observed in patients after bariatric surgery, similarly to obese non-HIV patients. However, ART treatment should be monitored to control HIV infection and some ART doses should be adjusted following this degree of weight loss (127).
Lipodystrophy associated changes in adipokine concentration could be the basis of future therapeutic options. Leptin and adiponectin decreases have been demonstrated in patients with lipodystrophy. Recombinant leptin therapy increases adiponectin levels and improve insulin sensitivity, glucose tolerance and dyslipidemia and decreases VAT without any change in SAT (128). Finally, therapy with locally injected fillers or autologous fat transplantation, are cosmetic therapies with positive results on patient wellbeing and therapy compliance (129).
Conclusion
Many frequently used medications can cause weight gain, preferential central (visceral) fat accumulation, ectopic fat accumulation in liver and muscle, and consequently have adverse glucolipid metabolic side effects that increase patient’s risk for type 2 diabetes and cardiovascular disease. In high-risk patients, use of alternative or less metabolically-active drugs can reverse or prevent unwanted weight gain and metabolic disturbances. In those patients who need to remain on medications that induce obesity and metabolic dysfunction, frequent monitoring and management of resulting weight gain, elevated blood pressure, dyslipidemia, and type 2 diabetes using standard therapies is warranted.
References
- 1.
- Domecq JP, Prutsky G, Leppin A, Sonbol MB, Altayar O, Undavalli C, Wang Z, Elraiyah T, Brito JP, Mauck KF, Lababidi MH, Prokop LJ, Asi N, Wei J, Fidahussein S, Montori VM, Murad MH. Clinical review: Drugs commonly associated with weight change: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2015;100(2):363–370. [PMC free article: PMC5393509] [PubMed: 25590213]
- 2.
- Verhaegen AA, Van Gaal LF. Drug-induced obesity and its metabolic consequences: a review with a focus on mechanisms and possible therapeutic options. J Endocrinol Invest. 2017;40(11):1165–1174. [PubMed: 28660606]
- 3.
- Van Gaal L, Scheen A. Weight management in type 2 diabetes: current and emerging approaches to treatment. Diabetes Care. 2015;38(6):1161–1172. [PubMed: 25998297]
- 4.
- Russell-Jones D, Khan R. Insulin-associated weight gain in diabetes--causes, effects and coping strategies. Diabetes, obesity & metabolism. 2007;9(6):799–812. [PubMed: 17924864]
- 5.
- Pontiroli AE, Miele L, Morabito A. Increase of body weight during the first year of intensive insulin treatment in type 2 diabetes: systematic review and meta-analysis. Diabetes, obesity & metabolism. 2011;13(11):1008–1019. [PubMed: 21645195]
- 6.
- Makimattila S, Nikkila K, Yki-Jarvinen H. Causes of weight gain during insulin therapy with and without metformin in patients with Type II diabetes mellitus. Diabetologia. 1999;42(4):406–412. [PubMed: 10230643]
- 7.
- Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):854–865. [PubMed: 9742977]
- 8.
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O'Neill MC, Zinman B, Viberti G, Group AS. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427–2443. [PubMed: 17145742]
- 9.
- Medici V, McClave SA, Miller KR. Common Medications Which Lead to Unintended Alterations in Weight Gain or Organ Lipotoxicity. Current gastroenterology reports. 2016;18(1):2. [PubMed: 26700070]
- 10.
- Cignarelli A, Giorgino F, Vettor R. Pharmacologic agents for type 2 diabetes therapy and regulation of adipogenesis. Archives of physiology and biochemistry. 2013;119(4):139–150. [PubMed: 23724947]
- 11.
- Aleman-Gonzalez-Duhart D, Tamay-Cach F, Alvarez-Almazan S, Mendieta-Wejebe JE. Current Advances in the Biochemical and Physiological Aspects of the Treatment of Type 2 Diabetes Mellitus with Thiazolidinediones. PPAR research. 2016;2016:7614270. [PMC free article: PMC4893583] [PubMed: 27313601]
- 12.
- Wilding J. Thiazolidinediones, insulin resistance and obesity: Finding a balance. Int J Clin Pract. 2006;60(10):1272–1280. [PubMed: 16981971]
- 13.
- Mills EP, Brown KPD, Smith JD, Vang PW, Trotta K. Treating nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: a review of efficacy and safety. Therapeutic advances in endocrinology and metabolism. 2018;9(1):15–28. [PMC free article: PMC5761952] [PubMed: 29344336]
- 14.
- Banini BA, Sanyal AJ. Current and future pharmacologic treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2017;33(3):134–141. [PMC free article: PMC5491795] [PubMed: 28346237]
- 15.
- Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, Sowers J. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment--a position paper of the The Obesity Society and The American Society of Hypertension. Obesity (Silver Spring, Md). 2013;21(1):8–24. [PubMed: 23401272]
- 16.
- Sharma AM, Pischon T, Hardt S, Kunz I, Luft FC. Hypothesis: Beta-adrenergic receptor blockers and weight gain: A systematic analysis. Hypertension (Dallas, Tex : 1979). 2001;37(2):250-254.
- 17.
- Newsom SA, Richards JC, Johnson TK, Kuzma JN, Lonac MC, Paxton RJ, Rynn GM, Voyles WF, Bell C. Short-term sympathoadrenal inhibition augments the thermogenic response to beta-adrenergic receptor stimulation. The Journal of endocrinology. 2010;206(3):307–315. [PubMed: 20603265]
- 18.
- Welle S, Schwartz RG, Statt M. Reduced metabolic rate during beta-adrenergic blockade in humans. Metabolism. 1991;40(6):619–622. [PubMed: 1650879]
- 19.
- Astrup A, Simonsen L, Bulow J, Madsen J, Christensen NJ. Epinephrine mediates facultative carbohydrate-induced thermogenesis in human skeletal muscle. Am J Physiol. 1989;257(3 Pt 1):E340–345. [PubMed: 2782399]
- 20.
- Koch G, Franz IW, Lohmann FW. Effects of short-term and long-term treatment with cardio-selective and non-selective beta-receptor blockade on carbohydrate and lipid metabolism and on plasma catecholamines at rest and during exercise. Clinical science (London, England : 1979). 1981;61 Suppl 7:433s-435s.
- 21.
- Arner P. Genetic variance and lipolysis regulation: implications for obesity. Annals of medicine. 2001;33(8):542–546. [PubMed: 11730161]
- 22.
- Hoffstedt J, Arner P, Hellers G, Lonnqvist F. Variation in adrenergic regulation of lipolysis between omental and subcutaneous adipocytes from obese and non-obese men. Journal of lipid research. 1997;38(4):795–804. [PubMed: 9144094]
- 23.
- Nicklas BJ, Rogus EM, Colman EG, Goldberg AP. Visceral adiposity, increased adipocyte lipolysis, and metabolic dysfunction in obese postmenopausal women. The American journal of physiology. 1996;270(1 Pt 1):E72–78. [PubMed: 8772476]
- 24.
- Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369(9557):201–207. [PubMed: 17240286]
- 25.
- Fonseca VA. Effects of beta-blockers on glucose and lipid metabolism. Curr Med Res Opin. 2010;26(3):615–629. [PubMed: 20067434]
- 26.
- Bordicchia M, Pocognoli A, D'Anzeo M, Siquini W, Minardi D, Muzzonigro G, Dessi-Fulgheri P, Sarzani R. Nebivolol induces, via beta3 adrenergic receptor, lipolysis, uncoupling protein 1, and reduction of lipid droplet size in human adipocytes. Journal of hypertension. 2014;32(2):389–396. [PubMed: 24256707]
- 27.
- Vecsei L, Majlath Z, Szok D, Csati A, Tajti J. Drug safety and tolerability in prophylactic migraine treatment. Expert opinion on drug safety. 2015;14(5):667–681. [PubMed: 25676133]
- 28.
- Manconi FM ZA, Piccardi MP, et al. Behavioural and biochemical effects of flunarizine on the dopaminergic system in rodents. . Cephalalgia. 1987;7:420–421.
- 29.
- Compton MT, Daumit GL, Druss BG. Cigarette smoking and overweight/obesity among individuals with serious mental illnesses: a preventive perspective. Harvard review of psychiatry. 2006;14(4):212–222. [PubMed: 16912007]
- 30.
- Milaneschi Y, Simmons WK, van Rossum EFC, Penninx BW. Depression and obesity: evidence of shared biological mechanisms. Mol Psychiatry. 2018 [PubMed: 29453413]
- 31.
- Menard C, Pfau ML, Hodes GE, Russo SJ. Immune and Neuroendocrine Mechanisms of Stress Vulnerability and Resilience. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2017;42(1):62-80.
- 32.
- Zimmermann U, Kraus T, Himmerich H, Schuld A, Pollmacher T. Epidemiology, implications and mechanisms underlying drug-induced weight gain in psychiatric patients. Journal of psychiatric research. 2003;37(3):193–220. [PubMed: 12650740]
- 33.
- Vandenberghe F, Gholam-Rezaee M, Saigi-Morgui N, Delacretaz A, Choong E, Solida-Tozzi A, Kolly S, Thonney J, Gallo SF, Hedjal A, Ambresin AE, von Gunten A, Conus P, Eap CB. Importance of early weight changes to predict long-term weight gain during psychotropic drug treatment. J Clin Psychiatry. 2015;76(11):e1417–1423. [PubMed: 26646038]
- 34.
- Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care. 2004;27(2):596–601. [PubMed: 14747245]
- 35.
- Serretti A, Mandelli L. Antidepressants and body weight: a comprehensive review and meta-analysis. The Journal of clinical psychiatry. 2010;71(10):1259–1272. [PubMed: 21062615]
- 36.
- Masand PS, Gupta S. Long-term side effects of newer-generation antidepressants: SSRIS, venlafaxine, nefazodone, bupropion, and mirtazapine. Annals of clinical psychiatry : official journal of the American Academy of Clinical Psychiatrists. 2002;14(3):175–182. [PubMed: 12585567]
- 37.
- Gadde KM, Xiong GL. Bupropion for weight reduction. Expert review of neurotherapeutics. 2007;7(1):17–24. [PubMed: 17187492]
- 38.
- Billes SK, Sinnayah P, Cowley MA. Naltrexone/bupropion for obesity: an investigational combination pharmacotherapy for weight loss. Pharmacol Res. 2014;84:1–11. [PubMed: 24754973]
- 39.
- Livingstone C, Rampes H. Lithium: a review of its metabolic adverse effects. J Psychopharmacol. 2006;20(3):347–355. [PubMed: 16174674]
- 40.
- Chen Y, Silverstone T. Lithium and weight gain. Int Clin Psychopharmacol. 1990;5(3):217–225. [PubMed: 2230066]
- 41.
- Sohn M, Moga DC, Blumenschein K, Talbert J. National trends in off-label use of atypical antipsychotics in children and adolescents in the United States. Medicine. 2016;95(23):e3784. [PMC free article: PMC4907659] [PubMed: 27281081]
- 42.
- Verdoux H, Tournier M, Begaud B. Antipsychotic prescribing trends: a review of pharmaco-epidemiological studies. Acta Psychiatr Scand. 2010;121(1):4–10. [PubMed: 20059452]
- 43.
- Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261(5562):717–719. [PubMed: 945467]
- 44.
- Allison DB, Casey DE. Antipsychotic-induced weight gain: a review of the literature. J Clin Psychiatry. 2001;62 Suppl 7:22–31. [PubMed: 11346192]
- 45.
- Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, Weiden PJ. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999;156(11):1686–1696. [PubMed: 10553730]
- 46.
- Correll CU, Manu P, Olshanskiy V, Napolitano B, Kane JM, Malhotra AK. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. Jama. 2009;302(16):1765–1773. [PMC free article: PMC3055794] [PubMed: 19861668]
- 47.
- Hoffmann VP, Case M, Stauffer VL, Jacobson JG, Conley RR. Predictive value of early changes in triglycerides and weight for longer-term changes in metabolic measures during olanzapine, ziprasidone or aripiprazole treatment for schizophrenia and schizoaffective disorder post hoc analyses of 3 randomized, controlled clinical trials. Journal of clinical psychopharmacology. 2010;30(6):656–660. [PubMed: 21105275]
- 48.
- Raben AT, Marshe VS, Chintoh A, Gorbovskaya I, Muller DJ, Hahn MK. The Complex Relationship between Antipsychotic-Induced Weight Gain and Therapeutic Benefits: A Systematic Review and Implications for Treatment. Frontiers in neuroscience. 2017;11:741. [PMC free article: PMC5786866] [PubMed: 29403343]
- 49.
- Ballon JS, Pajvani U, Freyberg Z, Leibel RL, Lieberman JA. Molecular pathophysiology of metabolic effects of antipsychotic medications. Trends Endocrinol Metab. 2014;25(11):593–600. [PubMed: 25190097]
- 50.
- Vestri HS, Maianu L, Moellering DR, Garvey WT. Atypical antipsychotic drugs directly impair insulin action in adipocytes: effects on glucose transport, lipogenesis, and antilipolysis. Neuropsychopharmacology. 2007;32(4):765–772. [PubMed: 16823387]
- 51.
- Mitchell AJ, Vancampfort D, Sweers K, van Winkel R, Yu W, De Hert M. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders--a systematic review and meta-analysis. Schizophr Bull. 2013;39(2):306–318. [PMC free article: PMC3576174] [PubMed: 22207632]
- 52.
- Holt RI, Peveler RC. Association between antipsychotic drugs and diabetes. Diabetes, obesity & metabolism. 2006;8(2):125–135. [PubMed: 16448516]
- 53.
- De Hert M, Detraux J, van Winkel R, Yu W, Correll CU. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2011;8(2):114–126. [PubMed: 22009159]
- 54.
- Henderson DC, Cagliero E, Gray C, Nasrallah RA, Hayden DL, Schoenfeld DA, Goff DC. Clozapine, diabetes mellitus, weight gain, and lipid abnormalities: A five-year naturalistic study. Am J Psychiatry. 2000;157(6):975–981. [PubMed: 10831479]
- 55.
- Balf G, Stewart TD, Whitehead R, Baker RA. Metabolic adverse events in patients with mental illness treated with antipsychotics: a primary care perspective. Prim Care Companion J Clin Psychiatry. 2008;10(1):15–24. [PMC free article: PMC2249834] [PubMed: 18311417]
- 56.
- Orsolini L, Tomasetti C, Valchera A, Vecchiotti R, Matarazzo I, Vellante F, Iasevoli F, Buonaguro EF, Fornaro M, Fiengo AL, Martinotti G, Mazza M, Perna G, Carano A, De Bartolomeis A, Di Giannantonio M, De Berardis D. An update of safety of clinically used atypical antipsychotics. Expert opinion on drug safety. 2016;15(10):1329–1347. [PubMed: 27347638]
- 57.
- Dwyer DS, Donohoe D. Induction of hyperglycemia in mice with atypical antipsychotic drugs that inhibit glucose uptake. Pharmacology, biochemistry, and behavior. 2003;75(2):255–260. [PubMed: 12873613]
- 58.
- Daumit GL, Goff DC, Meyer JM, Davis VG, Nasrallah HA, McEvoy JP, Rosenheck R, Davis SM, Hsiao JK, Stroup TS, Lieberman JA. Antipsychotic effects on estimated 10-year coronary heart disease risk in the CATIE schizophrenia study. Schizophr Res. 2008;105(1-3):175–187. [PMC free article: PMC2614656] [PubMed: 18775645]
- 59.
- Alvarez-Jimenez M, Hetrick SE, Gonzalez-Blanch C, Gleeson JF, McGorry PD. Non-pharmacological management of antipsychotic-induced weight gain: systematic review and meta-analysis of randomised controlled trials. The British journal of psychiatry : the journal of mental science. 2008;193(2):101–107. [PubMed: 18669990]
- 60.
- Mukundan A, Faulkner G, Cohn T, Remington G. Antipsychotic switching for people with schizophrenia who have neuroleptic-induced weight or metabolic problems. The Cochrane database of systematic reviews. 2010;(12):Cd006629. [PubMed: 21154372]
- 61.
- Gierisch JM, Nieuwsma JA, Bradford DW, Wilder CM, Mann-Wrobel MC, McBroom AJ, Wing L, Musty MD, Chobot MM, Hasselblad V, Williams JW, Jr. AHRQ Comparative Effectiveness Reviews. Interventions To Improve Cardiovascular Risk Factors in People With Serious Mental Illness. Rockville (MD): Agency for Healthcare Research and Quality (US); 2013.
- 62.
- Mizuno Y, Suzuki T, Nakagawa A, Yoshida K, Mimura M, Fleischhacker WW, Uchida H. Pharmacological strategies to counteract antipsychotic-induced weight gain and metabolic adverse effects in schizophrenia: a systematic review and meta-analysis. Schizophr Bull. 2014;40(6):1385–1403. [PMC free article: PMC4193713] [PubMed: 24636967]
- 63.
- Maayan L, Vakhrusheva J, Correll CU. Effectiveness of medications used to attenuate antipsychotic-related weight gain and metabolic abnormalities: a systematic review and meta-analysis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010;35(7):1520-1530.
- 64.
- Zheng W, Li XB, Tang YL, Xiang YQ, Wang CY, de Leon J. Metformin for Weight Gain and Metabolic Abnormalities Associated With Antipsychotic Treatment: Meta-Analysis of Randomized Placebo-Controlled Trials. Journal of clinical psychopharmacology. 2015;35(5):499–509. [PubMed: 26280837]
- 65.
- Hu Y, Young AJ, Ehli EA, Nowotny D, Davies PS, Droke EA, Soundy TJ, Davies GE. Metformin and berberine prevent olanzapine-induced weight gain in rats. PloS one. 2014;9(3):e93310. [PMC free article: PMC3965561] [PubMed: 24667776]
- 66.
- Ebdrup BH, Knop FK, Ishoy PL, Rostrup E, Fagerlund B, Lublin H, Glenthoj B. Glucagon-like peptide-1 analogs against antipsychotic-induced weight gain: potential physiological benefits. BMC Med. 2012;10:92. [PMC free article: PMC3573939] [PubMed: 22891821]
- 67.
- Lykkegaard K, Larsen PJ, Vrang N, Bock C, Bock T, Knudsen LB. The once-daily human GLP-1 analog, liraglutide, reduces olanzapine-induced weight gain and glucose intolerance. Schizophr Res. 2008;103(1-3):94–103. [PubMed: 18579346]
- 68.
- Sharma AN, Ligade SS, Sharma JN, Shukla P, Elased KM, Lucot JB. GLP-1 receptor agonist liraglutide reverses long-term atypical antipsychotic treatment associated behavioral depression and metabolic abnormalities in rats. Metab Brain Dis. 2015;30(2):519–527. [PubMed: 25023888]
- 69.
- Larsen JR, Vedtofte L, Jakobsen MSL, Jespersen HR, Jakobsen MI, Svensson CK, Koyuncu K, Schjerning O, Oturai PS, Kjaer A, Nielsen J, Holst JJ, Ekstrom CT, Correll CU, Vilsboll T, Fink-Jensen A. Effect of Liraglutide Treatment on Prediabetes and Overweight or Obesity in Clozapine- or Olanzapine-Treated Patients With Schizophrenia Spectrum Disorder: A Randomized Clinical Trial. JAMA psychiatry. 2017;74(7):719–728. [PMC free article: PMC5710254] [PubMed: 28601891]
- 70.
- Gross C, Blasey CM, Roe RL, Belanoff JK. Mifepristone reduces weight gain and improves metabolic abnormalities associated with risperidone treatment in normal men. Obesity (Silver Spring). 2010;18(12):2295–2300. [PubMed: 20339369]
- 71.
- Rimessi A, Pavan C, Ioannidi E, Nigro F, Morganti C, Brugnoli A, Longo F, Gardin C, Ferroni L, Morari M, Vindigni V, Zavan B, Pinton P. Protein Kinase C beta: a New Target Therapy to Prevent the Long-Term Atypical Antipsychotic-Induced Weight Gain. Neuropsychopharmacology. 2017;42(7):1491–1501. [PMC free article: PMC5436118] [PubMed: 28128334]
- 72.
- Capuron L, Lasselin J, Castanon N. Role of Adiposity-Driven Inflammation in Depressive Morbidity. Neuropsychopharmacology. 2017;42(1):115–128. [PMC free article: PMC5143483] [PubMed: 27402495]
- 73.
- Muller N. COX-2 inhibitors as antidepressants and antipsychotics: clinical evidence. Current opinion in investigational drugs (London, England : 2000). 2010;11(1):31-42.
- 74.
- Ben-Menachem E. Weight issues for people with epilepsy--a review. Epilepsia. 2007;48 Suppl 9:42–45. [PubMed: 18047602]
- 75.
- Verrotti A, D'Egidio C, Mohn A, Coppola G, Chiarelli F. Weight gain following treatment with valproic acid: pathogenetic mechanisms and clinical implications. Obes Rev. 2011;12(5):e32–43. [PubMed: 20880119]
- 76.
- Belcastro V, D'Egidio C, Striano P, Verrotti A. Metabolic and endocrine effects of valproic acid chronic treatment. Epilepsy Res. 2013;107(1-2):1–8. [PubMed: 24076030]
- 77.
- Aycicek A, Iscan A. The effects of carbamazepine, valproic acid and phenobarbital on the oxidative and antioxidative balance in epileptic children. Eur Neurol. 2007;57(2):65–69. [PubMed: 17179706]
- 78.
- Wong HY, Chu TS, Lai JC, Fung KP, Fok TF, Fujii T, Ho YY. Sodium valproate inhibits glucose transport and exacerbates Glut1-deficiency in vitro. J Cell Biochem. 2005;96(4):775–785. [PubMed: 16149077]
- 79.
- Farinelli E, Giampaoli D, Cenciarini A, Cercado E, Verrotti A. Valproic acid and nonalcoholic fatty liver disease: A possible association? World J Hepatol. 2015;7(9):1251–1257. [PMC free article: PMC4438499] [PubMed: 26019740]
- 80.
- Swann AC. Major system toxicities and side effects of anticonvulsants. J Clin Psychiatry. 2001;62 Suppl 14:16–21. [PubMed: 11469670]
- 81.
- Antel J, Hebebrand J. Weight-reducing side effects of the antiepileptic agents topiramate and zonisamide. Handb Exp Pharmacol. 2012;(209):433–466. [PubMed: 22249827]
- 82.
- Smith SM, Meyer M, Trinkley KE. Phentermine/topiramate for the treatment of obesity. The Annals of pharmacotherapy. 2013;47(3):340–349. [PubMed: 23482732]
- 83.
- Curtis JR, Westfall AO, Allison J, Bijlsma JW, Freeman A, George V, Kovac SH, Spettell CM, Saag KG. Population-based assessment of adverse events associated with long-term glucocorticoid use. Arthritis and rheumatism. 2006;55(3):420–426. [PubMed: 16739208]
- 84.
- Christ-Crain M, Kola B, Lolli F, Fekete C, Seboek D, Wittmann G, Feltrin D, Igreja SC, Ajodha S, Harvey-White J, Kunos G, Muller B, Pralong F, Aubert G, Arnaldi G, Giacchetti G, Boscaro M, Grossman AB, Korbonits M. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing's syndrome. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22(6):1672–1683. [PubMed: 18198220]
- 85.
- Larsen PJ, Jessop DS, Chowdrey HS, Lightman SL, Mikkelsen JD. Chronic administration of glucocorticoids directly upregulates prepro-neuropeptide Y and Y1-receptor mRNA levels in the arcuate nucleus of the rat. J Neuroendocrinol. 1994;6(2):153–159. [PubMed: 8049712]
- 86.
- Harfstrand A, Cintra A, Fuxe K, Aronsson M, Wikstrom AC, Okret S, Gustafsson JA, Agnati LF. Regional differences in glucocorticoid receptor immunoreactivity among neuropeptide Y immunoreactive neurons of the rat brain. Acta Physiol Scand. 1989;135(1):3–9. [PubMed: 2643254]
- 87.
- Soumano K, Desbiens S, Rabelo R, Bakopanos E, Camirand A, Silva JE. Glucocorticoids inhibit the transcriptional response of the uncoupling protein-1 gene to adrenergic stimulation in a brown adipose cell line. Molecular and cellular endocrinology. 2000;165(1-2):7–15. [PubMed: 10940478]
- 88.
- Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood JA, Li Z, Tamashiro KL, Schwartz GJ, Makriyannis AM, Kunos G, Hillard CJ, McEwen BS, Hill MN. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc Natl Acad Sci U S A. 2015;112(1):285–290. [PMC free article: PMC4291642] [PubMed: 25535367]
- 89.
- Fardet L, Feve B. Systemic glucocorticoid therapy: a review of its metabolic and cardiovascular adverse events. Drugs. 2014;74(15):1731–1745. [PubMed: 25204470]
- 90.
- Fardet L, Cabane J, Lebbe C, Morel P, Flahault A. Incidence and risk factors for corticosteroid-induced lipodystrophy: a prospective study. J Am Acad Dermatol. 2007;57(4):604–609. [PubMed: 17582650]
- 91.
- Dube S, Slama MQ, Basu A, Rizza RA, Basu R. Glucocorticoid Excess Increases Hepatic 11beta-HSD-1 Activity in Humans: Implications in Steroid-Induced Diabetes. J Clin Endocrinol Metab. 2015;100(11):4155–4162. [PMC free article: PMC4702452] [PubMed: 26308294]
- 92.
- Wolf G. Glucocorticoids in adipocytes stimulate visceral obesity. Nutr Rev. 2002;60(5 Pt 1):148–151. [PubMed: 12030279]
- 93.
- Karatasakis A, Danek BA, Karacsonyi J, Rangan BV, Roesle MK, Knickelbine T, Miedema MD, Khalili H, Ahmad Z, Abdullah S, Banerjee S, Brilakis ES. Effect of PCSK9 Inhibitors on Clinical Outcomes in Patients With Hypercholesterolemia: A Meta-Analysis of 35 Randomized Controlled Trials. J Am Heart Assoc. 2017;6(12) [PMC free article: PMC5779013] [PubMed: 29223954]
- 94.
- Baragetti A, Balzarotti G, Grigore L, Pellegatta F, Guerrini U, Pisano G, Fracanzani AL, Fargion S, Norata GD, Catapano AL. PCSK9 deficiency results in increased ectopic fat accumulation in experimental models and in humans. Eur J Prev Cardiol. 2017;24(17):1870–1877. [PubMed: 28758421]
- 95.
- Schmidt AF, Swerdlow DI, Holmes MV, Patel RS, Fairhurst-Hunter Z, Lyall DM, Hartwig FP, Horta BL, Hypponen E, Power C, Moldovan M, van Iperen E, Hovingh GK, Demuth I, Norman K, Steinhagen-Thiessen E, Demuth J, Bertram L, Liu T, Coassin S, Willeit J, Kiechl S, Willeit K, Mason D, Wright J, Morris R, Wanamethee G, Whincup P, Ben-Shlomo Y, McLachlan S, Price JF, Kivimaki M, Welch C, Sanchez-Galvez A, Marques-Vidal P, Nicolaides A, Panayiotou AG, Onland-Moret NC, van der Schouw YT, Matullo G, Fiorito G, Guarrera S, Sacerdote C, Wareham NJ, Langenberg C, Scott R, Luan J, Bobak M, Malyutina S, Pajak A, Kubinova R, Tamosiunas A, Pikhart H, Husemoen LL, Grarup N, Pedersen O, Hansen T, Linneberg A, Simonsen KS, Cooper J, Humphries SE, Brilliant M, Kitchner T, Hakonarson H, Carrell DS, McCarty CA, Kirchner HL, Larson EB, Crosslin DR, de Andrade M, Roden DM, Denny JC, Carty C, Hancock S, Attia J, Holliday E, O'Donnell M, Yusuf S, Chong M, Pare G, van der Harst P, Said MA, Eppinga RN, Verweij N, Snieder H, Christen T, Mook-Kanamori DO, Gustafsson S, Lind L, Ingelsson E, Pazoki R, Franco O, Hofman A, Uitterlinden A, Dehghan A, Teumer A, Baumeister S, Dorr M, Lerch MM, Volker U, Volzke H, Ward J, Pell JP, Smith DJ, Meade T, Maitland-van der Zee AH, Baranova EV, Young R, Ford I, Campbell A, Padmanabhan S, Bots ML, Grobbee DE, Froguel P, Thuillier D, Balkau B, Bonnefond A, Cariou B, Smart M, Bao Y, Kumari M, Mahajan A, Ridker PM, Chasman DI, Reiner AP, Lange LA, Ritchie MD, Asselbergs FW, Casas JP, Keating BJ, Preiss D, Hingorani AD, Sattar N. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. The lancet Diabetes & endocrinology. 2017;5(2):97–105. [PMC free article: PMC5266795] [PubMed: 27908689]
- 96.
- Norata GD, Tavori H, Pirillo A, Fazio S, Catapano AL. Biology of proprotein convertase subtilisin kexin 9: beyond low-density lipoprotein cholesterol lowering. Cardiovasc Res. 2016;112(1):429–442. [PMC free article: PMC5031950] [PubMed: 27496869]
- 97.
- Roubtsova A, Munkonda MN, Awan Z, Marcinkiewicz J, Chamberland A, Lazure C, Cianflone K, Seidah NG, Prat A. Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler Thromb Vasc Biol. 2011;31(4):785–791. [PubMed: 21273557]
- 98.
- Wierzbicki AS, Viljoen A. Anti-sense oligonucleotide therapies for the treatment of hyperlipidaemia. Expert Opin Biol Ther. 2016;16(9):1125–1134. [PubMed: 27248482]
- 99.
- Hashemi N, Odze RD, McGowan MP, Santos RD, Stroes ES, Cohen DE. Liver histology during Mipomersen therapy for severe hypercholesterolemia. J Clin Lipidol. 2014;8(6):606–611. [PMC free article: PMC4268487] [PubMed: 25499943]
- 100.
- Berberich AJ, Hegele RA. Lomitapide for the treatment of hypercholesterolemia. Expert Opin Pharmacother. 2017;18(12):1261–1268. [PubMed: 28598687]
- 101.
- Alger HM, Brown JM, Sawyer JK, Kelley KL, Shah R, Wilson MD, Willingham MC, Rudel LL. Inhibition of acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) prevents dietary cholesterol-associated steatosis by enhancing hepatic triglyceride mobilization. J Biol Chem. 2010;285(19):14267–14274. [PMC free article: PMC2863169] [PubMed: 20231283]
- 102.
- Meuwese MC, Franssen R, Stroes ES, Kastelein JJ. And then there were acyl coenzyme A:cholesterol acyl transferase inhibitors. Current opinion in lipidology. 2006;17(4):426–430. [PubMed: 16832167]
- 103.
- Chen D, Misra A, Garg A. Clinical review 153: Lipodystrophy in human immunodeficiency virus-infected patients. J Clin Endocrinol Metab. 2002;87(11):4845–4856. [PubMed: 12414837]
- 104.
- Mirza FS, Luthra P, Chirch L. Endocrinological aspects of HIV infection. J Endocrinol Invest. 2018;41(8):881–899. [PubMed: 29313284]
- 105.
- Hulgan T, Haubrich R, Riddler SA, Tebas P, Ritchie MD, McComsey GA, Haas DW, Canter JA. European mitochondrial DNA haplogroups and metabolic changes during antiretroviral therapy in AIDS Clinical Trials Group Study A5142. AIDS. 2011;25(1):37–47. [PMC free article: PMC2995830] [PubMed: 20871389]
- 106.
- de Waal R, Cohen K, Maartens G. Systematic review of antiretroviral-associated lipodystrophy: lipoatrophy, but not central fat gain, is an antiretroviral adverse drug reaction. PloS one. 2013;8(5):e63623. [PMC free article: PMC3665842] [PubMed: 23723990]
- 107.
- McComsey GA, Moser C, Currier J, Ribaudo HJ, Paczuski P, Dube MP, Kelesidis T, Rothenberg J, Stein JH, Brown TT. Body Composition Changes After Initiation of Raltegravir or Protease Inhibitors: ACTG A5260s. Clin Infect Dis. 2016;62(7):853–862. [PMC free article: PMC4787610] [PubMed: 26797215]
- 108.
- Erlandson KM, Lake JE. Fat Matters: Understanding the Role of Adipose Tissue in Health in HIV Infection. Curr HIV/AIDS Rep. 2016;13(1):20–30. [PMC free article: PMC4779424] [PubMed: 26830284]
- 109.
- Scherzer R, Heymsfield SB, Lee D, Powderly WG, Tien PC, Bacchetti P, Shlipak MG, Grunfeld C. Study of Fat R, Metabolic Change in HIVI. Decreased limb muscle and increased central adiposity are associated with 5-year all-cause mortality in HIV infection. AIDS. 2011;25(11):1405–1414. [PMC free article: PMC3933309] [PubMed: 21572308]
- 110.
- Lundgren JD, Battegay M, Behrens G, De Wit S, Guaraldi G, Katlama C, Martinez E, Nair D, Powderly WG, Reiss P, Sutinen J, Vigano A, Committee EE. European AIDS Clinical Society (EACS) guidelines on the prevention and management of metabolic diseases in HIV. HIV Med. 2008;9(2):72–81. [PubMed: 18257770]
- 111.
- Caron-Debarle M, Lagathu C, Boccara F, Vigouroux C, Capeau J. HIV-associated lipodystrophy: from fat injury to premature aging. Trends in molecular medicine. 2010;16(5):218–229. [PubMed: 20400373]
- 112.
- Giralt M, Domingo P, Guallar JP, Rodriguez de la Concepcion ML, Alegre M, Domingo JC, Villarroya F. HIV-1 infection alters gene expression in adipose tissue, which contributes to HIV- 1/HAART-associated lipodystrophy. Antiviral therapy. 2006;11(6):729–740. [PubMed: 17310817]
- 113.
- Koczor CA, Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin Drug Metab Toxicol. 2010;6(12):1493–1504. [PubMed: 20929279]
- 114.
- Giralt M, Domingo P, Villarroya F. Adipose tissue biology and HIV-infection. Best Pract Res Clin Endocrinol Metab. 2011;25(3):487–499. [PubMed: 21663842]
- 115.
- Flint OP, Noor MA, Hruz PW, Hylemon PB, Yarasheski K, Kotler DP, Parker RA, Bellamine A. The role of protease inhibitors in the pathogenesis of HIV-associated lipodystrophy: cellular mechanisms and clinical implications. Toxicol Pathol. 2009;37(1):65–77. [PMC free article: PMC3170409] [PubMed: 19171928]
- 116.
- Diaz-Delfin J, del Mar Gutierrez M, Gallego-Escuredo JM, Domingo JC, Gracia Mateo M, Villarroya F, Domingo P, Giralt M. Effects of nevirapine and efavirenz on human adipocyte differentiation, gene expression, and release of adipokines and cytokines. Antiviral research. 2011;91(2):112–119. [PubMed: 21619898]
- 117.
- Hruz PW. Molecular mechanisms for insulin resistance in treated HIV-infection. Best Pract Res Clin Endocrinol Metab. 2011;25(3):459–468. [PMC free article: PMC3115529] [PubMed: 21663839]
- 118.
- Rietschel P, Hadigan C, Corcoran C, Stanley T, Neubauer G, Gertner J, Grinspoon S. Assessment of growth hormone dynamics in human immunodeficiency virus-related lipodystrophy. J Clin Endocrinol Metab. 2001;86(2):504–510. [PubMed: 11158000]
- 119.
- Dube MP, Stein JH, Aberg JA, Fichtenbaum CJ, Gerber JG, Tashima KT, Henry WK, Currier JS, Sprecher D, Glesby MJ. Guidelines for the evaluation and management of dyslipidemia in human immunodeficiency virus (HIV)-infected adults receiving antiretroviral therapy: recommendations of the HIV Medical Association of the Infectious Disease Society of America and the Adult AIDS Clinical Trials Group. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2003;37(5):613–627. [PubMed: 12942391]
- 120.
- Schmitz M, Michl GM, Walli R, Bogner J, Bedynek A, Seidel D, Goebel FD, Demant T. Alterations of apolipoprotein B metabolism in HIV-infected patients with antiretroviral combination therapy. J Acquir Immune Defic Syndr. 2001;26(3):225–235. [PubMed: 11242195]
- 121.
- Herink M, Ito MK. Medication Induced Changes in Lipid and Lipoproteins. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2000.
- 122.
- Martin A, Smith DE, Carr A, Ringland C, Amin J, Emery S, Hoy J, Workman C, Doong N, Freund J, Cooper DA. Mitochondrial Toxicity Study G. Reversibility of lipoatrophy in HIV-infected patients 2 years after switching from a thymidine analogue to abacavir: the MITOX Extension Study. AIDS. 2004;18(7):1029–1036. [PubMed: 15096806]
- 123.
- Norwood J, Turner M, Bofill C, Rebeiro P, Shepherd B, Bebawy S, Hulgan T, Raffanti S, Haas DW, Sterling TR, Koethe JR. Brief Report: Weight Gain in Persons With HIV Switched From Efavirenz-Based to Integrase Strand Transfer Inhibitor-Based Regimens. J Acquir Immune Defic Syndr. 2017;76(5):527–531. [PMC free article: PMC5680113] [PubMed: 28825943]
- 124.
- Rochira V, Guaraldi G. Growth hormone deficiency and human immunodeficiency virus. Best Pract Res Clin Endocrinol Metab. 2017;31(1):91–111. [PubMed: 28477736]
- 125.
- Edgeworth A, Treacy MP, Hurst TP. Thiazolidinediones in the treatment of HIV/HAART-associated lipodystrophy syndrome. AIDS Rev. 2013;15(3):171–180. [PubMed: 24002201]
- 126.
- Raboud JM, Diong C, Carr A, Grinspoon S, Mulligan K, Sutinen J, Rozenbaum W, Cavalcanti RB, Wand H, Costagliola D, Walmsley S. Glitazone, Lipoatrophy Meta-Analysis Working G. A meta-analysis of six placebo-controlled trials of thiazolidinedione therapy for HIV lipoatrophy. HIV Clin Trials. 2010;11(1):39–50. [PMC free article: PMC3733492] [PubMed: 20400410]
- 127.
- Amouyal C, Buyse M, Lucas-Martini L, Hirt D, Genser L, Torcivia A, Bouillot JL, Oppert JM, Aron-Wisnewsky J. Sleeve Gastrectomy in Morbidly Obese HIV Patients: Focus on Anti-retroviral Treatment Absorption After Surgery. Obesity surgery. 2018;28(9):2886–2893. [PubMed: 29781044]
- 128.
- Tsoukas MA, Farr OM, Mantzoros CS. Leptin in congenital and HIV-associated lipodystrophy. Metabolism. 2015;64(1):47–59. [PubMed: 25267014]
- 129.
- Jagdeo J, Ho D, Lo A, Carruthers A. A systematic review of filler agents for aesthetic treatment of HIV facial lipoatrophy (FLA). J Am Acad Dermatol. 2015;73(6):1040-1054 e1014.
- Review Drug-induced obesity and its metabolic consequences: a review with a focus on mechanisms and possible therapeutic options.[J Endocrinol Invest. 2017]Review Drug-induced obesity and its metabolic consequences: a review with a focus on mechanisms and possible therapeutic options.Verhaegen AA, Van Gaal LF. J Endocrinol Invest. 2017 Nov; 40(11):1165-1174. Epub 2017 Jun 28.
- Review Drugs Affecting Body Weight, Body Fat Distribution, and Metabolic Function-Mechanisms and Possible Therapeutic or Preventive Measures: an Update.[Curr Obes Rep. 2021]Review Drugs Affecting Body Weight, Body Fat Distribution, and Metabolic Function-Mechanisms and Possible Therapeutic or Preventive Measures: an Update.Verhaegen AA, Van Gaal LF. Curr Obes Rep. 2021 Mar; 10(1):1-13. Epub 2021 Jan 5.
- Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.[Cochrane Database Syst Rev. 2022]Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Crider K, Williams J, Qi YP, Gutman J, Yeung L, Mai C, Finkelstain J, Mehta S, Pons-Duran C, Menéndez C, et al. Cochrane Database Syst Rev. 2022 Feb 1; 2(2022). Epub 2022 Feb 1.
- Review Diabetes in People Living with HIV.[Endotext. 2000]Review Diabetes in People Living with HIV.Sarkar S, Brown TT. Endotext. 2000
- Review Thin Fat Obesity: The Tropical Phenotype of Obesity.[Endotext. 2000]Review Thin Fat Obesity: The Tropical Phenotype of Obesity.Kapoor N. Endotext. 2000
- Drugs That Affect Body Weight, Body Fat Distribution, and Metabolism - EndotextDrugs That Affect Body Weight, Body Fat Distribution, and Metabolism - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...