3.1. Adhesion to Subendothelium Following Vascular Injury
Under physiologic conditions, platelets circulate preferentially in close proximity to vascular walls [80,81]. However, they do not interact with endothelial cells, which provide a natural resistance to thrombosis. When the continuity of endothelial layer is disrupted and the underlying subendothelial matrix is exposed, a coordinated series of events are set in motion to seal the defect, as depicted in Figure 3.1. Platelets play the primary role in this process, and various substrates may mediate their adhesion to the vascular wall in response to injury.
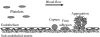
Figure 3.1
Cartoon illustrating the sequence of events involved in platelet-vessel wall interactions following vascular injury. Resting platelets circulating in discoid shape are shown on the left; vascular injury (depicted as endothelial denudation) results in (more...)
3.1.1. von Willebrand factor (vWF)
A key initial step in platelet adhesion to the site of injury involves interactions between the GP Ib-IX-V complex and the A1 domain of vWF in the exposed subendothelium [1,82,83]. vWF is a multimeric protein synthesized by endothelial cells and megakaryocytes; it is contained in Weibel-Palade bodies of endothelial cells, α-granules of platelets, in soluble form in plasma, as well as in the subendothelial matrix [84]. Immobilized vWF is sufficient to initiate platelet adhesion under flow, though the kinetics of these interactions vary according the hydrodynamic conditions [85]. A main determinant of the role of vWF on platelet adhesion is shear rate [85], which is a measure of the gradient of flow velocity relative to the distance from the vascular wall. Shear rate is a direct function of blood flow velocity and an inverse function of cross-sectional area, and is expressed in units of inverse seconds (s−1). Ex vivo studies with human blood demonstrate that GPIb-vWF is the primary adhesive interaction initiating platelet adhesion at high shear rates (> 1000 s−1 [85]), as occur in arterial microvessels, or arterioles [86]. Analogous studies in mice (an increasingly studied experimental model of thrombosis) reveal comparable shear rate-dependence of GPIb-vWF-mediated adhesion occurring at higher shear rates [87]. Of note, in the presence of a significant reduction in vascular cross-sectional area (as may result from thrombus, atherosclerotic plaque, vasoconstriction, etc.), wall shear rates may increase substantially and GPIb-vWF interactions may predominate [88,89]. Shear rate is a determinant of shear stress, a force per unit area applied parallel to the vascular wall (frequently described in units of dyn/cm2). The relevance of shear stress to platelet-endothelial interactions is discussed in Section 3.2.
In addition to its role in mediating platelet-vessel wall interactions, vWF also serves as the carrier molecule for coagulation factor VIII. The relevance of vWF in hemostasis and thrombosis is illustrated in humans by von Willebrand disease, a group of bleeding disorders characterized by varying degrees of vWF deficiency [90]. Similarly, mice with genetically induced vWF deficiency demonstrate delayed platelet interactions with vascular walls following injury [91,92].
3.1.2. Collagen
Endothelial cells provide a barrier to the interaction of platelets in flowing blood with various types of collagen present in the subendothelial matrix [93]. The platelet receptors GPVI and α2β1 mediate the interaction with collagen, though these interactions require platelet capture mediated by GPIb-vWF interactions. Both GPVI and α2β1 mediate collagen-induced platelet activation under flow conditions, though the precise sequence of these interactions is not entirely clear. For example, some studies on human platelets ex vivo suggest that initial binding of GPIb complex to vWF results in a conformational change of the α subunit of collagen receptor α2β1, enhancing its affinity to collagen [94], and subsequently enabling GPVI-collagen interaction. Others suggest an essential, cooperative role for both GPVI and α2β1 in mediating platelet adhesion at the site of injury, with GPVI signaling first and subsequently activating α2β1 [95]. Further, studies in mice with genetic deficiency of α2β1 reveal discordant results, with reports of prolonged [95] as well as normal [96] bleeding response following transection of the tail (tail bleeding time). The relative contributions of GPVI and α2β1 to platelet interactions with collagen, and how they relate to GPIb-vWF mediated adhesion in vivo remain to be elucidated fully.
3.1.3. Other Subendothelial Components
The subendothelium contains various other substrates capable of binding to and activating platelets, including laminin, thrombospondin, fibronectin, and vitronectin, among others [54,97]. These substrates may bind to the integrins described earlier (αIIbβ3 and α2β1) as well as other β1 and β3 integrins. The relative role of each of these subendothelial matrix components in hemostasis and thrombosis is incompletely understood.
3.2. Endogenous Mechanisms Preventing Platelet Adhesion to Endothelium
The integrity of the endothelial lining provides a barrier preventing platelet adhesion to subendothelial substrates, and focal removal of endothelium (denudation) has long been studied as a mechanism of platelet adhesion to vascular walls. However, increasing evidence both in vitro and in vivo demonstrates that in response to various stimuli, platelets may adhere to the vascular endothelium in the absence of endothelial denudation. Various mechanisms prevent adhesion of platelets to endothelial cells under normal conditions; these are outlined below.
3.2.1. Nitric Oxide
Nitric oxide (NO) was initially discovered as an endothelial-derived relaxing factor [98,99]; it is now recognized to mediate a plethora of physiologic processes throughout a broad range of cells. NO is derived from L-arginine and oxygen in the presence of several cofactors; endothelial NO synthase (eNOS) is one of the three isoforms of NOS [100]. eNOS is expressed constitutively on endothelial cells though is activity may be regulated actively by a variety of mechanisms [101]. Endothelial cells may release NO in response to agonists binding to endothelial receptors (e.g., bradykinin, histamine, ATP, etc.) or by blood flow-induced shear stress. Many of the biological actions of NO released by endothelial cells, including vasodilatation, are mediated by activation of soluble guanylate cyclase resulting in increased intracellular cyclic guanosine monophosphate (GMP) [102]. NO has also been shown to exert physiologic roles via cyclic GMP-independent mechanisms, including nitrosylation of a host of target proteins [103]. The role of NO as an inhibitor of platelet adhesion and activation has been described in a number of models [100,104–107], although the mechanisms involved remain to be fully clarified. Both cyclic GMP-dependent and – independent mechanisms have been reported to contribute to the NO-induced inhibition of platelet adhesion and activation [105,108]. Although the antiplatelet actions of NO are well documented from experiments on isolated platelets, the relative contribution of endogenous NO as an inhibitor of platelet adhesion in vivo is not straightforward. Experimental models have examined this question using a variety of approaches, including inhibitors of NOS, pharmacologic agents that release NO, and mice with genetic deficiency of eNOS under various conditions, with evidence of discordant findings [107,109–111]. The role of NO on platelet adhesion in vivo, particularly under conditions associated with endothelial dysfunction, are likely highly complex. For example, the balance between NO and reactive oxygen species has been suggested as a key determinant of platelet adhesion in some experimental models [112]. Further, NO may also inhibit leukocyte adhesion to vascular endothelium [113], and this may impact leukocyte-dependent platelet adhesion to endothelium, as outlined in Section 3.3. Understanding the role of NO in platelet-endothelial adhesion in vivo is complicated further by conflicting reports on whether platelets express a functional form of eNOS [114–116].
3.2.2. Prostacyclin
Prostacyclin (PGI2) is the main product of arachidonic acid metabolism on endothelial cells, and shares some functional similarities with nitric oxide [117]. PGI2 is also an endothelial-derived relaxing factor and is synthesized in response to various stimuli that release NO, including shear stress. Endothelial prostacyclin synthesis depends on cyclooxygenase-1 (COX-1) and prostacyclin synthase [118]. In addition to being a vasodilator, PGI2 has long been known to be a potent inhibitor of platelet aggregation [119,120]. While the role of PGI2 as inhibitor of aggregation is well established, fewer reports suggest an inhibitory effect on platelet adhesion on injured endothelium in vivo or in ex vivo experiments [121–123]. While NO signaling involves cyclic GMP, prostacyclin signaling involves cyclic adenosine monophosphate (AMP); NO and prostacyclin seem to have synergistic effects as inhibitors of platelet activation [118]. The effects of prostacyclin on platelets and blood vessels contrasts with that of another cyclooxygenase metabolite, thromboxane A2, a vasoconstrictor and stimulator of platelet activation. A balance between these metabolites is presumed to be relevant for platelet activation in vivo and to mediate the beneficial effects of low-dose aspirin used clinically as an antithrombotic [118].
3.2.3. Ecto-adenosine diphosphatase (ADPase)
Endothelial ecto-ADPase (CD39/nucleoside triphosphate diphosphohydrolase) is an integral component of the endothelial cell surface membrane; its antiplatelet effects are mediated by metabolizing ADP, which is a potent stimulator of platelet aggregation [124]. CD39 seems to inhibit platelet activation indirectly, and not via a direct interaction with platelets; this characteristic differs from the roles of NO and PGI2 outlined above. A soluble form of CD39 has been demonstrated to exert antithrombotic effects in experimental models and has been proposed as a novel therapeutic approach in patients with thrombotic disorders [125].
3.2.4. Endothelial Surface Layer
The interface between flowing blood and endothelial cell membrane contains a hydrodynamically significant layer, composed mainly of glycoproteins and proteoglycans. This layer, known as glycocalyx, is increasingly recognized as a physiologically relevant structure in regulation of endothelial permeability, responses to shear stress, and leukocyte-endothelial interactions [126]. In addition, the glycocalyx may represent another endothelial mechanism preventing platelet adhesion, given its physical location, and experimental evidence of platelet adhesion to endothelium following disruption of the glycocalyx [127,128]. Precise determinations of glycocalyx dimensions in the vasculature are limited by the physical characteristics of the layer, which may be subject to alteration during processing for ultrastructural studies [129], such as those depicted in Figure 3.2. These characteristics likely result in the broad range of reported values, ranging from approximately 20 nm to nearly 1 μm [130–132]. The endothelial glycocalyx is currently an area of active investigation in endothelial biology and may become a target for therapeutic manipulations, including for its potential effects on platelet adhesion [127,133].

Figure 3.2
Transmission electron microscopy image of rat mesenteric microvascular endothelium, following perfusion with Alcian Blue, using techniques based on those described by van den Berg, et al. [129]. An electron dense layer is evident adjacent to the endothelial (more...)
3.3. Mechanisms of Platelet Adhesion to Endothelium
A variety of inflammatory states may result in platelet adhesion to endothelial cells in the absence of denudation, and without evidence of significant alterations in the endothelial integrity. These processes may result from inhibition of the endogenous mechanisms preventing platelet adhesion and/or by inducing endothelial release of molecules that promote platelet adhesion. Further, several of these inflammatory conditions result in leukocyte adhesion to endothelial cells, primarily in the post-capillary venules of the microcirculation, with presence of leukocyte-platelet adhesion at those sites.
Platelet adhesion to endothelial cells has been documented experimentally in a number of inflammatory states, including ischemia/reperfusion injury, exposure to endotoxin, sickle cell disease, and hypercholesterolemia, among others [39]. These studies frequently involve the technique of intravital video microscopy, by monitoring trafficking of exogenously administered fluorescent-labeled platelets in the microcirculation [134], as shown in Figure 3.3. Several mechanisms for platelet adhesion to endothelial cells have been suggested.

Figure 3.3
Adhesion of fluorescently-labeled platelets to endothelial cells of a mouse cremaster venule, under endotoxin-induced inflammation. Exogenous platelets were labeled with a fluorescein-based dye (green) and infused into a recipient mouse for monitoring (more...)
3.3.1. Endothelial P-Selectin-Dependent Mechanisms
Many of the inflammatory conditions noted above are associated with endothelial activation and release of P-selectin stored in granules known as Weibel-Palade bodies. Exposure of P-selectin on the surface of endothelium may promote platelet adhesion by binding to GPIb complex on platelets. This mechanism appears to mediate platelet adhesion in experimental models of sickle cell disease and endotoxemia [135,136].
Endothelial cell P-selectin also mediates the initial transient interactions of leukocytes with endothelial cells (rolling), which precedes their firm adhesion to endothelium. A counter-ligand on leukocytes is P-selectin glycoprotein ligand-1 (PSGL-1). In some inflammatory states, platelet adhesion to endothelium depends largely on leukocyte adhesion, since depletion of circulating neutrophils prevents adhesion of platelets. Leukocyte-dependent platelet adhesion has been documented in various models including ischemia/reperfusion injury, endotoxemia, hypercholesterolemia, and corneal wound injury [39,137–139]. In many of these models, recruitment of leukocytes and platelets to vascular walls reveal marked temporal and spatial correlation, as shown in Figure 3.4. The adhesive mechanisms mediating platelet-leukocyte interactions in the various inflammatory states remain to be characterized fully. Platelet P-selectin-Leukocyte PSGL-1 is a likely mediator of platelet-leukocyte adhesive interactions [140]. However, several other mechanisms mediating these interactions have been proposed, including platelet GPIbα-leukocyte CD11b/CD18, platelet intercellular adhesion molecule-2 (ICAM-2)-leukocyte lymphocyte function-associated antigen-1 (LFA-1), and platelet junctional adhesion molecule-3 (JAM-3) and leukocyte macrophage antigen-1 (Mac-1) [43,140,141]. The relative contribution of the multiple potential mediators of platelet-leukocyte adhesive interactions in inflammatory disorders is unclear. Of interest, while leukocytes promote platelet adhesion in these models, evidence of platelets promoting leukocyte adhesion has been demonstrated experimentally, since depletion of circulating platelets markedly reduces leukocyte recruitment in various models [139,142]. These findings suggest an elaborate inter-dependence between platelet and leukocyte adhesion to endothelium in inflammation, and represents an area of active investigation.

Figure 3.4
Platelet and leukocyte adhesion to endothelial cells in vivo in a mouse model of inflammation induced by corneal injury. A) Immunofluorescence of a venule in the corneal limbus, with abundant neutrophils (green) and platelets (red); the arrows depict (more...)
3.3.2. Endothelial vWF-Dependent Mechanisms
Endothelial vWF is also stored in Weibel-Palade bodies, and is released in response to similar stimuli as those that induce endothelial P-selectin release. The newly released form of vWF is larger and more adhesive than vWF present in plasma, also known as ultralarge (UL) vWF. This multimeric form is normally cleaved by the plasma metalloprotease, ADAMTS-13 [143]. The ULVWF anchors to the endothelial surface in part by P-selectin, and promotes platelet adhesion via interaction with GPIb complex on platelets [36,143,144]. In the absence of cleavage, the ULVWF multimers result in long string-like structures, which support platelet adhesion with a “beads-on-a-string” appearance [145]. Figure 3.5 illustrates platelets on ULVWF on endothelial surface in vitro; similar findings have been demonstrated in vivo [146]. As discussed in more detail with regards to microvascular thrombosis (Section 6.3), deficiency of ADAMTS-13 is associated with some clinical cases of severe thrombosis, including thrombotic thrombocytopenic purpura [147], and severe forms of infections [148,149]. Conceivably, ADAMTS-13 and vWF may become future therapeutic targets in thrombosis associated with inflammation [150].

Figure 3.5
Platelet adhesion on ULVWF strings on endothelial cells in culture in a parallel-plate flow chamber, demonstrating a characteristic “bead-on-a-string” appearance (arrows). Bar = 10 μm. Figure courtesy of Jing-Fei Dong, M.D., (Baylor (more...)
3.3.3. Other Mechanisms
While PSGL-1 expression on leukocytes is well characterized as a mediator of leukocyte-endothelial and leukocyte-platelet interactions, some reports suggest that endothelial cells may express functional PSGL-1 [151,152]. By binding to P-selectin on the surface of activated platelets, endothelial PSGL-1 may represent another mechanism of platelet adhesion to endothelium during inflammation [151]. Further, some data suggest that platelets may also express functional PSGL-1 [153]. The physiologic role of the putative platelet PSGL-1 and of endothelial PSGL-1 on platelet-vessel wall interactions remains to be established.
Another proposed mechanism of platelet adhesion to endothelium in inflammation is via platelet GPIIb/IIIa, through its known interaction with fibrinogen [85,154]. In an experimental model of ischemia-reperfusion injury, platelet GPIIb/IIIa mediates platelet adhesion to fibrinogen deposited on the endothelial surface. In these models, fibrinogen appears to bind to endothelium via intercellular adhesion molecule-1 (ICAM-1) on the surface of inflamed endothelial cells [154,155].
Adhesive interactions of platelets with endothelial cells are increasingly recognized in inflammatory states, and they provide insight into the known links between inflammation and thrombosis. Further, adhesion of platelets to endothelium (and to leukocytes) contributes to the roles of platelets beyond their role in hemostasis and thrombosis, including their function as mediators of acute inflammation. Our knowledge of this field is evolving.
Publication Details
Copyright
Publisher
Morgan & Claypool Life Sciences, San Rafael (CA)
NLM Citation
Rumbaut RE, Thiagarajan P. Platelet-Vessel Wall Interactions in Hemostasis and Thrombosis. San Rafael (CA): Morgan & Claypool Life Sciences; 2010. Chapter 3, Platelet Adhesion to Vascular Walls.




.&p=BOOKS&id=53456_fig3.5.jpg "Click on image to zoom")