

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Annual Reviews Collection [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2002 Nov.

Reproduced from Annu. Rev. Biophys. Biomol. Struct. 2000. 29:327-359.
This review describes how kinetic experiments using techniques with dramatically improved time resolution have contributed to understanding mechanisms in protein folding. Optical triggering with nanosecond laser pulses has made it possible to study the fastest-folding proteins as well as fundamental processes in folding for the first time. These include formation of α-helices, β-sheets, and contacts between residues distant in sequence, as well as overall collapse of the polypeptide chain. Improvements in the time resolution of mixing experiments and the use of dynamic nuclear magnetic resonance methods have also allowed kinetic studies of proteins that fold too fast (≳103 s-1) to be observed by conventional methods. Simple statistical mechanical models have been extremely useful in interpreting the experimental results. One of the surprises is that models originally developed for explaining the fast kinetics of secondary structure formation in isolated peptides are also successful in calculating folding rates of single domain proteins from their native three-dimensional structure.
Introduction and Overview
Protein folding is a subject that is attracting scientists from a wide range of disciplines (34). The interest arises from the explosion of information on protein sequences and the challenge of understanding one of the most fundamental biochemical processes. The protein-folding problem is generally divided into two parts. In the first the goal is to predict the three-dimensional structure from the amino acid sequence. The second part is the no less daunting task of understanding the relation between protein sequences and mechanisms of folding. In protein folding, the mechanism is the distribution of microscopic pathways that connect the myriad of structures of the denatured state with the unique structure of the native state.
Over the past decade major advances have occurred in both theoretical (7, 19, 29, 37, 50, 83, 85, 103, 115) and experimental approaches to investigating protein-folding mechanisms. A turning point in experimental studies was the recognition that it is essential to first characterize the kinetics and thermodynamics of folding in the simplest systems. The idea of studying small, single domain proteins without additional chemical complexity from disulfide bridges, metals, or other cofactors began with the equilibrium and kinetic experiments on chymotrypsin inhibitor 2 (CI2) (56). CI2 was found to fold and unfold as a simple two-state system with no kinetic intermediates. In subsequent work, the participation of individual residues in the transition state for folding was investigated by studying the relative effects of mutations on folding rates and equilibrium constants (35, 54). The studies on CI2 sparked considerable interest among experimentalists in carefully characterizing the kinetics, thermodynamics, and effects of mutations in other small, single domain proteins (34, 55).
A second major advance in experimental studies has resulted from the introduction of a new generation of experiments with dramatically improved time resolution. The contribution of these studies to our understanding of protein folding is the subject of this review. Until just a few years ago, folding kinetics were studied almost exclusively using stopped-flow techniques. In this experiment, protein folding is initiated by rapidly mixing a chemically denatured protein solution with a buffer to dilute the denaturant. The kinetics of folding are then monitored with one of several optical spectroscopic methods. Similarly, unfolding can be initiated by mixing a native protein solution with concentrated denaturant. Stopped flow experiments have yielded an enormous amount of valuable information that has provided the basis for much of what we know about the kinetics of folding and unfolding. It does, however, have a fundamental limitation—poor time resolution. The time required to mix solutions and move them into an observation cuvette is generally at least a few milliseconds. This is the so-called “dead time” of the instrument. A typical observation in many experiments has been that much, and in some cases all, of the spectroscopic changes associated with folding already occur within this dead time (64, 72, 95, 98). Furthermore, it was known for some time from studies on synthetic polymers that elementary processes such as α-helix formation are too fast to be observed in stopped-flow experiments (44).
A second motivation for developing fast kinetic methods has been to provide a much-needed reality check on computer simulations, which are flooding the protein-folding literature. Our conceptualization of the folding process is strongly influenced by the results of simulations, so it is important to perform real experiments on time scales that can be directly compared with the computer experiments. So far the most important insights have come from simulations of simplified representations of proteins in lattice and off-lattice models (19, 29, 83, 85, 103, 115). Such models provide simple, concrete examples that can be extremely helpful in clarifying both theoretical and experimental issues. However, the most detailed and potentially realistic kinetic simulations employ all-atom molecular dynamics calculations. With few exceptions (23, 24, 75), these calculations have been restricted by computer time to one or just a few trajectories of tens of nanoseconds or less. They are therefore not yet capable of direct simulation of protein folding (see, however, Ref. 30) but are rapidly approaching time scales long enough to simulate secondary structure formation.
Finally, development of fast methods has been motivated by theoretical studies that have introduced an energy landscape approach to protein folding (9). A particularly interesting result from calculations of free energy surfaces is that the barriers to protein folding should be quite small, suggesting that for the fastest folding proteins the barrier might disappear altogether. This barrierless or so-called downhill folding could produce unusual (i.e. nonexponential) kinetics (8). The energy landscape perspective also suggested that in many proteins the multiphasic kinetics observed in stopped-flow experiments arise from the escape of misfolded or partially folded molecules from traps in the landscape and not from a stepwise formation of native structure (8, 9). For such proteins the fast productive routes to the native state were yet to be resolved (61). A notable example is cytochrome c in which folding is multiexponential and requires hundreds of milliseconds (33, 110). In this case kinetic complexity results from transient covalent binding to the heme iron of a nonnative histidine almost 50 residues distant along the sequence from the native methionine ligand. Once binding of this histidine is blocked, folding is submillisecond, too fast to be observed by the stopped-flow method (33, 110).
With this background several groups of biophysical scientists working in the area of time-resolved spectroscopy were attracted to the protein-folding problem. Time-resolved spectroscopy with pulsed lasers had been quite successful in functional studies on proteins, so it seemed natural to take advantage of this powerful technology to investigate protein folding. It was, moreover, apparent that finding ways to initiate protein folding with laser pulses would provide dramatically improved time resolution. Recognizing the importance of experiments in which the concentration of chemical denaturant is rapidly changed to initiate folding or unfolding, a second line of investigation has been directed toward the development of methods to improve the time resolution in mixing experiments. These new, fast-folding techniques have permitted the investigation of fundamental processes in protein folding, including formation of α-helices, β-sheets, and contacts between residues distant in sequence as well as overall collapse of the polypeptide chain and folding of the very fastest proteins.
Although these new techniques have allowed the study of previously unobserved processes in protein folding, it is only quite recently that they have provided major new insights into folding mechanisms. These include a much deeper understanding of the mechanism of secondary structure formation, the introduction of the notion of an upper limit on the rate of protein folding (a “speed limit”), the discovery of unusual kinetics suggesting that very fast folding is continuously downhill in free energy, and direct observation of polypeptide collapse prior to formation of the native structure. One of the surprises from this work is that simple statistical mechanical models, originally developed for explaining the fast kinetics of secondary structure formation in isolated peptides, are remarkably successful in quantitatively explaining the kinetic behavior of small proteins. This finding has several important implications, including the possibility that the underlying physics of folding may be much simpler than previously thought.
Basic Ideas of Fast-Folding Methods
There have been several recent reviews on fast-folding techniques, so here we only present a very brief description of the basic physical ideas underlying these methods (14, 43, 52, 99). Methods for rapid initiation of protein folding can be roughly classified into three categories—photochemical triggering, temperature or pressure jump, and ultrarapid mixing methods. The first fast-folding study employed a photochemical trigger—the photodissociation of carbon monoxide from denatured cytochrome c (17, 59). This experiment takes advantage of the fact that CO binds much more strongly to the heme of the denatured protein than to the heme of the native protein. Photodissociation of CO initiates folding because the CO-free protein is much more stable (Figure 1). The experiment has unlimited time resolution for folding experiments because photodissociation occurs in less than one picosecond. A conceptually similar, but more general, method utilizes a photo-induced electron transfer reaction to initiate folding (89) (Figure 1). The first such experiment was also performed on cytochrome c (20, 73, 89, 113). The reduced form of cytochrome c is more stable than the oxidized form, primarily due to the increased stability of the bond between methionine 80 and the heme iron in the native protein. Optical excitation of a ruthenium bL-pyridine complex or NADH produces a long-lived excited state that is a powerful reductant. Injection of an electron reduces the heme iron of cytochrome c from ferric to ferrous in a few microseconds in a diffusion-limited bimolecular reaction and initiates folding. This technique has also been used to trigger folding of cytochrome b562 (121, 122) and myoglobin (123).
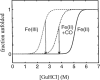
A different kind of photochemical trigger uses a photolabile disulfide (70, 118), which does not require a metal site, so in principle it can be used as a trigger for a much wider range of proteins. In this method, segments of a polypeptide chain are cross-linked by an aromatic disulfide. An appropriately engineered cross-link constrains the peptide or protein in a misfolded conformation, allowing folding to be triggered by sub-picosecond photo-cleavage of the disulfide bond.
A second class of optical triggers uses an intense laser pulse to produce a rapid temperature jump. This is a much more generally applicable method because it can be used to perturb the folding/unfolding equilibrium for any process that produces a significant enthalpy change. This was first done in a protein-folding experiment by heating a solution of a dye with a mode-locked picosecond laser operating at 532 nm (90). The time resolution of this experiment was ~70 ps—the time required for thermal diffusion from the hot dye molecule to the solvent. A much more commonly employed method has been to directly heat water 10–20°C by shifting the fundamental of a Q-switched Nd:YAG laser at 1064 nm to a longer wavelength where water strongly absorbs (3, 4, 116, 120). The solution is heated during the ~5 ns duration of the pulse by vibrational excitation of an O-H stretching overtone of water. The Eigen T-jump method of resistive heating with an electrical discharge has also been used to study folding kinetics with ~10 μs time resolution (81, 82). Another perturbation method, pressure jump, has recently been employed to study folding kinetics (57). In this experiment, a stack of piezo-electric crystals is used to change the pressure 100–200 bar in 50–100 μs. The pressure change alters the folding/unfolding equilibrium because of the associated volume change.
Use of chemical denaturants, such as urea and guanidinium chloride, has played a central role in protein-folding experiments. Almost all proteins can be unfolded and remain soluble at sufficiently high denaturant concentrations, so dilution of denaturants continues to be an extremely important way of initiating protein folding. For this reason, there has been considerable effort to improve the time-resolution in mixing experiments by using continuous flow methods. Two such methods have recently been applied to protein folding, one based on turbulent mixing (18, 88, 104–106, 112, 124) and another based on hydrodynamic focusing (63, 93). In the turbulent mixing method liquids are forced through a small gap at high velocities (96). The turbulence created by the high shear forces “breaks” the liquids into very small volume elements called turbulent eddies. Mixing is rapid (1–10 μs) because diffusion of the denaturant occurs over short distances. In this experiment the mixed liquids flow continuously at constant velocity. The kinetics are monitored at various positions along the jet emerging from the mixer, time being measured by the distance from the mixing region and the flow velocity. The first time point in the kinetic measurements is significantly longer than the mixing time because the mixed solution must typically flow through a volume that is inaccessible to the monitoring probe. Nevertheless, the dead time of these instruments is only 50–200 μs, an improvement of more than an order of magnitude over the stopped flow method.
The second rapid mixing method employs hydrodynamic focusing to create a micron or submicron-wide stream of protein solution flowing at constant velocity in contact with a surrounding flowing reservoir (63, 93). Mixing occurs by diffusion into and out of the narrow stream. Solutions have been focused to diameters as small as 50 nm, corresponding to ~1 μs mixing times (63). It should therefore be possible to obtain significant reduction in effective dead time compared to the turbulent mixing methods. An advantage of the hydrodynamic focusing method is that the flow is laminar. Consequently, the concentration of all species at all positions in the mixing device, including the mixing region, can in principle be calculated theoretically and also determined by experiments. This technique has allowed measurements of submillisecond, time-resolved small angle X-ray scattering for the determination of the radius of gyration of transient structures in protein folding (93).
Finally, protein-folding kinetics can be studied at equilibrium using dynamic nuclear magnetic resonance (NMR) methods (12, 13, 53). For a protein undergoing a simple two-state folding/unfolding transition, both the folding and unfolding rates can be derived from the measured lineshape if the resonant frequencies and transverse relaxation times for the two states are known. This method is useful for determining folding rates in the 10 μs-10 ms range under conditions where there is a significant population of both folded and unfolded states.
Elementary Processes in Folding
Contact Formation
Perhaps the simplest elementary process in protein folding is the formation of a contact between two residues of an unfolded polypeptide chain. In spite of its importance, this process was not investigated until the photodissociation experiments on cytochrome c (59). Time-resolved spectroscopy with nanosecond lasers was used to monitor heme absorption following photodissociation of the carbon monoxide complex of denatured reduced cytochrome c (Figure 1). The first change in absorption occurs with a relaxation time of ~3 μs and was identified as binding of methionine to the heme. Detailed kinetic modeling led to an estimate of the time constant for methionine binding and dissociation of ~40 μs and ~4 μs, respectively. In subsequent experiments, the binding of free methionine to the heme attached to a small fragment of the cytochrome c polypeptide (the 11–21 undecapeptide) was studied, and it was found that the bimolecular rate is ~2 × 108 M-1 s-1, close to the diffusion limit (48, 49). Analysis of a simple two-step mechanism, in which binding occurs by first forming an encounter complex followed by formation of the heme-methionine covalent bond, showed that the observed unimolecular rate is almost purely diffusion limited. The heme iron is covalently bonded to a histidine at position 18, while the two methionines in the sequence are located at positions 65 and 80. The measured time of 40 μs is therefore the time required for diffusion-controlled contact between regions of the polypeptide separated by ~50 residues. Together with the length scaling from polymer theory (15, 45, 111), this number could be used to estimate the formation rate of a contact between residues separated by any number of residues. For example, ~3 μs was estimated for the formation of a short contact between residues separated by ~10 peptide bonds using the n-3/2 scaling for a random walk chain (see below). A second important result from these experiments was the calculation of the effective diffusion coefficient ≈5 × 10-7 cm2 s-1 for the relative motion of heme and methionines, assuming a Gaussian distance distribution (111). This value is comparable to the diffusion constant ≈(1.5–8) × 10-7 cm2 s-1 for the relative motion of a donor and acceptor in unfolded ribonuclease A, determined from the effect of Förster excitation energy transfer on the donor fluorescence decay kinetics (11).
The cytochrome c experiments indicated that it would be interesting to develop a more generic method for investigating contact formation in unfolded polypeptides. One approach to this problem has been to label the ends of small peptides with probes that undergo triplet-triplet energy transfer upon contact (5). In this experiment the triplet donor was a three-ring organic compound, thioxanthone, attached to the N-terminus of peptide, while the triplet acceptor was naphthalene attached to the peptide as 1-naphthyl alanine (Figure 2). The triplet state of thioxanthone has a lifetime of ~30 μs and transfers its energy in a diffusion-limited process to the triplet state of naphthalene upon contact. A nice feature of this experiment is that energy transfer was clearly demonstrated by the disappearance of the thiox-anthone triplet-triplet absorption spectrum simultaneously with the appearance of the naphthalene triplet-triplet absorption spectrum. Peptides were investigated that contain 1–4 glycine/serine pairs, corresponding to a spacing of 3–9 peptide bonds between the donor and acceptor. Exponential kinetics was found in all cases, with time constants varying from 20 ns for the shortest peptide to 50 ns for the longest.
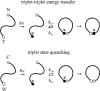
One limitation of this triplet-triplet energy transfer method is that, with the current probes, positioning of the triplet states for efficient energy transfer requires the use of ethanol/water mixtures. Also, the use of extrinsic probes makes the study of proteins and the exploitation of site-directed mutagenesis more difficult. Another method has been developed for measuring contact formation rates that does not suffer from either of these limitations (67). In this method, contact formation was measured from the quenching of the triplet state of tryptophan at the C-terminus of a peptide by cysteine at the N-terminus (Figure 2). Earlier work had suggested that cysteine is a much more efficient triplet quencher than any other amino acid, exhibiting a near diffusion-limited bimolecular rate for the quenching of free tryptophan (42). The tryptophan triplet state is most probably depopulated by transfer of an electron to the sulfur of the quencher at distances close to van der Waals contact. The long lifetime (> 50 μs) of the tryptophan triplet state, moreover, permits quenching rates to be measured over a relatively wide dynamic range by measuring the decay of the triplet-triplet absorption spectrum. The peptides studied contained the repeating amino acid triplet—alanine/glycine/glutamine—to avoid any specific secondary structure formation. Varying the number of peptide bonds between tryptophan and cysteine from 4 to 19 yielded contact time constants of 30 ns to 150 ns, consistent with the values found in the triplet-triplet energy transfer study.
Although both kinds of triplet state experiments yield times ~50-fold smaller than the earlier estimate of ~3 μs for contact between residues separated by ~10 peptide bonds (49), they are not inconsistent with this prediction. At least two factors contribute to this difference. For an idealized chain with a Gaussian end-to-end equilibrium distribution, this rate is given by: 3Da/[(π/6)1/2 〈r2〉3/2]; where D is the effective relative diffusion constant of the two ends, a is the contact radius, and 〈r2〉 is the mean squared end-to-end distance (111). The contact radius for the tryptophan-cysteine interaction is larger than for the methionine-heme encounter complex of cytochrome c, and the heme is only accessible to the methionine on one side, so the effective value of a is larger in the quenching experiments. Also, the peptides are rich in glycine and therefore have smaller 〈r2〉 compared to cytochrome c, which has only an average of 1 glycine per 6–7 residues between the heme and methionines.
The above results suggest that tryptophan triplet lifetime measurements can provide a useful method for investigating the kinetics of forming a specific intramolecular contact in an unfolded or folding protein. They also show that this method can be used to address fundamental questions on the dynamical properties of polypeptides. Foremost among these are the length and composition dependence of contact formation rates. A maximum in the loop probability as a function of chain length has been predicted at about 10 residues for a generic polypeptide (15, 45, 114). In these calculations, formation of an end-to-end contact in longer chains is less probable because of the larger entropy decrease, while forming a contact in shorter chains is disfavored because of chain stiffness. In the simplest picture, the maximum in the loop probability should be reflected as a maximum in the contact formation rate [the rate of dissociation of the contacting polymer ends ( =3 D / a2) is independent of chain length]. No such maximum was observed in the glycine-containing peptides, presumably because of their larger flexibility compared to the generic peptide considered in the theoretical calculations. This question is now being studied using less flexible peptides (LJ Lapidus, unpublished results). Another important aspect of studies of contact formation, at least in small glycine-containing peptides, is that it occurs on a time scale that can be directly simulated by all-atom molecular dynamics calculations, simulations that have yet to be performed.
Helix-Coil Transition
Although both the thermodynamics and kinetics of the helix-coil transition have been studied experimentally and theoretically for over 40 years, it was not until the late 1980s that a significant number of peptides of the size and composition found in proteins were shown to be helical in pure aqueous solvents. Since that time extensive equilibrium studies of α-helix formation in isolated peptides have been carried out and theoretically analyzed (16, 71, 78). Peptides rich in alanine are particularly suitable because of their high helix-forming propensity. This together with their ready availability have made them prime candidates for kinetic investigations using fast kinetic techniques.
In the first such study a 21-residue peptide having the sequence X-(A)5 (AAARA)3-A-NH2 (X = succinyl) was investigated (120). A nanosecond laser temperature jump was used to perturb the helix-coil equilibrium, and infrared absorption measurements in the amide I region were used to monitor the decrease in helix content. The relaxation following the temperature jump was found to be biphasic with an unresolved (< 10 ns) amplitude and a larger amplitude 160 ns relaxation. Subsequent laser T-jump measurements with a fluorescent probe at the N-terminus (methyl amino benzoic acid instead of the succinyl group) showed only a single fast relaxation at ~20 ns (116). In this study a simple nucleation/propagation model was developed to explain both the infrared and fluorescence experiments. This model suggested that the fast relaxation results from helix propagation and partial melting of stretches of helix, while the slow relaxation corresponds to crossing a helix nucleation barrier (Figure 3). This barrier crossing occurs in both directions, corresponding to nucleation and growth of a new stretch of helix in one direction, and complete melting of a preexisting stretch of helix in the reverse direction. However, the model predicted that the N-terminal fluorescent probe would also show a comparable amplitude for the slow barrier crossing process, which was not observed.

This problem prompted further investigation of helix-coil transition kinetics in a very similar peptide, but one having an intrinsic fluorescent probe (117). In this peptide, with sequence Ac-WAAAH(AAARA)3-A-NH2, formation of the first turn of helix is signaled by quenching of tryptophan (W) fluorescence by the protonated histidine (H) (Figure 3). Both the temperature (117) and viscosity dependence (GS Jas, unpublished results) of the relaxation kinetics were studied. Again, only a single relaxation was observed, but with a rate (1/(220 ns)) very close to the slower phase observed in the infrared experiments on a very similar peptide described above. To understand this result, the model developed in the study of a β-hairpin was used (77, 80) (Figure 4), which takes into account both the side chain interactions and the secondary structure propensities of individual residues (117). According to the model, in phases corresponding to both, helix propagation and partial melting are slowed in this peptide. The much lower helical propensities of histidine and tryptophan, together with the histidine-arginine (i, i + 4) repulsion, slow incorporation of the 5 N-terminal residues into a helix growing from the C-terminal side, while melting of the N-terminus is slowed by the stabilizing tryptophan-histidine (i, i + 4) interaction. The net result is that the fast relaxation is slower and with a much smaller amplitude, so that the observed kinetics are dominated by crossing the nucleation barrier (117).
A similar peptide without the tryptophan-histidine probe has also recently been studied using time-resolved resonance Raman spectra measured with far UV (204 nm) excitation to probe helix content (68). The Raman spectrum of the helical peptide exhibits much weaker intensities for the amide II, amide III, and Cα-H bending bands than does the random coil, so the Raman spectrum provides several independent measures of helical content. A single exponential relaxation was observed over the temperature range studied (310 K–337 K), with τ = 250 ns at 300 K and an apparent activation energy of 7 kcal/mol. These results are very similar to those obtained from the fluorescence and infrared absorption experiments. No fast relaxation could be detected in these experiments even though the time resolution was ~3 ns.
With a statistical mechanical model, quantitative predictions of helix-coil kinetics in other peptides are straightforward. Using the same parameters from fitting the equilibrium and kinetic data on the tryptophan/histidine peptide, the kinetics of the infrared experiments were simulated (117). Excellent agreement with experiment was obtained, with calculated relaxation times of 4 ns and 220 ns and an amplitude ratio for the fast and slow relaxations of approximately 1:4. More recently the model has been modified to predict the kinetics of peptides having the length and composition found in helices of proteins (V Muñoz, unpublished results) incorporating the more detailed equilibrium description of the helix-coil transition contained in the program AGADIR (78, 79). These calculations show that relaxation times for 15–20 residue peptides span the range from a few nanoseconds for peptides having less than 1% helix at equilibrium to ~1 μs for peptides containing > 75% helix. The relaxation time is much shorter for very unstable peptides because melting, which is dominating the relaxation in these peptides, is downhill in free energy. On the other hand, the relaxation time for stable, protein-like sequences is longer than that of alanine-based peptides of comparable stability. This is a consequence of two factors. The entropy loss associated with forming a turn is greater for nonalanine peptides (alanine has the highest helical propensity), producing a larger nucleation barrier. To compensate for the lower helical propensity, stable helices require stronger side chain interactions, and these interactions must be broken for complete melting to occur, increasing the barrier in the reverse direction.
A surprising result has recently been reported from stopped-flow kinetic studies of helix formation monitored by circular dichroism (CD) in a 16-residue alanine-based peptide (21). In this study helix formation was initiated by dilution from 6 M guanidinium chloride. All of the equilibrium circular dichroism change was found to occur in a ~100 ms relaxation. To reconcile these results with the T-jump experiments, it was argued that at this high concentration of denaturant the peptide is completely unfolded and that the 100 ms relaxation corresponds to helix nucleation, while the much faster relaxation observed in the T-jump experiments arises from unzipping of existing helical sequences. If this explanation were correct, then the helix nucleation rate would be several orders of magnitude slower than previously thought. In an attempt to reconcile these discrepant observations, we reexamined the trypophan-histidine peptide using both stopped-flow and T-jump experiments. The stopped-flow experiments showed that helix formation as monitored by the tryptophan fluorescence is complete within the 3 millisecond dead time, while precise measurements of the kinetic amplitudes in T-jump experiments confirm that the fluorescence reaches its equilibrium value in less than a microsecond (117; GS Jas, unpublished results). Similar results were also obtained in the infrared (120) and Raman (68) studies. We should also point out that the statistical mechanical model predicts that the kinetics of helix formation from the completely unfolded state exhibit almost exactly the same relaxation time as that observed in the T-jump perturbation experiment. The origin of the 100 ms circular dichroism change remains unclear.
Helix-coil kinetics have also been observed in laser T-jump experiments on proteins. An interesting example is the study on apomyoglobin using infrared spectroscopy (31, 40, 41). By monitoring the infrared absorption at different wavelengths, it was possible not only to determine the change in total helix content, but also to distinguish between changes in solvent exposed helices and helices that are packed against other helices. Following a temperature jump, partially unfolded apomyoglobin exhibits a multiphasic relaxation. A rapid helix melting/formation relaxation at 50 ns has maximal amplitude at wavelengths characteristic of solvent-exposed helix (possibly helices C, D, or E in the denatured state). A slower relaxation at 120 μs has maximal amplitude at wavelengths for packed helices and may represent the cooperative unfolding and refolding of the core of the protein containing helices A, G, and H (31, 40, 41).
What is the relation between real and computer experiments on helix-coil kinetics? As mentioned in the introduction, one of the motivations for studying the kinetics of the helix-coil transition is that it occurs on time scales that are becoming accessible to all-atom molecular dynamics simulations. Ideally one would like to observe the helix form and completely melt many times in a single trajectory. To have adequate statistics to obtain equilibrium populations and rate constants, a large number of such trajectories would be required. Such simulations have recently been carried out on a β heptapeptide in methanol, which forms a left-handed 31-helix (in a β peptide a methylene group is inserted in the backbone between Cα and NH). The helix melts and reforms several times during the course of the 50 ns simulation (23, 24). The analysis of the residence time in helix and coil states yields an equilibrium constant close to the experimental value. Unfortunately, the kinetics of helix formation in this heptapeptide have not yet been studied experimentally.
No kinetic study by molecular dynamics is available yet for alanine peptides, but there is a hint from a single trajectory on a 13-residue alanine peptide in explicit water that accurate rates can be calculated (22). This alanine helix unfolds in the simulation at 373 K in 200 ps, compared to 700 ps obtained by extrapolating the experimental data to 373 K using the statistical mechanical model for this peptide (117). Another fundamental kinetic quantity that could be readily obtained by molecular dynamics is the time to add an alanine residue to a growing helix. This time comes directly from the modeling of the kinetic data and is found to be ~600 ps at 300 K, varying from ~50 ps at 373 K to ~2 ns at 273 K (117).
A number of important issues concerning the kinetics of the helix-coil transition remain to be resolved. These include the direct observation of the rate of helix growth and the exploration of the effects on the kinetics of amino acid substitution and chain length. While estimates of the growth rate can be obtained from models, it should be possible to directly observe this process by using time-resolved infrared or Raman measurements and a T-jump method with subnanosecond time resolution. Such studies might be facilitated by using peptides constrained by chemical modification to have prenucleated helices.
β -Hairpin Formation
Studies on β-sheets have not been nearly as extensive as those of the α-helix. The reason is that until recently it has not been possible to study β-sheets in isolation. The simplest β-sheet is one with just two antiparallel strands—a so-called β-hairpin. Isolated β-hairpins tend to aggregate at the high concentrations necessary to characterize their structure by nuclear magnetic resonance because the hydrophobic side chains that are required for stability also make them sticky. Over the past several years, however, several peptides have been shown to form β-hairpins that do not aggregate (38, 65). In the first kinetic investigation of β-hairpins, the 16-residue C-terminal fragment of the protein GB1 was studied (80) (Figure 4a). This peptide had been shown by NMR to have a high population of β-hairpin structure at room temperature (6). The folded conformation of the GB1 hairpin is stabilized by a hydrophobic cluster of 4 residues, including a tryptophan that can be used as an intrinsic fluorescent probe (Figure 4a). Upon formation of this cluster, the quantum yield of tryptophan fluorescence increases because the indole side chain becomes partially buried and is less accessible to quenching by water. To obtain an essentially independent probe, a dansylated lysine was attached to the C-terminus of the peptide, 13 residues distant in the sequence from the tryptophan. The absorption of the dansyl group occurs in the same wavelength range as the tryptophan emission and therefore acts as an efficient acceptor of excitation energy from the tryptophan. In the dansylated peptide, the tryptophan quantum yield is sharply reduced and changes in fluorescence are dominated by the energy transfer rate, which depends on the distance between the tryptophan and the dansyl groups.
Interestingly, the hairpin was found to fold in a few microseconds, more than an order of magnitude slower than α-helix formation in the alanine peptides (51). The surprising result was that, except for less cooperative thermal unfolding, the thermodynamic and kinetic behavior of this β-hairpin is very much like that of a small protein (80). In both the unmodified and dansylated peptide, the thermal unfolding curve could be explained with just two thermodynamic states—a folded state with an intact hydrophobic cluster and an unfolded state without the cluster and with a much larger average separation between the tryptophan and the dansyl group. In the laser temperature jump experiments, a single exponential relaxation was observed in both peptides with the same time constants of 3.5 μs at 288 K. The finding of single exponential progress curves with the same relaxation rates using essentially different probes is the hallmark of a two-state kinetic system. Another interesting finding in the kinetics was that the two-state analysis yields an apparently negative activation energy for hairpin folding (the observed relaxation rate, which is the sum of the folding and unfolding rates, shows a positive activation energy because unfolding of the hairpin has a positive and larger activation energy).
To explain these results a simple statistical mechanical model was developed (77, 80), which was subsequently used in the helix-coil study discussed above (117). An interesting aspect of the model is that it contains much of the underlying physics considered in the folding of a protein, i.e. the competition between loss of conformational entropy and stabilization from hydrophobic interactions and hydrogen bonds. Indeed, we shall see later that the model can be successfully applied to proteins. In this model, structures are classified according to their backbone conformation, with each peptide bond having either native or nonnative values for the ψ, φ dihedral angles. This idea was used previously in a simple model for protein folding (125). A natural reaction coordinate in such a model is the number of native peptide bonds (10). As can be readily seen from the free energy calculated as a function of this coordinate in the single sequence approximation (Figure 4b), there are only two regions of stability separated by a substantial barrier. One corresponds to the unfolded hairpin and one to the hairpin with the hydrophobic cluster intact but the ends frayed.
This immediately suggests that the peptide will behave like a two-state system both thermodynamically and kinetically. Indeed, solution of the differential equations with parameters that fit the experimental data shows a single exponential relaxation. This result is obtained with both the single sequence approximation (121 states) and the complete model (32,768 states). The projection of the free energy as a function of the number of native peptide bonds in the complete model also exhibits a barrier, showing the two-state behavior (ER Henry, V Muñoz, unpublished results). At the top of the free energy barrier, which is just before the transition state, the turn has formed, and the hairpin contains two of the seven hydrogen bonds connecting opposite strands (Figure 4b). The lower enthalpy of the transition state compared to the unfolded state (which has no stabilizing interactions in the model) explains the apparent negative activation energy for folding. Another interesting feature of the free energy profile (Figure 4b) is that hairpin formation is uphill in free energy until the onset of hydrophobic interaction between residues on opposite strands. Repositioning of the cluster closer to the turn would therefore be expected to speed up folding, without changing the difference in free energies between the unfolded and completely folded (i.e. unfrayed) peptide (Figure 4b). The idea of speeding up folding by decreasing the distance in sequence between stabilizing interactions will resurface when we discuss the calculation of protein folding rates (see above).
The statistical mechanical model developed to describe the β-hairpin kinetics imposes a mechanism in which the β-turn and proximal hydrogen bonds must form before the hydrophobic cluster. An alternative mechanism, which could also explain the data, was considered in the experimental study, but no detailed model was formulated (80). In this other mechanism, the molecule first forms a closed loop by docking of the hydrophobic cluster. The formation of the loop is rate-limiting and is followed by very fast formation of interstrand hydrogen bonds and the turn. Because the hairpin forms with two-state kinetics, it is not possible to distinguish between these two mechanisms experimentally. It may also not be realistic to answer this question with mutation experiments (φ analysis) because the stability of the hairpin is marginal (~1 kcal/mol at 273 K). All-atom molecular dynamics simulations might help to distinguish between the two mechanisms (27, 87). Two different strategies have been employed to overcome the limitation that folding and unfolding of the hairpin are too slow for currently available molecular dynamics simulations at experimental temperatures. In one, transition states found in high-temperature unfolding simulations were shown to behave like transition states at ambient temperature (87). The mechanism at the two temperatures was therefore assumed to be identical. In the other, Monte Carlo sampling was used to generate a large number of conformations that were then grouped according to their structure to produce free energy surfaces (27). In both studies, it was concluded that the hydrophobic cluster forms first. However, as with many simulation studies, structures were examined in great detail, but experimentally measured properties were not calculated. It was therefore not shown that the simulations are consistent with the major kinetic results, namely: the observation of a single exponential relaxation, the observed rates and apparent activation energies, and the observed changes in fluorescence quantum yields upon folding with and without Forster excitation energy transfer. It would also be important to know how the mechanism obtained from the simulations depends on the choice of force fields and parameters.
The laser temperature-jump experiments on the protein GB1 hairpin have provided the first glimpse of the time scales for formation of β-structure in isolation. These experimental results and their analysis with a simple statistical mechanical model, moreover, have exposed important issues that also arise in investigating the mechanism of protein folding. At this stage it is crucial to investigate other β-hairpin forming peptides to determine experimentally their range of kinetic and thermodynamic behavior. To this end a variant of a β-hairpin-forming peptide from ubiquitin (102) has been studied in which a tryptophan has been inserted in place of a tyrosine and a dansylated lysine added to the C-terminus. Folding in this system is more complex because of self-assembly into a trimer of hairpins (V Muñoz , FJ Blanco, R Ghirlando, unpublished results). However, at low concentrations where both the unfolded and folded peptide remain monomeric, the β-hairpin structure forms in ~30 μs at 30°C, only five times slower than the GB1 hairpin.
Fast Protein Folding
Speed Limit
An interesting issue raised from measurements of elementary processes in protein folding is the question of a speed limit (49). How fast can a protein possibly fold? Is there an upper limit for the rate of protein folding as there is for a bimolecular reaction in solution? For a bimolecular reaction, the upper limit is determined by the rate at which the reactants come together by diffusion. This problem was solved theoretically more than 80 years ago by Smoluchowski, who derived a simple analytical formula. No such theory has been developed yet for protein folding, so a semi-empirical approach was taken (49). It was reasoned that the rate of folding would be limited by the rate of polypeptide collapse, which would in turn be limited by the time required to form a small loop. From the experiment on cytochrome c discussed above, this minimum time for protein folding was estimated to be ~1 μs (49). We now know that this is comparable to the time required to make a hairpin or a stable natural helix, so it remains a reasonable estimate. This notion of a speed limit for protein folding of ~(1 μs)-1 has stimulated experimentalists to look for fast folding proteins and redesign them to fold even faster.
Several proteins fold with rates approaching this speed limit. The single chain arc repressor folds with a rate of ~104 s-1 obtained by extrapolating millisecond stopped flow data to zero denaturant concentration (97). Folding times as little as 600 μs were observed for oxidized cytochrome c (with misfolding blocked by binding to the extrinsic ligand, imidazole) using a rapid mixing, continuous flow method (18). The fastest measured folding rates have been determined for monomeric λ repressor using dynamic NMR methods (12, 13, 53). This small (80 residue), single domain protein exhibits two-state behavior (39). From lineshape analysis, rates were determined as a function of urea concentration, the shortest time constant being ~1 ms in 1.4 M urea solution. In a later study (12), rates at lower urea concentration were measured using the contribution of the exchange between folded and unfolded structures to the transverse relaxation time, T2. In the absence of urea, a folding rate of 5 × 103 s-1 was determined. This is the current speed record for a measured folding rate (from the denatured state, vide infra). A fast-folding mutant was also investigated (12), in which two of the glycine residues were replaced by alanine (G46A/G48A). In this case, however, even the T2 measurements were limited by the lack of population of the unfolded state at low urea concentrations. In 2.1 M urea the folding rate was already 3 × 103 s-1, with a rate extrapolated to zero urea concentration of ~105 s-1.
The electron transfer triggering method has been used to show that several heme containing proteins fold very fast. Extrapolation to zero denaturant concentration has led to an estimate for reduced cytochrome c of ~104 s-1 (73, 89, 113). Cytochrome b562 is a particularly striking case. This protein consists of a bundle of four helices wrapped around a heme group. Reduction of the heme iron from ferric to ferrous leads to a marked increase in stability, the concentration of guanidinium chloride for 50% unfolding increasing from ~1.8 M to ~6 M. Triggering folding by reducing the oxidized protein, a folding rate of 1.5 × 103 s-1 in 2.2 M guanidinium chloride was measured (122). Extrapolation to zero denaturant yielded a folding time constant of 5 μs, very close to the speed limit!
Downhill Folding
Measurements of fast folding have additional physical significance. Theoretical calculations of free energy surfaces for folding predict that the barriers to folding are quite small. Barriers of only 5–7 kBT have been calculated for monomeric λ repressor (94) under conditions where folding occurs in ~1 ms. This indicates a pre-exponential factor k0 is an expression k0 exp(-Δ G‡ / kBT) of only 105 s-1 to 106 s-1, compared to kBT / h = 6 × 1012 s-1 of gas phase chemical reaction kinetics. The calculation also suggests that, in the absence of a free energy barrier, the folding rate would be of the order of 106 s-1, providing yet another estimate of an upper limit for protein folding. More importantly, the absence of a barrier could lead to non-exponential kinetics because folding is continuously downhill in free energy.
The ideal demonstration of downhill folding would be one in which the barrier of a two-state protein is made to decrease and then disappear as the stability of the native state is increased, switching the time course monotonically from exponential to non-exponential as folding speeds up. Conversion from exponential to non-exponential kinetics with an increased folding rate has recently been observed, but in a somewhat more complicated situation (32, 100). A nanosecond laser T-jump instrument was used to study the folding kinetics of a 76-residue protein, ubiquitin, from the cold denatured state (100). At 2°C, folding occurs with a relaxation time ≈5 ms (τ1/2 ≈ 3 ms), while at 8°C, where the protein is more stable, the half-time is dramatically reduced (τ1/2 ≈ 100 μs) and the time course becomes non-exponential. Interpretation of this result was not obvious because NMR data showed that ubiquitin exhibits two state equilibrium behavior at 8°C, indicating a barrier between cold-denatured and native states. An intriguing explanation of these results was proposed (100). It was suggested that the shape of the free energy surface changes with temperature, so that at 8°C the barrier is shifted significantly toward the unfolded state. In this way cold denatured molecules prior to the temperature jump find themselves on the native side of the barrier at the elevated temperature. Folding is now downhill in free energy, making it much faster and non-exponential. The movement of the barrier top is attributed to the strengthening of the hydrophobic interactions at the elevated temperature, overcoming the entropy decrease earlier along the reaction coordinate, reminiscent of our earlier discussion on the barrier to β-hairpin formation.
In this work the possible contribution of landscape ruggedness to the non-exponential kinetics was also discussed (100). In describing folding as diffusion on a low-dimensional free energy surface, the effective diffusion coefficient reflects the underlying energetic roughness that corresponds to transient trapping in local minima (108). Very hydrophobic sequences, for example, may cause transient formation of numerous nonnative interactions, resulting in a smaller effective diffusion coefficient. If this effective diffusion coefficient decreases as folding proceeds, the kinetics would become more extended in time—producing so-called “stretched” exponential kinetics having the form: exp[‐(ktβ]) with β < 1 (46). One interesting speculation from these considerations is the possibility of observing a maximum in the folding rate as the free energy bias toward the native state is increased because of an even larger effect on the effective diffusion constant from increased ruggedness (32)
Downhill folding could be of great importance for obtaining the ultimate experimental description of protein folding, namely the microscopic paths that individual denatured molecules take to find the unique conformation of the native structure. In contrast to two-state folders, in which only the population of the native and denatured states can be observed, intermediate structures on the paths from the denatured to native state are populated in downhill folding. Downhill folders would be excellent candidates for study using the rapidly emerging technology of single molecule spectroscopy (32). There is the exciting prospect that by increasing the viscosity to slow the downhill kinetics, it will be possible to resolve the sequence of structural events as a single molecule progresses from a denatured to native conformation.
Polypeptide Collapse
If solvent conditions are changed from strongly denaturing to those that favor folding, i.e. from a good to a poor solvent, an unfolded protein is expected to collapse to form more compact structures. Collapse may occur simultaneously with folding, or it may occur in a prior phase that involves the formation of compact, but still-denatured structures. Resolving the kinetics of collapse has been one of the major objectives of fast-folding studies. The focus has initially been on cytochrome c because it has a readily accessible probe and is known to exhibit a collapse process that cannot be resolved by stopped flow methods (98, 109, 110). Horse cytochrome c contains a single tryptophan (W59) about 40 residues distant in sequence from the point of covalent attachment of the heme to the polypeptide chain (H18). In the native structure the fluorescence of this tryptophan is completely quenched by excitation energy transfer to the strongly absorbing heme chromophore. At high concentrations of chemical denaturant, the protein is unfolded and the tryptophan quantum yield is only 20–30% reduced compared to free tryptophan. Assuming that the heme-tryptophan distance reflects the overall size of the protein, the decrease in fluorescence is a sensitive indicator of polypeptide collapse.
Using a rapid mixing/continuous flow method with a dead time of ~70 μs, the folding of cytochrome c complexed with imidazole was studied following dilution of guanidinium chloride (18). Imidazole blocks binding of nonnative ligands, with the result that folding becomes too fast to be observed by the stopped flow method (98, 110). Although submillisecond folding to the native conformation was completely resolved, the collapse process was still too fast for this technique (18). In a subsequent study using a continuous flow instrument with improved signal-to-noise and a dead time of only ~45 μs, the collapse process was partially resolved and shown to have a time-constant of 40–60 μs (105). The partially resolved kinetic progress curve is consistent with an exponential form, suggesting a barrier crossing process. More recently, the collapse process has been completely resolved using nanosecond laser T-jump (47). These measurements confirm that collapse has an exponential time course and is an activated process.
A three-state analysis of the kinetics suggests that tryptophan fluorescence in the initial collapsed state is almost completely quenched, indicating that this state is compact (105). The radius of gyration of this transient state has been subsequently measured in a submillisecond, time-resolved small angle X-ray scattering experiment. These experiments used synchrotron radiation and a continuous flow method that employs hydrodynamic focusing to achieve rapid mixing (93). A value of Rg < 1.8 nm was determined, consistent with the fluorescence quenching and comparable to the equilibrium radius of gyration of cytochrome c at acid pH in the presence of high salt (the so-called “acid molten globule” state).
The finding of a free energy barrier between two denatured states in these experiments on cytochrome c may have greater significance than simply an interesting observation on a specific protein. Theoretical considerations suggest that proteins may generically have two denatured states—an expanded denatured state and a compact denatured state (25, 26, 86). If there is no barrier between expanded and compact denatured states, theoretical estimates suggest that collapse would be submicrosecond (91). It will be important to further investigate this question in other proteins using Förster excitation energy transfer and time-resolved X-ray scattering measurements. There are now several cases in which submillisecond kinetic processes have been attributed to polypeptide collapse (3, 4, 88), but the probes in these experiments prevent a completely unambiguous interpretation.
Another interesting aspect of the collapse process that merits further study by fast-folding methods is the question of the overall topology of transient compact denatured states. Is the overall topology similar to the final native conformation, as suggested for the compact denatured state which has been called the “molten globule state” (95), or is it widely distributed in a “random globule state” (26), as found in many lattice simulations? In the latter case, the slower process in protein folding corresponds to the search for the correct topology. In the former, the protein-folding problem, as far as overall topology is concerned, is “solved” in the very fast initial process, the slow process corresponding mainly to the annealing of side chains to reach the final native conformation.
Protein-Folding Rates from a Simple Model
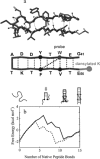
What factors determine whether a protein will be a fast or slow folder? Is there any relation between folding rate and the final native structure? It is now possible to address these questions because of the availability of a sufficiently large set of data on the folding of small single domain proteins. We pointed out in the introduction to this review that the study of CI2 marked a turning point in experimental studies on protein folding (56). The 64-residue version of this protein, which has neither disulfide bridges nor cis-prolines, behaves like a perfect two-state system. For a two-state protein, only two populations of molecules are detected in both equilibrium and kinetic experiments (126). One state corresponds to molecules having the native structure and the second to the ensemble of conformations that make up the denatured state. Over 20 proteins are now known to exhibit two-state behavior, and the results of these studies have been summarized in a recent review (55). Their folding rates span more than four orders of magnitude, from (200 μs)-1 for monomeric λ repressor to (4 s)-1 for muscle acyl phosphatase (Figure 5).
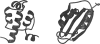
Figure 5
Schematic structures of monomeric λ repressor (left) and muscle acyl phosphatase (right).
In searching for empirical correlations in this set of kinetic data, a key observation was made: The rates of folding show a significant correlation with a simple measure of protein topology, the so-called “contact order” (1, 92). The relative contact order is the mean separation in sequence between residues that are in contact in the three-dimensional structure, normalized by the total number of amino acids in the protein. For proteins with many long-range interactions, such as muscle acyl phosphatase, the contact order is large and folding is relatively slow. For α-helical proteins, such as monomeric λ repressor, local (i, i + 4) interactions are much more abundant The contact order is therefore small and folding is observed to be relatively much faster.
The observation of a correlation between folding rates and such a simple measure of structure suggested that a simple physical model may be capable of calculating the rates of protein folding. A simple statistical mechanical model had already been developed for explaining the kinetics of β-hairpin formation, which as pointed out earlier, contains most of the basic features of folding a small protein. It was therefore logical to apply this model to two-state proteins (76). The analysis of the β-hairpin revealed that for this molecule the single sequence approximation, which allows only one stretch of native structure simultaneously in each molecule, produces results very close to the complete model, which imposes no restrictions on the number of stretches (125). To account for the larger size of the molecule in applying the model to proteins, this approximation was relaxed to allow two and three simultaneous stretches. For 18 proteins that exhibit two-state behavior experimentally the free energy as a function of the number of native peptide bonds, the natural reaction coordinate in this model, shows only two deep minima separated by a free energy barrier. Furthermore, free energy profiles for four other proteins, known to populate more than two states in kinetic experiments, show additional deep minima. The striking finding was that the calculated free energy barriers to folding are small for fast-folding proteins and large for slow-folding proteins (Figure 6a,b). The rates calculated for the 18 two-state proteins from diffusion on these free energy profiles, moreover, show a remarkably good correlation with the experimentally determined rates (Figure 6c). It is perhaps surprising that even approximate relative rates can be calculated using a single reaction coordinate for such a complex system. However, the absolute folding rate of a lattice protein with ~1016 possible structures was accurately calculated from diffusion on a one-dimensional free energy profile (108).
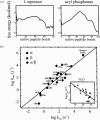
In addition to ignoring much polymer physics, there are several obvious weaknesses to these simple calculations. One is a restrictive assumption of the model that does not allow native interactions between residues unless the intervening chain has its native conformation. A second is the naive treatment of the entropy of the denatured state. The denatured state appears as a sharp minimum in the free energy profiles (Figure 6a,b), instead of the broad minimum that would appear with no restriction on the distribution of native peptide bonds (125). Other weaknesses are the use of a simplified free energy function in which native propensities and contact energies are sequence independent and solvation effects are ignored. Nevertheless, the success of the simple model in calculating rates is an important result, for it suggests that the underlying physics determining folding rates is simpler than previously thought. It appears that the folding rate is determined largely by the distribution and strength of contacts in the native three-dimensional structure. As pointed out earlier in our discussion of β-hairpin formation (Figure 4b), compensation of the conformational entropy loss by strong stabilizing interactions earlier along the reaction coordinate lowers the free energy barrier and allows faster folding.
What about mechanism? Calculations with this simple model contain an enormous amount of detail on microscopic structural pathways between the native and denatured state. For these pathways it is possible to identify the structures that, once formed, rapidly proceed to the native state. This is the so-called transition state ensemble. Detailed structural information on this transition state ensemble can in principle be obtained from experimental measurements of the relative effects of mutations on the rates and equilibrium constants for folding (so-called φ values, defined as Δlnkfolding/ ΔlnKequilibrium)(34, 35, 54, 84). This model, like two other simple models with similar approximations (2, 36), shows qualitative agreement with mutation experiments. However, thus far, these simple models do not give good quantitative agreement between the observed and calculated φ values, so it is somewhat premature to use them to describe structural pathways in any detail. Accurate calculation of φ values remains an important goal, and more realistic theoretical models (107) as well as knowledge of the detailed structure of the mutated protein may be necessary. The task would also be made easier by experimental studies in which there are several different mutations at each of the studied positions. Predicting φ values theoretically is a crucial issue because the mutation experiments probably hold the key to elucidating mechanism in structural detail.
Biological Relevance of Folding Kinetics
From the preceding discussion we have seen that kinetics is important for a physical understanding of how proteins fold. However, is there any direct biological implication of folding kinetics? We should say at the outset that there are no answers to this question that can be rigorously supported, but there are a number of interesting speculations. One subject under study is whether proteins with similar structure but no sequence homology have the same folding mechanism. This is, in principle, independent of whether these proteins are products of divergent or convergent evolution. To explore this idea, sequence and structure alignments have been used to search for clusters of conserved residues that are not necessary for either function or stability. The assumption is that these residues, if they appear, correspond to “nuclei” that determine the folding rate (74).
A more general question is whether there is any pressure on folding rates other than that required for the appropriate stability. There must be some constraint, for newly synthesized polypeptide chains must be able to fold within a certain time to be able to perform their biological function and to avoid proteolysis and aggregation. What is the minimal folding time required for biological function? In Escherichia coli, proteins are synthesized at a rate of 10–15 amino-acids per second. Assuming an average molecular mass of 40,000 daltons (~350 amino-acids) for E. coli proteins, the ribosomes require on average 30 sec to synthesize a protein molecule (69). One could argue that folding speed will not be a limiting factor as long as it is comparable to the rate of protein synthesis. If there is indeed biological pressure, we should expect proteins to fold no slower than 1/minute. All known folding rates for two-state proteins are faster than this minimum speed (55) (Figure 6). Other processes coupled to protein folding, such as proline isomerization and disulfide formation, could slow folding considerably, but specialized enzymes catalyze these reactions in vivo (101). The role of molecular chaperones, such as GroEL and GroES, can also be rationalized in this light. Theoretical estimates indicate that the known chaperones only assist folding of ~5% of the existing proteins (69). One of their roles might be to ensure folding of a small subset of biologically critical proteins that would otherwise fold with rates slower than the one-minute biological minimum.
A still open issue, however, is whether it is difficult for evolution to find sequences that can form stable 3-D structures faster than this biological minimum speed. This question has been approached using combinatorial chemistry. Heavily mutated variants of the IgG binding domain of protein L were found to fold at similar, or even faster, rates than the wild-type protein (62). This result has been used to suggest that for single domain proteins, it is not difficult to find sequences that fold “biologically fast.” It would be important to further pursue this idea with experiments on other proteins, and, ideally, to explore large regions of sequence space that are not limited to the neighborhood of naturally occurring sequences. A potential drawback of combinatorial methods is that they generally involve biological selection. It is therefore possible that the method is finding fast folding proteins because they are the only ones that are biologically viable.
Does protein function ever require folding much faster than one minute? For λ repressor, which folds in 200 μs, it has been argued that very fast folding is required for stability to compensate fast unfolding, fast unfolding being required for efficient regulation by proteolysis (13). We do not know the rate of protease binding to unfolded states of proteins, but it could be argued that what really determines the rate of proteolysis of a target protein is the residence time in the unfolded state. Fast folding could then be a strategy to avoid proteolysis while maintaining the low stability necessary for its function (e.g. binding to DNA). Related to this is the interesting suggestion that there is evolutionary pressure for proteins to be marginally stable in order to have the conformational flexibility required for function (58).
A corollary to the idea of folding speed as an evolutionary pressure is the suggestion that to avoid aggregation in the cell, compact states may be required to form much more quickly than the final native structure (49). This idea is consistent with the finding that polypeptide collapse is submillisecond (see above). In a typical E. coli cell in logarithmic phase with a volume of ~10-15 liter, there are ~35,000 ribosomes (69), which are producing ~1000 protein molecules per second. We can make a rough estimate of the maximum folding time that is compatible with minimal aggregation. If we assume that aggregation is nonspecific, irreversible, and diffusion limited (~108 M-1 s-1) (119), folding in less than ~10 milliseconds would result in aggregation of less than ~1% of newly synthesized proteins. Although this calculation is very approximate for many reasons, it does support the idea that competition between aggregation and folding may provide additional selection pressure for fast-folding sequences or for sequences that collapse quickly to form compact denatured states that do not aggregate. Understanding this competition in more detail may be important for investigations of the pathophysiology of several human diseases (28, 60, 66).
Finally, is there any connection between evolutionary pressure on folding speed and the catalogue of naturally occurring protein structures? The model for protein folding discussed above would suggest that topologically simple structures are expected to fold faster than complicated structures. A large number of geometrically feasible and stable structures may not be observed in nature because they would fold too slowly to be consistent with the constraints of synthesis and aggregation discussed above. This could be a factor responsible for the limited number and distribution of distinct protein folds that have been observed so far (74, 127, 128).
Literature Cited
- 1.
- Alm E, Baker D. Matching theory and experiment in protein folding. Curr. Opin. Struct. Biol. 1999;9:189–96. [PubMed: 10322214]
- 2.
- Alm E, Baker D. Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. USA. 1999;96:11305–10. [PMC free article: PMC18029] [PubMed: 10500172]
- 3.
- Ballew R M, Sabelko J, Gruebele M. Direct observation of fast protein folding: the initial collapse of apomyoglobin. Proc. Natl. Acad. Sci. USA. 1996;93:5759–64. [PMC free article: PMC39134] [PubMed: 8650166]
- 4.
- Ballew R M, Sabelko J, Gruebele M. Observation of distinct nanosecond and microsecond protein folding events. Nat. Struct. Biol. 1996;3:923–26. [PubMed: 8901868]
- 5.
- Bieri O, Wirz J, Hellrung B, Schutkowski M, Drewello M, Kiefhaber T. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc. Natl. Acad. Sci. USA. 1999;96:9597–601. [PMC free article: PMC22254] [PubMed: 10449738]
- 6.
- Blanco F J, Rivas G, Serrano L. A short linear peptide that folds into a native stable β-hairpin in aqueous solution. Nat. Struct. Biol. 1994;1:584–90. [PubMed: 7634098]
- 7.
- Brooks C L. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 1998;8:222–26. [PubMed: 9631297]
- 8.
- Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins: Struct. Funct. Genet. 1995;21:167–95. [PubMed: 7784423]
- 9.
- Bryngelson J D, Wolynes P G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA. 1987;84:7524–28. [PMC free article: PMC299331] [PubMed: 3478708]
- 10.
- Bryngelson J D, Wolynes P G. Intermediates and barrier crossing in a random energy model (with applications to protein folding). J. Phys. Chem. 1989;93:6902–15.
- 11.
- Buckler D R, Haas E, Scheraga H A. Analysis of the structure of ribonuclease A in native and partially denatured states by time-resolved nonradiative dynamic excitation energy transfer. Biochemistry. 1995;34:15965–78. [PubMed: 8519753]
- 12.
- Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T G. The energy landscape of a fast-folding protein mapped by Ala → Gly substitutions. Nat. Struct. Biol. 1997;4:305–10. [PubMed: 9095199]
- 13.
- Burton R E, Huang G S, Daugherty M A, Fullbright P W, Oas T G. Microsecond protein folding through a compact transition state. J. Mol. Biol. 1996;263:311–22. [PubMed: 8913309]
- 14.
- Callender R H, Dyer R B, Gilmanshin R, Woodruff W H. Fast events in protein folding: the time evolution of primary processes. Annu. Rev. Phys. Chem. 1998;49:173–202. [PubMed: 9933907]
- 15.
- Camacho J, Thirumalai D. Theoretical predictions of folding pathways by using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 1995;92:1277–81. [PMC free article: PMC42502] [PubMed: 7533290]
- 16.
- Chakrabartty A, Baldwin R L. Stability of α-helices. Adv. Protein Chem. 1995;46:141–76. [PubMed: 7771317]
- 17.
- Chan C -K, Hofrichter J, Eaton W A. Optical triggers in protein folding. Science. 1996;274:628–29. [PubMed: 8928010]
- 18.
- Chan C -K, Hu Y, Takahashi S, Rousseau D L, Eaton W A, Hofrichter J. Sub-millisecond protein folding kinetics studied by ultrarapid mixing. Proc. Natl. Acad. Sci. USA. 1997;94:1779–84. [PMC free article: PMC19993] [PubMed: 9050855]
- 19.
- Chan H S, Dill K A. Protein folding in the landscape perspective: chevron plots and non-arrhenius kinetics. Proteins. 1998;30:2–33. [PubMed: 9443337]
- 20.
- Chen E, Wittung-Stafshede P, Kliger D S. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc. 1999;121:3811–17.
- 21.
- Clarke D T, Doig A J, Stapley B J, Jones G R. The alpha-helix folds on the millisecond time scale. Proc. Natl. Acad. Sci. USA. 1999;96:7232–37. [PMC free article: PMC22062] [PubMed: 10377397]
- 22.
- Daggett V, Levitt M. Molecular-dynamics simulation of helix denaturation. J. Mol. Biol. 1992;223:1121–38. [PubMed: 1538392]
- 23.
- Daura X, Jaun B, Seebach D, van Gunsteren W F, Mark A E. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 1998;280:925–32. [PubMed: 9671560]
- 24.
- Daura X, van Gunsteren W F, Mark A E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins. 1999;34:269–80. [PubMed: 10024015]
- 25.
- Dill K A, Shortle D. Denatured states of proteins. Annu. Rev. Biochem. 1991;60:795–825. [PubMed: 1883209]
- 26.
- Dill K A, Stigter D. Modeling protein stability as heteropolymer collapse. Adv. Protein Chem. 1995;46:59–104. [PubMed: 7771323]
- 27.
- Dinner A R, Lazaridis T, Karplus M. Understanding β-hairpin formation. Proc. Natl. Acad. Sci. USA. 1999;96:9068–73. [PMC free article: PMC17733] [PubMed: 10430896]
- 28.
- Dobson C M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999;24:329–32. [PubMed: 10470028]
- 29.
- Dobson C M, Sali A, Karplus M. Protein folding: a perspective from theory and experiment. Angew. Chem. Int. Edit. 1998;37:868–93. [PubMed: 29711488]
- 30.
- Duan Y, Kollman P A. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–44. [PubMed: 9784131]
- 31.
- Dyer R B, Gai F, Woodruff W H, Gilman-shin R, Callender R H. Infrared studies of fast events in protein folding. Acc. Chem. Res. 1998;31:709–16.
- 32.
- Eaton W A. Commentary: searching for “downhill scenarios” in protein folding. Proc. Natl. Acad. Sci. USA. 1999;96:5897–99. [PMC free article: PMC34202] [PubMed: 10339514]
- 33.
- Elöve G A, Bhuyan A K, Roder H. Kinetic mechanism of cytochrome c folding: involvement of the heme and its ligands. Biochemistry. 1994;33:6925–35. [PubMed: 8204626]
- 34.
- Fersht AR. 1998. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. San Francisco: Freeman .
- 35.
- Fersht A R, Matouschek A, Serrano L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992;224:771–82. [PubMed: 1569556]
- 36.
- Galzitskaya O V, Finkelstein A. A theoretical search for folding/unfolding nuclei in three-dimensional protein structures. Proc. Natl. Acad. Sci. USA. 1999;96:11299–304. [PMC free article: PMC18028] [PubMed: 10500171]
- 37.
- Garel T, Orland H, Pitard E. 1998. Protein folding and heteropolymers. In Spin Glasses and Random Fields, ed. AP Young, pp. 387–443. Singapore: World Scientific .
- 38.
- Gellman S H. Minimal model systems for beta sheet secondary structure in proteins. Curr. Opin. Chem. Biol. 1998;2:717–25. [PubMed: 9914187]
- 39.
- Ghaemmaghami S, Word J M, Burton R E, Richardson J S, Oas T G. Folding kinetics of a fluorescent variant of monomeric lambda repressor. Biochemistry. 1998;37:9179–85. [PubMed: 9636065]
- 40.
- Gilmanshin R, Callender R H, Dyer R B. The core of apomyoglobin E-form folds at the diffusion limit. Nat. Struct. Biol. 1998;5:363–65. [PubMed: 9586997]
- 41.
- Gilmanshin R, Williams S, Callender R H, Woodruff W H, Dyer R B. Fast events in protein folding: relaxation dynamics of secondary and tertiary structure in native apomyoglobin. Proc. Natl. Acad. Sci. USA. 1997;94:3709–13. [PMC free article: PMC20505] [PubMed: 9108042]
- 42.
- Gonnelli M, Strambini G. Phosphorescence lifetime of tryptophan in proteins. Biochemistry. 1995;34:13847–57. [PubMed: 7577979]
- 43.
- Gruebele M. The fast protein folding problem. Annu. Rev. Phys. Chem. 1999;50:485–516. [PubMed: 15012420]
- 44.
- Gruenewald B, Nicola C U, Lustig A, Schwarz G, Klump H. Kinetics of the helix-coil transition of a polypeptide with non-ionic side groups, derived from ultrasonic relaxation measurements. Biophys. Chem. 1979;9:137–47. [PubMed: 427245]
- 45.
- Guo Z, Thirumalai D. Kinetics of protein folding—nucleation mechanism, time scales, and pathways. Biopolymers. 1995;36:83–102.
- 46.
- Hagen S J, Eaton W A. Nonexponential structural relaxations in proteins. J. Chem. Phys. 1996;104:3395–98.
- 47.
- Hagen S J, Eaton W A. Two-state expansion and collapse of a polypeptide. J. Mol. Biol. 2000;301:1019–27. [PubMed: 10966803]
- 48.
- Hagen S J, Hofrichter J, Eaton W A. Rate of intrachain diffusion of unfolded cytochrome-c. J. Phys. Chem. B. 1997;101:2352–65.
- 49.
- Hagen S J, Hofrichter J, Szabo A, Eaton W A. Diffusion-limited contact formation in unfolded cytochrome-c: estimating the maximum rate of protein folding. Proc. Natl. Acad. Sci. USA. 1996;93:11615–17. [PMC free article: PMC38106] [PubMed: 8876184]
- 50.
- Hao M -H, Scheraga H A. Theory of two-state cooperative folding of proteins. Acc. Chem. Res. 1998;31:433–40.
- 51.
- Hardin C, Luthey-Schulten Z, Wolynes P G. Backbone dynamics, fast folding, and secondary structure formation in helical proteins and peptides. Proteins. 1999;34:281–94. [PubMed: 10024016]
- 52.
- Hofrichter J, Thompson PA. 2000. Laser temperature jump methods for studying folding dynamics. In Protein Stability and Folding, ed. KP Murphy. Totowa, NJ: Humana. 2nd ed. In press .
- 53.
- Huang G S, Oas T G. Submillisecond folding of monomeric λ repressor. Proc. Natl. Acad. Sci. USA. 1995;92:6878–82. [PMC free article: PMC41433] [PubMed: 7624336]
- 54.
- Itzhaki L S, Otzen D E, Fersht A R. The structure of the transition state for folding of chymotrypsin inhibitor-2 analyzed by protein engineering methods—evidence for a nucleation condensation mechanism for protein folding. J. Mol. Biol. 1995;254:260–88. [PubMed: 7490748]
- 55.
- Jackson S E. How do small single-domain proteins fold? Folding Design. 1998;3:R81–R91. [PubMed: 9710577]
- 56.
- Jackson S E, Fersht A R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 1991;30:10428–35. [PubMed: 1931967]
- 57.
- Jacob M, Holterman G, Perl D, Reinsten J, Schindler T, Geeves, Schmid F X. Microsecond folding of the cold shock protein measured by a pressure-jump technique. Biochemistry. 1999;38:2882–91. [PubMed: 10074340]
- 58.
- Jaenicke J. Protein stability and molecular adaptation to extreme conditions. Eur. J. Biochem. 1991;202:715–28. [PubMed: 1765088]
- 59.
- Jones C M, Henry E R, Hu Y, Chan C K, Luck S D, Bhuyan A, Roder H, Hofrichter J, Eaton W A. Fast events in protein folding initiated by nanosecond laser photolysis. Proc. Natl. Acad. Sci. USA. 1993;90:11860–64. [PMC free article: PMC48084] [PubMed: 8265638]
- 60.
- Kelly J W. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 1998;8:101–6. [PubMed: 9519302]
- 61.
- Kiefhaber T. Kinetic traps in lysozyme folding. Proc. Natl. Acad. Sci. USA. 1995;92:9029–33. [PMC free article: PMC40917] [PubMed: 7568066]
- 62.
- Kim D E, Gu H D, Baker D. The sequences of small proteins are not extensively optimized for rapid folding by natural selection. Proc. Natl. Acad. Sci. USA. 1998;95:4982–86. [PMC free article: PMC20199] [PubMed: 9560214]
- 63.
- Knight J B, Vishwanath A, Brody J P, Austin R H. Hydrodynamic focusing on a silicon chip: mixing nanoliters in microseconds. Phys. Rev. Lett. 1998;80:3863–66.
- 64.
- Kuwajima K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6:87–103. [PubMed: 2695928]
- 65.
- Lacroix E, Kortemme T, de la Paz M L, Ser-rano L. The design of linear peptides that fold as monomeric beta-sheet structures. Curr. Opin. Struct. Biol. 1999;9:487–93. [PubMed: 10449370]
- 66.
- Lansbury P T. Evolution of amy-loid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA. 1999;96:3342–44. [PMC free article: PMC34271] [PubMed: 10097040]
- 67.
- Lapidus L J, Eaton W A, Hofrichter J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA. 2000;97:7220–25. [PMC free article: PMC16526] [PubMed: 10860987]
- 68.
- Lednev I K, Karnoup A S, Sparrow M C, Asher S A. Alpha-helix peptide folding and unfolding activation barriers: a nanosecond UV resonance raman study. J. Am. Chem. Soc. 1999;121:8074–86.
- 69.
- Lorimer G H. A quantitative assessment of the role of chaperonin proteins in protein folding in vivo. FASEB J. 1996;10:5–9. [PubMed: 8566548]
- 70.
- Lu H S M, Volk M, Kholodenko Y, Gooding E, Hochstrasser R M, DeGrado W F. Aminothiotyrosine disulfide, an optical trigger for initiation of protein folding. J. Am. Chem. Soc. 1997;119:7173–80.
- 71.
- Marqusee S, Robbins V H, Baldwin R L. Unusually stable helix formation in short alanine based peptides. Proc. Natl. Acad. Sci. USA. 1989;86:5286–90. [PMC free article: PMC297606] [PubMed: 2748584]
- 72.
- Matthews C R. Pathways of protein folding. Annu. Rev. Biochem. 1993;62:653–83. [PubMed: 8352599]
- 73.
- Mines G A, Pascher T, Lee S C, Winkler J R, Gray H B. Cytochrome c folding triggered by electron transfer. Chem. Biol. 1996;3:491–97. [PubMed: 8807879]
- 74.
- Mirny L A, Shakhnovich E I. Universally conserved positions in protein folds: Reading evolutionary signals about stability, folding kinetics and function. J. Mol. Biol. 1999;291:177–96. [PubMed: 10438614]
- 75.
- Mohanty D, Elber R, Thirumalai D, Beglov D, Roux B. Kinetics of peptide folding: Computer simulations of SYPFDV and peptide variants in water. J. Mol. Biol. 1997;272:423–42. [PubMed: 9325101]
- 76.
- Muñoz V, Eaton W A. A simple model for calculating the kinetics of protein folding from three-dimensional structures. Proc. Natl. Acad. Sci. USA. 1999;96:11311–16. [PMC free article: PMC18030] [PubMed: 10500173]
- 77.
- Muñoz V, Henry E R, Hofrichter J, Eaton W A. A statistical mechanical model for β-hairpin kinetics. Proc. Natl. Acad. Sci. USA. 1998;95:5872–79. [PMC free article: PMC34489] [PubMed: 9600886]
- 78.
- Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1994;1:399–409. [PubMed: 7664054]
- 79.
- Muñoz V, Serrano L. Elucidating the folding problem of helical peptides using empirical parameters. II. Helix macrodipole effects and rational modification of the helical content of natural peptides. J. Mol. Biol. 1995;245:275–296. [PubMed: 7844817]
- 80.
- Muñoz V, Thompson P A, Hofrichter J, Eaton W A. Folding dynamics and mechanism of β-hairpin formation. Nature. 1997;390:196–99. [PubMed: 9367160]
- 81.
- Nölting B. Temperature-jump induced fast refolding of cold-unfolded protein. Biochem. Biophys. Res. Commun. 1996;227:903–8. [PubMed: 8886028]
- 82.
- Nölting B, Golbik R, Fersht A R. Submillisecond events in protein folding. Proc. Natl. Acad. Sci. USA. 1995;92:10668–72. [PMC free article: PMC40673] [PubMed: 7479862]
- 83.
- Onuchic J, Luthey-Schulten A, Wolynes P G. Theory of protein folding: the energy landscape perspective. Annu. Rev. Phys. Chem. 1997;48:545–600. [PubMed: 9348663]
- 84.
- Onuchic J, Socci N D, Luthey-Schulten Z, Wolynes P G. Protein folding funnels: the nature of the transition state ensemble. Folding Design. 1996;1:441–50. [PubMed: 9080190]
- 85.
- Pande V S, Grosberg A Y, Tanaka T, Rok-hsar D S. Pathways for protein folding: Is a new view needed? Curr. Opin. Struct. Biol. 1998;8:68–79. [PubMed: 9519298]
- 86.
- Pande V S, Rokhsar D S. Is the molten globule a third phase of proteins? Proc. Natl. Acad. Sci. USA. 1998;95:1490–94. [PMC free article: PMC19058] [PubMed: 9465042]
- 87.
- Pande V S, Rokhsar D S. Molecular dynamics simulations of unfolding and refolding of a β-hairpin fragment of protein G. Proc. Natl. Acad. Sci. USA. 1999;96:9062–67. [PMC free article: PMC17732] [PubMed: 10430895]
- 88.
- Park S -H, Shastry M C R, Roder H. Folding dynamics of the B1 domain of protein G explored by ultrarapid mixing. Nature Struct. Biol. 1999;6:943–47. [PubMed: 10504729]
- 89.
- Pascher T, Chesick J P, Winkler J R, Gray H B. Protein folding triggered by electron transfer. Science. 1996;271:1558–60. [PubMed: 8599112]
- 90.
- Phillips C M, Mizutani Y, Hochstrasser R M. Ultrafast thermally induced unfolding of RNase A. Proc. Natl. Acad. Sci. USA. 1995;92:7292–96. [PMC free article: PMC41325] [PubMed: 7638183]
- 91.
- Pitard E, Orland H. Dynamics of the swelling or collapse of a homopolymer. Europhys. Lett. 1998;41:467–72.
- 92.
- Plaxco K W, Simons K T, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–94. [PubMed: 9545386]
- 93.
- Pollack L, Tate M W, Darnton N C, Knight J B, Gruner S, Eaton W A, Austin R H. Compactness of the denatured state of a fast-folding protein measured by sub-millisecond small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA. 1999;96:10115–17. [PMC free article: PMC17851] [PubMed: 10468571]
- 94.
- Portman J J, Takada S, Wolynes P G. Variational theory for site resolved protein folding free energy surfaces. Phys. Rev. Lett. 1998;81:5237–40.
- 95.
- Ptitsyn O B. Molten globule and protein folding. Adv. Protein Chem. 1995;47:83–229. [PubMed: 8561052]
- 96.
- Regenfuss P, Clegg R M, Fulwyler M J, Barrantes F J, Jovin T M. Mixing liquids in microseconds. Rev. Sci. Instrum. 1985;56:283–90.
- 97.
- Robinson C R, Sauer R T. Equilibrium stability and sub-millisecond refolding of a designed single-chain arc repressor. Biochemistry. 1996;35:13878–84. [PubMed: 8909284]
- 98.
- Roder H, Colón W. Kinetic role of early intermediates in protein folding. Curr. Opin. Struct. Biol. 1997;7:15–28. [PubMed: 9032062]
- 99.
- Roder H, Shastry M C R. Methods for exploring early events in protein folding. Curr. Opin. Struct. Biol. 1999;9:620–26. [PubMed: 10508774]
- 100.
- Sabelko J, Ervin J, Gruebele M. Observation of strange kinetics in protein folding. Proc. Natl. Acad. Sci. USA. 1999;96:6031–36. [PMC free article: PMC26830] [PubMed: 10339536]
- 101.
- Schmid F X. Protein-folding-prolyl isomerases join the fold. Curr. Biol. 1995;5:993–94. [PubMed: 8542292]
- 102.
- Searle M S, Williams D H, Packman L C. A short linear peptide derived from the N-terminal sequence of ubiquitin folds into a water-stable non-native β-hairpin. Nat. Struct. Biol. 1995;2:999–1006. [PubMed: 7583674]
- 103.
- Shakhnovich E I. Theoretical studies of protein-folding thermodynamics and kinetics. Curr. Opin. Struct. Biol. 1997;7:29–40. [PubMed: 9032061]
- 104.
- Shastry M C R, Luck S D, Roder H. A continuous-flow capillary mixing method to monitor reactions on the microsecond time scale. Biophys. J. 1998;74:2714–21. [PMC free article: PMC1299611] [PubMed: 9591695]
- 105.
- Shastry M C R, Roder H. Evidence for barrier-limited protein folding kinetics on the microsecond time scale. Nat. Struct. Biol. 1998;5:385–92. [PubMed: 9587001]
- 106.
- Shastry M C R, Sauder J M, Roder H. Kinetic and structural analysis of submil-lisecond folding events in cytochrome-c. Acc. Chem. Res. 1998;31:717–25.
- 107.
- Shoemaker B A, Wang J, Wolynes P G. Exploring structures in protein folding funnels with free energy functionals: the transition state ensemble. J. Mol. Biol. 1999;287:657–94. [PubMed: 10092467]
- 108.
- Socci N D, Onuchic J N, Wolynes P G. Diffusive dynamics of the reaction coordinate for protein folding funnels. J. Chem. Phys. 1996;104:5860–68.
- 109.
- Sosnick T R, Mayne L, Englander S W. Molecular collapse: the rate-limiting step in two-state cytochrome c folding. Proteins. 1996;24:413–26. [PubMed: 9162942]
- 110.
- Sosnick T R, Mayne L, Hiller R, Englander S W. The barriers in protein folding. Nat. Struct Biol. 1994;1:149–56. [PubMed: 7656032]
- 111.
- Szabo A, Schulten K, Schulten Z. The first passage time approach to diffusion-controlled reactions. J. Chem. Phys. 1980;72:4350–57.
- 112.
- Takahashi S, Yeh S R, Das T K, Chan C K, Gottfried D S, Rousseau D L. Folding of cytochrome c initiated by submil-lisecond mixing. Nat. Struct. Biol. 1997;4:44–50. [PubMed: 8989323]
- 113.
- Telford J R, Tezcan A, Gray H B, Winkler J R. Role of ligand substitution in ferrocytochrome c folding. Biochemistry. 1999;38:1944–49. [PubMed: 10026276]
- 114.
- Thirumalai D. Time scales for the formation of the most probable tertiary contacts in proteins with applications to cytochrome c. J. Phys. Chem. B. 1999;103:608–10.
- 115.
- Thirumalai D, Klimov D. Deciphering the time scales and mechanisms of protein folding using minimal off-lattice models. Curr. Opin. Struct. Biol. 1999;9:197–207. [PubMed: 10322218]
- 116.
- Thompson P A, Eaton W A, Hofrichter J. Laser temperature-jump study of the helix-coil kinetics of an alanine peptide interpreted with a ‘kinetic zipper’ model. Biochemistry. 1997;36:9200–10. [PubMed: 9230053]
- 117.
- Thompson P A, Muñoz V, Jas G S, Henry E R, Eaton W A. The helix-coil kinetics of a heteropeptide. J. Phys. Chem. B. 2000;104:378–89.
- 118.
- Volk M, Lu H S M, Kholodenko Y, Gooding E, DeGrado W F, Hochstrasser R M. Peptide conformational dynamics and vibrational Stark effects following photinitiated disulfide cleavage. J. Phys. Chem. B. 1997;101:8607–16.
- 119.
- Waldburger C D, Jonsson T, Sauer R T. Barriers to protein folding: formation of buried polar interactions is a slow step in acquisition of structure. Proc. Natl. Acad. Sci. USA. 1996;93:2629–34. [PMC free article: PMC39681] [PubMed: 8610092]
- 120.
- Williams K, Causgrove T P, Gilmanshin R, Fang K S, Callender R H, Woodruff W H, Dyer R B. Fast events in protein folding: helix melting and formation in a small peptide. Biochemistry. 1996;35:691–97. [PubMed: 8547249]
- 121.
- Wittung-Stafshede P, Gray H B, Winkler J R. Rapid formation of a four-helix bundle Cytochrome b562 folding triggered by electron transfer. J. Am. Chem. Soc. 1997;119:9562–63. [PMC free article: PMC21958] [PubMed: 10359755]
- 122.
- Wittung-Stafshede P, Lee J C, Winkler J R, Gray H B. Cytochrome b562 folding triggered by electron transfer: approaching the speed limit for formation of a four-helix-bundle protein. Proc. Natl. Acad. Sci. USA. 1999;96:6587–90. [PMC free article: PMC21958] [PubMed: 10359755]
- 123.
- Wittung-Stafshede P, Malmström B G, Winkler J R, Gray H B. Folding of deoxymyoglobin triggered by electron transfer. J. Phys. Chem. A. 1998;102:5599–1.
- 124.
- Yeh S R, Takahashi S, Fan B, Rousseau D L. Ligand exchange during cytochrome c folding. Nat. Struct. Biol. 1997;4:51–56. [PubMed: 8989324]
- 125.
- Zwanzig R. A simple model of protein folding kinetics. Proc. Natl. Acad. Sci. USA. 1995;92:9801–4. [PMC free article: PMC40890] [PubMed: 7568221]
- 126.
- Zwanzig R. Two-state models of protein folding kinetics. Proc. Natl. Acad. Sci. USA. 1997;94:148–50. [PMC free article: PMC19262] [PubMed: 8990176]
- 127.
- Thirumalai D, Klimov DK. 2000. Emergence of stable and fast folding protein structures. In Stochastic Dynamics and Pattern Formation in Biological and Complex Systems, eds. S Kim, KJ Lee, W. Sung. pp. 95–111. Proceedings #501 American Institute of Physics, Melvelle, New York.
- 128.
- Govindarajan S, Goldstein R A. Estimating the total number of protein folds. Proteins: Struct. Funct. Genet. 1999;35:408–14. [PubMed: 10382668]
- *
Present address: Department of Chemistry and Biochemistry, and Center for Biological Structure and Molecular Organization, University of Maryland, College Park, MD 20742.
- †
Present address: Department of Physics, University of Florida, Gainesville, FL 32611-8440.
- Kinetics and motional dynamics of spin-labeled yeast iso-1-cytochrome c: 1. Stopped-flow electron paramagnetic resonance as a probe for protein folding/unfolding of the C-terminal helix spin-labeled at cysteine 102.[Biochemistry. 1997]Kinetics and motional dynamics of spin-labeled yeast iso-1-cytochrome c: 1. Stopped-flow electron paramagnetic resonance as a probe for protein folding/unfolding of the C-terminal helix spin-labeled at cysteine 102.Qu K, Vaughn JL, Sienkiewicz A, Scholes CP, Fetrow JS. Biochemistry. 1997 Mar 11; 36(10):2884-97.
- Protein folding kinetics: timescales, pathways and energy landscapes in terms of sequence-dependent properties.[Fold Des. 1997]Protein folding kinetics: timescales, pathways and energy landscapes in terms of sequence-dependent properties.Veitshans T, Klimov D, Thirumalai D. Fold Des. 1997; 2(1):1-22.
- Lattice models for proteins reveal multiple folding nuclei for nucleation-collapse mechanism.[J Mol Biol. 1998]Lattice models for proteins reveal multiple folding nuclei for nucleation-collapse mechanism.Klimov DK, Thirumalai D. J Mol Biol. 1998 Sep 18; 282(2):471-92.
- Review Polypeptide chain collapse and protein folding.[Arch Biochem Biophys. 2013]Review Polypeptide chain collapse and protein folding.Udgaonkar JB. Arch Biochem Biophys. 2013 Mar; 531(1-2):24-33. Epub 2012 Oct 19.
- Review Fast events in protein folding.[Structure. 1996]Review Fast events in protein folding.Eaton WA, Thompson PA, Chan CK, Hage SJ, Hofrichter J. Structure. 1996 Oct 15; 4(10):1133-9.
- Fast Kinetics and Mechanisms in Protein Folding - Annual Reviews CollectionFast Kinetics and Mechanisms in Protein Folding - Annual Reviews Collection
Your browsing activity is empty.
Activity recording is turned off.
See more...