

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.

It is possible to make replication-competent retroviral vectors by adding sequences to existing viruses, but a more common design involves the replacement of retroviral sequences to create replication-defective vectors. In addition, the amount of foreign DNA that can be accommodated in replication-competent vectors is much smaller than can be accommodated in replication-defective vectors. Expression of retroviral proteins in most of the naturally occurring oncogenic retroviruses is driven by a single promoter in the 5′long terminal repeat (LTR), and the expression of multiple viral coding regions is achieved by alternative splicing. However, vector design is not limited to the use of the single retroviral promoter with alternative splicing. Other strategies include the use of multiple promoters, insertion of genes in the reverse orientation, and the use of internal ribosome entry sites (IRESs).
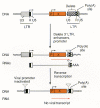
Efficient gene transduction and integration depend on the inclusion in the retroviral vector of a number of cis-acting viral elements (Fig. 1). These include (1) a promoter and polyadenylation signal in the viral genome; (2) a viral packaging signal (ψ or E) to direct incorporation of vector RNA into virions; (3) signals required for reverse transcription, including a transfer RNA-binding site (PBS) and polypurine tract (PPT) for initiation of first- and second-strand DNA synthesis, and a repeated (R) region at both ends of the viral RNA required for transfer of DNA synthesis between templates; and (4) short, partially inverted repeats located at the termini of the viral LTRs required for integration. For a discussion of these elements, and their roles in viral replication, see Chapters 4, 5, and 6. An important general consideration in the design of retroviral vectors is the effect of viral replication on vector structure. After one round of viral replication (Fig. 1), the U3 regions in both LTRs are derived from the U3 region originally present in the 3′LTR in the plasmid form of the vector, and both U5 regions are derived from the U5 region originally present in the 5′LTR in the plasmid. Ordinarily, R sequences should arise primarily from the 5′plasmid LTR, but they may also include 3′plasmid LTR sequences.

Because retroviral replication involves an RNA intermediate, all strategies for inserting genes into a retroviral vector must permit synthesis of full-length copies of the vector genome. For example, insertion of a polyadenylation signal between the LTRs should be avoided because premature polyadenylation within the vector will reduce (if not prevent) full-length vector transcription which interferes with the production of vector-containing virions (Shimotohno and Temin 1981; Miller et al. 1983). In addition, introns contained within the insert may be removed during vector replication (Shimotohno and Temin 1982; Sorge and Hughes 1982; Cepko et al. 1984; Kriegler et al. 1984; McIvor 1990). This property allows the use of retroviral vectors to generate complementary DNAs from genes with introns, but it can also be a disadvantage if an intron contains information that is required for the appropriate regulation of gene expression. Vectors that rely on alternative splicing for gene expression (Fig. 2C) retain their introns because critical elements of the packaging signal are contained within the intron in these vectors, and RNAs without this signal are not efficiently encapsidated into virions (Mann and Baltimore 1985).

Replication-competent Vectors
Avian Viruses
Rous sarcoma virus (RSV) carries the src oncogene in addition to a full complement of genes required for replication, and thus provides the earliest example of a replication-competent retroviral vector (Fig. 3). RSV has been exploited as a vector by replacement of the src gene with other cDNAs (Hughes and Kosik 1984; Hughes et al. 1987). Early attempts to use RSV as a vector were hindered by the presence of direct repeats at either end of the src gene, which led to frequent deletion of new inserts (as well as the original src gene). These problems were solved, however, by removal of the upstream repeat (Hughes and Kosik 1984). Vectors derived from RSV efficiently infect avian cells, and although they can infect mammalian cells, the efficiency of infection is quite low. Two solutions to this problem now exist. Recently, the receptor for subgroup A RSV has been cloned (Bates et al. 1993). Expression of this cloned receptor in mammalian cells allows the efficient infection of those cells by the RSV(A)-based vectors. Avian viruses are naturally replication-defective in mammalian cells; no viral spread is seen after infection. The replication-competent vector can be grown to high titer on avian cells and then used like a replication-defective vector in mammalian cells or in transgenic mice expressing the RSV(A) receptor (Federspiel et al. 1994, 1996).

The alternative approach is to replace the env gene in the RSV-based vector with the env gene from an amphotropic murine leukemia virus (MLV). The resulting vector is then able to replicate efficiently in chicken cells and to infect a variety of mammalian cells efficiently; however, it is still replication-defective in mammalian cells (Barsov and Hughes 1996).
Murine Viruses
Replication-competent vectors based on MLVs have been described which include a tRNA suppressor gene (Lobel et al. 1985; Reik et al. 1985), a mutant dihydrofolate reductase (dhfr*) gene (Stuhlmann et al. 1989), or an HIV tat gene (Dillon et al. 1991). For construction of the dhfr* and tat vectors, the respective cDNAs were linked to an SV40 promoter and inserted in the forward orientation into the U3 region of the 3′viral LTR. The vectors containing dhfr* expression cassettes rearranged frequently during viral production (Stuhlman et al. 1989), but high-titer replication-competent virus that expressed dhfr* could be obtained. High-titer stocks of vectors expressing tat and the tRNA suppressor gene could be prepared without rearrangement.
Human Immunodeficiency Virus
A replication-competent HIV vector that encodes chloramphenicol acetyltransferase has been described (Terwilliger et al. 1989). Although it seems unlikely that replication-competent HIV vectors will gain wide acceptance for gene transfer experiments, the inclusion of a marker will facilitate the study of HIV biology.
Foamy Virus
Replication-competent human foamy virus (HFV) vectors that express chloramphenicol acetyltransferase, luciferase, or the mouse hepatitis virus surface protein have been described (Schmidt and Rethwilm 1995). The cDNAs for these proteins were inserted in place of the bel-2- and bel-3-coding regions at the 3′end of the viral genome. Although expression of bel-1 is essential for HFV replication in cultured cells, deletion of bel-2 and bel-3 has little effect on viral replication. Several strategies were used for transcription and translation of the cDNAs, including in-frame fusion of the cDNAs with upstream HFV-coding regions with or without a self-cleaving foot-and-mouth disease virus (FMDV) protease cleavage site, by use of an IRES or by use of an internal SV40 promoter. HFV vectors with inserted sequences of at least 1.3 kb could be produced at titers similar to those of wild-type HFV.
Replication-defective Vectors
In most applications, the retroviral vector should not continue to spread after the initial infection. This can be accomplished by replacement of most or all of the coding regions of a retrovirus with the gene(s) or sequence elements to be transferred, so that the vector by itself is incapable of making proteins required for additional rounds of replication. Viral proteins needed for the initial infection can be provided in trans by a retroviral packaging cell; there is no need for retroviral protein synthesis in recipient cells for proviral integration. The process of gene transfer and expression by retroviral vectors is often referred to as transduction rather than infection to differentiate this process from productive viral infection by a replication-competent virus.
Retroviral Vector Derivation
Most replication-defective retroviral vectors have been derived from the murine and avian retroviruses, but vectors have also been derived from bovine leukemia virus (Derse and Martarano 1990; Milan and Nicolas 1991), simian retroviruses (Rizvi and Panganiban 1992; Vile et al. 1992), HIV (Terwilliger et al. 1989; Page et al. 1990; Poznansky et al. 1991; Shimada et al. 1991; Buchschacher and Panganiban 1992; Carroll et al. 1994; Richardson et al. 1995; Akkina et al. 1996; Naldini et al. 1996), virus-like 30S (VL30, Chapter 8 elements found in mouse cells (Cook et al. 1991), and HFV (Russell and Miller 1996). Vector design involves the construction of a “provirus” that contains all of the signals needed in cis for vector packaging, reverse transcription, and integration, but which lacks the coding regions for most or all of the viral proteins, and a corresponding packaging cell line to produce the viral proteins required for viral assembly and transduction. Although it is in principle possible to derive vectors from any retrovirus, the complexity, toxicity, and regulatory intricacies of some retroviruses, especially the lentiviruses such as HIV, make construction of vectors more difficult, and the titers of such vectors are typically low. New vector systems are constantly being developed to take advantage of particular properties of the parent retroviruses, such as host range, usage of alternative cell surface receptors, and levels of tissue-specific expression. Since lentivirus can infect nondividing cells, the additional work needed to develop efficient vectors based on these viruses appears to be justified (Akkina et al. 1996; Naldini et al. 1996).
Design of Retroviral Vectors for Protein Expression
Retroviral vectors are typically used to induce the production of a specific protein in transduced cells. The simplest approach is to use the promoter in the retroviral LTR to control the expression of a cDNA encoding the protein of interest (see Fig. 2A). Changes can be made in the enhancer/promoter of the LTR to provide tissue-specific expression or inducibility. The presence of retroviral sequences between the viral promoter and the translational start codon of the inserted gene appears not to markedly affect gene expression, even though several ATG start codons are present in the 5′-untranslated regions of commonly used vectors (Adam et al. 1995). Alternatively, a single coding region can be expressed by using an internal promoter (Fig. 2B) which allows more flexibility in promoter selection. These strategies for expression can be most easily implemented when the gene of interest is also a selectable marker, as in the case of hypoxanthine-guanine phosphoribosyl transferase (hprt) (Miller et al. 1983), which allows facile selection of vector-transduced cells. If the vector contains a gene that is not a selectable marker, the vector can be introduced into packaging cells by cotransfection with a selectable marker present on a separate plasmid. This strategy has the appealing advantage for gene therapy that a single protein is expressed in the ultimate target cells, and possible toxicity or antigenicity of a selectable markers is avoided. When the inserted gene is not selectable, this approach has the disadvantage that it is more difficult to generate cells that produce a high-titer vector stock; in addition, it is usually more difficult to determine the titer of the vector.
Three general strategies have been used to design retroviral vectors that express two or more proteins: (1) expression of different proteins from alternatively spliced messenger RNAs transcribed from one promoter (Fig. 2C), (2) use of the promoter in the LTR and internal promoters to drive transcription of different cDNAs (Fig. 2D,E,F,G), and (3) use of IRES elements to allow translation of multiple coding regions from a single mRNA (Fig. 2H). An alternative approach is to design fusion proteins that can then be expressed from a single open reading frame (Fig. 2I). The latter approach mirrors the fusion proteins containing viral and oncogene sequences that are found in naturally occurring oncogenic viruses (Van Beveren and Verma 1986).
A widely used vector that employs alternative splicing to express genes from the viral LTR (Fig. 2C), SV(X) (Cepko et al. 1984), contains the neomycin phosphotransferase gene as a selectable marker. The model for this type of vector is the parental virus, Mo-MLV, in which the Gag and Gag-Pol proteins are translated from the full-length viral mRNA and the Env protein is made from a spliced mRNA. One of the proteins encoded by the vector is translated from the full-length vector RNA, whereas splicing that links a splice donor near the 5′LTR to a splice acceptor just upstream of the second gene produces an RNA from which the second gene product can be translated. One drawback of this strategy is that foreign sequences are inserted into the intron of the spliced gene. This can affect the ratio of spliced to unspliced RNAs or provide alternative splice acceptors that interfere with production of the spliced RNA encoding the second gene product (Korman et al. 1987). These effects are unpredictable and can affect the production of the encoded proteins.
Vectors containing internal promoters have been widely used to express multiple genes. An internal promoter makes it simple to exploit promoter/enhancer combinations other than the viral LTR for driving gene expression (Fig. 2D,E,F). Multiple internal promoters can be included in a retroviral vector; it is possible to express at least three different cDNAs, each from its own promoter (Overell et al. 1988). It has been reported, in vectors with internal promoters, that selection for the expression of one gene resulted in reduced expression from the other gene also present in the vector (Emerman and Temin 1984). This effect was called promoter suppression. However, this effect is highly dependent on vector construction (Emerman and Temin 1986a, 1986b) and, at least in some cases, is insignificant (Palmer et al. 1987). In addition, because expression of the “suppressed” gene was always compared to expression of the same gene in cells selected for expression of the gene (which must express the gene at a high level in order to survive selection), it is difficult to determine whether low expression can really be attributed to promoter suppression or is simply a result of the particular design of these experiments.
A variation on the general theme of multiple promoters in retroviral vectors is the insertion of a promoter-cDNA combination (minigene) in the U3 region of the retroviral LTR. This strategy produces two copies of the minigene in the resulting provirus (Fig. 2F). Minigene expression in these “double-copy” vectors has been reported to be much higher than that obtained when the minigene is inserted internally in the vector (Hantzopoulos et al. 1989; Chen et al. 1992). It has been argued that expression is enhanced because one of the minigenes is located outside of the retroviral transcription unit, which could reduce any negative effect of viral transcription (Hantzopoulos et al. 1989). However, the performance of double-copy vectors was not compared to vectors containing cDNAs driven by the viral LTR, which are usually quite efficient, and it is unclear whether double-copy vectors offer higher levels of expression than simple vectors in which cDNA expression is driven by the viral LTR.
Double-copy vectors have also been made by inserting cDNAs into the R region of the viral LTR (Fig. 2G). When such vectors were compared to vectors in which the same cDNAs are transcribed from the LTR promoter but are located downstream from the LTR, there was a small (up to fivefold) increase in protein expression, but the vector titer was also reduced about sevenfold (Adam et al. 1995). A disadvantage of this type of double-copy vector is that the RNA form of the vector genome contains two copies of the cDNA, and since the size of the vector RNA is limited to about 10 kb, the size of the cDNA that can be accommodated is about half of that possible in other retroviral vectors.
Insertion of IRES elements into retroviral vectors is compatible with the retroviral replication cycle and allows expression of multiple coding regions from a single promoter (Adam et al. 1991; Koo et al. 1992; Chen et al. 1993). IRES elements were first found in the nontranslated 5′ends of picornaviruses where they promote cap-independent translation of viral proteins (Jang et al. 1990; Sonenberg 1990). When located between open reading frames in an RNA, IRES elements allow efficient translation of the downstream open reading frame by promoting entry of the ribosome at the IRES element followed by downstream initiation of translation. IRES elements from poliovirus, encephalomyocarditis virus, and swine vesicular disease virus have been used in retroviral vectors. Expression of cDNAs situated downstream from IRES elements in a vector of the type shown in Figure 2H can be as high as that of cDNAs located upstream of the IRES element. Expression of two proteins from a single RNA ensures coordinate expression of both proteins. If one of the two proteins is a selectable marker, selection should guarantee expression of the other protein.
Of the methods used to express two proteins in a retroviral vector, the best concordance of expression can be obtained by expression of a chimeric protein that combines the functions of both proteins (Fig. 2I). The chimeric protein is encoded by an in-frame fusion of the cDNAs encoding the proteins. Retroviral vectors have been constructed that transduce chimeric combinations of multidrug resistance and adenosine deaminase (Germann et al. 1990) or hygromycin phosphotransferase (hph) and thymidine kinase (tk) (Lupton et al. 1991). The latter vector provides the opportunity for either positive (by using hygromycin) or negative (by using ganciclovir) selection. Expression of the two functions can become discordant as a result of mutations within the chimeric gene, but the rate of these events is less than 0.1% for the hph-tk vector (Lupton et al. 1991).
Selectable Markers
Many different selectable markers have been used successfully in retroviral vectors. These include the bacterial neomycin and hygromycin phosphotransferase genes which confer resistance to G418 and hygromycin, respectively (Palmer et al. 1987; Yang et al. 1987); a mutant mouse dihydrofolate reductase gene (dhfr*) which confers resistance to methotrexate (Miller et al. 1985); the bacterial gpt gene which allows cells to grow in medium containing mycophenolic acid, xanthine, and aminopterin (Mann et al. 1983); the bacterial hisD gene which allows cells to grow in medium without histidine but containing histidinol (Danos and Mulligan 1988); the multidrug resistance gene (mdr) which confers resistance to a variety of drugs (Guild et al. 1988; Pastan et al. 1988); and the bacterial genes which confer resistance to puromycin or phleomycin (Morgenstern and Land 1990). All of these markers are dominant selectable markers and allow chemical selection of most cells expressing these genes.
β-galactosidase can also be considered a dominant marker; cells expressing β-galactosidase can be selected by using the fluorescence-activated cell sorter (Nolan et al. 1988). In fact, any cell surface protein can provide a selectable marker for cells not already making the protein. Cells expressing the protein can be selected by using a fluorescent antibody to the protein and a cell sorter.
Other selectable markers that have been included in vectors include the hprt (Miller et al. 1983) and HSV thymidine kinase (Shimotohno and Temin 1981; Wei et al. 1981), which allow cells to grow in medium containing hypoxanthine, amethopterin, and thymidine. For these selectable markers to be useful, however, the target cells must initially be hprt- or tk-deficient.
Self-inactivating Vectors
The first self-inactivating retroviral vectors were constructed by deleting the transcriptional enhancers or the enhancers and promoter in the U3 region of the 3′ LTR. After one round of vector replication, these changes are copied into both the 5′and the 3′LTRs producing an inactive provirus (Fig. 4) (Yu et al. 1986; Dougherty and Temin 1987; Hawley et al. 1987; Yee et al. 1987). However, any promoter(s) internal to the LTRs in such vectors will still be active. This strategy has been employed to eliminate effects of the enhancers and promoters in the viral LTRs on transcription from internally placed genes. Such effects include increased transcription (Jolly et al. 1983) or suppression of transcription (Emerman and Temin 1984). This strategy can also be used to eliminate downstream transcription from the 3′LTR into genomic DNA (Herman and Coffin 1987). This is of particular concern in human gene therapy where it is of critical importance to prevent the adventitious activation of an endogenous oncogene. Drawbacks of this strategy include the lower titer of self-inactivating vectors in comparison with vectors having intact LTRs (at least tenfold lower) and the propensity of the current vectors to arrange to produce viruses with intact LTRs, presumably by recombination of the vector with itself or with viral sequences in the retroviral packaging cells used to produce the vector stocks.
Another type of self-inactivating vector has been constructed that has direct repeats flanking the packaging signal such that the packaging signal is frequently deleted during reverse transcription, producing virus defective for packaging (Julias et al. 1995). With sufficiently long direct repeats, a majority of resultant proviruses lose their packaging sequences. The rate of deletion could be increased to 100% by designing the vector so that packaging signal deletion reconstituted the neo marker and by selecting the vector-infected cells in G418. This strategy may be particularly useful for gene therapy applications where any spread of the vector following gene transfer is undesirable.
High-titer Vector Production
Helper-virus-free vector titers of 107 cfu/ml are obtainable with currently available vectors. Often, experiments can be done with much lower-titer vector stocks, but for practical reasons, high-titer virus is desirable, especially when a large number of cells must be infected. In addition, high titers are a requirement for transduction of a large percentage of certain cell types. For example, the frequency of human hematopoietic progenitor cell infection is strongly dependent on vector titer, and useful frequencies of infection occur only with very high-titer stocks (Hock and Miller 1986; Hogge and Humphries 1987). In these cases, it is not sufficient simply to expose the cells to a larger volume of virus to compensate for a low virus titer, instead, the concentration of infectious vector virions seems to be critical to promote efficient transduction.
Comparison of different vector designs allowed the definition of essential elements for high-titer viral production (see Fig. 1). Early work on vector design showed that almost all of the internal protein-encoding regions of MLVs could be deleted without abolishing the infectivity of the vector (Miller et al. 1983). These early vectors retained only a small portion of the 3′ end of the env-coding region. Subsequent work has shown that all of the env-gene-coding sequences can be removed without further reduction in vector titer (Miller and Rosman 1989; Morgenstern and Land 1990). Only the viral LTRs and short regions adjoining the LTRs, including the segments needed for plus- and minus-strand DNA priming and a region required for packaging of viral RNA into virions (the ψ site; Mann et al. 1983), were needed for vector transmission. However, viral titers obtained with these early vectors were still about tenfold lower than the parental helper virus titer. Additional experiments showed that retention of sequences at the 5′end of the gag gene significantly raised viral titers and that this was due to an increase in the packaging efficiency of viral RNA into virions (Armentano et al. 1987; Bender et al. 1987; Adam and Miller 1988). This effect was not due to viral protein synthesis from the gag region of the vector because disruption of the gag reading frame or mutating the gag start codon to a TAG stop codon had no effect on vector titer (Bender et al. 1987). These experiments have demonstrated that the sequences required for efficient packaging of genomic RNA in MLV are larger than the ψ signal previously defined by deletion analysis (Mann et al. 1983). To obtain high titers (106 to >107), it is important that this larger signal, called ψ+, be included in retroviral vectors.
Several methods for concentration of retroviral vectors have been developed, including the use of centrifugation (Fekete and Cepko 1993a), hollow fiber filtration (Paul et al. 1993), and tangential flow filtration (Kotani et al. 1994). About a 20-fold increase in viral titer can be achieved. The relative fragility of retroviral Env protein limits the ability to concentrate retroviral vectors, and concentrating the virus usually results in a poor recovery of infectious virions. This problem can be overcome by substitution of the retroviral Env protein with the more stable VSV-G protein, allowing more effective vector concentration with better yields (see below, Production by Transient Transfection).
In addition to high-titer vectors, methods to facilitate vector infection of cells are also important for maximizing the efficiency of gene transfer. Polybrene and protamine sulfate are typically used to facilitate binding and entry of retroviruses, and various cationic lipids can result in further improvements (Hodgson and Solaiman 1996). Cell transduction is limited by viral diffusion in solution, and passage of the virus through a porous membrane on which target cells have been placed can improve transduction rates (Chuck and Palsson 1996). Centrifugation of vectors onto cells can increase transduction rates, and this increase may also be related to bringing the virus to the cells or preventing the virus from diffusing away from the cells (Bahnson et al. 1995).
Limitations
Sequences inserted into retroviral vectors must be compatible with the retroviral life cycle and, in particular, must allow the efficient transcription of the complete retroviral genome. This means that, ordinarily, a polyadenylation signal cannot be inserted into the virus in a forward orientation. This is a major constraint for the insertion of AT-rich DNA, which can have multiple polyadenylation signals in both orientations. Because the vector replicates via an RNA intermediate, introns will be removed, including novel introns generated in vector construction (McIvor 1990). There are also sequences within the coding regions of some cDNAs, for example, in the cDNA for clotting factor VIII, that inhibit RNA accumulation by more than 100-fold and reduce vector titer proportionally (Lynch et al. 1993).
Although there appears to be no lower limit on retroviral vector size, there do appear to be upper limits. The genome of a typical replication-competent murine retrovirus is about 8.3 kb, whereas that of RSV, which contains src sequences in addition to the normal complement of viral genes, is about 9.3 kb. The maximum size for a replication-competent spleen necrosis virus vector is similar, about 10 kb (Gelinas and Temin 1986). Although this precludes vector transmission of large genes, most cDNAs can be accommodated.
Genomic rearrangements and more subtle mutations can occur during the generation of retroviral vector stocks. Both problems can be minimized by harvesting virus from packaging cells that contain a single integrated provirus of the correct structure. The mutation rate for a single round of viral replication has been estimated to be on the order of 10−4 to 10−5 per base pair per replication cycle (Dougherty and Temin 1988) (see Chapter 4.
Retroviral vectors based on murine viruses appear to transduce only dividing cells (Miller et al. 1990), limiting the utility of these vectors. Lentiviruses such as HIV are able to infect nondividing cells (Lewis et al. 1992; Lewis and Emerman 1994), and with the use of VSV-G proteins in place of the normal Env protein, relatively high-titer HIV-1 vectors have been created (Akkina et al. 1996; Naldini et al. 1996). Vectors based on HFV have been developed and have an ability to infect nondividing cells that is intermediate between the murine and HIV vectors (Russell and Miller 1996). The ability to transduce nondividing cells would be particularly useful for gene transfer to somatic tissues of humans for gene therapy.
Retroviral vector expression is subject to suppression, particularly in embryonic cells (Barklis et al. 1986; Mitrani et al. 1987). The retroviral LTRs and the tRNA-binding site have been clearly implicated in this effect, but other elements may also be involved (Weiher et al. 1987; Akgun et al. 1991; Kempler et al. 1993). Alterations have been made in retroviral vectors to overcome these effects (Hawley et al. 1989; Grez et al. 1990), but the possibilities for vector alteration are more limited than in simple plasmid-based vectors.
Retroviral Packaging Cells
Unlike bacteriophage assembly which can be accomplished in a cell-free system, production of retroviral virions has been accomplished only in intact cells (see Chapter 7. To make replication-defective vectors, retroviral packaging cells have been designed to provide all viral proteins but not to package or transmit the RNAs encoding these functions. Retroviral vectors produced by packaging cells can transduce cells but cannot replicate further.
Packaging Cell Design and Evolution
The earliest example of a retroviral packaging cell line is the SE21Q1b cell line, which expressed a naturally occurring avian sarcoma virus mutant that failed to package viral genomic RNA (Linial et al. 1978). The virus contained two mutations—a deletion of sequences near the 5′end of the viral RNA required for packaging into virions and another mutation that allowed nonspecific packaging of cellular RNA instead of viral RNA (Shank and Linial 1980). This cell line provided stimulus for construction of synthetic packaging cell lines.
The design of retroviral packaging cell lines has evolved to address the problem of the spontaneous production of helper virus that was frequently encountered with early designs. Early packaging cells (Fig. 5A) contained replication-competent retroviral genomes from which a portion of the packaging signal of the virus, lying between the 5′LTR and the gag-coding region, had been deleted. These deleted viruses produced all of the retroviral proteins, but genomic RNA was poorly encapsidated into virions and the virus spread very slowly. However, a single recombination event between the deleted retrovirus contained in the helper cells and the retroviral vector could result in the production of wild-type virus (Fig. 5A) which spreads very efficiently. The viral genome contained in later packaging cell lines was modified so that two recombination events are required to yield wild-type virus (Fig. 5B), which reduces the potential for the production of a replication-competent helper virus. Additional alterations in the genome of the helper virus have included deletion of portions of the 5′LTR or substitution of other promoters for the viral LTR promoter and replacement of the 3′LTR with an alternative polyadenylation signal such as that from SV40. More recently, packaging cells have been developed in which the Gag-Pol and Env viral protein-coding regions are carried on separate expression plasmids that are independently transfected into the packaging cells (Fig. 5C), so that three recombination events are necessary for wild-type viral production. Recombination is greatly facilitated by homology; reducing or eliminating homology between the genomes of the vector and the helper reduces the problem of helper virus production. A summary of the available packaging lines in these different classes is presented in Table 1.
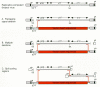
Table 1
Retroviral Packaging Cell Lines.
It is difficult to obtain a reliable rate for helper virus production from a particular packaging cell line because helper virus generation is dependent on both the cell line and the vector. In addition, helper virus assays differ considerably in their sensitivities, and helper virus production can be the result of contamination of packaging cells with exogenous helper virus. However, use of several packaging cell lines (e.g., PA317, ψCRIP, PG13, and GP+envAm12) for human gene therapy has shown that helper virus generation occurs at a very low rate in the newer packaging cell lines, and it is possible to make hundreds of liters of helper-free stocks. Even the older packaging cell lines such as ψ-2 can be kept free of helper by freezing vector-containing cells soon after their construction, passaging the cells for relatively short periods of time, and then discarding the cells and thawing a new sample.
For gene therapy, it has become apparent that both the cells and the virus used to make a packaging cell line influence the sensitivity of vectors produced by the cells to human complement (Takeuchi et al. 1994). Vectors produced from mouse cells were sensitive regardless of the virus used, whereas vectors produced from particular human cells were less sensitive. The endogenous cat virus RD114 was more resistant to human complement than amphotropic MLV in all cells studied. Sensitivity to complement is due to direct lysis of virions and to indirect lysis involving anti-O-galactosyl antibodies prevalent in human serum (Rother et al. 1995). It remains to be seen whether the use of packaging cells derived using human cells and RD114 virus (FLYRD18 cells; Cosset et al. 1995) improves gene transfer in human trials.
Nonreciprocal interference between retroviruses suggested that some retroviruses can use more than one cell surface receptor for entry (see Chapter 3. In particular, the 10A1 strain of MLV can use either the amphotropic receptor Ram-1 or the gibbon ape leukemia virus (GALV) receptor Glvr-1 for cell entry (Rein and Schultz 1984; Miller and Miller 1994). Packaging cells based on the 10A1 virus are available and may be useful for gene transfer into cells where the relative levels of Ram-1 and Glvr-1 are not known (Miller and Chen 1996).
The packaging line GP7C-tTA-G10 is designed to express the VSV-G protein. VSV-G increases the host range of the vector and, because of stability imparted by VSV-G, makes it possible to concentrate viral stocks (Yang et al. 1995). Because VSV-G protein is toxic to cells, a tetracycline-repressible promoter has been used to control the expression of VSV-G. Cells can be grown in the presence of tetracycline where the expression of VSV-G is suppressed. Removal of the tetracycline induces VSV-G synthesis for vector production.
Host Range
A primary consideration in the selection of an appropriate packaging cell line is the host range of vectors produced by the cells, which is determined by the particular retrovirus used in the construction of the packaging cells. Host range has been constantly expanding with the development of new packaging lines. The host range of vectors with various envelope proteins is shown in Table 2. These host-range classes should be used only as a general guide, since there are exceptions to the rules. For example, some sublines of Chinese hamster ovary (CHO) cells can be transduced by amphotropic vectors, but most are completely resistant and also show partial resistance to transduction by vectors with a GALV host range (Miller et al. 1991). Similarly, amphotropic vectors can transduce some bovine and chicken cells but not others. These examples are denoted by +/– in Table 2, but other exceptions also exist.
Table 2
Host Range of Vectors with Various Pseudotypes.
Vector Production Techniques
Two methods have been used to introduce vectors into packaging cells: direct transfection of DNA into the cells or infection with virus produced by DNA transfection of other packaging cells. Whereas the first method can result in the insertion of multiple copies of the vector into the recipient cells and may lead to the production of both unrearranged and rearranged viruses, the latter method usually results in the insertion of only single integrated proviruses, which can be screened for the proper structure, that generally yields unrearranged virus (Bender et al. 1988). One might expect that direct transfection of vector sequences might lead to higher viral titers because of the presence of multiple copies of the vector. However, the titer of virus from infected cells containing single proviruses is often as high if not higher (Hwang and Gilboa 1984; Miller et al. 1986). Plasmids have been made that include the origin of replication and T-antigen-coding region from polyomavirus in an attempt to increase viral titer by generating multiple copies of the plasmid in transfected cells, but again the titers usually are no higher than those of packaging cells containing single integrated proviruses (Korman et al. 1987).
Once a packaging cell line has been isolated that produces helper-free unrearranged vector at high titer, this line provides a continuous source of the vector. However, some instability of viral production by packaging cell lines has been observed (Bender et al. 1987). For example, after growth of PA317 cells for 4 months in culture, only 20% of clones isolated following transduction with a retroviral vector produced the vector at high titer. In addition, many of the PA317 cells did not express the HSV thymidine kinase gene that was cotransfected into the cells with the helper virus DNA. Selection for the TK+ cells with HAT restored packaging function in the majority of cells, i.e., infection of these cells with a retroviral vector resulted in a high percentage of clones that secreted high-titer virus. This illustrates the importance of using early passage stocks of cells, which should be stored as frozen stocks. Prolonged passage of packaging cells and vector-producing packaging cells should be avoided. This practice also minimizes the possibility of helper virus production due to recombination of helper sequences with vector or endogenous viral sequences, which is more likely with longer periods of cell culture.
Packaging cells are resistant to infection by vectors produced by the same cell line because the endogenously produced Env blocks the cell surface receptors required for retroviral entry (see Chapter 3. Thus, the vector copy number in a vector-producing packaging cell line remains relatively constant, although it does increase slowly. However, if vector-producing packaging cells are cocultivated with a packaging cell line that expresses an env gene which does not interfere with the env gene in the first helper cell line, the vector can “ping-pong” between cells, resulting in multiple reinfection events. An increase in the number of copies of the genome of the vector per cell results in an increase in the titer of produced vectors (Bestwick et al. 1988; Bodine et al. 1990; Kozak and Kabat 1990; Lynch and Miller 1991). Although vector titer can be increased by using this ping-pong amplification procedure, there is also a greater chance that the vector will rearrange and that helper virus will be produced during the amplification process (Lynch and Miller 1991). A report of a very high-titer murine retroviral vector (1010 cfu/ml; Bodine et al. 1990) produced by using this technique was not reproducible (Lynch and Miller 1991). An increase in titer of up to tenfold is typical.
Transduceable Cells
The only limitation to transduction of avian and mammalian cells with currently available retroviral vectors based on avian and murine viruses is the need for cell division (Fritsch and Temin 1977; Varmus et al. 1977; Harel et al. 1981; Miller et al. 1990). Otherwise, all cell types tested can be transduced. In particular, vectors made by using GALV and amphotropic MLV packaging cell lines can transduce a broad range of cell types from different species (Table 2). If the retroviral env gene is replaced with the VSV-G protein, the host range of retroviral vectors can be extended to other vertebrate species, for example, zebrafish (Burns et al. 1993; Lin et al. 1994).
Most cultured mammalian cell lines can be transduced with retroviral vectors. However, the efficiency of transduction for some cell types can be low; for example, murine erythroleukemia cells and HL-60 promyelocytic leukemia cells are inefficiently transduced (Bender et al. 1987; Collins 1988). Some of the widely used hamster cell lines, such as CHO cells, are relatively difficult to transduce; in this case, the difficulty is at the level of viral entry (Miller and Miller 1992).
Primary cells can also be transduced, sometimes with efficiencies greater than those observed in immortal cell lines. Human skin fibroblasts can be transduced at about 50% efficiency (Palmer et al. 1987) and human keratinocytes at up to 0.5% efficiency (Morgan et al. 1987). Primary hepatocytes can be transduced at efficiencies of up to 25% (Wilson et al. 1988). Many types of cells capable of further differentiation, such as mouse, dog, and human hematopoietic progenitor cells, can be transduced at efficiencies from 5% to 20% (Dick et al. 1985; Hock and Miller 1986; Kwok et al. 1986). Pluripotent hematopoietic stem cells from mice can be transduced with greater than 10% efficiency (Dick et al. 1985; Keller et al. 1985; Lemischka et al. 1986; Bowtell et al. 1987). Transduction of stem cells has been demonstrated by showing that specific vector integrants can be detected in all myeloid and lymphoid cell lineages in a mouse receiving transduced bone marrow cells and by showing that these integrants can be detected over long periods of time in vivo. The percentage of transplanted cells that contain a provirus carrying the neo gene can be substantially improved by preselecting the bone marrow cells with G418 for 24–48 hours before transplanting the cells into animals; this eliminates cells that do not express the vector (Dick et al. 1985; Keller et al. 1985).
Recombinant Helper Virus Detection Techniques
Packaging cell lines can produce replication-competent helper viruses as a result of recombination between the defective helper virus used to construct the cells and an introduced retroviral vector, by recombination of the defective helper and/or retroviral vector with endogenous sequences, or by activation of endogenous replication-competent retroviruses present in the cells. The frequency of helper virus production appears to be related to overlap between the defective helper virus and retroviral vector sequences, supporting the hypothesis that recombination between defective helper and vector sequences is the primary cause of helper virus production (Miller et al. 1986; Miller and Buttimore 1986; Lynch and Miller 1991; Otto et al. 1994). Recombinant helper viruses that arise in a vector-producing packaging cell line can be the result of recombination between the defective helper and vector sequences (Otto et al. 1994).
Because there are several possible sources of helper virus packaging cells, techniques should be used that are capable of detecting all of the possible helper viruses, including recombinants derived from endogenous viruses. For example, a packaging cell line producing amphrotropic viruses, derived from a mouse cell line, might release amphotropic murine helper virus, but might also release xenotropic murine helper virus. Assays for helper virus based on laboratory mouse cell lines could be used to detect the amphotropic helper virus but not the xenotropic helper virus; multiple assays must be used to detect all possible helper viruses.
The most sensitive cell culture assay for the production of helper virus by packaging cell lines is a functional assay involving marker rescue. Cell lines carrying a selectable retroviral vector are exposed to a large amount of the virus to be tested and passaged for more than 2 weeks to allow spread of the helper virus (if one is present), and the medium from the infected cells is tested for the presence of virus that can confer drug resistance to naive cells. If transfer of drug resistance is detected, the selectable provirus was rescued because replication-competent virus was present in the initial viral stock. It is important to be sure that the cells used in tests of this type allow replication of likely helper virus contaminants. For example, human cells do not support the replication of ecotropic murine viruses and cannot be used to test for these viruses. It is also essential to validate the sensitivity of these assays by adding limiting dilutions of standard helper virus stocks to parallel assay dishes to show that the assay is capable of detecting low numbers of infectious virions.
An assay that is commonly used for helper virus detection is the S+L− assay (Bassin et al. 1971; Peebles 1975), which is a marker rescue assay based on a defective oncogenic retrovirus. In this case, rescue of the defective oncogenic retrovirus by helper virus produces a transformed focus on indicator cells. The transformed cells used in this assay were prepared by infecting cells with a mixture of transforming (sarcomagenic) virus plus helper (leukemogenic) virus at low multiplicity and by isolating transformed individual clones that were contained only by the defective sarcomagenic virus (Sarcomagenic+ Leukemogenic− cells). Subclones of these cells were then isolated that showed revertant flat morphology but still carried the transforming virus. Upon infection with helper virus, the transforming virus spreads to nearby cells and causes a transformed focus of cells in an otherwise flat cell layer. In a variation of this assay, the S+L− cells are exposed to virus to be assayed, and then another nontransformed cell line is added to detect rescue of the transforming virus (see, e.g., Miller et al. 1985).
It is appropriate not only to monitor vector stocks for the presence of a helper virus before they are used to infect target cells (or experimental animals), but also to monitor the experiments themselves. In animal experiments, it is sometimes possible to test for recombinant virus using a sensitive biological assay for the presence of antibodies directed against viral proteins. Although an antibody test does not provide definitive proof that a replication-competent virus is present, it is indicative, and since the test is simple, it can be used as a sensitive screen.
Vector Production by Transient Transfection
Transient transfection can be used to measure vector production when vectors are being developed, because it avoids the longer time required to generate stable vector-producing cell lines and is used if the vector or retroviral packaging components are toxic to cells. Components typically used to generate retroviral vectors include a plasmid encoding the Gag-Pol proteins, a plasmid encoding the Env protein, and a plasmid containing the retroviral vector itself. Vector production involves transient transfection of one or more of these components into cells containing the other components. If the vector encodes toxic genes or genes that interfere with the replication of the host cell, such as inhibitors of the cell cycle or genes that induce apoptosis, it may be difficult to generate stable vector-producing cell lines, but transient transfection can be used to produce the vector before the cells die. The VSV-G protein, which can be used in place of the retroviral env gene, is quite toxic to cells, making transient transfection of this component a useful strategy for generation of high-titer vectors (Yee et al. 1994).
Cell lines have been developed that produce vector titers following transient transfection that are comparable to titers obtained from stable vector-producing cell lines (Pear et al. 1993). A key feature is the use of the highly transfectable 293T cells. Retroviral vectors have also been produced at high titer following transient transfection of a retroviral vector and a construct that expresses the packaging functions into cells that express the SV40 large T antigen, either COS-7 (Landau and Littman 1992) or 293T cells (Finer et al. 1994; Soneoka et al. 1995). An SV40 origin is included in the plasmids used in the transfections to induce episomal replication.
HIV vectors are usually made by transient transfection of vector and helper virus because the toxicity of some HIV proteins makes it difficult to generate stable HIV-based packaging cells. Replacement of the HIV Env protein with that of VSV greatly expands the host range of HIV vectors and facilitates vector concentration, but as has already been stated several times, the VSV-G protein is also cytotoxic. HIV/VSV-G vectors with titers of 5 × 105 (108 after concentration) have been generated by transient transfection (Naldini et al. 1996).
Alteration of Viral Tropism
Significant progress has been made in the alteration of retroviral Env proteins. This procedure can be used to target vector infection to cells bearing specific cell surface molecules or, alternatively, to expand the host range of retroviral vectors. Targeting may be useful for human gene therapy where it may be desirable to transduce specific tissues or cell types.
Chimeric Retroviral Envelope Proteins
A general approach to the targeting of specific cell surface proteins involves the incorporation of single-chain antibody fragments into the amino terminus of retroviral Env proteins. Specific targeting has been demonstrated using antibodies directed against an antigen expressed on tumor cells (Chu and Dornburg 1995), the human low-density lipoprotein receptor (Somia et al. 1995), and a determinant on human major histocompatibility complex (MHC) class I molecules (Marin et al. 1996). In two cases, the production of infectious virus depended on the incorporation of both the wild-type envelope glycoprotein and the hybrid protein. Vector titers of 103 to 104 cfu/ml were reported (Chu and Dornburg 1995; Somia et al. 1995). In one case, infectivity was reported to be independent of coexpressed wild-type Env, but the vector titer was very low, averaging about 50 cfu/ml (Marin et al. 1996). Although these experiments demonstrate that the technique is feasible, improvements in titer will be required for in vivo tissue targeting for gene therapy.
Chimeric Env proteins containing ligands that bind to specific cell surface receptors have been tested for their ability to target retroviral vectors to cells expressing the respective receptors. Specific targeting has been shown for a chimeric Env protein containing FLA16, a 16-amino-acid peptide that binds integrin receptors (Valsesia-Wittmann et al. 1994); an Env protein containing erythropoietin, the ligand for the erythropoietin receptor (Kasahara et al. 1994); and an Env protein containing human heregulin, a ligand for the human epidermal growth factor receptor and two other receptors in the same family (Han et al. 1995). The vector titers ranged from 103 to 104. Production of infectious vectors with chimeric envelopes made with erythropoietin or heregulin required incorporation of wild-type Env protein, whereas those made with FLA16 did not. As is the case for the single-chain antibody-envelope chimeras, these results represent a good first attempt, but improvements in vector titer will be needed for in vivo gene therapy.
Envelope Proteins from Other Viruses
Retroviral Env proteins replaced with the VSV-G protein produce retroviral vectors with broad host range and improved virion stability which allows concentration of the vectors to high titer. The Env protein of RSV has also been replaced with the hemagglutinin protein of influenza virus, resulting in expanded host range but also very low-titer (100 cfu/ml) vector production (Dong et al. 1992). The use of envelope proteins from other viruses has increased the range of cells that can be transduced with retroviral vectors.
Chemical and Physical Modification of the Envelope Glycoprotein
Efforts to alter receptor utilization by retroviral vectors can also involve the direct modification of vector virions. Modification of an ecotropic vector by chemical addition of lactose resulted in virions that could transduce human hepatoma cells which expressed asialoglycoprotein receptors but not human hepatoma cells which did not express these receptors (Neda et al. 1991). Although the titer of the modified vector was not determined, the efficiency of transfer appeared to be high, suggesting that this technique might be used to direct vectors to the liver in humans. Mixing an ecotropic retroviral vector with replication-defective adenovirus can also promote efficient infection of human cells by the vector (Adams et al. 1995). Although this approach appears to have little direct application, a further examination of the mechanism may suggest methods that could be used to improve vectors for gene therapy applications.
Another method for direct modification of vector virions (Roux et al. 1989) involved incubation of the virions with biotinylated antibody directed against the retroviral envelope protein. A different biotinylated antibody reactive with histocompatibility antigens on human cells was bound to cells, and streptavidin was used to link the vector virions to the human cells. Although this method promoted infection of human cells by an ecotropic vector, the efficiency of infection was very low, and the technique would not be useful in practice without further improvement.
Safety in the Laboratory
Safety issues that arise from the use of retroviral vectors in the laboratory involve potential toxicities of both the vectors and replication-competent viruses, particularly viruses that can infect human cells. Because retroviruses are surrounded by a lipid membrane derived from the virus-producing cell, they are readily inactivated by detergents or ethanol, and thus environmental hazards can be easily controlled. Many retroviruses, including amphotropic MLV, are inactivated by human serum by antibody-independent (Welsh et al. 1975, 1976; Cooper et al. 1976) and antibody-dependent (Rother et al. 1995) mechanisms, providing a natural barrier against human infection by standard retroviral vectors or associated helper virus. However, human serum does not inactivate human T-cell leukemia virus type 1 (HTLV-1) (Hoshino et al. 1984) or HIV (Banapour et al. 1986), and vectors produced by some packaging cells are resistant to inactivation by human serum (Cosset et al. 1995). Retroviral vectors are generally made in the absence of helper virus, so there should be no spread after the initial infection. Care should be taken that such helper-free viral stocks are truly free of helper virus; it is possible to generate a complex array of replication-competent recombinant viruses using some of the standard packaging cell lines (Vanin et al. 1994; Purcell et al. 1996). Moreover, some of these recombinant viruses have been shown to cause T-cell lymphomas in rhesus macaques (Donahue et al. 1992). Since the activation of an endogenous oncogene by integration is a rare event (see Chapter 10, there would appear to be little, if any, consequence of transduction of a few somatic cells in a human with a replication-defective retroviral vector encoding a selectable marker or a normal cellular gene. Oncogenic retroviruses present a different concern in that, in theory, a small number of cells infected with such a virus might lead to neoplastic growth; thus, more care should be exercised with oncogenic retroviruses to prevent contact with virus-containing medium or production of virus-containing aerosols.
- Principles of Retroviral Vector Design - RetrovirusesPrinciples of Retroviral Vector Design - Retroviruses
Your browsing activity is empty.
Activity recording is turned off.
See more...