In this and the preceding three chapters, we discussed the structure of genes, the way they are arranged in chromosomes, the intricate cellular machinery that converts genetic information into functional protein and RNA molecules, and the many ways in which gene expression is regulated by the cell. In this section, we discuss some of the ways that genes and genomes have evolved over time to produce the vast diversity of modern-day life forms on our planet. Genome sequencing has revolutionized our view of this process of molecular evolution, uncovering an astonishing wealth of information about the family relationships among organisms and evolutionary mechanisms.
It is perhaps not surprising that genes with similar functions can be found in a diverse range of living things. But the great revelation of the past 20 years has been the discovery that the actual nucleotide sequences of many genes are sufficiently well conserved that homologous genes—that is, genes that are similar in their nucleotide sequence because of a common ancestry—can often be recognized across vast phylogenetic distances. For example, unmistakable homologs of many human genes are easy to detect in such organisms as nematode worms, fruit flies, yeasts, and even bacteria.
As discussed in Chapter 3 and again in Chapter 8, the recognition of sequence homology has become a major tool for inferring gene and protein function. Although finding such a homology does not guarantee similarity in function, it has proven to be an excellent clue. Thus, it is often possible to predict the function of a gene in humans for which no biochemical or genetic information is available simply by comparing its sequence to that of an intensively studied gene in another organism.
Gene sequences are often far more tightly conserved than is overall genome structure. As discussed in Chapter 4, features of genome organization such as genome size, number of chromosomes, order of genes along chromosomes, abundance and size of introns, and amount of repetitive DNA are found to differ greatly among organisms, as does the actual number of genes.
The number of genes is only very roughly correlated with the phenotypic complexity of an organism. Thus, for example, current estimates of gene number are 6,000 for the yeast Saccharomyces cerevisiae, 18,000 for the nematode Caenorhabditis elegans, 13,000 for Drosophila melanogaster, and 30,000 for humans (see Table 1-1). As we shall soon see, much of the increase in gene number with increasing biological complexity involves the expansion of families of closely related genes, an observation that establishes gene duplication and divergence as major evolutionary processes. Indeed, it is likely that all present-day genes are descendants—via the processes of duplication, divergence, and reassortment of gene segments—of a few ancestral genes that existed in early life forms.
Genome Alterations are Caused by Failures of the Normal Mechanisms for Copying and Maintaining DNA
With a few exceptions, cells do not have specialized mechanisms for creating changes in the structures of their genomes: evolution depends instead on accidents and mistakes. Most of the genetic changes that occur result simply from failures in the normal mechanisms by which genomes are copied or repaired when damaged, although the movement of transposable DNA elements also plays an important role. As we discussed in Chapter 5, the mechanisms that maintain DNA sequences are remarkably precise—but they are not perfect. For example, because of the elaborate DNA-replication and DNA-repair mechanisms that enable DNA sequences to be inherited with extraordinary fidelity, only about one nucleotide pair in a thousand is randomly changed every 200,000 years. Even so, in a population of 10,000 individuals, every possible nucleotide substitution will have been “tried out” on about 50 occasions in the course of a million years—a short span of time in relation to the evolution of species.
Errors in DNA replication, DNA recombination, or DNA repair can lead either to simple changes in DNA sequence—such as the substitution of one base pair for another—or to large-scale genome rearrangements such as deletions, duplications, inversions, and translocations of DNA from one chromosome to another. It has been argued that the rates of occurrence of these mistakes have themselves been shaped by evolutionary processes to provide an acceptable balance between genome stability and change.
In addition to failures of the replication and repair machinery, the various mobile DNA elements described in Chapter 5 are an important source of genomic change. In particular, transposable DNA elements (transposons) play a major part as parasitic DNA sequences that colonize a genome and can spread within it. In the process, they often disrupt the function or alter the regulation of existing genes; and sometimes they even create altogether novel genes through fusions between transposon sequences and segments of existing genes. Examples of the three major classes of transposons were presented in Table 5-3, p. 287. Over long periods of evolutionary time, these transposons have profoundly affected the structure of genomes.
The Genome Sequences of Two Species Differ in Proportion to the Length of Time That They Have Separately Evolved
The differences between the genomes of species alive today have accumulated over more than 3 billion years. Lacking a direct record of changes over time, we can nevertheless reconstruct the process of genome evolution from detailed comparisons of the genomes of contemporary organisms.
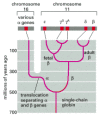
The basic tool of comparative genomics is the phylogenetic tree. A simple example is the tree describing the divergence of humans from the great apes (). The primary support for this tree comes from comparisons of gene and protein sequences. For example, comparisons between the sequences of human genes or proteins and those of the great apes typically reveal the fewest differences between human and chimpanzee and the most between human and orangutan.
A phylogenetic tree showing the relationship between the human and the great apes based on nucleotide sequence data. As indicated, the sequences of the genomes of all four species are estimated to differ from the sequence of the genome of their last common (more...)
For closely related organisms such as humans and chimpanzees, it is possible to reconstruct the gene sequences of the extinct, last common ancestor of the two species (). The close similarity between human and chimpanzee genes is mainly due to the short time that has been available for the accumulation of mutations in the two diverging lineages, rather than to functional constraints that have kept the sequences the same. Evidence for this view comes from the observation that even DNA sequences whose nucleotide order is functionally unconstrained—such as the sequences that code for the fibrinopeptides (see p. 236) or the third position of “synonymous” codons (codons specifying the same amino acid—see )—are nearly identical.
Tracing the ancestor sequence from a sequence comparison of the coding regions of human and chimpanzee leptin genes. Leptin is a hormone that regulates food intake and energy utilization in response to the adequacy of fat reserves. As indicated by the (more...)
For less closely related organisms such as humans and mice, the sequence conservation found in genes is largely due to purifying selection (that is, selection that eliminates individuals carrying mutations that interfere with important genetic functions), rather than to an inadequate time for mutations to occur. As a result, protein-coding sequences and regulatory sequences in the DNA that are constrained to engage in highly specific interactions with conserved proteins are often remarkably conserved. In contrast, most DNA sequences in the human and mouse genomes have diverged so far that it is often impossible to align them with one another.
Integration of phylogenetic trees based on molecular sequence comparisons with the fossil record has led to the best available view of the evolution of modern life forms. The fossil record remains important as a source of absolute dates based on the decay of radioisotopes in the rock formations in which fossils are found. However, precise divergence times between species are difficult to establish from the fossil record even for species that leave good fossils with distinctive morphology. Populations may be small and geographically localized for long periods before a newly arisen species expands in numbers sufficiently to leave a fossil record that is detectable. Furthermore, even when a fossil closely resembles a contemporary species, it is not certain that it is ancestral to it—the fossil may come from an extinct lineage, while the true ancestors of the contemporary species may remain unknown.
The integrated phylogenetic trees support the basic idea that changes in the sequences of particular genes or proteins occur at a constant rate, at least in the lineages of organisms whose generation times and overall biological characteristics are quite similar to one another. This apparent constancy in the rates at which sequences change is referred to as the molecular-clock hypothesis. As described in Chapter 5, the molecular clock runs most rapidly in sequences that are not subject to purifying selection—such as intergenic regions, portions of introns that lack splicing or regulatory signals, and genes that have been irreversibly inactivated by mutation (the so-called pseudogenes). The clock runs most slowly for sequences that are subject to strong functional constraints—for example, the amino acid sequences of proteins such as actin that engage in specific interactions with large numbers of other proteins and whose structure, therefore, is highly constrained (see, for example, ).
Because molecular clocks run at rates that are determined both by mutation rates and by the amount of purifying selection on particular sequences, a different calibration is required for genes replicated and repaired by different systems within cells. Most notably, clocks based on functionally unconstrained mitochondrial DNA sequences run much faster than clocks based on functionally unconstrained nuclear sequences because of the high mutation rate in mitochondria.
Molecular clocks have a finer time resolution than the fossil record and are a more reliable guide to the detailed structure of phylogenetic trees than are classical methods of tree construction, which are based on comparisons of the morphology and development of different species. For example, the precise relationship among the great-ape and human lineages was not settled until sufficient molecular-sequence data accumulated in the 1980s to produce the tree that was shown in .
The Chromosomes of Humans and Chimpanzees Are Very Similar
We have just seen that the extent of sequence similarity between homologous genes in different species depends on the length of time that has elapsed since the two species last had a common ancestor. The same principle applies to the larger scale changes in genome structure.
The human and chimpanzee genomes—with their 5-million-year history of separate evolution—are still nearly identical in overall organization. Not only do humans and chimpanzees appear to have essentially the same set of 30,000 genes, but these genes are arranged in nearly the same way along the chromosomes of the two species (see ). The only substantial exception is that human chromosome 2 arose by a fusion of two chromosomes that are separate in the chimpanzee, the gorilla, and the orangutan.
Even the massive resculpting of genomes that can be produced by transposon activity has had only minor effects on the 5-million-year time scale of the human-chimpanzee divergence. For example, more than 99% of the one million copies of the Alu family of retrotransposons that are present in both genomes are in corresponding positions. This observation indicates that most of the Alu sequences in our genome underwent duplication and transposition before the divergence of the human and chimpanzee lineages. Nevertheless, the Alu family is still actively transposing. Thus, a small number of cases have been observed in which new Alu insertions have caused human genetic disease; these cases involve transposition of this DNA into sites unoccupied in the genomes of the patient's parents. More generally, there exists a class of “human-specific” Alu sequences that occupy sites in the human genome that are unoccupied in the chimpanzee genome. Since perfect-excision mechanisms for Alu sequences appear to be lacking, these human-specific Alu sequences most likely reflect new insertions in the human lineage, rather than deletions in the chimpanzee lineage. The close sequence similarity among all of the human-specific Alu sequences suggests that they have a recent common ancestor; it may even be that only a single “master” Alu sequence remains capable of spawning new copies of itself in humans.
A Comparison of Human and Mouse Chromosomes Shows How The Large-scale Structures of Genomes Diverge
The human and chimpanzee genomes are much more alike than are the human and mouse genomes. Although the size of the mouse genome is approximately the same and it contains nearly identical sets of genes, there has been a much longer time period over which changes have had a chance to accumulate—approximately 100 million years versus 5 million years. It may also be that rodents have significantly higher mutation rates than humans; in this case the great divergence of the human and mouse genomes would be dominated by a high rate of sequence change in the rodent lineage. Lineage-specific differences in mutation rates are, however, difficult to estimate reliably, and their contribution to the patterns of sequence divergence observed among contemporary organisms remains controversial.
As indicated by the DNA sequence comparison in , mutation has led to extensive sequence divergence between humans and mice at all sites that are not under selection—such as the nucleotide sequences of introns. Indeed, human-mouse-sequence comparisons are much more informative of the functional constraints on genes than are human-chimpanzee comparisons. In the latter case, nearly all sequence positions are the same simply because not enough time has elapsed since the last common ancestor for large numbers of changes to have occurred. In contrast, because of functional constraints in human-mouse comparisons the exons in genes stand out as small islands of conservation in a sea of introns.
Comparison of a portion of the mouse and human leptin genes. Positions where the sequences differ by a single nucleotide substitution are boxed in green, and positions that differ by the addition or deletion of nucleotides are boxed in yellow. Note that (more...)
As the number of sequenced genomes increases, comparative genome analysis is becoming an increasingly important method for identifying their functionally important sites. For example, conservation of open-reading frames between distantly related organisms provides much stronger evidence that these sequences are actually the exons of expressed genes than does a computational analysis of any one genome. In the future, detailed biological annotation of the sequences of complex genomes—such as those of the human and the mouse—will depend heavily on the identification of sequence features that are conserved across multiple, distantly related mammalian genomes.
In contrast to the situation for humans and chimpanzees, local gene order and overall chromosome organization have diverged greatly between humans and mice. According to rough estimates, a total of about 180 break-and-rejoin events have occurred in the human and mouse lineages since these two species last shared a common ancestor. In the process, although the number of chromosomes is similar in the two species (23 per haploid genome in the human versus 20 in the mouse), their overall structures differ greatly. For example, while the centromeres occupy relatively central positions on most human chromosomes, they lie next to an end of each chromosome in the mouse. Nonetheless, even after the extensive genomic shuffling, there are many large blocks of DNA in which the gene order is the same in the human and the mouse. These regions of conserved gene order in chromosomes are referred to as synteny blocks (see ).
Analysis of the transposon families in the human and the mouse provide additional evidence of the long divergence time separating the two species. Although the major retrotransposon families in the human have counterparts in the mouse—for example, human Alu repeats are similar in sequence and transposition mechanism to the mouse B1 family—the two families have undergone separate expansions in the two lineages. Even in regions where human and mouse sequences are sufficiently conserved to allow reliable alignment, there is no correlation between the positions of Alu elements in the human genome and the B1 elements in corresponding segments of the mouse genome ().
A comparison of the β-globin gene cluster in the human and mouse genomes, showing the location of transposable elements. This stretch of human genome contains five functional β-globin-like genes (orange); the comparable region from the (more...)
It Is Difficult to Reconstruct the Structure of Ancient Genomes
The genomes of ancestral organisms can be inferred, but never directly observed: there are no ancient organisms alive today. Although a modern organism such as the horseshoe crab looks remarkably similar to fossil ancestors that lived 200 million years ago, there is every reason to believe that the horseshoe-crab genome has been changing during all that time at a rate similar to that occurring in other evolutionary lineages. Selective constraints must have maintained key functional properties of the horseshoe-crab genome to account for the morphological stability of the lineage. However, genome sequences reveal that the fraction of the genome subject to purifying selection is small; hence the genome of the modern horseshoe crab must differ greatly from that of its extinct ancestors, known to us only through the fossil record.
It is difficult to infer even gross features of the genomes of long-extinct organisms. An important example is the so-called introns-early versus introns-late controversy. Soon after the discovery in 1977 that the coding regions of most genes in metazoan organisms are interrupted by introns, a debate arose about whether introns reflect a late acquisition during the evolution of life on earth or whether they were instead present in the earliest genes. According to the introns-early model, fast-growing organisms such as bacteria lost the introns present in their ancestors because they were under selection for a compact genome adapted for rapid replication. This view is contested by an introns-late model, in which introns are viewed as having been inserted into intronless genes long after the evolution of single-cell organisms, perhaps through the agency of certain types of transposons.
There is presently no reliable way of resolving this controversy. Comparative studies of existing genomes provide estimates of rates of intron gain and loss in various evolutionary lineages. However, these estimates bear only indirectly on the question of how genomes were organized billions of years ago. Bacteria and humans are equally “modern” organisms, both of whose genomes differ so greatly from that of their last common ancestor that we can only speculate about the properties of this very ancient, ancestral genome.
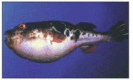
When two modern organisms share nearly identical patterns of intron positions in their genes, we can be confident that the introns were present in the last common ancestor of the two species. An illuminating comparison involves humans and the puffer fish, Fugu rubripes (). The Fugu genome is remarkable in having an unusually small size for a vertebrate (0.4 billion nucleotide pairs compared to 1 billion or more for many other fish and 3 billion for typical mammals). The small size of the Fugu genome is due almost entirely to the small size of its introns. Specifically, Fugu introns, as well as other non-coding segments of the Fugu genome, lack the repetitive DNA that makes up a large portion of the genomes of most well studied vertebrates. Nevertheless, the positions of Fugu introns are nearly perfectly conserved relative to their positions in mammalian genomes ().
The puffer fish, Fugu rubripes. (Courtesy of Byrappa Venkatesh.)
Comparison of the genomic sequences of the human and Fugu genes encoding the protein huntingtin. Both genes (indicated in red) contain 67 short exons that align in 1:1 correspondence to one another; these exons are connected by curved lines. The human (more...)
The question of why Fugu introns are so small is reminiscent of the introns-early versus introns-late debate. Obviously, either introns grew in many lineages while staying small in the Fugu lineage, or the Fugu lineage experienced massive loss of repetitive sequences from its introns. We have a clear understanding of how genomes can grow by active transposition since most transposition events are duplicative [i.e., the original copy stays where it was while a copy inserts at the new site (see and )]. There is considerably less evidence in well-studied organisms for mutational processes that would efficiently delete transposons from immense numbers of sites without also deleting adjacent functionally critical sequences at rates that would threaten the survival of the lineage. Nonetheless, the origin of Fugu's unusually small introns remains uncertain.
Gene Duplication and Divergence Provide a Critical Source of Genetic Novelty During Evolution
Much of our discussion of genome evolution so far has emphasized neutral change processes or the effects of purifying selection. However, the most important feature of genome evolution is the capacity for genomic change to create biological novelty that can be positively selected for during evolution, giving rise to new types of organisms.
Comparisons between organisms that seem very different illuminate some of the sources of genetic novelty. A striking feature of these comparisons is the relative scarcity of lineage-specific genes (for example, genes found in primates but not in rodents, or those found in mammals but not in other vertebrates). Much more prominent are selective expansions of preexisting gene families. The genes encoding nuclear hormone receptors in humans, a nematode worm, and a fruit fly, all of which have fully sequenced genomes, illustrate this point (). Many of the subtypes of these nuclear receptors (also called intracellular receptors) have close homologs in all three organisms that are more similar to each other than they are to other family subtypes present in the same species. Therefore, much of the functional divergence of this large gene family must have preceded the divergence of these three evolutionary lineages. Subsequently, one major branch of the gene family underwent an enormous expansion only in the worm lineage. Similar, but smaller lineage-specific expansions of particular subtypes are evident throughout the gene family tree, but they are particularly evident in the human—suggesting that such expansions offer a path toward increased biological complexity.
A phylogenetic tree based on the inferred protein sequences for all nuclear hormone receptors encoded in the genomes of human (H. sapiens), a nematode worm (C. elegans), and a fruit fly (D. melanogaster).
Triangles represent protein subfamilies that (more...)
Gene duplication appears to occur at high rates in all evolutionary lineages. An examination of the abundance and rate of divergence of duplicated genes in many different eucaryotic genomes suggests that the probability that any particular gene will undergo a successful duplication event (i.e., one that spreads to most or all individuals in a species) is approximately 1% every million years. Little is known about the precise mechanism of gene duplication. However, because the two copies of the gene are often adjacent to one another immediately following duplication, it is thought that the duplication frequently results from inexact repair of double-strand chromosome breaks (see ).
Duplicated Genes Diverge
A major question in genome evolution concerns the fate of newly duplicated genes. In most cases, there is presumed to be little or no selection—at least initially—to maintain the duplicated state since either copy can provide an equivalent function. Hence, many duplication events are likely to be followed by loss-of-function mutations in one or the other gene. This cycle would functionally restore the one-gene state that preceded the duplication. Indeed, there are many examples in contemporary genomes where one copy of a duplicated gene can be seen to have become irreversibly inactivated by multiple mutations. Over time, the sequence similarity between such a pseudogene and the functional gene whose duplication produced it would be expected to be eroded by the accumulation of many mutational changes in the pseudogene—eventually becoming undetectable.
An alternative fate for gene duplications is for both copies to remain functional, while diverging in their sequence and pattern of expression and taking on different roles. This process of “duplication and divergence” almost certainly explains the presence of large families of genes with related functions in biologically complex organisms, and it is thought to play a critical role in the evolution of increased biological complexity.
Whole-genome duplications offer particularly dramatic examples of the duplication-divergence cycle. A whole-genome duplication can occur quite simply: all that is required is one round of genome replication in a germline cell lineage without a corresponding cell division. Initially, the chromosome number simply doubles. Such abrupt increases in the ploidy of an organism are common, particularly in fungi and plants. After a whole-genome duplication, all genes exist as duplicate copies. However, unless the duplication event occurred so recently that there has been little time for subsequent alterations in genome structure, the results of a series of segmental duplications—occurring at different times—are very hard to distinguish from the end product of a whole-genome duplication. In the case of mammals, for example, the role of whole genome duplications versus a series of piecemeal duplications of DNA segments is quite uncertain. Nevertheless, it is clear that a great deal of gene duplication has ocurred in the distant past.
Analysis of the genome of the zebrafish, in which either a whole-genome duplication or a series of more local duplications occurred hundreds of millions of years ago, has cast some light on the process of gene duplication and divergence. Although many duplicates of zebrafish genes appear to have been lost by mutation, a significant fraction—perhaps as many as 30–50%—have diverged functionally while both copies have remained active. In many cases, the most obvious functional difference between the duplicated genes is that they are expressed in different tissues or at different stages of development (see ). One attractive theory to explain such an end result imagines that different, mildly deleterious mutations quickly occur in both copies of a duplicated gene set. For example, one copy might lose expression in a particular tissue due to a regulatory mutation, while the other copy loses expression in a second tissue. Following such an occurrence, both gene copies would be required to provide the full range of functions that were once supplied by a single gene; hence, both copies would now be protected from loss through inactivating mutations. Over a longer period of time, each copy could then undergo further changes through which it could acquire new, specialized features.
The Evolution of the Globin Gene Family Shows How DNA Duplications Contribute to the Evolution of Organisms
The globin gene family provides a particularly good example of how DNA duplication generates new proteins, because its evolutionary history has been worked out particularly well. The unmistakable homologies in amino acid sequence and structure among the present-day globins indicate that they all must derive from a common ancestral gene, even though some are now encoded by widely separated genes in the mammalian genome.
We can reconstruct some of the past events that produced the various types of oxygen-carrying hemoglobin molecules by considering the different forms of the protein in organisms at different positions on the phylogenetic tree of life. A molecule like hemoglobin was necessary to allow multicellular animals to grow to a large size, since large animals could no longer rely on the simple diffusion of oxygen through the body surface to oxygenate their tissues adequately. Consequently, hemoglobin-like molecules are found in all vertebrates and in many invertebrates. The most primitive oxygen-carrying molecule in animals is a globin polypeptide chain of about 150 amino acids, which is found in many marine worms, insects, and primitive fish. The hemoglobin molecule in higher vertebrates, however, is composed of two kinds of globin chains. It appears that about 500 million years ago, during the evolution of higher fish, a series of gene mutations and duplications occurred. These events established two slightly different globin genes, coding for the α- and β-globin chains in the genome of each individual. In modern higher vertebrates each hemoglobin molecule is a complex of two α chains and two β chains (). The four oxygen-binding sites in the α2β2 molecule interact, allowing a cooperative allosteric change in the molecule as it binds and releases oxygen, which enables hemoglobin to take up and to release oxygen more efficiently than the single-chain version.
A comparison of the structure of one-chain and four-chain globins. The four-chain globin shown is hemoglobin, which is a complex of two α- and β-globin chains. The one-chain globin in some primitive vertebrates forms a dimer that dissociates (more...)
Still later, during the evolution of mammals, the β-chain gene apparently underwent duplication and mutation to give rise to a second β-like chain that is synthesized specifically in the fetus. The resulting hemoglobin molecule has a higher affinity for oxygen than adult hemoglobin and thus helps in the transfer of oxygen from the mother to the fetus. The gene for the new β-like chain subsequently mutated and duplicated again to produce two new genes, ε and γ, the ε chain being produced earlier in development (to form α2ε2) than the fetal γ chain, which forms α2γ2. A duplication of the adult β-chain gene occurred still later, during primate evolution, to give rise to a δ-globin gene and thus to a minor form of hemoglobin (α2δ2) found only in adult primates ().
An evolutionary scheme for the globin chains that carry oxygen in the blood of animals. The scheme emphasizes the β-like globin gene family. A relatively recent gene duplication of the γ-chain gene produced γG and γA, which (more...)
Each of these duplicated genes has been modified by point mutations that affect the properties of the final hemoglobin molecule, as well as by changes in regulatory regions that determine the timing and level of expression of the gene. As a result, each globin is made in different amounts at different times of human development (see ).
The end result of the gene duplication processes that have given rise to the diversity of globin chains is seen clearly in the human genes that arose from the original β gene, which are arranged as a series of homologous DNA sequences located within 50,000 nucleotide pairs of one another. A similar cluster of α-globin genes is located on a separate human chromosome. Because the α- and β-globin gene clusters are on separate chromosomes in birds and mammals but are together in the frog Xenopus, it is believed that a chromosome translocation event separated the two gene clusters about 300 million years ago (see ).
There are several duplicated globin DNA sequences in the α- and β-globin gene clusters that are not functional genes, but pseudogenes. These have a close homology to the functional genes but have been disabled by mutations that prevent their expression. The existence of such pseudogenes make it clear that, as expected, not every DNA duplication leads to a new functional gene. We also know that nonfunctional DNA sequences are not rapidly discarded, as indicated by the large excess of noncoding DNA that is found in mammalian genomes.
Genes Encoding New Proteins Can Be Created by the Recombination of Exons
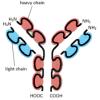
The role of DNA duplication in evolution is not confined to the expansion of gene families. It can also act on a smaller scale to create single genes by stringing together short, duplicated segments of DNA. The proteins encoded by genes generated in this way can be recognized by the presence of repeating, similar protein domains, which are covalently linked to one another in series. The immunoglobulins () and albumins, for example, as well as most fibrous proteins (such as collagens) are encoded by genes that have evolved by repeated duplications of a primordial DNA sequence.
Schematic view of an antibody (immunoglobulin) molecule. This molecule is a complex of two identical heavy chains and two identical light chains. Each heavy chain contains four similar, covalently linked domains, while each light chain contains two such (more...)
In genes that have evolved in this way, as well as in many other genes, each separate exon often encodes an individual protein folding unit, or domain. It is believed that the organization of DNA coding sequences as a series of such exons separated by long introns has greatly facilitated the evolution of new proteins. The duplications necessary to form a single gene coding for a protein with repeating domains, for example, can occur by breaking and rejoining the DNA anywhere in the long introns on either side of an exon encoding a useful protein domain; without introns there would be only a few sites in the original gene at which a recombinational exchange between DNA molecules could duplicate the domain. By enabling the duplication to occur by recombination at many potential sites rather than just a few, introns increase the probability of a favorable duplication event.
More generally, we know from genome sequences that component parts of genes—both their individual exons and their regulatory elements—have served as modular elements that have been duplicated and moved about the genome to create the present great diversity of living things. As a result, many present-day proteins are formed as a patchwork of domains from different domain families, reflecting their long evolutionary history ().
Domain structure of a group of evolutionary related proteins that are thought to have a similar function. In general, there is a tendency for the proteins in more complex organisms, such as ourselves, to contain additional domains—as is the case (more...)
Genome Sequences Have Left Scientists with Many Mysteries to Be Solved
Now that we know from genome sequences that a human and a mouse contain essentially the same genes, we are forced to confront one of the major problems that will challenge cell biologists throughout the next century. Given that a human and a mouse are formed from the same set of proteins, what has happened during the evolutionary process to make a mouse and a human so different? Although the answer is present somewhere among the three billion nucleotides in each sequenced genome, we do not yet know how to decipher this type of information—so that the answer to this critical, most fundamental question is not known.
Despite our ignorance, it is perhaps worth engaging in a bit of speculation, if only to help point the way forward to some of the hard problems ahead. In biology, timing is everything, as will become clear when we examine the elaborate mechanisms that allow a fertilized egg to develop into an embryo, and the embryo to develop into an adult (discussed in Chapter 21). The human body is formed as the result of many billions of decisions that are made during our development as to which RNA molecule and which protein are to be made where, as well as exactly when and in what amount each is to be produced. These decisions are different for a human than for a chimpanzee or a mouse. The coding sequences of genomes represent a more or less standard set of the 30,000 or so basic parts from which all three organisms are made. It is therefore the many different types of controls on gene expression described in this Chapter that must largely create the difference between a human and other mammals.
Given these assumptions, it would be reasonable to expect genomes to have evolved in a way that allows organisms to experiment with altered gene timing and expression patterns in selected cells. We have already seen some evidence that this is so, when we discussed alternative RNA splicing and RNA editing mechanisms. There also appear to be mechanisms—some based on the movements of transposable DNA elements—that allow modules to be readily added to and subtracted from the regulatory regions of genes, so as to produce changes in the pattern of their transcription as organisms evolve. In fact, an analysis of these regulatory regions provides evidence to support the claim that most gene regulatory regions have been formed by the evolutionary mixing and matching of the DNA-binding sites that are recognized by gene regulatory proteins ().
Gene control regions for mouse and chicken eye lens crystallins. Crystallins make up the bulk of the lens and are responsible for refracting and focusing light onto the retina. Many proteins in the cell have properties (high solubility, proper refractive (more...)
Genetic Variation within a Species Provides a Fine-Scale View of Genome Evolution
In comparisons between two species that have diverged from one another by millions of years, it makes little difference which individuals from each species are compared. For example, typical human and chimpanzee DNA sequences differ from one another by 1%. In contrast, when the same region of the genome is sampled from two different humans, the differences are typically less than 0.1%. For more distantly related organisms, the inter-species differences overshadow intra-species variation even more dramatically. However, each “fixed difference” between the human and the chimpanzee (i.e., each difference that is now characteristic of all or nearly all individuals of each species) started out as a new mutation in a single individual. If the size of the interbreeding population in which the mutation occurred is N, the initial allele frequency of a new mutation would be ½N for a diploid organism. How does such a rare mutation become fixed in the population, and hence become a characteristic of the species rather than of a particular individual genome?
The answer to this question depends on the functional consequences of the mutation. If the mutation has a significantly deleterious effect, it will simply be eliminated by purifying selection and will not become fixed. (In the most extreme case, the individual carrying the mutation will die without producing progeny.) Conversely, the rare mutations that confer a major reproductive advantage on individuals who inherit them will spread rapidly in the population. Because humans reproduce sexually and genetic recombination occurs each time a gamete is formed, the genome of each individual who has inherited the mutation will be a unique recombinational mosaic of segments inherited from a large number of ancestors. The selected mutation along with a modest amount of neighboring sequence—ultimately inherited from the individual in which the mutation occurred—will simply be one piece of this huge mosaic.
The great majority of mutations that are not harmful are not beneficial either. These selectively neutral mutations can also spread and become fixed in a population, and they make a large contribution to the evolutionary change in genomes. Their spread is not as rapid as the spread of the rare strongly advantageous mutations. The process by which such neutral genetic variation is passed down through an idealized interbreeding population can be described mathematically by equations that are surprisingly simple. The idealized model that has proven most useful for analyzing human genetic variation assumes a constant population size, and random mating, as well as selective neutrality for the mutations. While neither of these assumptions is a good description of human population history, they nonetheless provide a useful starting point for analyzing intra-species variation.
When a new neutral mutation occurs in a constant population of size N that is undergoing random mating, the probability that it will ultimately become fixed is approximately ½N. For those mutations that do become fixed, the average time to fixation is approximately 4N generations. A detailed analysis of data on human genetic variation suggests an ancestral population size of approximately 10,000 during the period when the current pattern of genetic variation was largely established. Under these conditions, the probability that a new, selectively neutral mutation would become fixed was small (5 × 10–5), while the average time to fixation was on the order of 800,000 years. Thus, while we know that the human population has grown enormously since the development of agriculture approximately 15,000 years ago, most human genetic variation arose and became established in the human population much earlier than this, when the human population was still small.
Even though most of the variation among modern humans originates from variation present in a comparatively tiny group of ancestors, the number of variations encountered is very large. Most of the variations take the form of single-nucleotide polymorphisms (SNPs). These are simply points in the genome sequence where one large fraction of the human population has one nucleotide, while another large fraction has another. Two human genomes sampled from the modern world population at random will differ at approximately 2.5 × 106 sites (1 per 1300 nucleotide pairs). Mapped sites in the human genome that are polymorphic—meaning that there is a reasonable probability that the genomes of two individuals will differ at that site—are extremely useful for genetic analyses, in which one attempts to associate specific traits (phenotypes) with specific DNA sequences for medical or scientific purposes (see p. 531).
Against the background of ordinary SNPs inherited from our prehistoric ancestors, certain sequences with exceptionally high mutation rates stand out. A dramatic example is provided by CA repeats, which are ubiquitous in the human genome and in the genomes of other eucaryotes. Sequences with the motif (CA)n are replicated with relatively low fidelity because of a slippage that occurs between the template and the newly synthesized strands during DNA replication; hence, the precise value of n can vary over a considerable range from one genome to the next. These repeats make ideal DNA-based genetic markers, since most humans are heterozygous—carrying two values of n at any particular CA repeat, having inherited one repeat length (n) from their mother and a different repeat length from their father. While the value of n changes sufficiently rarely that most parent-child transmissions propagate CA repeats faithfully, the changes are sufficiently frequent to maintain high levels of heterozygosity in the human population. These and other simple repeats that display exceptionally high variability provide the basis for identifying individuals by DNA analysis in crime investigations, paternity suits, and other forensic applications (see ).
While most of the SNPs and other common variations in the human genome sequence are thought to have no effect on phenotype, a subset of them must be responsible for nearly all of the heritable aspects of human individuality. A major challenge in human genetics is to learn to recognize those relatively few variations that are functionally important—against the large background of neutral variation that distinguishes the genomes of any two human beings.
Summary
Comparisons of the nucleotide sequences of present-day genomes have revolutionized our understanding of gene and genome evolution. Due to the extremely high fidelity of DNA replication and DNA repair processes, random errors in maintaining the nucleotide sequences in genomes occur so rarely that only about 5 nucleotides in 1000 are altered every million years. Not surprisingly, therefore, a comparison of human and chimpanzee chromosomes—which are separated by about 5 million years of evolution—reveals very few changes. Not only are our genes essentially the same, but their order on each chromosome is almost identical. In addition, the positions of the transposable elements that make up a major portion of our noncoding DNA are mostly unchanged.
When one compares the genomes of two more distantly related organisms—such as a human and a mouse, separated by about 100 million years—one finds many more changes. Now the effects of natural selection can be clearly seen: through purifying selection, essential nucleotide sequences—both in regulatory regions and coding sequences (exon sequences)—have been highly conserved. In contrast, nonessential sequences (for example, intron sequences) have been altered to such an extent that an accurate alignment according to ancestry is often not possible.
Because of purifying selection, homologous genes can be recognized over large phylogenetic distances, and it is often possible to construct a detailed evolutionary history of a particular gene, tracing its history back to common ancestors of present-day species. We can thereby see that a great deal of the genetic complexity of present-day organisms is due to the expansion of ancient gene families. DNA duplication followed by sequence divergence has thus been a major source of genetic novelty during evolution.