Enhanced excitatory connectivity and decreases in GABAergic inhibition are important mechanisms underlying injury-induced epileptogenesis in many animal models and in humans. Sprouting of excitatory axons and establishment of new synapses is a ubiquitous epileptogenic response to cortical injury. In the rodent chronic partial isolation (undercut) model of posttraumatic epilepsy, tetrodotoxin treatment of undercut cortex during a critical period decreases this axonal response to injury and blocks epileptogenesis. Gabapentin, an agonist that competes with glial-derived thrombospondins at the α2δ-1 calcium subunit receptor, is antiepileptogenic in neocortical slices from undercut rats and decreases injury-induced excitatory synapse formation, cell death, and neurofilament immunoreactivity. GABAergic interneurons become atrophic and dysfunctional in undercuts, resulting in decreases in inhibitory connectivity and the strength of inhibition on pyramidal cells. A potential underlying mechanism is loss of trophic support from brain derived neurotrophic factor (BDNF) released by pyramidal neurons acting on interneuronal TrkB receptors. Treatment of undercut rats after injury with agents that mimic activation of TrkB receptors by BDNF may reduce signs of injury and dysfunction of interneurons and provide a second promising antiepileptogenic approach. A focus on limiting new excitatory connectivity and providing trophic support for injured GABAergic interneurons may allow development of effective prophylactic measures for posttraumatic epilepsy.
The epidemiology of posttraumatic epilepsy (PTE) has been extensively analyzed and reviewed in a number of studies of both civilian and military brain injuries1,2; reviewed in3. Several conclusions from this research are relevant to considerations of the potential mechanisms and prophylaxis of PTE. Results clearly show that the incidence of PTE is related to the severity of injury, and is therefore significantly higher in the military during wartime than in the civilian population, ranging up to 53 % with penetrating wounds1,2; reviewed in3. Both the increased incidence at older ages, and the potential development of PTE by the large number of individuals who have survived severe concussive injury during recent conflicts, suggest that the size of the affected population will increase in coming years, emphasizing the need for understanding the underlying pathophysiological processes and the development of prophylactic strategies.4,5 Although initial seizures in those who develop epilepsy most commonly have a focal origin in neocortex, both partial neocortical and temporal lobe epilepsy can follow traumatic brain injury (TBI) in man6. One remarkable feature of PTE is the variable, often very prolonged latency from injury to epilepsy which can range from weeks to years1,2,6. This provides a possible window for prophylactic intervention, once more information regarding the underlying pathophysiological processes and strategies for modifying them is available. However the long latency also represents a potential therapeutic problem, particularly in the absence of reliable biomarkers of “epileptogenesis in progress”. This chapter will focus on examples of aberrant excitatory and inhibitory processes in injured epileptogenic cortex and potential approaches to prevention of epileptogenesis that are focused on these pathophysiological mechanisms. Some of the challenges for development of prophylactic therapies are also discussed. Readers are referred to a number of reviews and papers published very recently that deal with various aspects of the basic mechanisms, pathogenesis and potential prophylaxis of PTE, and complement the areas covered in this chapter6–18.
Spectrum of potential epileptogenic mechanisms induced by traumatic brain injury
A large number of alterations in gene expression19 and a variety of pathophysiological processes occur in parallel following a brain injury20–22, reviewed in7,23, making it unlikely that an intervention focused on any one of these, in isolation, will emerge as a prophylactic “silver bullet”. The situation is further complicated by the likelihood that variables such as the level of brain maturation, site and distribution of injury (focal vs. multifocal vs. diffuse), type of trauma (e.g. concussive versus penetrating), presence or absence of significant bleeding, and other factors may affect the underlying type and sequence of epileptogenic events and the optimal timing of a potentially successful intervention in a given individual. Do different combinations of pathophysiological mechanisms underlie human epileptogenesis that follows different types of cortical injuries such as those due to stroke with cortical infarction, penetrating vs. closed concussive head injuries, focal infections or other etiologies? The same question is relevant to potential similarities or differences in events underlying chronic epileptogenesis in various models of TBI such as fluid percussion injury24 versus controlled cortical impact25 versus neocortical partial isolation or “undercut”26. Are underlying mechanisms in these models in neocortex the same as those in posttraumatic temporal lobe epilepsy models, or when hippocampal damage is induced by status epilepticus rather than direct trauma? These are critical questions because they bear on potential prophylactic therapies and, unfortunately, the detailed data required for answers are incomplete.
A survey of the limited cellular results from neocortical injury models, and from animals whose temporal lobes are injured in the course of experimental status epilepticus, as well as from available human material, indicates that two pathophysiological processes are prominent in focal epileptogenesis, namely, enhanced excitatory connectivity10,27–34, and alterations in GABAergic inhibitory mechanisms33,35–40. But even within these broad categories, different types of abnormalities may be present that will require different prophylactic or therapeutic approaches. For example, disinhibition might involve alterations in gamma-aminobutyric acid A (GABAA) receptor subunits41,42, decreases in voltage dependent calcium channels40 or Na+/K+ adenosine triphosphatase (ATPase) at inhibitory terminals43,44, shifts in the chloride gradient due to changes in expression of chloride transporters KCC2 or NKCC45–47, loss of inhibitory connectivity due to structural changes in interneurons16,48 or actual loss of interneurons of various subtypes36,38.
Subsets of abnormalities can also affect the mechanisms controlling excitation, such as alterations in the probability of release (Pr) at terminals49, burst firing in axons50, reviewed in51, receptor efficacy or number52–56, and dysfunction of ion or transmitter transport57–62. In addition to alterations in inhibitory efficacy and enhanced excitation, many other potentially epileptogenic changes are present following injury such as alterations in voltage dependent ion channels63–67, blood brain barrier disturbances68, inflammatory responses and release of cytokines21,69, alterations in glia70,71, and so on. In terms of evaluation of therapeutic trials of potential prophylactic agents, this plethora of abnormalities raises a difficult issue: a single agent may fail to prevent PTE, even though it is effective at its intended target, that is a false negative result may be obtained due to the presence of other epileptogenic mechanisms acting in parallel.
Choice of models for research on posttraumatic epilepsy
There is no perfect model of human posttraumatic epilepsy. The advantages and disadvantages of acute and chronic models of epilepsy have recently been reviewed in detail72. Fluid percussion injury, controlled cortical impact and undercut models each have their place in advancing our understanding of PTE. Valuable information has also been obtained from status epilepticus temporal lobe injury models, although direct traumatic injury is not present and the resulting epileptogenesis represents a different epilepsy syndrome that may involve a somewhat different spectrum of underlying mechanisms. Discussions about the merits of one model versus another thus are only useful in the context of the particular pathophysiological process or event to be investigated. Obviously, to determine whether a drug will be prophylactic against seizures in vivo, a model in which there might be extensive injury and an expected high incidence of electrographic and behavioral posttraumatic seizures at relatively short latency after injury (i.e. high throughput), would be most practical and desirable.73–75 However, this might not be the model of choice for investigation of the details of functional or structural alterations in neocortical GABAergic interneurons or pyramidal cells that occur at a site of stereotyped restricted epileptogenic focal injury, and the potential prophylactic effects induced by the same drug on these alterations. Such a question would be better addressed with a more reductionistic approach using a model that would facilitate detailed cellular in vitro experiments and avoid the complications of widespread damage and variability.30,32,76 Both kinds of experiments are critical for progress, and fitting the preparation used to the question posed is certainly not a new concept in neurobiological research. There is no one best approach to unraveling the mechanisms underlying the pathogenesis and prophylaxis of PTE.
Partial neocortical isolation (“undercut”) model
The authors’ familiarity with this model, the significant amount of anatomical and cellular electrophysiological data available (references below and in77) and the fact that this is the first case in which prophylaxis of epileptogenesis after local cortical injury has been demonstrated (discussion below), has lead us to focus on the undercut model in this review. The advantages of this model have been detailed elsewhere.77 Most important is the relatively short interval between injury and epileptiform activity that is present in a high proportion of neocortical slices cut through the damaged area and maintained in vitro 78–80 (). This has allowed the detailed examination of epileptogenic cellular structural and functional alterations in pyramidal (Pyr) cells and GABAergic interneurons detailed below. We have also obtained in vivo video/electroencephalographic (EEG) recordings that show electrographic and behavioral seizures beginning with focal discharge in the undercut cortex, spreading across the cortex on the injured side and propagating contralaterally ( in77).
Undercut cortex in rats and humans. A: Coronal section of human brain in patient who underwent undercutting surgery of right frontal lobe for intractable pain. Dashed white lines drawn through the undercut here and in C. (Modified from Scoville WB. Selective (more...)
Structural alterations in fast-spiking interneurons in undercut cortex. A,B: Images of single layer V fast-spiking interneurons filled with biocytin and processed in control (A) and undercut slice (B). The cell from the undercut has thinner dendrites (more...)
Partially isolated neocortical islands with intact pial circulation (“undercuts” below) are an established in vivo and in vitro model for development of chronic post-traumatic hyperexcitability and epileptogenesis32,78,79,81,82, and partially isolated neocortex is also epileptogenic in man83; ). The undercut cortex retains normal laminations (), although it becomes thinner with modest cell loss and obvious structural alterations in deep lying pyramidal (Pyr) cells78,84, (K Graber and DA Prince, unpublished). Disinhibition, increases in neuronal membrane excitability, and increases in excitatory synaptic coupling have been suggested as potential mechanisms in this chronic epilepsy model.32,78,85–87 The undercut cortex becomes progressively more epileptogenic over several weeks82,88, and spontaneous interictal discharges can persist for at least 1 year in the monkey26. The time of onset of epileptogenesis, or the “critical period” in rats occurs during the first 3 days after injury89 and recent data suggest that epileptogenic activity is already present at 3 days after the undercut (DK Takahashi and DA Prince, unpublished results). Isolated islands of neocortical gray matter, with neuropathological evidence of substantial axonal reorganization, are also present in postmortem specimens from epileptic children who developed extensive underlying white matter lesions as infants90. Interictal epileptiform activity can be recorded within partially isolated cortex of anesthetized rats, and c-fos immunoreactivity (IR) is increased for weeks in the injured cortex, suggesting ongoing abnormal activity91. Behavioral and electrographic seizures occur in in vivo experiments on monkey, cat and rodents in this model (see above and references in77). Areas of partially isolated cortex with underlying loss of white matter are also present in the cortical contusion ( in25 and fluid percussion injury models ( in23), although it is not clear whether seizure activity originates from the cortex above these sites. Of interest are reports of chronic electrographic and clinical seizures in humans as a complication of psychosurgery in which connections from portions of frontal lobes were severed, essentially producing large partially isolated neocortical slabs 92,93 ().
Abnormalities in excitatory mechanisms in the undercut cortex
The capacity of injured brain to make new connections has been known since the groundbreaking anatomical studies of Cajal94 who described sprouting of injured Pyr cell axons in neocortex. Maladaptive axonal sprouting and establishment of new excitatory connections occur in mature undercut neocortex32 and as early as 2 days after injury in isolated immature neocortex95. Excitatory sprouting also occurs following injury to the hippocampus in other animal models of epileptogenesis27,28,96–102; and in epileptic human temporal lobes103–107. The ubiquitous nature of this phenomenon is shown by its occurrence in other models of cortical trauma such as stroke108,109, thermo-ischemic lesions110 and fluid percussion injury111, where it may begin hours after the trauma112. The onset of an axonal reaction and sprouting, as signaled by increases in immunoreactivity for growth-associated protein (GAP)43, may begin in as early as 12h after lesions in culture and these alterations are well established by 3 days after injury30,113. Sprouting occurs 3-4 days after injury to dissociated neocortical neurons in culture114. Hyperexcitability due to synaptic innervation by sprouted axons has been shown in experiments in hippocampus29,30,76 and neocortex34. Activation of brain-derived neurotrophic factor (BDNF) may be an important mechanism underlying injury-induced sprouting and hyperactivity in hippocampus.115,116
Results from whole cell recordings of layer V pyramidal (Pyr) neurons done 2–3 weeks after injury in the undercut cortex model support the conclusion that there is enhanced synaptic excitatory connectivity by showing (1) an increased frequency of miniature (m) excitatory postsynaptic currents (EPSCs); (2) a steeper input/output relationship for evoked EPSCs; and (3) an increased probability of release of glutamate from excitatory terminals33,49. The latter finding suggests intrinsic abnormalities in the terminals of Pyr cells. In addition, anatomical studies of biocytin-filled layer V Pyr cell axons showed evidence of significant sprouting, mainly in layer V32, where the epileptogenic field potentials were initiated78,79. These functional and structural abnormalities presumably contribute to the large polysynaptic excitatory currents in Pyr cells that occur synchronously with field potential epileptiform bursts () and propagate across the cortex ().
More recently, laser scanning photostimulation of caged glutamate in epileptogenic slices from undercuts allowed detailed mapping of excitatory and inhibitory connectivity34,48. Results showed that the excitatory “map” was significantly expanded, particularly in layer V, and that both Pyr cells and fast-spiking inhibitory interneurons were targets of presumed sprouted axons and terminals from other nearby Pyr neurons32. These alterations in excitatory synaptic connectivity and strength, together with abnormalities in inhibitory circuits discussed below may contribute to the development and increased conduction of epileptiform activity across cortex in the in vivo undercut model in cat117 (reviewed in118).
It is interesting that hyperexcitability closely resembling that recorded in layer V of undercut cortex has also been shown in neocortical slices from the fluid percussion injury model. Rats from both models have epileptiform seizures in vivo77,119, and there is also a similarity in the morphology of field potentials that are evoked or occur spontaneously in slices versus those in in vivo EEG recordings in these two models. Also, recent recordings from another TBI model studied in vitro clearly show repetitive bursts of EPSCs that coincide with epileptiform field potentials which would be termed “ictal” EEG discharges if they occurred in vivo10.
Abnormalities in GABAergic inhibitory mechanisms in undercut cortex
A variety of structural and electrophysiological evidence shows that GABAergic inhibition is compromised in undercut cortex. Recent experiments in the cat suggest that glutamic acid decarboxylase (GAD-) or GABA- positive neocortical interneurons are selectively and progressively reduced in density in cat undercut cortex84. Although our initial cell counts in undercut rat cortex have not shown a selective decrease in density of parvalbumin (PV)-immunoreactive interneurons120, we have found significant structural changes in biocytin-filled fast-spiking PV-containing cells, including marked decreases in axonal lengths and dendritic volume (), giving them an appearance similar to that seen in immature PV interneurons (cf of121 with ). Further, the axons of these interneurons in the undercut cortex have a significant increase in the proportion of small (<1 um in diameter) boutons and a decrease in numbers of larger (>1 um in diameter) boutons (see in16), changes that would be associated with altered pre- and postsynaptic structures at GABAergic synapses and with less effective inhibitory transmission122,123.
Loss of perisomatic α3Na+K+ ATPase in undercut cortex. A: Immumoreactivity (IR) for α3Na+K+ATPase in layer V of control cortex contralateral and homotopic to undercut on rat 21d after lesion. IR is localized around the somata of pyramidal (more...)
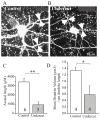
Gabapentin (GBP) in vivo reduces epileptogenesis and excitatory synapse density in undercut slices. A–B: Field potentials evoked in layer V of undercut slice by stimuli in partial cortical isolations 14 d after injury. A: Rat was treated with (more...)
Results of several electrophysiological experiments confirm a decreased efficacy of GABAergic inhibitory transmission in the undercut cortex. Whole cell recordings in rat undercut slices showed a decreased frequency of mIPSCs in Pyr cells33, and quantitative electronmicroscopic experiments confirmed a decreased density of symmetrical (inhibitory) synapses on somata of layer V Pyr cells (J. Wenzel, PA Schwartzkroin and DA Prince, unpublished results) as one potential mechanism for decreased miniature inhibitory postsynaptic current (mIPSC) frequency. More recently, we have also shown that the axonal terminals of layer V interneurons in undercuts are abnormal in that they have a decreased probability of GABA release and increased failure rate40 due in part to a downregulation of N-type calcium channels in terminals.123a Dual recordings from synaptically coupled FS-Pyr or FS-spiny stellate pairs in layer IV of undercuts showed a decrease in Pr, a large reduction in the amplitude of unitary IPSCs, increased coefficient of variation and increased failures, indicating alterations in presynaptic terminals of the largest subgroup of GABAergic neurons in cortex, FS cells 123b. Neuronal injury can also decrease the efficacy of postsynaptic inhibition by decreasing expression of KCC2 and impairing outward chloride transport124,125. In the undercut, there are also decreases in KCC2 and in the outward transport of chloride in postsynaptic Pyr cells that would make GABAergic inhibition less effective at times of high frequency activity126. Recent results, obtained with laser scanning photostimulation of caged glutamate in combination with whole cell recordings, have shown that the net effects of some of the above-mentioned anatomical and electrophysiological abnormalities are to reduce the spatial extent of inhibitory inputs onto both Pyr cells and FS interneurons in the chronic undercut127.
Fast-spiking interneurons in neocortex normally have a high density of Na+-K+ ATPase (“sodium pump”) in their membranes44 and particularly in their axonal terminals surrounding Pyr cell somata43,128; (). Sodium pump activation would be important in fast-firing neurons to prevent excessive increases in [Na+]i that might depolarize terminals and decrease GABA release. There is a significant loss of Na pump immunoreactivity in undercut cortex surrounding Pyr cells (), similar to that previously found in the freeze-microgyrus model of epileptogenesis43, suggesting another potential mechanism that would lead to terminal dysfunction and decreased GABA release.
When does posttraumatic epileptogenesis begin?
Answers to this critical question would influence decisions about the timing of potential antiepileptogenic treatment. From the available data, it appears likely that processes eventually leading to hyperexcitability in cortical networks and to seizures may be set into motion at the time of the TBI, although the latency to the first behavioral spontaneous seizure is highly variable. Seizures in the first week after injury are usually not followed by epilepsy in man; however they are associated with an increased statistical risk of subsequent epilepsy3, indicating that, at least in some individuals, an epileptogenic process is initiated early. Epileptiform activity may be initially undetectable by surface EEG, making this an unreliable marker of the onset of epileptogenesis129. There is evidence for early emergence of epileptogenesis in a variety of experimental data. In models of acute neocortical trauma, epileptiform activity may develop within minutes or hours after injury in vitro130,131 and results from acute partial isolation experiments done under ketamine anesthesia in cats do show that acute epileptiform discharges are generated in cortex near the isolation132. However the underlying mechanisms in these early seizures may be different from those in more chronic models in that they might involve acute alterations such as release of excitatory amino acids131, spreading depression, large increases in [K+]o and blood-brain barrier disruption.
Recently we recorded in vitro from undercut slices obtained 3 days after the injury and found that they generate robust prolonged spontaneous and evoked epileptiform field potentials associated with large amplitude EPSCs. Confocal images obtained after immunocytochemical processing for GAP43, vesicular glutamate transporter 1 (vGLUT1) and postsynaptic density (PSD)95 have suggested that there is significant sprouting of excitatory axons onto pyramidal somata within days after the undercut is placed. Other electrophysiological data suggest that these sprouted terminals are functional and contribute to epileptogenesis (D.K. Takahashi and D.A. Prince, unpublished).
Axonal sprouting and excitatory synapse formation occur in parallel with a number of other pathophysiological events following TBI (e.g. alterations in GABAergic inhibition discussed above, intrinsic changes in membrane excitability78), so it is difficult to determine whether hyperconnectivity alone would be sufficient to induce posttraumatic epileptogenesis. A potential answer to this question comes from recent results in C1q knockout mice that have behavioral and electrographic seizures resulting from failure to prune excitatory cortical synapses during development.133
In vitro slices from these animals are epileptogenic due to increased excitatory connectivity without apparent alterations in inhibitory events.
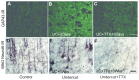
In experiments using controlled cortical impact (CCI), epileptiform activity and electrographic seizures are present in vitro at the first week after injury10 and appear to progress with generation of “ictal” discharges lasting for many seconds by the second week. In other experiments such as those involving kainate kindling, epileptogenic activity is present early in hippocampus but goes undetected in in vivo recordings from the usual skull electrodes134. Our previous results in the undercut model provided the first proof-in principle that posttraumatic epileptogenesis, as gauged by the occurrence of epileptiform activity in in vitro slices, begins shortly after injury and can be prevented80. However this prophylaxis was only effective if the treatment, namely topical exposure of the injured cortex to tetrodotoxin (TTX) in a slow release resin, was administered for the first 3 days after injury. Later applications were ineffective at limiting the proportion of slices that were epileptogenic89. Thus the results revealed a brief critical period of a few days beginning after the partial isolation when the seeds for subsequent epileptogenesis are sown in the undercut cortex model. Tetrodotoxin has also been effective in decreasing axonal sprouting and rhythmic neocortical burst discharges that begin by the second day after thermocoagulation lesions in rat neocortex, although the relationship to later epileptogenesis is unclear.110 These results with TTX appear contrary to the hypothesis that the enhanced excitability and epileptogenesis in the undercut may be due to activation of homeostatic increases in excitatory neurotransmission due to deafferentation118,135. It is possible that homeostatic compensatory increases in alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors do occur, but are offset by decreases in innervation of postsynaptic targets induced by the TTX treatment. Immunocytochemical analysis of undercut cortex shows that TTX blockade of activity down-regulates anatomical markers of the axonal and terminal sprouting response that are evident as early as 3 days after injury () and are long-lasting (). Other as yet unexplored results of silencing injured cortex may account for the blockade of hyperexcitability in the undercut that outlasts the TTX treatment by many months (K.D. Graber and D.A. Prince, unpublished results). It is important to note that TTX may produce quite different (opposite) effects on epileptogenesis when given during early development in hippocampus136, a result that emphasizes the difficulty of generalizing results from one model to another in terms of potential prophylactic approaches.
Immunoreactivity (IR) of axons and terminals in partially isolated neocortex. A–C: Sections through layer V of rat sensorimotor cortex reacted with growth-associated protein (GAP) 43 antibody. D–F: Comparable sections from rats reacted (more...)
Prophylaxis of posttraumatic epileptogenesis
Potential approaches to modification of the increased excitatory sprouting and synapse formation and decreased GABAergic interneuronal structure/function, are suggested by results of experiments dealing with normal development of excitatory synapses and interneurons.
Limiting excitatory connectivity
Reactive astrogliosis is a ubiquitous pathological finding following TBI and is present in all of the models discussed above, including the undercut. Release of thrombospondins (TSPs) by astrocytes provides an important signal for excitatory synapse formation early in development137,138 and following injury to the mature central nervous system (CNS).139,140 The α2δ-1 voltage-gated calcium channel subunit, that is up-regulated in peripheral pain models and after brain injury, is the receptor for the antiallodynic/antiepileptic drug gabapentin (GBP)141,142, reviewed in143,144 and is also the receptor for TSPs137. GBP can block excitatory synapse formation in the developing retinogeniculate pathway by interfering with TSP actions137. The increased axonal sprouting and synapse formation are reduced in TSP knockout animals in a stroke model, leading to the hypothesis that GBP would have similar actions and might be an antiepileptogenic agent in the undercut model. In recent experiments, GBP, given by sc infusion or ip for 2-3d up to 14d following the day of the undercut, decreased the proportion of slices that subsequently generated evoked epileptiform activity (; H Li, KD Graber, DA Prince. Soc Neurosci abstr, 2009). In addition, dual immunocytochemical processing of sections from the animals treated with GBP showed significantly fewer presumptive excitatory synapses (i.e. close appositions between pre- [vGLUT1] and postsynaptic markers [PSD95] ()). GBP also reduced expression of 200 kD neurofilament-IR and the numbers of neurons stained with fluorojade C (not shown), suggesting potential neuroprotective effects.
Preventing structural/functional alterations in GABAergic interneurons
The above structural changes in FS interneurons gave them an appearance that resembled, in some respects, that seen in immature GABAergic cells121, prompting us to assess expression of BDNF in neurons of the undercut, as this trophic factor is a key molecule in regulating development and maintenance of both interneuronal and Pyr cell structure and function121,135,145–147, reviewed in145. Immunoreactivity for BDNF in Pyr cells and its TrkB receptor on parvalbumin-containing interneurons and the associated mRNAs were significantly down-regulated as early as 3d after the undercut, suggesting that supplying this or another trophic factor after injury might be an approach to prevention of trauma-induced alterations in these cells (Figure 6 in16; see also17. BDNF has many potential actions including both enhancement of network excitation and inhibition150,151, so that it is unclear whether the net effect of BDNF or other TrkB receptor agonists will be anti- or pro-epileptogenic at this time. Variables such as dose level and timing or choice of mimetic molecule might allow differentiation of beneficial vs. detrimental effects.
Important unresolved issues affecting application of antiepileptogenic therapies for PTE
The question of adaptive vs. maladaptive changes in connectivity following injury is a key one that must be considered in approaching potential preventative treatments that decrease epileptogenic sprouting. A number of reports implicate axonal sprouting and new connections as major adaptive plastic events in recovery of function after cortical lesions
108,109. In recent experiments in a stroke model where middle cerebral artery occlusion induces expression of TSPs in astrocytes, TSP1–2 knock-out mice showed significant defects in the axonal sprouting and synaptic density compared to wild type animals, together with defects in functional recovery
140. The post-stroke incidence of epilepsy was not studied in these experiments; however the results, and those in the above references, provide a cautionary note.
A number of pathophysiological processes occur in parallel after a serious epileptogenic brain injury. Although any one of these in isolation might not induce seizure activity, in combination their effects on excitability would summate and epileptogenesis could result. Thus, a single prophylactic approach might be ineffective and a “prophylactic cocktail” might be required.
Two key elements in developing epileptogenesis in a variety of injury models are reductions in functional GABAergic inhibition and enhanced new excitatory connectivity. Although attempts to reverse such alterations may be effective, the relationships between both excitatory and inhibitory circuit function, circuit repair and epileptiform activity are complex. GABAergic synchronization of cortical networks occurs in epileptogenic cortical lesions
152, and in both acute
153 and genetic models of epileptiform discharge
154. Also, depolarizing GABA responses due to altered chloride gradients occur in excitatory cells during development
155 and after injury
125,156. These factors make the net effect of enhanced interneuronal output hard to predict. Antiepileptogenesis, through decreases in excitatory circuit activities might also have obverse effects such as decreased activation of interneurons
157; but see
158–160 or reduced activity-dependent axonal sprouting, pathfinding and circuit repair
110,161,162.
As more becomes known about processes controlling excitation and inhibition during cortical development or following injury, it is possible that prophylactic therapies selectively affecting maladaptive processes might be applied. One important obstacle at this time is the unavailability of a reliable biological marker that would select for individuals who will go on to develop post-traumatic epilepsy, although it is clear that the incidence increases with the severity of brain injury (reviewed in
163).
We know little about the temporal extent of critical periods in man when prophylactic intervention would be effective, or how to identify epileptogenesis “in progress”. The latent period may be very long between injury and expression of behavioral seizures
1; however the critical period for intervention could closely follow injury
89,164.
Finally, multiple offsetting potential effects of a given intervention are possible, such as both enhancement of excitatory connectivity together with “rescue” of inhibitory interneurons by TrkB receptor agonists (e.g.
165,166).
The chapters in this volume suggest that significant progress is being made in understanding the basic mechanisms leading to epilepsy, and that we may have potential prophylactic therapies available in the years to come, providing that some of the issues mentioned above are settled by detailed basic and clinical investigations.
Acknowledgements
Supported by NIH grants NS12151, NS39579 and NS06477 from the NINDS and a grant from Citizens United for Research in Epilepsy.
Reference List
- 1.
Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study.
Neurology. 1985;35:1406–1414. [
PubMed: 3929158]
- 2.
Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries.
N Engl J Med. 1998;338:20–24. [
PubMed: 9414327]
- 3.
Frey LC. Epidemiology of posttraumatic epilepsy: a critical review.
Epilepsia. 2003;44(Suppl 10):11–17. [
PubMed: 14511389]
- 4.
- 5.
Dichter MA. Posttraumatic epilepsy: the challenge of translating discoveries in the laboratory to pathways to a cure.
Epilepsia. 2009;50(Suppl 2):41–45. [
PubMed: 19187293]
- 6.
Raymont V, Salazar AM, Lipsky R, Goldman D, Tasick G, Grafman J. Correlates of posttraumatic epilepsy 35 years following combat brain injury.
Neurology. 2010;75(3):224–229. [
PMC free article: PMC2906177] [
PubMed: 20644150]
- 7.
- 8.
Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy.
J Neurophysiol. 2010;103(3):1490–1500. [
PMC free article: PMC2887624] [
PubMed: 20089815]
- 9.
Kharatishvili I, Pitkanen A. Posttraumatic epilepsy.
Curr Opin Neurol. 2010;23(2):183–188. [
PubMed: 20125011]
- 10.
Yang L, Afroz S, Michelson HB, Goodman JH, Valsamis HA, Ling DS. Spontaneous epileptiform activity in rat neocortex after controlled cortical impact injury.
J Neurotrauma. 2010;27(8):1541–1548. [
PubMed: 20504156]
- 11.
D’Ambrosio R, Hakimian S, Stewart T, Verley DR, Fender JS, Eastman CL, et al. Functional definition of seizure provides new insight into post-traumatic epileptogenesis.
Brain. 2009;132(Pt 10):2805–2821. [
PMC free article: PMC2759339] [
PubMed: 19755519]
- 12.
Dichter MA. Emerging concepts in the pathogenesis of epilepsy and epileptogenesis.
Arch Neurol. 2009;66(4):443–447. [
PubMed: 19364928]
- 13.
Israelsson C, Wang Y, Kylberg A, Pick CG, Hoffer B, Ebendal T. Closed head injury in a mouse model results in molecular changes indicating inflammatory responses.
J Neurotrauma. 2009 [
PMC free article: PMC2989856] [
PubMed: 19317611]
- 14.
Lowenstein DH. Epilepsy after head injury: an overview.
Epilepsia. 2009;50(Suppl 2):4–9. [
PubMed: 19187288]
- 15.
Pitkanen A, Lukasiuk K. Molecular and cellular basis of epileptogenesis in symptomatic epilepsy.
Epilepsy Behav. 2009;14(Suppl 1):16–25. [
PubMed: 18835369]
- 16.
Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis.
Epilepsia. 2009;50(Suppl 2):30–40. [
PMC free article: PMC2710960] [
PubMed: 19187292]
- 17.
Paradiso B, Marconi P, Zucchini S, Berto E, Binaschi A, Bozac A, et al. Localized delivery of fibroblast growth factor-2 and brain-derived neurotrophic factor reduces spontaneous seizures in an epilepsy model.
Proc Natl Acad Sci U S A. 2009;106(17):7191–7196. [
PMC free article: PMC2678472] [
PubMed: 19366663]
- 18.
Temkin NR. Preventing and treating posttraumatic seizures: the human experience.
Epilepsia. 2009;50(Suppl 2):10–13. [
PubMed: 19187289]
- 19.
Raghavendra RV, Dhodda VK, Song G, Bowen KK, Dempsey RJ.
Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis, J Neurosci Res. 2. Vol. 71. 2003. pp. 208–219. [
PubMed: 12503083]
- 20.
David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, et al. Astrocytic Dysfunction in Epileptogenesis: Consequence of Altered Potassium and Glutamate Homeostasis.
J Neurosci. 2009;29:10588–10599. [
PMC free article: PMC2875068] [
PubMed: 19710312]
- 21.
- 22.
Hicks RR, Martin VB, Zhang L, Seroogy KB. Mild experimental brain injury differentially alters the expression of neurotrophin and neurotrophin receptor mRNAs in the hippocampus.
Exp Neurol. 1999;160(2):469–478. [
PubMed: 10619564]
- 23.
Pitkanen A, Immonen RJ, Grohn OH, Kharatishvili I. From traumatic brain injury to posttraumatic epilepsy: what animal models tell us about the process and treatment options.
Epilepsia. 2009;50(Suppl 2):21–29. [
PubMed: 19187291]
- 24.
McIntosh TK, Vink R, Yamakami I, Faden AI. Magnesium protects against neurological deficit after brain injury.
Brain Res. 1989;482(2):252–260. [
PubMed: 2784989]
- 25.
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to Cortical Injury: I. Methodology and Local Effects of Contusions in the Rat.
Brain Res. 1981;211:67–77. [
PubMed: 7225844]
- 26.
Echlin FA, Battista A. Epileptiform seizures from chronic isolated cortex.
Arch Neurol. 1963;9:154–170. [
PubMed: 14048164]
- 27.
- 28.
Cronin J, Dudek FE. Chronic seizures and collateral sprouting of dentate mossy fibers after kainic acid treatment in rats.
Brain Res. 1988;474(1):181–184. [
PubMed: 3214710]
- 29.
Molnar P, Nadler JV. Mossy fiber-granule cell synapses in the normal and epileptic rat dentate gyrus studied with minimal laser photostimulation.
J Neurophysiol. 1999;82(4):1883–1894. [
PubMed: 10515977]
- 30.
McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of post-traumatic epilepsy.
Nature Med. 1997;3:990–996. [
PubMed: 9288725]
- 31.
Esclapez M, Hirsch JC, Ben Ari Y, Bernard C. Newly formed excitatory pathways provide a substrate for hyperexcitability in experimental temporal lobe epilepsy.
J Comp Neurol. 1999;408(4):449–460. [
PubMed: 10340497]
- 32.
Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex.
J Neurosci. 1995;15:8234–8245. [
PMC free article: PMC6577943] [
PubMed: 8613757]
- 33.
Li H, Prince DA. Synaptic activity in chronically injured, epileptogenic sensory-motor neocortex.
J Neurophysiol. 2002;88(1):2–12. [
PubMed: 12091528]
- 34.
Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats.
J Neurosci. 2006;26(18):4891–4900. [
PMC free article: PMC6674164] [
PubMed: 16672663]
- 35.
Ribak CE, Bradburne M, Harris AB. A preferential loss of GABAergic, symmetric synapses in epileptic foci: a quantitative ultrastructural analysis of monkey neocortex.
J Neurosci. 1982;2:1725–1735. [
PMC free article: PMC6564370] [
PubMed: 6815309]
- 36.
Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben Ari Y, et al. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy.
Nat Neurosci. 2001;4(1):52–62. [
PubMed: 11135645]
- 37.
Magloczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus.
Trends Neurosci. 2005;28:334–340. [
PubMed: 15927690]
- 38.
Rosen GD, Jacobs KM, Prince DA. Effects of neonatal freeze lesions on expression of parvalbumin in rat neocortex.
Cereb Cortex. 1998;8(8):753–761. [
PubMed: 9863702]
- 39.
Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy.
J Neurosci. 2006;26(17):4613–4623. [
PMC free article: PMC6674073] [
PubMed: 16641241]
- 40.
- 41.
Kharlamov EA, Downey KL, Jukkola PI, Grayson DR, Kelly KM. Expression of GABA A receptor alpha1 subunit mRNA and protein in rat neocortex following photothrombotic infarction.
Brain Res. 2008;1210:29–38. [
PMC free article: PMC2587253] [
PubMed: 18407248]
- 42.
Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABA
A receptor subunit expression and function in temporal lobe epilepsy.
Nature Med. 1998;4:1166–1172. [
PubMed: 9771750]
- 43.
Chu Y, Parada I, Prince DA. Temporal and topographic alterations in expression of the alpha3 isoform of Na+, K(+)-ATPase in the rat freeze lesion model of microgyria and epileptogenesis.
Neuroscience. 2009;162(2):339–348. [
PMC free article: PMC2706117] [
PubMed: 19362129]
- 44.
- 45.
Aronica E, Boer K, Redeker S, Spliet WG, van Rijen PC, Troost D, et al. Differential expression patterns of chloride transporters, Na+-K+-2Cl--cotransporter and K+-Cl--cotransporter, in epilepsy-associated malformations of cortical development.
Neuroscience. 2007;145(1):185–196. [
PubMed: 17207578]
- 46.
Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain.
Nat Med. 2005;11(11):1205–1213. [
PubMed: 16227993]
- 47.
Shimizu-Okabe C, Okabe A, Kilb W, Sato K, Luhmann HJ, Fukuda A. Changes in the expression of cation-Cl- cotransporters, NKCC1 and KCC2, during cortical malformation induced by neonatal freeze-lesion.
Neurosci Res. 2007;59(3):288–295. [
PubMed: 17904674]
- 48.
- 49.
Li H, Bandrowski AE, Prince DA. Cortical injury affects short-term plasticity of evoked excitatory synaptic currents.
J Neurophysiol. 2005;93(1):146–156. [
PubMed: 15342719]
- 50.
Gutnick MJ, Prince DA. Thalamocortical relay neurons: antidromic invasion of spikes from cortical epileptogenic focus.
Science. 1972;176:424–426. [
PubMed: 4337289]
- 51.
Pinault D. Backpropagation of action potentials generated at ectopic axonal loci: Hypothesis that axon terminals integrate local environmental signals.
Brain Res Reviews. [
PubMed: 8547954]
- 52.
De Lanerolle NC, Eid T, Von Campe G, Kovacs I, Spencer DD, Brines M. Glutamate receptor subunits GluR1 and GluR2/3 distribution shows reorganization in the human epileptogenic hippocampus.
Eur J Neurosci. 1998;10(5):1687–1703. [
PubMed: 9751141]
- 53.
Blumcke I, Beck H, Scheffler B, Hof PR, Morrison JH, Wolf HK, et al. Altered distribution of the alpha-amino-3-hydroxy-5-methyl-4- isoxazole propionate receptor subunit GluR2(4) and the N-methyl-D- aspartate receptor subunit NMDAR1 in the hippocampus of patients with temporal lobe epilepsy.
Acta Neuropathol (Berl). 1996;92:576–587. [
PubMed: 8960315]
- 54.
Chen HX, Xiang H, Roper SN. Impaired Developmental Switch of Short-term Plasticity in Pyramidal Cells of Dysplastic Cortex.
Epilepsia. 2007;48(1):141–148. [
PubMed: 17241221]
- 55.
Wong RK, Bianchi R, Taylor GW, Merlin LR. Role of metabotropic glutamate receptors in epilepsy.
Adv Neurol. 1999;79:685–698. [
PubMed: 10514855]
- 56.
Gold SJ, Hennegriff M, Lynch G, Gall CM. Relative concentrations and seizure-induced changes in mRNAs encoding three AMPA receptor subunits in hippocampus and cortex.
J Comp Neurol. 1996;365:541–555. [
PubMed: 8742301]
- 57.
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1.
Science. 1997;276:1699–1702. [
PubMed: 9180080]
- 58.
- 59.
Boulland JL, Ferhat L, Tallak ST, Ferrand N, Chaudhry FA, Storm-Mathisen J, et al. Changes in vesicular transporters for gamma-aminobutyric acid and glutamate reveal vulnerability and reorganization of hippocampal neurons following pilocarpine-induced seizures.
J Comp Neurol. 2007;503(3):466–485. [
PubMed: 17503488]
- 60.
Brines ML, Tabuteau H, Sundaresan S, Kim J, Spencer DD, De Lanerolle N. Regional distributions of hippocampal Na+,K(+)-ATPase, cytochrome oxidase, and total protein in temporal lobe epilepsy.
Epilepsia. 1995;36(4):371–383. [
PubMed: 7607116]
- 61.
Grisar T. Glial and neuronal Na+-K+ pump in epilepsy.
Ann Neurol. 1984;16(Suppl):S128–134. [
PubMed: 6095737]
- 62.
Vaillend C, Mason SE, Cuttle MF, Alger BE. Mechanisms of neuronal hyperexcitability caused by partial inhibition of Na+-K+-ATPases in the rat CA1 hippocampal region.
J Neurophysiol. 2002;88(6):2963–2978. [
PubMed: 12466422]
- 63.
Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability.
Nat Med. 2001;7(3):331–337. [
PMC free article: PMC3382967] [
PubMed: 11231632]
- 64.
Beck H, Steffens R, Elger CE, Heinemann U. Voltage-dependent Ca2+ currents in epilepsy.
Epilepsy Res. 1998;32(1–2):321–332. [
PubMed: 9761331]
- 65.
Kohling R. Voltage-gated sodium channels in epilepsy.
Epilepsia. 2002;43(11):1278–1295. [
PubMed: 12423377]
- 66.
Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, et al. A potassium channel mutation in neonatal human epilepsy.
Science. 1998;279:403–406. [
PubMed: 9430594]
- 67.
Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, et al. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice.
J Physiol (Lond). 1998;20:809–819. [
PubMed: 9581771]
- 68.
Ivens S, Kaufer D, Flores LP, Bechmann I, Zumsteg D, Tomkins O, et al. TGF-beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis.
Brain. 2007;130:535–547. [
PubMed: 17121744]
- 69.
Marcon J, Gagliardi B, Balosso S, Maroso M, Noe F, Morin M, et al. Age-dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis.
Neurobiol Dis. 2009;34(1):121–132. [
PubMed: 19320047]
- 70.
Takahashi DK, Vargas JR, Wilcox KS. Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus.
Neurobiol Dis. 2010 [
PMC free article: PMC3031166] [
PubMed: 20691786]
- 71.
- 72.
Pitkanen A, McIntosh TK. Animal models of post-traumatic epilepsy.
J Neurotrauma. 2006;23(2):241–261. [
PubMed: 16503807]
- 73.
Kharatishvili I, Nissinen JP, McIntosh TK, Pitkanen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats.
Neuroscience. 2006;140(2):685–697. [
PubMed: 16650603]
- 74.
Kharlamov EA, Jukkola PI, Schmitt KL, Kelly KM. Electrobehavioral characteristics of epileptic rats following photothrombotic brain infarction.
Epilepsy Res. 2003;56(2–3):185–203. [
PubMed: 14643003]
- 75.
D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, et al. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat.
Brain. 2005;128(Pt 1):174–188. [
PMC free article: PMC2696356] [
PubMed: 15563512]
- 76.
Dudek FE, Staley KJ. Laser scanning photostimulation: new evidence for enhanced recurrent excitation in a model of posttraumatic epilepsy.
Epilepsy Curr. 2006;6(6):215–216. [
PMC free article: PMC1783486] [
PubMed: 17260064]
- 77.
Graber KD, PD . Chronic partial cortical isolation. In: Pitkanen ASPaMS, editor. Models of Seizures and Epilepsy. San Diego: Elsevier Academic Press; 2006. pp. 477–493.
- 78.
Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies.
J Neurophysiol. 1993;69:1276–1291. [
PubMed: 8492163]
- 79.
Hoffman SN, Salin PA, Prince DA. Chronic neocortical epileptogenesis in vitro.
J Neurophysiol. 1994;71:1762–1773. [
PubMed: 8064347]
- 80.
Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats.
Ann Neurol. 1999;46(2):234–242. [
PubMed: 10443889]
- 81.
Halpern LM. Chronically isolated aggregates of mammalian cerebral cortical neurons studied in situ. In: Purpura DP, Penry JK, Tower D, Woodbury DM, Walter R, editors. Experimental models of epilepsy--a manual for the laboratory worker. New York: Raven; 1972. pp. 197–221.
- 82.
Sharpless SK, Halpern LM. The electrical excitability of chronically isolated cortex studied by means of permanently implanted electrodes.
Electroenceph clin Neurophysiol. 1962;14:244–255. [
PubMed: 13911417]
- 83.
Echlin FA, Arnett V, Zoll J. Paroxysmal high-voltage and rhythmic low-voltage discharges from isolated and partially isolated human cortex.
AMA Arch Neurol Psychiatry. 1952;67(5):692–693. [
PubMed: 14914253]
- 84.
- 85.
Ribak CE, Reiffenstein RJ. Selective inhibitory synapse loss in chronic cortical slabs: a morphological basis for epileptic susceptibility.
Can J Physiol Pharmacol. 1982;60(6):864–870. [
PubMed: 7116231]
- 86.
Bush PC, Prince DA, Miller KD. Increased pyramidal excitability and NMDA conductance can explain posttraumatic epileptogenesis without disinhibition: a model.
J Neurophysiol. 1999;82(4):1748–1758. [
PubMed: 10515964]
- 87.
Prince DA. Epileptogenic neurons and circuits.
Adv Neurol. 1999;79:665–684. [
PubMed: 10514854]
- 88.
Grafstein B, Sastry P. Some preliminary electrophysiological studies on chronically neuronally isolated cerebral cortex.
Electroencephalogr Clin Neurophysiol. 1957;9:723–725. [
PubMed: 13480247]
- 89.
Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats.
Ann Neurol. 2004;55(6):860–870. [
PubMed: 15174021]
- 90.
Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex.
J Neuropathol Exp Neurol. 1997;56(3):219–235. [
PubMed: 9056536]
- 91.
Jacobs KM. PIPD. Enhanced c-fos staining in two post-lesional models of cortical hyperexcitability: neonatal freeze lesions and partial cortical isolations. Epilepsia. 2001;42(Suppl 7):221. Ref Type: Abstract.
- 92.
Scoville WB. Selective cortical undercutting as a means of modifying and studying frontal lobe function in man; preliminary report of 43 operative cases.
J Neurosurg. 1949;6(1):65–73. [
PubMed: 18106833]
- 93.
Scoville WB. Late results of orbital undercutting. Report of 76 patients undergoing quantitative selective lobotomies.
Am J Psychiatry. 1960;117:525–532. [
PubMed: 13749417]
- 94.
Cajal SRy. Study of traumatic degeneration in the cerebral cortex. In: May RM, editor. Degeneration and regeneration of the nervous system. London: Oxford Univ. Press; 1928. pp. 656–692.
- 95.
Purpura DP, Housepian EM. Morphological and physiological properties of chronically isolated immature neocortex.
Exp Neurol. 1961;4:377–401. [
PubMed: 14037425]
- 96.
Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity.
Science. 1988;239:1147–1150. [
PubMed: 2449733]
- 97.
Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, et al. Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting.
Epilepsia. 1993;34:985–995. [
PubMed: 7694849]
- 98.
Represa A, Jorquera I, Le Gal La, Salle G, Ben-Ari Y. Epilepsy induced collateral sprouting of hippocampal mossy fibers: does it induce the development of ectopic synapses with granule cell dendrites.
Hippocampus. 1993;3:257–268. [
PubMed: 8353609]
- 99.
Perez Y, Morin F, Beaulieu C, Lacaille JC. Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats.
Eur J Neurosci. 1996;8:736–748. [
PubMed: 9081625]
- 100.
Buckmaster PS, Zhang GF, Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit.
J Neurosci. 2002;22(15):6650–6658. [
PMC free article: PMC6758164] [
PubMed: 12151544]
- 101.
Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats.
J Comp Neurol. 1997;385(3):385–404. [
PubMed: 9300766]
- 102.
Smith BN, Dudek FE. Network Interactions Mediated by New Excitatory Connections Between CA1 Pyramidal Cells in Rats With Kainate-Induced Epilepsy.
J Neurophysiol. 2002;87(3):1655–1658. [
PubMed: 11877537]
- 103.
Babb TL, Kupfer WR, Pretorius JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata.
Neuroscience. 1991;42:351–363. [
PubMed: 1716744]
- 104.
Babb TL, Pretorius JK, Kupfer WR, Mathern GW, Crandall PH, Levesque MF. Aberrant synaptic reorganization in human epileptic hippocampus: evidence for feedforward excitation. Dendron. 1992;1:7–25.
- 105.
Masukawa LM, Uruno K, Sperling M, O’Connor MJ, Burdette LJ. The functional relationship between antidromically evoked field responses of the dentate gyrus and mossy fiber reorganization in temporal lobe epileptic patients.
Brain Res. 1992;579:119–127. [
PubMed: 1623399]
- 106.
Isokawa M, Levesque MF, Babb TL, Engel J Jr. Single mossy fiber axonal systems of human dentate granule cells studied in hippocampal slices from patients with temporal lobe epilepsy.
J Neurosci. 1993;13:1511–1522. [
PMC free article: PMC6576742] [
PubMed: 8463831]
- 107.
Marco P, DeFelipe J. Altered synaptic circuitry in the human temporal neocortex removed from epileptic patients.
Exp Brain Res. 1997;114:1–10. [
PubMed: 9125446]
- 108.
- 109.
- 110.
- 111.
Christman CW, Salvant JB Jr, Walker SA, Povlishock JT. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in adult cat: a light and electron microscopic immunocytochemical study.
Acta Neuropathol (Berl ). 1997;94:329–337. [
PubMed: 9341933]
- 112.
Povlishock JT, Kontos HA. Continuing axonal and vascular change following experimental brain trauma.
Cent Nerv Syst Trauma. 1985;2(4):285–298. [
PubMed: 3836013]
- 113.
Dickson TC, Adlard PA, Vickers JC. Sequence of cellular changes following localized axotomy to cortical neurons in glia-free culture.
J Neurotrauma. 2000;17(11):1095–1103. [
PubMed: 11101211]
- 114.
Chuckowree JA, Vickers JC. Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron Axons in vitro.
J Neurosci. 2003;23:3715–3725. [
PMC free article: PMC6742187] [
PubMed: 12736342]
- 115.
Dinocourt C, Gallagher SE, Thompson SM. Injury-induced axonal sprouting in the hippocampus is initiated by activation of trkB receptors.
Eur J Neurosci. 2006;24(7):1857–1866. [
PubMed: 17040478]
- 116.
Scharfman HE, Goodman JH, Sollas AL, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor.
Exp Neurol. 2002;174(2):201–214. [
PubMed: 11922662]
- 117.
Nita DA, Cisse Y, Timofeev I, Steriade M. Waking-sleep modulation of paroxysmal activities induced by partial cortical deafferentation.
Cereb Cortex. 2007;17(2):272–283. [
PubMed: 16495431]
- 118.
- 119.
- 120.
Graber KDKVNPIaPDA. Parvalbumin-containing interneurons are spared in the undercut model of posttraumatic epileptogenesis., 1999. Epilepsia. 1999;40(Suppl 7):31. Ref Type: Abstract.
- 121.
Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures.
J Neurosci. 2003;23(13):5662–5673. [
PMC free article: PMC6741232] [
PubMed: 12843269]
- 122.
Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses.
Can J Physiol Pharmacol. 1995;34(11):1387–1395. [
PubMed: 8606788]
- 123.
Pierce JP, Lewin GR. An ultrastructural size principle.
Neuroscience. 1994;58(3):441–446. [
PubMed: 8170532]
- 123a.
- 123b.
Ma Y, Prince DA. Functional alterations in GABAergic fast-spiking interneurons in chronically injured epileptogenic neocortex.
Neurobiol. Dis. 2012. doi:10.1016/j.nbd.2012.03.027. Epub. [
PMC free article: PMC3416869] [
PubMed: 22484482]
- 124.
Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, et al. Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury.
J Neurosci. 2002;22(11):4412–4417. [
PMC free article: PMC6758784] [
PubMed: 12040048]
- 125.
- 126.
Jin XM, Huguenard JR, Prince DA. Impaired Cl- extrusion in layer V pyramidal neurons of chronically injured epileptogenic neocortex.
J Neurophysiol. 2005;93:2117–2126. [
PubMed: 15774713]
- 127.
Jin X, Huguenard JR, Prince DA. Reorganization of Inhibitory Synaptic Circuits in Rodent Chronically Injured Epileptogenic Neocortex.
Cereb Cortex. 2010 Sep 20; [Epub ahead of print] [
PMC free article: PMC3077430] [
PubMed: 20855494]
- 128.
Lee L, Parada I, Prince DA. Downregulation of the α3 subunit of NA+K+ ATPAse in a model of post-traumatic epileptogenesis. Epilepsia. 2006;47:3.058.
- 129.
Spencer SS, Sperling MRSDAKP. Intracranial electrodes. In: Engel T, JraTAP, editors. Epilepsy, a comprehensive textbook. Philadelphia: Lippencott, Williams & Wilkins; 2010. pp. 1791–1815.
- 130.
Yang L, Benardo LS. Epileptogenesis following neocortical trauma from two sources of disinhibition.
J Neurophysiol. 1997;78:2804–2810. [
PubMed: 9356429]
- 131.
Nilsson P, Ronne-Engström E, Flink R, Ungerstedt U, Carlson H, Hillered L. Epileptic seizure activity in the acute phase following cortical impact trauma in rat.
Brain Res. 1994;637:227–232. [
PubMed: 8180800]
- 132.
Topolnik L, Steriade M, Timofeev I. Partial cortical deafferentation promotes development of paroxysmal activity.
Cereb Cortex. 2003;13(8):883–893. [
PubMed: 12853375]
- 133.
Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice.
Proc Natl Acad Sci U S A. 2010;107(17):7975–7980. [
PMC free article: PMC2867906] [
PubMed: 20375278]
- 134.
Hellier JL, Patrylo PR, Dou P, Nett M, Rose GM, Dudek FE. Assessment of inhibition and epileptiform activity in the septal dentate gyrus of freely behaving rats during the first week after kainate treatment.
J Neurosci. 1999;19(22):10053–10064. [
PMC free article: PMC6782973] [
PubMed: 10559413]
- 135.
Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same.
Trends Neurosci. 1999;22:221–227. [
PubMed: 10322495]
- 136.
Galvan CD, Hrachovy RA, Smith KL, Swann JW. Blockade of neuronal activity during hippocampal development produces a chronic focal epilepsy in the rat.
J Neurosci. 2000;20:2904–2916. [
PMC free article: PMC6772221] [
PubMed: 10751443]
- 137.
Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis.
Cell. 2009;139(2):380–392. [
PMC free article: PMC2791798] [
PubMed: 19818485]
- 138.
Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis.
Cell. 2005;120(3):421–433. [
PubMed: 15707899]
- 139.
Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion.
Stroke. 2003;34(1):177–186. [
PubMed: 12511771]
- 140.
Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke.
J Cereb Blood Flow Metab. 2008;28(10):1722–1732. [
PubMed: 18594557]
- 141.
Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats.
J Neurosci. 2001;21(6):1868–1875. [
PMC free article: PMC6762626] [
PubMed: 11245671]
- 142.
Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, et al. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin.
J Pharmacol Exp Ther. 2002;303(3):1199–1205. [
PubMed: 12438544]
- 143.
Maneuf YP, Luo ZD, Lee K. alpha2delta and the mechanism of action of gabapentin in the treatment of pain.
Semin Cell Dev Biol. 2006;17(5):565–570. [
PubMed: 17067834]
- 144.
Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, et al. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin.
Proc Natl Acad Sci U S A. 2006;103(46):17537–17542. [
PMC free article: PMC1859964] [
PubMed: 17088553]
- 145.
McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity.
Annu Rev Neurosci. 1999;22:295–318. [
PubMed: 10202541]
- 146.
Marty S, Berzaghi M, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons.
Trends Neurosci. 1997;20(5):198–202. [
PubMed: 9141194]
- 147.
Croll SD, Wiegand SJ, Anderson KD, Lindsay RM, Nawa H. Regulation of neuropeptides in adult rat forebrain by the neurotrophins BDNF and NGF.
Eur J Neurosci. 1994;6:1343–1353. [
PubMed: 7981876]
- 148.
Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons.
J Neurosci. 2003;23(23):8212–8220. [
PMC free article: PMC6740684] [
PubMed: 12967982]
- 149.
Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents.
J Clin Invest. 2010;120(5):1774–1785. [
PMC free article: PMC2860903] [
PubMed: 20407211]
- 150.
Danzer SC, McNamara JO. Localization of brain-derived neurotrophic factor to distinct terminals of mossy fiber axons implies regulation of both excitation and feedforward inhibition of CA3 pyramidal cells.
J Neurosci. 2004;24(50):11346–11355. [
PMC free article: PMC1351361] [
PubMed: 15601941]
- 151.
Koyama R, Ikegaya Y. To BDNF or not to BDNF: that is the epileptic hippocampus.
Neuroscientist. 2005;11(4):282–287. [
PubMed: 16061515]
- 152.
D’Antuono M, Louvel J, Kohling R, Mattia D, Bernasconi A, Olivier A, et al. GABAA receptor-dependent synchronization leads to ictogenesis in the human dysplastic cortex.
Brain. 2004;127(Pt 7):1626–1640. [
PubMed: 15175227]
- 153.
Marchionni I, Maccaferri G. Quantitative dynamics and spatial profile of perisomatic GABAergic input during epileptiform synchronization in the CA1 hippocampus.
J Physiol. 2009;587(Pt 23):5691–5708. [
PMC free article: PMC2805379] [
PubMed: 19840998]
- 154.
Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy.
Proc Natl Acad Sci U S A. 2006;103(50):19152–19157. [
PMC free article: PMC1681351] [
PubMed: 17146052]
- 155.
Tyzio R, Minlebaev M, Rheims S, Ivanov A, Jorquera I, Holmes GL, et al. Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus.
Eur J Neurosci. 2008;27(10):2515–2528. [
PubMed: 18547241]
- 156.
Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu FC, Moss SJ, et al. Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy.
J Neurosci. 2007;27(51):14012–14022. [
PMC free article: PMC2211568] [
PubMed: 18094240]
- 157.
Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy.
Hippocampus. 1991;1:41–66. [
PubMed: 1688284]
- 158.
Zhang W, Yamawaki R, Wen X, Uhl J, Diaz J, Prince DA, et al. Surviving hilar somatostatin interneurons enlarge, sprout axons, and form new synapses with granule cells in a mouse model of temporal lobe epilepsy.
J Neurosci. 2009;29(45):14247–14256. [
PMC free article: PMC2802278] [
PubMed: 19906972]
- 159.
- 160.
Sloviter RS, Zappone CA, Harvey BD, Bumanglag AV, Bender RA, Frotscher M. “Dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat.
J Comp Neurol. 2003;459(1):44–76. [
PubMed: 12629666]
- 161.
Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits.
Science. 1996;274(5290):1133–1138. [
PubMed: 8895456]
- 162.
Ming G, Henley J, Tessier-Lavigne M, Song H, Poo M. Electrical activity modulates growth cone guidance by diffusible factors.
J Physiol (Lond). 2001;29(2):441–452. [
PubMed: 11239434]
- 163.
Chen JW, Ruff RL, Eavey R, Wasterlain CG. Posttraumatic epilepsy and treatment.
J Rehabil Res Dev. 2009;46(6):685–696. [
PubMed: 20104398]
- 164.
Yang L, Benardo LS. Valproate prevents epileptiform activity after trauma in an in vitro model in neocortical slices.
Epilepsia. 2000;41(12):1507–1513. [
PubMed: 11114207]
- 165.
Binder DK, Croll SD, Gall CM, Scharfman HE. BDNF and epilepsy: too much of a good thing.
Trends Neurosci. 2001;24:47–53. [
PubMed: 11163887]
- 166.
Reibel S, Depaulis A, Larmet Y. BDNF and epilepsy--the bad could turn out to be good.
Trends Neurosci. 2001;24(6):318–319. [
PubMed: 11421232]